

Abstract
Kidney diseases such as AKI, CKD, and GN can lead to dialysis and the need for kidney transplantation. The pathologies for kidney diseases are extremely complex, progress at different rates, and involve several cell types and cell signaling pathways. Complex kidney diseases require therapeutics that can act on multiple targets. In the past 10 years, in silico design of drugs has allowed for multi-target drugs to progress quickly from concept to reality. Several multi-target drugs have been made successfully to target AA pathways and transcription factors for the treatment of inflammatory, fibrotic, and metabolic diseases. Multi-target drugs have also demonstrated great potential to treat diabetic nephropathy and fibrotic kidney disease. These drugs act by decreasing renal TGF-β signaling, inflammation, mitochondrial dysfunction, and oxidative stress. There are several other recently developed multi-target drugs that have yet to be tested for their ability to combat kidney diseases. Overall, there is excellent potential for multi-target drugs that act on several cell types and signaling pathways to treat kidney diseases.
Keywords: nephro-pharmacology, basic science, chronic kidney disease, diabetes, drug delivery systems, eicosanoids, fatty acids, hypertension, kidney diseases, multi-ligand drugs, pharmaceutical preparations, transcription factors
Introduction
Kidney disease afflicts an estimated 37 million people in the United States, and the Medicare costs for treating kidney diseases exceed $130 billion (1–3). Types of kidney diseases include AKI, CKD, and GN. Major risk factors for kidney diseases are hypertension, diabetes, and family history (1–3). Cardiovascular disease and high morbidity and mortality are associated with kidney diseases (2,4). Despite the seriousness of kidney diseases, there are limited treatment options, with many patients requiring dialysis and kidney transplantation.
AKI, CKD, and glomerular diseases have varied and multifaceted etiologies. Causes for AKI can range from drug toxicity, ischemia during thoracic surgeries, and septic infections (5,6). Drug toxicities that cause AKI include nonsteroidal anti-inflammatory drugs, anticancer drugs, antibacterial drugs, and immunosuppressant drugs (5,6). The pathophysiology of AKI includes decreased blood and oxygen supply from systemic hypotension, systemic hypoxia, and disrupted regional oxygen delivery in the kidney (5,6). Signaling and metabolic pathways in renal tubular segments and epithelial cells that contribute to AKI include activation of hypoxia-inducible factor, activation of the peroxisome proliferator-activated receptor γ (PPARγ)–PPARγ coactivator 1α (PGC-1α) pathway, mitochondrial signaling, and fructokinase activation (5–7). CKD is largely due to hypertension and diabetes that cause progressive renal damage via very different cellular mechanisms (2,3,8). Diabetic nephropathy results from oxidative stress, inflammation, mitochondrial dysfunction, and fatty acid metabolism that contributes to kidney fibrosis and impairs tubular transport, renal hemodynamics, and glomerular filtration (8,9). Glomerular diseases, such as GN, start because of damage to the glomerulus that can then lead to renal damage outside the glomerular structure (10,11). Immune complexes and complement components, such as C3 and C5, lead to glomerular inflammatory cell infiltration (11,12). Glomerular mesangial cells, podocytes, and endothelial cells become damaged, with increased extracellular matrix leading to glomerulosclerosis (10,11). Subsequently, nonimmune mechanisms result in progressive renal damage leading to interstitial fibrosis (11,13). Taken as a whole, kidney diseases are extremely complex because they involve several cell types, multiple cell signaling pathways, and progress at different rates.
The complexity of kidney diseases requires the development of therapeutics that can act on multiple targets (Figure 1). Kidney diseases—such as AKI, diabetic nephropathy, CKD, FSGS, GN, and ESKD—involve several renal cell types and disease progression depends on changes in many cellular signaling pathways. The emergence of multi-target drugs in recent years has resulted in novel therapeutics that could treat kidney diseases.
Figure 1.
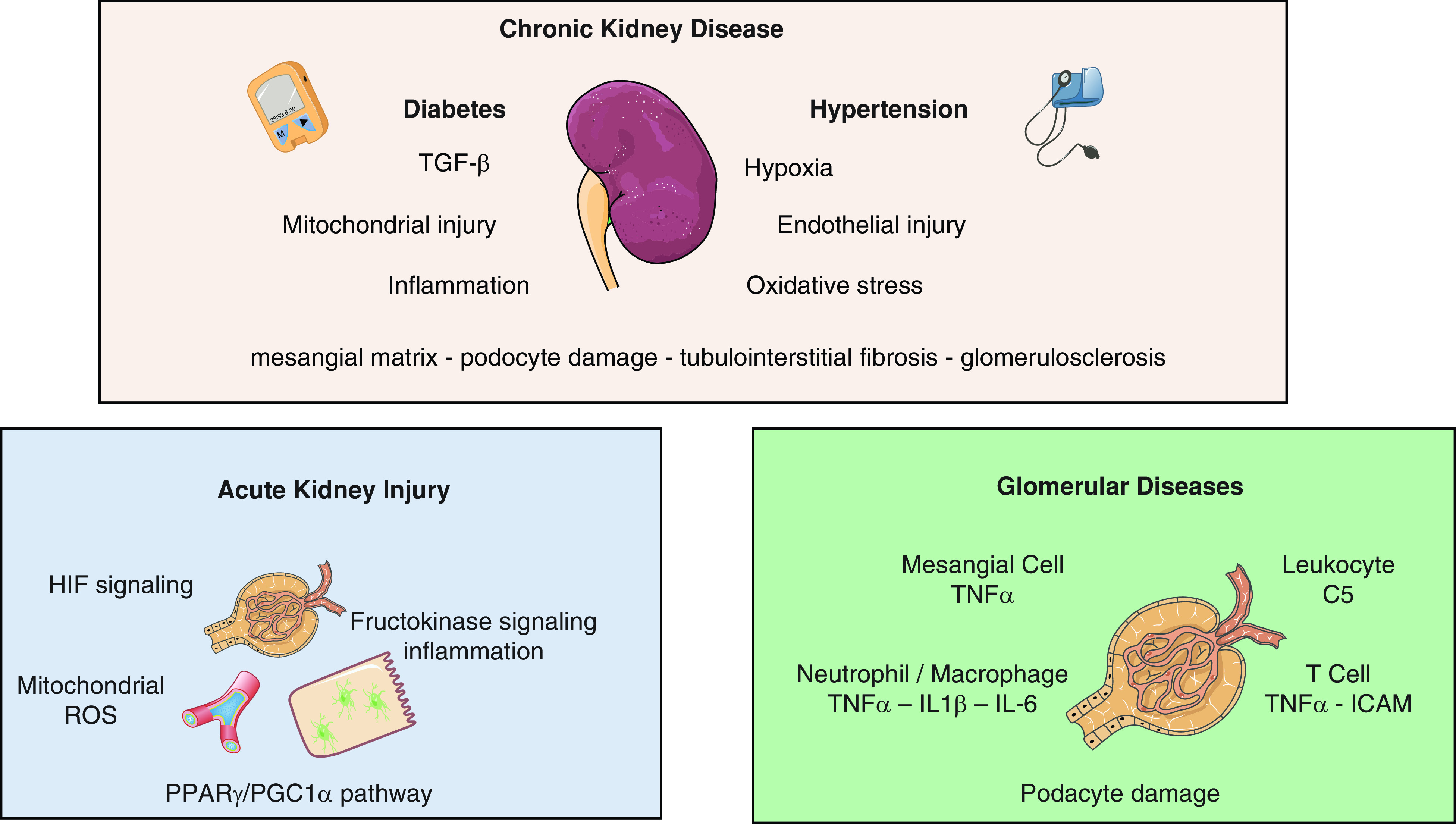
Kidney diseases have several targets for multi-target drugs. In CKD, diabetes and hypertension contribute to 70%–80% of all patients with CKD (top). Factors contributing to CKD include endothelial dysfunction, TGF-β signaling, inflammation, hypoxia, mitochondrial injury, and oxidative stress. Kidney injury in CKD involves the mesangial matrix, podocyte damage, tubulointerstitial fibrosis, and glomerulosclerosis. AKI can be caused by drug toxicity, ischemia during thoracic surgeries, and septic infections (bottom left). Therapeutic targets for AKI include hypoxia-inducible factor (HIF) signaling, mitochondrial function and reactive oxygen species (ROS), peroxisome proliferator-activated receptor γ (PPARγ)/PPARγ coactivator 1α (PGC-1α) pathway, and fructokinase signaling and inflammation. For glomerular diseases, therapeutic targets include mesangial cells and TNFα; leukocytes and complement 5 (C5); neutrophils and macrophages and TNFα, IL-1β, and IL-6 signaling; T cells and TNFα/intracellular adhesion molecule (ICAM), and signaling pathways resulting in podocyte damage (bottom right).
Designing Multi-Target Small-Molecule Drugs for Kidney Diseases
The deliberate and rational design of drugs which act on multiple targets has gained momentum over the past decade (14–16). Compensatory mechanisms and redundant functions built into biologic systems make them resistant to single-point perturbations; therefore, diseases are often caused by multiple genetic and/or environmental factors that result in failure of physiologic systems. Complex disorders—such as metabolic diseases, fibrotic diseases, AKI, CKD, and glomerular diseases—are more likely to be treated through simultaneous modulation of multiple targets.
The concept that bifunctional molecules could be developed to treat kidney diseases has been energized by the approval of sacubitril/valsartan, a combined neprilysin and angiotensin type 1 (AT1) receptor inhibitor, for heart failure (17,18). Our research group and others have focused on dual-acting small molecules to target molecular pathways for organ fibrosis and life-threatening kidney diseases (19,20). These multi-target drugs have much more potential than single-target and highly specific agents due to (1) better disease-modifying actions, (2) additive and/or synergistic therapeutic actions, (3) more predictable pharmacokinetics than combination therapies, (4) prolonged duration of effectiveness, and (5) lower probability for drug interactions (Table 1). As a result, it is increasingly recognized that a balanced modulation of two targets can provide a superior therapeutic effect and side effect profile.
Table 1.
Advantages of multi-target drugs
| Advantage |
|---|
| Complex disease-modifying actions Synergistic or additive therapeutic actions Predictable pharmacokinetics Prolonged duration of effectiveness Decreased drug interactions |
Types of Multi-Target Drugs
A major challenge for developing multi-target drugs, also known as multiple ligand drugs, is the need to optimize the drugs against multiple biologic targets while maintaining proper drug properties (14–16). On average, multi-target drugs have larger mol wts and are more lipophilic than compounds designed to modulate a single target. Although multi-target drugs have been developed with appropriate druglike properties, a critical aspect of the drug design and development process is the selection of the biologic targets. Computational tools and structural information that allows for pharmacophore modeling enables the design of multi-target drugs that show selectivity for the intended biologic targets (14–16). Defining the desired activity balance and balancing pharmacologic properties and selectivity for biologic targets is another major challenge in developing multi-target drugs.
Multi-target drugs can be classified into three categories: linked, fused, and merged pharmacophore drugs (Figure 2). Linked multi-target drugs comprise two distinct pharmacophores for each target that are connected by a linker. These linked multi-target drugs tend to have a larger mol wt. sacubitril/valsartan is an example of a linked multi-target drug that has a distinct pharmacophore for inhibiting the enzyme neprilysin, and another distinct pharmacophore for antagonizing the AT1 receptor (17,18). Decreasing the linker size of linked multi-target drugs will eventually lead to a point where the pharmacophores are essentially touching, which results in a fused multi-target drug. Thus, fused multi-target drugs have distinct pharmacophores that are not separated by a linker. Disadvantages of linked and fused multi-target drugs include large mol wt and extensive lipophilicity. Merged multi-target drugs are based on a common, merged pharmacophore that is designed to engage both biologic targets of interest while possessing a low mol wt and fulfilling other aspects of the Lipinski rule of five (14–16). Multi-target merged pharmacophore drugs are, by far, the most challenging to design and optimize. The advent of well-defined structure-activity relationships for drugs via x-ray structural information for protein targets and in silico design can help find starting points for merged multi-target drugs (14–16). Several multi-target drugs with linked, fused, or merged pharmacophores have been developed and tested for their ability to combat kidney diseases using cell-based and animal models.
Figure 2.

PTUPB and RB394 represent different types of multi-target drugs. PTUPB is an example of a linked multi-target drug with a distinct pharmacophore for cyclooxygenase-2 (COX-2) inhibition and soluble epoxide hydrolase (sEH) inhibition (left). RB394 is an example of a merged multi-target drug that unites the pharmacophore features required for sEH inhibition and PPARγ activation (right). EC50, half maximal effective concentration; IC50, half maximal inhibitory concentration
Progress with Multi-Target Drugs for Kidney Diseases
A critical step in designing multi-target drugs for kidney diseases is deciding on the molecular targets. The targets must be disease modifying and attack different signaling pathways or the same signaling pathway from different angles. For example, in kidney diseases, one mechanism of interest is blocking the TGF-β signaling that leads to fibrosis (13,21,22). The critical signaling cascades that are initiated primarily by TGF-β but also involve inflammatory cytokines and signaling molecules, which stimulate profibrotic reactions in myofibroblasts, are potential therapeutic targets (13,22). Because TGF-β plays a pivotal role in fibrogenesis, it was originally thought that therapeutic targeting of TGF-β would control organ fibrosis (21,22). Unfortunately, this approach failed because TGF-β also plays a crucial role in a number of important biologic processes, such as immunity and cellular growth (21,22). Therefore, an ideal approach for a therapy to treat fibrotic kidney disease would be to modulate several mechanisms downstream, without blocking important TGF-β–regulated biologic processes. Multi-target drugs for kidney diseases have targeted transcription factors, AA metabolites, incretins, G-protein coupled receptors, and the renin-angiotensin system (19,20).
The evaluation of multi-target drugs for kidney diseases has been expanding over the past 5 years. These efforts have largely focused on diabetic and hypertensive CKD and kidney fibrosis (19,20). Multi-target drugs have been designed not only to treat kidney disease, but also to combat diabetes, metabolic disease, and hypertension at the same time (23,24). This has been achieved by initially comparing multi-target drugs to the respective single-target approach in enzymatic or cell-based systems (23,24). Because the target combinations for CKD involve individual targets that are expressed in different tissues of the body, an in vivo approach is required for validation of an anti-CKD multi-target drug. This is a significant obstacle because reaching this step requires significant medicinal chemistry efforts to develop multi-target drugs to selectively modulate the individual targets of interest while maintaining suitable pharmacokinetic properties. Excitingly, the high-risk, high-reward development of multi-target drugs to treat kidney diseases has resulted in promising drugs that are close to, or are already in, human clinical trials (20,24).
Drugs that modulate the AA pathway have anti-inflammatory, antifibrotic, antihypertensive, and antidiabetic actions with the potential to treat kidney diseases (19,24). A major pathway that has been exploited in multi-target drugs is the epoxygenase pathway (19,24). Inhibition of the enzyme soluble epoxide hydrolase (sEH) has been combined with cyclooxygenase (COX) inhibition or transcription factor agonism (25,26). The sEH enzyme promotes the hydrolysis of AA metabolites, epoxyeicosatrienoic acids (EETs), to their corresponding, less bioactive diols (DHETEs) (27,28). Through sEH inhibition, EET levels are increased (27). EETs are a major eicosanoid in human kidneys and have anti-inflammatory properties (28). Increased sEH expression or decreased EET levels in the kidney are involved in hypertension, diabetes, and kidney diseases (27,28). Inhibition of sEH also prevents renal inflammation and interstitial fibrosis by inhibiting the endothelial to mesenchymal transition and expression of α-smooth muscle actin and TGF-β (29,30). Thus, sEH inhibition combined with another therapeutic target could treat hypertension or diabetes while combating kidney disease.
PTUPB
A linked multi-target drug that acts as a dual COX-2 and sEH inhibitor (PTUPB) was developed and tested in Zucker diabetic fatty rats (Figure 3) (26). Combining sEH inhibition with COX-2 inhibition can limit COX-2 side effects while increasing therapeutic potential to combat kidney fibrosis (19,31,32). The ability for PTUPB to increase EETs and lower COX-2 metabolites, without significantly altering other AA metabolites, has been demonstrated in multiple studies (33–35). These studies have not extensively evaluated the effects of PTUPB on mRNA or protein expression of lipoxygenase, CYP4A, or CYP2C enzymes. The effects of PTUPB on glucose homeostasis and kidney injury were first evaluated in Zucker diabetic fatty rats treated in a preventive manner for 2 months (26). PTUPB lowered fasting blood glucose, improved glucose tolerance, improved pancreatic islet morphology, and improved the plasma lipid profile (26). Diabetic kidney injury was significantly reduced and associated with a reduction in inflammation and oxidative stress (26). Interestingly, studies in the obese diabetic Zucker fatty/spontaneously hypertensive heart failure F1 hybrid (ZSF1) rats demonstrated that interventional PTUPB treatment did not lower blood glucose, but could effectively alleviate hypertension, hyperlipidemia, and diabetic nephropathy (33). PTUPB also decreased inflammation and fibrosis in the kidney and liver of obese ZSF1 hypertensive and diabetic rats (33). PTUPB treatment exhibited anti-inflammatory and antifibrotic actions in animal models of sepsis, nonalcoholic fatty liver disease, and pulmonary fibrosis (36–39). Taken together, these studies demonstrate the potential for PTUPB to decrease inflammation and oxidative stress and combat fibrosis to slow the progression of diabetic kidney injury.
Figure 3.
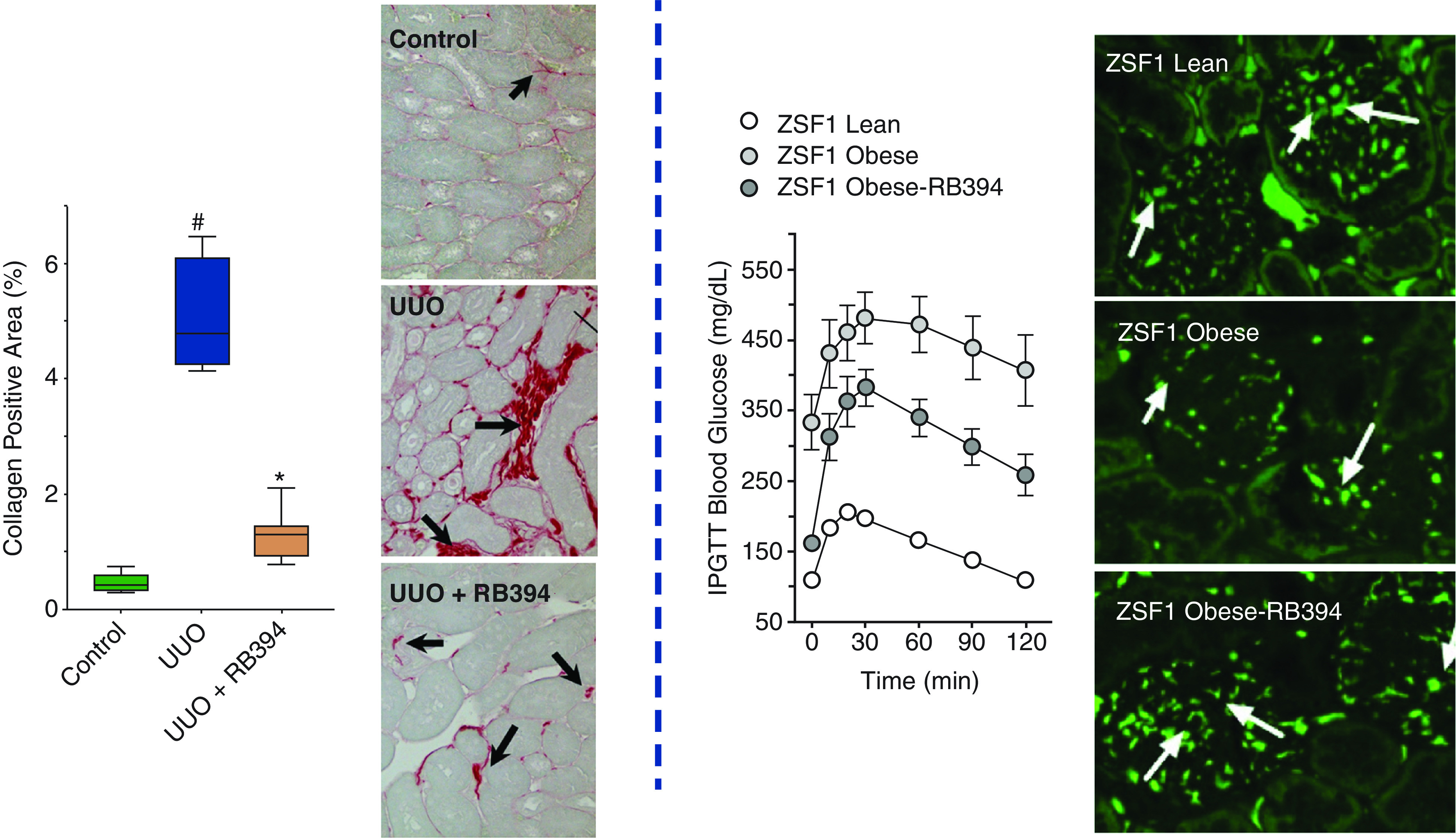
Kidney and systemic protective actions of multi-target drugs. RB394 has direct actions on the kidney in mice with unilateral ureter obstruction (UUO) (left). RB394 decreased the collagen area, as determined by kidney histology using Picrosirius red staining. ZSF1 obese diabetic rats treated with RB394 had improved metabolic status, as assessed by intraperitoneal glucose tolerance test (IPGTT) (right). RB394 also decreased kidney injury and nephrin expression was preserved in ZSF1 obese diabetic rats. Left panel is adapted from ref. 48, with permission. Right panel is adapted from ref. 46, with permission. # = P<0.05 versus control, * = P<0.05 versus UUO.
Other multi-target drugs that manipulate two enzymes in the AA pathway are in development and being tested for therapeutic potential in animal models. Recently, dual sEH and 5-lipoxygenase (5-LOX) inhibition, which results in synergistic anti-inflammatory effects, has been targeted to treat CKD (40). An initial optimization of the sEH and 5-LOX inhibitor lead candidate (7ad) included cellular testing in human PMNs, determination of oral bioavailability, and assessment of target engagement (40). The ability for 7ad to combat kidney disease was tested in the unilateral ureter obstruction (UUO) CKD model. Treatment with 7ad mitigated tubulointerstitial fibrosis and ameliorated macrophage infiltration into the obstructed kidney (40). Additional studies with combined sEH and 5-LOX inhibitors are needed to determine the therapeutic potential for combating kidney diseases.
RB394
The combination of a dual PPARγ agonist and sEH inhibitor (RB394) to treat diabetic complications has also demonstrated great potential (19,24). The combination of sEH inhibition and PPARγ agonism reduces side effects while increasing efficacy against kidney fibrosis (41). PPARγ activation by thiazolidinediones, such as rosiglitazone and pioglitazone, induces beneficial effects on insulin action and blood glucose levels (42–44). However, the clinical use of thiazolidinediones is limited because of excessive weight gain, fluid retention, elevated cardiovascular risk, and increased risk of osteoporosis in treated patients (42,43). Fortunately, sEH inhibition and EETs are natriuretic and positively influence water and electrolyte homeostasis (27,28). Therefore, a merged multi-target drug, RB394, was developed and initially tested for sEH enzymatic inhibitor activity and ability to selectively activate PPARγ target genes in differentiated murine and human adipocytes (45,46).
Animal studies evaluated RB394 in rat models of the metabolic syndrome—the spontaneously hypertensive obese rat and the ZSF rat (46). RB394 was administered orally to spontaneously hypertensive obese rats in a preventive manner, which attenuated the development of hypertension, insulin resistance, hyperlipidemia, and kidney injury (46). ZSF1 rats became diabetic, hypertensive, and demonstrated albuminuria as an index of kidney injury before administering RB394 (46). Interventional treatment with RB394 to ZSF1 rats for 2 months reduced hemoglobin A1c levels, improved glucose tolerance, reduced BP, and improved lipid profiles (Figure 3) (46). One mechanism that could contribute to the improved metabolic status is the ability of RB394 to induce browning in human white adipocytes (47). In addition, RB394 ameliorated liver fibrosis, hepatosteatosis, and diabetic nephropathy by reducing renal fibrosis and tubular and glomerular injury in obese diabetic ZSF1 rats (46). The decrease in kidney injury with RB394 treatment can occur independently of decreases in BP, blood glucose, and lipid levels. RB394 administered 3 days after the induction of UUO attenuated renal fibrosis by reducing inflammation and oxidative stress (Figure 3) (48). RB394 demonstrated a superior decrease in kidney fibrosis, TGF-β, inflammation, and endothelial to mesenchymal transition gene expression when compared with the PPARγ agonist, rosiglitazone; the sEH inhibitor, t-AUCB; or the combination of rosiglitazone and t-AUCB (48). Importantly, the dual PPARγ agonist and sEH inhibitor RB394 did not lead to excessive weight gain or fluid retention (46,48). These findings demonstrate the potential therapeutic benefit for RB394 in metabolic and kidney diseases and highlight the concept of designed polypharmacology, because the dual ligand was superior to the combination of selective drugs for the same targets (Figure 4).
Figure 4.

Mechanisms by which multi-target drugs combat kidney injury. PTUPB is a combined sEH inhibitor (sEHi) and COX-2 inhibitor (COX-2i) (top left). PTUPB decreases kidney injury by combating diabetes, and decreasing inflammation, plasma lipid levels, reactive oxygen species (ROS), and fibrosis. RB394 is an sEHi and PPARγ agonist that combats hypertension and diabetes, and decreases TGF-β inflammation signaling, epithelial to mesenchymal transition (EMT), and fibrosis (top right). DM509 is an sEHi and farnesoid X receptor (FXR) agonist. DM509 decreases kidney injury by lowering lipids, decreasing TNFα inflammatory signaling, and decreasing plasma cholesterol (bottom left). INT-767 is a combined FXR and Takeda G protein receptor 5 (TGR5) agonist that combats kidney injury through several mechanisms, including TGF-β inflammation signaling, lowering plasma lipids and cholesterol, decreasing PGC-1α, and nuclear respiratory factor 1 (Nrf-1), resulting in decreased endoplasmic reticulum (ER) stress and podocyte damage (bottom right).
DM509
The combination of sEH inhibition and the farnesoid X receptor (FXR) agonism has been developed as the merged multi-target drug DM509 (Figure 4). FXR agonism and sEH inhibition were combined because they are validated targets for nonalcoholic steatohepatitis (NASH) and kidney fibrosis (29,30,49,50). Bile acids are the endogenous ligands for FXR and obeticholic acid (OCA) is an FXR agonist that has reached clinical trials for fibrotic liver disease; however, strong FXR activation leads to serious disturbances in cholesterol homeostasis (51,52). By design, DM509 activates FXR only partially while potently inhibiting sEH (53,54). Initial studies in two liver fibrosis models demonstrated that orally administered DM509 is superior to the FXR agonist OCA (54). In addition, the dual FXR agonist/sEH inhibitor DM509 had positive actions on cholesterol homeostasis by reducing triglyceride levels and increasing the HDL-cholesterol/non-HDL-cholesterol ratio in NASH mice (54). Moreover, curative treatment with DM509 counteracted pre-established NASH in diet-induced obese mice with anti-inflammatory and remarkable antifibrotic effects widely exceeding OCA as standard of care (54). Experimental studies in UUO mice revealed that DM509 decreased renal fibrosis and injury (55). DM509 had anti-inflammatory actions with reductions in TGF-β, TNFα, IL-1β, and IL-6 levels in the UUO mice (55). With this highly promising in vivo profile, DM509 stands as a lead potential candidate for preclinical evaluation to combat organ fibrosis and kidney diseases.
INT-767
FXR agonism has also been combined with Takeda G protein receptor 5 (TGR5) agonism in the semisynthetic bile acid derivative INT-767 (Figure 4) (56). INT-767’s ability to target FXR and TGR5 in cellular settings was validated by FXR-dependent lipid uptake by adipocytes and TGR5-dependent glucagon-like peptide-1 (GLP-1) secretion by enteroendocrine cells (56,57). Initial animal studies in metabolic diseases, such as NASH, found that INT-767 treatment resulted in beneficial metabolic and liver effects (57–59). INT-767’s positive metabolic actions are mediated, in part, through brown adipogenesis and improved mitochondrial function (60). Animal studies in db/db diabetic mice determined that INT-767 decreased cholesterol and triglyceride levels in streptozotocin diabetic and db/db diabetic mice (61). Diabetic nephropathy was also evaluated in streptozotocin-induced diabetes, db/db diabetic mice, and high-fat diet–induced obese mice after 2 months of oral treatment with INT-767 (61). INT-767 decreased proteinuria, prevented glomerular injury, and decreased tubulointerstitial fibrosis (61). The effects of the FXR and TGR5 dual agonist were exerted through multiple pathways, including stimulation of a signaling cascade involving AMP-activated protein kinase, sirtuins, PGC-1α, and nuclear respiratory factor 1, resulting in decreased endoplasmic reticulum stress and inhibition of enhanced renal fatty acid and cholesterol metabolism (61). Kidney disease in aging was evaluated by treating 22-month-old mice with INT-767 for 2 months. This study found that INT-767 reversed age-related kidney disease, with decreased proteinuria and podocyte injury (62). INT-767 decreased kidney TGF-β expression, improved mitochondrial function, and prevented escalation of inflammation (62). More recently, INT-767 counteracted the TGF-β–induced increase in phosphorylated mothers against decapentaplegic homolog 3 and transcription factor tafazzin to prevent fibrosis programming in renal organoids (X. Yang et al., unpublished data; https://doi.org/10.1101/2021.04.15.440011). There is a high therapeutic potential for INT-767 to treat kidney disease in the future because this drug has completed phase 1 clinical safety trials.
Other Potential Multi-Target Drugs for Kidney Diseases
Several multi-target drugs have been developed that act on the AA cascade, transcription factors, incretin signaling, and renin-angiotensin system which have the potential to combat kidney diseases (14,24,25). These multi-target drugs include combining sEH inhibition with 5-LOX inhibition and fatty acid amide hydrolase (25,36). COX-2 inhibition has been combined with thromboxane A2 inhibition and 5-LOX inhibition (25,36). The multi-target drugs that affect two AA pathways will have anti-inflammatory actions and other organ protective actions; however, several of these drugs have not been tested for their ability to combat kidney diseases (14,24,36).
Multi-target drugs that have the potential to treat metabolic diseases and diabetes have also emerged (23,24). For example, FXR agonism has been combined with PPARδ and combined PPARα/δ agonism (64,64). These combined FXR and PPAR agonists have demonstrated promise in liver diseases, but have yet to be tested in kidney diseases (63,64). PPARγ agonism has also been coupled to glucokinase activation and AT1 receptor inhibition (23,24). Renin-angiotensin system angiotensin-converting enzyme inhibition has been combined with dipeptidyl peptidase-4 inhibition (23,24). Lastly, dipeptidyl peptidase-4 inhibition, which increases plasma GLP-1 levels, has been combined with GPR119 activation, which modulates insulin release by pancreatic β-cells and GLP-1 secretion by gut enteroendocrine cells, to combat diabetes (23,24). Although the primary focus for these multi-target drugs are metabolic diseases and diabetes, these drugs also have potential to combat kidney disease via actions on kidney cell signaling pathways.
Although great strides have been made in developing multi-target drugs for CKD and associated conditions, a need still exists to develop multi-target drugs that can combat glomerular diseases and AKI. Several pathways could be targeted by multi-target drugs to combat AKI. These pathways include hypoxia-inducible factor signaling, fructokinase signaling, inflammation, mitochondrial function, oxidative stress, and the PPARγ/PGC-1α pathway (5–7). Targeting multiple immune pathways—such as neutrophil/macrophage TNFα and IL-6, leukocyte complement C3 and C5, and T-cell TNFα and intracellular adhesion molecule—could be beneficial in treating glomerular diseases (10–12). Current multi-target drugs for CKD can affect several of these pathways, but these drugs have not been tested for their ability to effectively combat glomerular diseases and AKI.
Initial evaluations of target engagement, administration routes, and pharmacokinetics have been conducted with PTUPB, RB394, DM509, and INT-767 (Table 2). Nevertheless, these multi-target drugs could have side effects that are associated with small molecules. These include the potential for off-target side effects and unwanted systemic actions. Extensive evaluation of the multi-target drugs is needed to limit the potential for off-target side effects. As for systemic actions, multi-target drugs have been designed in many cases to have beneficial effects on systemic disease in addition to combating kidney diseases (Figure 3) (33,46,52,54). Kidney-targeting strategies can be used to limit systemic drug side effects when direct kidney actions are desired (65,66). Kidney targeting with folate conjugation has been successfully demonstrated using EET analogues (67). An alternative strategy for limiting unwanted side effects of small molecules is to develop multi-target biologics and RNA-based therapies for kidney diseases. Biologics such as mAbs and nanobodies, which act on a single target, are advancing the treatment of kidney diseases with improvements in bioavailability and tissue targeting (68,69). Likewise, RNA-based short-interfering RNAs and long-coding RNAs are improving the treatment of kidney diseases. Developing biologics and RNA-based drugs with multi-target activities for kidney and glomerular diseases could have kidney and cell-targeting advantages; however, several challenges remain that would need to be overcome (70–71). Consequently, there are ample avenues to be investigated in developing multi-target therapies for kidney diseases.
Table 2.
Multi-target drug targets, dosages, pharmacokinetics, and references
| Multi-target Drug | Targets | Dosages and Administration Routes | Pharmacokinetics | References |
|---|---|---|---|---|
| PTUPB | sEH | 5–10 mg/kg per d s.c. | Data in references (31,34) | (26,31,33–35,37–39) |
| 10–30 mg/kg per d p.o. | ||||
| COX-2 | ||||
| 30–60 mg/kg per d i.p. | ||||
| RB394 | sEH | 10 mg/kg per d p.o. | Data in reference (45) | (45–48) |
| PPARγ | ||||
| DM509 | sEH | 10 mg/kg per d p.o. | Data in reference (53) | (53–55) |
| FXR | ||||
| INT-767 | FXR | 10–30 mg/kg per d p.o. | Data in references (56,72) | (56–62,72) |
| TGR5 |
sEH, soluble epoxide hydrolase; s.c., subcutaneous; COX-2, cyclooxygenase-2; p.o., by mouth; i.p., intraperitoneal; PPARγ, peroxisome proliferator-activated receptor γ; FXR, farnesoid X receptor; TGR5, Takeda G protein receptor 5.
Conclusion
Multi-target drugs have advantages for treating complex, multifactorial diseases such as AKI, CKD, and glomerular diseases. These drugs have anti-inflammatory, antidiabetic, antihypertensive, and antifibrotic actions to combat kidney diseases. Targets that have been combined and investigated for kidney diseases include sEH, COX-2, 5-LOX, FXR, PPAR, and TGR5. The design of these multi-target drugs has involved balancing the activity of each target of interest while maintaining suitable pharmacokinetic profiles. Multi-target drugs include PTUPB, 7ad, RB394, DM509, and INT-767, and these drugs all affect kidney TGF-β signaling, inflammation, and fibrosis to decrease kidney disease. Their therapeutic potential has been demonstrated in various animal models for diabetes, hypertension, aging, and UUO, where kidney damage is relevant to CKD. However, the effects of these multi-target drugs for the treatment of AKI and glomerular diseases are unknown. In addition, there are several other multi-target drugs in development that have the potential to treat kidney diseases. Overall, the future is bright for multi-target drugs to become therapeutics for kidney diseases.
Disclosures
J.D. Imig reports serving as a scientific advisor for, or member of, American Heart Association, American Physiological Society, and Biochemical Society; having ownership interest in DIPLOS Therapeutics, Nephraegis, and OROX Bio; having patents and inventions with the Medical College of Georgia and Medical College of Wisconsin; and receiving research funding from National Institutes of Health. J.D. Imig, D. Merk, and E. Proschak report having multiple patents and patent applications related to dual PPARγ/sEH and FXR/sEH ligands. D. Merk reports receiving research funding from Aventis Foundation, Else Kröner-Fresenius Foundation, and German Research Foundation; having patents and inventions with Goethe University Frankfurt. E. Proschak reports serving on the Journal of Medicinal Chemistry scientific advisory board.
Funding
J.D. Imig was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant DK103616 and NIDDK Diabetic Complications Consortium (RRID:SCR_001415, www.diacomp.org, accessed on February 1, 2021) grants DK076169 and DK115255.
Acknowledgments
Servier Medical Art was used to generate Figures 1 and 3 and is licensed by Servier under a Creative Commons Attribution 3.0 Unported License.
Author Contributions
J.D. Imig conceptualized the study and wrote the original draft; and J.D. Imig, D. Merk, and E. Proschak were responsible for visualization and reviewed and edited the manuscript.
References
- 1.Foley RN, Collins AJ: End-stage renal disease in the United States: An update from the United States renal data system. J Am Soc Nephrol 18: 2644–2648, 2007 [DOI] [PubMed] [Google Scholar]
- 2.US Department for Health and Human Services Centers for Disease Control and Prevention : Chronic Kidney Disease in the United States, 2019. Available at: https://www.cdc.gov/kidneydisease/pdf/2019_National-Chronic-Kidney-Disease-Fact-Sheet.pdf. Accessed May 18, 2021
- 3.National Kidney Foundation : Chronic kidney disease (CKD) symptoms and causes, 2017. Available at: https://www.kidney.org/atoz/content/about-chronic-kidney-disease. Accessed September 30, 2021
- 4.Loftus TJ, Filiberto AC, Ozrazgat-Baslanti T, Gopal S, Bihorac A: Cardiovascular and renal disease in chronic critical illness. J Clin Med 10: 1601, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scholz H, Boivin FJ, Schmidt-Ott KM, Bachmann S, Eckardt KU, Scholl UI, Persson PB: Kidney physiology and susceptibility to acute kidney injury: Implications for renoprotection. Nat Rev Nephrol 17: 335–349, 2021 [DOI] [PubMed] [Google Scholar]
- 6.Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, Kellum JA, Ronco C; ADQI XIII Work Group : Progression after AKI: Understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol 27: 687–697, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basile DP, Anderson MD, Sutton TA: Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Agati VD, Chagnac A, de Vries APJ, Levi M, Porrini E, Herman-Edelstein M, Praga M: Obesity-related glomerulopathy: Clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol 12: 453–471, 2016 [DOI] [PubMed] [Google Scholar]
- 9.Zoja C, Xinaris C, Macconi D: Diabetic nephropathy: Novel molecular mechanisms and therapeutic targets. Front Pharmacol 11: 586892, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Floege J, Barbour SJ, Cattran DC, Hogan JJ, Nachman PH, Tang SCW, Wetzels JFM, Cheung M, Wheeler DC, Winkelmayer WC, Rovin BH, Adler SG, Alpers CE, Ayoub I, Bagga A, Barratt J, Caster DJ, Chan DTM, Chang A, Choo JCJ, Cook HT, Coppo R, Fervenza FC, Fogo AB, Fox JG, Gibson KL, Glassock RJ, Harris D, Hodson EM, Hoxha E, Iseki K, Jennette JC, Jha V, Johnson DW, Kaname S, Katafuchi R, Kitching AR, Lafayette RA, Li PKT, Liew A, Lv J, Malvar A, Maruyama S, Mejía-Vilet JM, Moeller MJ, Mok CC, Nester CM, Noiri E, O'Shaughnessy MM, Özen S, Parikh SM, Park H-C, Peh CA, Pendergraft WF, Pickering MC, Pillebout E, Radhakrishnan J, Rathi M, Roccatello D, Ronco P, Smoyer WE, Tesař V, Thurman JM, Trimarchi H, Vivarelli M, Walters GD, Wang AY-M, Wenderfer SE; for Conference Participants : Management and treatment of glomerular diseases (part 1): Conclusions from a kidney disease: Improving global outcomes (KDIGO) controversies conference. Kidney Int 95: 268–280, 2019 [DOI] [PubMed] [Google Scholar]
- 11.Rovin BH, Caster DJ, Cattran DC, Gibson KL, Hogan JJ, Moeller MJ, Roccatello D, Cheung M, Wheeler DC, Winkelmayer WC, Floege J, Adler SG, Alpers CE, Ayoub I, Bagga A, Barbour SJ, Barratt J, Chan DTM, Chang A, Choo JCJ, Cook HT, Coppo R, Fervenza FC, Fogo AB, Fox JG, Glassock RJ, Harris D, Hodson EM, Hogan JJ, Hoxha E, Iseki K, Jennette JC, Jha V, Johnson DW, Kaname S, Katafuchi R, Kitching AR, Lafayette RA, Li PKT, Liew A, Lv J, Malvar A, Maruyama S, Mejía-Vilet JM, Mok CC, Nachman PH, Nester CM, Noiri E, O'Shaughnessy MM, Özen S, Parikh SM, Park H-C, Peh CA, Pendergraft WF, Pickering MC, Pillebout E, Radhakrishnan J, Rathi M, Ronco P, Smoyer WE, Tang SCW, Tesař V, Thurman JM, Trimarchi H, Vivarelli M, Walters GD, Wang AY-M, Wenderfer SE, Wetzels JFM;. Conference Participants: Management and treatment of glomerular diseases (part 2): Conclusions from a kidney disease: Improving global outcomes (KDIGO) controversies conference. Kidney Int 95: 281–295, 2019 [DOI] [PubMed] [Google Scholar]
- 12.Holdsworth SR, Gan PY, Kitching AR: Biologics for the treatment of autoimmune renal diseases. Nat Rev Nephrol 12: 217–231, 2016 [DOI] [PubMed] [Google Scholar]
- 13.Meng XM, Nikolic-Paterson DJ, Lan HY: Inflammatory processes in renal fibrosis. Nat Rev Nephrol 10: 493–503, 2014 [DOI] [PubMed] [Google Scholar]
- 14.Proschak E, Stark H, Merk D: Polypharmacology by design: A medicinal chemist’s perspective on multitargeting compounds. J Med Chem 62: 420–444, 2019 [DOI] [PubMed] [Google Scholar]
- 15.Bansal Y, Silakari O: Multifunctional compounds: Smart molecules for multifactorial diseases. Eur J Med Chem 76: 31–42, 2014 [DOI] [PubMed] [Google Scholar]
- 16.Talevi A: Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front Pharmacol 6: 205, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chalikias G, Tziakas D: Angiotensin receptor neprilysin inhibitors-2019 update. Cardiovasc Drugs Ther 34: 707–722, 2020 [DOI] [PubMed] [Google Scholar]
- 18.Gori M, D’Elia E, Senni M: Sacubitril/valsartan therapeutic strategy in HFpEF: Clinical insights and perspectives. Int J Cardiol 281: 158–165, 2019 [DOI] [PubMed] [Google Scholar]
- 19.Imig JD: Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol Ther 192: 1–19, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Libby AE, Jones B, Lopez-Santiago I, Rowland E, Levi M: Nuclear receptors in the kidney during health and disease. Mol Aspects Med 78: 100935, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng XM, Nikolic-Paterson DJ, Lan HY: TGF-β: The master regulator of fibrosis. Nat Rev Nephrol 12: 325–338, 2016 [DOI] [PubMed] [Google Scholar]
- 22.Branton MH, Kopp JB: TGF-beta and fibrosis. Microbes Infect 1: 1349–1365, 1999 [DOI] [PubMed] [Google Scholar]
- 23.Gattrell W, Johnstone C, Patel S, Smith CS, Scheel A, Schindler M: Designed multiple ligands in metabolic disease research: From concept to platform. Drug Discov Today 18: 692–696, 2013 [DOI] [PubMed] [Google Scholar]
- 24.Lillich FF, Imig JD, Proschak E: Multi-target approaches in metabolic syndrome. Front Pharmacol 11: 554961, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Proschak E, Heitel P, Kalinowsky L, Merk D: Opportunities and challenges for fatty acid mimetics in drug discovery. J Med Chem 60: 5235–5266, 2017 [DOI] [PubMed] [Google Scholar]
- 26.Hye Khan MA, Hwang SH, Sharma A, Corbett JA, Hammock BD, Imig JD: A dual COX-2/sEH inhibitor improves the metabolic profile and reduces kidney injury in Zucker diabetic fatty rat. Prostaglandins Other Lipid Mediat 125: 40–47, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imig JD, Hammock BD: Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8: 794–805, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imig JD, Jankiewicz WK, Khan AH: Epoxy fatty acids: From salt regulation to kidney and cardiovascular therapeutics: 2019 Lewis K. Dahl memorial lecture. Hypertension 76: 3–15, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J, Imig JD, Yang J, Hammock BD, Padanilam BJ: Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J Physiol Renal Physiol 307: F971–F980, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Yoon SP, Toews ML, Imig JD, Hwang SH, Hammock BD, Padanilam BJ: Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am J Physiol Renal Physiol 308: F131–F139, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang SH, Wagner KM, Morisseau C, Liu JY, Dong H, Wecksler AT, Hammock BD: Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J Med Chem 54: 3037–3050, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dogné JM, Hanson J, Supuran C, Pratico D: Coxibs and cardiovascular side-effects: From light to shadow. Curr Pharm Des 12: 971–975, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Hye Khan MA, Hwang SH, Barnett SD, Stavniichuk A, Jankiewicz WK, Hammock BD, Imig JD: Multi-target molecule to treat diabetic nephropathy in rats [published online ahead of print July 13, 2021]. Br J Pharmacol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang G, Panigrahy D, Hwang SH, Yang J, Mahakian LM, Wettersten HI, Liu JY, Wang Y, Ingham ES, Tam S, Kieran MW, Weiss RH, Ferrara KW, Hammock BD: Dual inhibition of cyclooxygenase-2 and soluble epoxide hydrolase synergistically suppresses primary tumor growth and metastasis. Proc Natl Acad Sci USA 111: 11127–11132, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang F, Zhang H, Ma AH, Yu W, Zimmermann M, Yang J, Hwang SH, Zhu D, Lin TY, Malfatti M, Turteltaub KW, Henderson PT, Airhart S, Hammock BD, Yuan J, de Vere White RW, Pan CX: COX-2/sEH dual inhibitor PTUPB potentiates the antitumor efficacy of cisplatin. Mol Cancer Ther 17: 474–483, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hiesinger K, Wagner KM, Hammock BD, Proschak E, Hwang SH: Development of multitarget agents possessing soluble epoxide hydrolase inhibitory activity. Prostaglandins Other Lipid Mediat 140: 31–39, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang HH, Duan JX, Liu SK: A COX-2/sEH dual inhibitor PTUPB alleviates lipopolysaccharide-induced acute lung injury in mice by inhibiting NLRP3 inflammasome activation. Theranostics 10: 4749–4761, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun CC, Zhang CY, Duan JX, Guan XX, Yang HH, Jiang HL, Hammock BD, Hwang SH, Zhou Y, Guan CX, Liu SK, Zhang J: PTUPB ameliorates high-fat diet-induced non-alcoholic fatty liver disease via inhibiting NLRP3 inflammasome activation in mice. Biochem Biophys Res Commun 523: 1020–1026, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang CY, Duan JX, Yang HH, Sun CC, Zhong WJ, Tao JH, Guan XX, Jiang HL, Hammock BD, Hwang SH, Zhou Y, Guan CX: COX-2/sEH dual inhibitor PTUPB alleviates bleomycin-induced pulmonary fibrosis in mice via inhibiting senescence. FEBS J 287: 1666–1680, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiesinger K, Kramer JS, Beyer S, Eckes T, Brunst S, Flauaus C, Wittmann SK, Weizel L, Kaiser A, Kretschmer SBM, George S, Angioni C, Heering J, Geisslinger G, Schubert-Zsilavecz M, Schmidtko A, Pogoryelov D, Pfeilschifter J, Hofmann B, Steinhilber D, Schwalm S, Proschak E: Design, synthesis, and structure-activity relationship studies of dual inhibitors of soluble epoxide hydrolase and 5-lipoxygenase. J Med Chem 63: 11498–11521, 2020 [DOI] [PubMed] [Google Scholar]
- 41.Imig JD, Walsh KA, Hye Khan MA, Nagasawa T, Cherian-Shaw M, Shaw SM, Hammock BD: Soluble epoxide hydrolase inhibition and peroxisome proliferator activated receptor γ agonist improve vascular function and decrease renal injury in hypertensive obese rats. Exp Biol Med (Maywood) 237: 1402–1412, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM: PPARγ signaling and metabolism: The good, the bad and the future. Nat Med 19: 557–566, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klinkhammer BM, Goldschmeding R, Floege J, Boor P: Treatment of renal fibrosis-turning challenges into opportunities. Adv Chronic Kidney Dis 24: 117–129, 2017 [DOI] [PubMed] [Google Scholar]
- 44.Staels B, Fruchart JC: Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes 54: 2460–2470, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Blöcher R, Lamers C, Wittmann SK, Merk D, Hartmann M, Weizel L, Diehl O, Brüggerhoff A, Boß M, Kaiser A, Schader T, Göbel T, Grundmann M, Angioni C, Heering J, Geisslinger G, Wurglics M, Kostenis E, Brüne B, Steinhilber D, Schubert-Zsilavecz M, Kahnt AS, Proschak E: N-Benzylbenzamides: A novel merged scaffold for orally available dual soluble epoxide hydrolase/peroxisome proliferator-activated receptor γ modulators. J Med Chem 59: 61–81, 2016 [DOI] [PubMed] [Google Scholar]
- 46.Khan MAH, Kolb L, Skibba M, Hartmann M, Blöcher R, Proschak E, Imig JD: A novel dual PPAR-γ/sEH inhibitor treats type 2 diabetic complications. Diabetologia 61: 2235–2246, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hartmann M, Bibli SI, Tews D, Ni X, Kircher T, Kramer JS, Kilu W, Heering J, Hernandez-Olmos V, Weizel L, Scriba GKE, Krait S, Knapp S, Chaikuad A, Merk D, Fleming I, Fischer-Posovszky P, Proschak E: Combined cardioprotective and adipocyte browning effects promoted by the eutomer of dual sEH/PPARγ modulator. J Med Chem 64: 2815–2828, 2021 [DOI] [PubMed] [Google Scholar]
- 48.Stavniichuk A, Hye Khan MA, Yeboah MM, Chesnik MA, Jankiewicz WK, Hartmann M, Blöcher R, Kircher T, Savchuk O, Proschak E, Imig JD: Dual soluble epoxide hydrolase inhibitor/PPAR-γ agonist attenuates renal fibrosis. Prostaglandins Other Lipid Mediat 150: 106472, 2020 [DOI] [PubMed] [Google Scholar]
- 49.Zhao K, He J, Zhang Y, Xu Z, Xiong H, Gong R, Li S, Chen S, He F: Activation of FXR protects against renal fibrosis via suppressing Smad3 expression. Sci Rep 6: 37234, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han CY: Update on FXR biology: Promising therapeutic target? Int J Mol Sci 19: 2069, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E; NASH Clinical Research Network : Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 385: 956–965, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D: Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145: 574–582.e1, 2013 [DOI] [PubMed] [Google Scholar]
- 53.Schmidt J, Rotter M, Weiser T, Wittmann S, Weizel L, Kaiser A, Heering J, Goebel T, Angioni C, Wurglics M, Paulke A, Geisslinger G, Kahnt A, Steinhilber D, Proschak E, Merk D: A dual modulator of farnesoid X receptor and soluble epoxide hydrolase to counter nonalcoholic steatohepatitis. J Med Chem 60: 7703–7724, 2017 [DOI] [PubMed] [Google Scholar]
- 54.Hye Khan MA, Schmidt J, Stavniichuk A, Imig JD, Merk D: A dual farnesoid X receptor/soluble epoxide hydrolase modulator treats non-alcoholic steatohepatitis in mice. Biochem Pharmacol 166: 212–221, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stavniichuk A, Savchuk O, Khan AH, Jankiewicz WK, Imig JD, Merk D: The effect of compound DM509 on kidney fibrosis in the conditions of the experimental mode. Visnyk Kyivskoho Natsionalnoho Universytetu Imeni Tarasa Shevchenka Biolohiia 80: 10–15, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G, Orlandi S, Sadeghpour B, Wang XX, Jiang T, Levi M, Pruzanski M, Adorini L: Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmacol 78: 617–630, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jadhav K, Xu Y, Xu Y, Li Y, Xu J, Zhu Y, Adorini L, Lee YK, Kasumov T, Yin L, Zhang Y: Reversal of metabolic disorders by pharmacological activation of bile acid receptors TGR5 and FXR. Mol Metab 9: 131–140, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anfuso B, Tiribelli C, Adorini L, Rosso N: Obeticholic acid and INT-767 modulate collagen deposition in a NASH in vitro model. Sci Rep 10: 1699, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu YB, Liu XY, Zhan W: Farnesoid X receptor agonist INT-767 attenuates liver steatosis and inflammation in rat model of nonalcoholic steatohepatitis. Drug Des Devel Ther 12: 2213–2221, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Comeglio P, Cellai I, Mello T, Filippi S, Maneschi E, Corcetto F, Corno C, Sarchielli E, Morelli A, Rapizzi E, Bani D, Guasti D, Vannelli GB, Galli A, Adorini L, Maggi M, Vignozzi L: INT-767 prevents NASH and promotes visceral fat brown adipogenesis and mitochondrial function. J Endocrinol 238: 107–127, 2018 [DOI] [PubMed] [Google Scholar]
- 61.Wang XX, Wang D, Luo Y, Myakala K, Dobrinskikh E, Rosenberg AZ, Levi J, Kopp JB, Field A, Hill A, Lucia S, Qiu L, Jiang T, Peng Y, Orlicky D, Garcia G, Herman-Edelstein M, D’Agati V, Henriksen K, Adorini L, Pruzanski M, Xie C, Krausz KW, Gonzalez FJ, Ranjit S, Dvornikov A, Gratton E, Levi M: FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity. J Am Soc Nephrol 29: 118–137, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang XX, Luo Y, Wang D, Adorini L, Pruzanski M, Dobrinskikh E, Levi M: A dual agonist of farnesoid X receptor (FXR) and the G protein-coupled receptor TGR5, INT-767, reverses age-related kidney disease in mice. J Biol Chem 292: 12018–12024, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schierle S, Neumann S, Heitel P, Willems S, Kaiser A, Pollinger J, Merk D: Design and structural optimization of dual FXR/PPARδ activators. J Med Chem 63: 8369–8379, 2020 [DOI] [PubMed] [Google Scholar]
- 64.Heitel P, Faudone G, Helmstädter M, Schmidt J, Kaiser A, Tjaden A, Schröder M, Müller S, Schierle S, Pollinger J, Merk D: A triple farnesoid X receptor and peroxisome proliferator-activated receptor α/δ activator reverses hepatic fibrosis in diet-induced NASH in mice. Commun Chem 3: 174, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang X, Ma Y, Li Y, Han F, Lin W: Targeted drug delivery systems for kidney diseases. Front Bioeng Biotechnol 9: 683247, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Z, Peng H, Zhang C: Advances in kidney-targeted drug delivery systems. Int J Pharm 587: 119679, 2020 [DOI] [PubMed] [Google Scholar]
- 67.Imig JD, Hye Khan MA, Burkhan A, Chen G, Adebesin AM, Falck JR: Kidney-targeted epoxyeicosatrienoic acid analog, EET-F01, reduces inflammation, oxidative stress, and cisplatin-induced nephrotoxicity. Int J Mol Sci 22: 2793, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wanner N, Eden T, Liaukouskaya N, Koch-Nolte F: Nanobodies: New avenue to treat kidney disease [published online ahead of print June 16, 2021]. Cell Tissue Res 10.1007/s00441-021-03479-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karras A, Jayne D: New biologics for glomerular disease on the horizon. Nephron Clin Pract 128: 283–291, 2014 [DOI] [PubMed] [Google Scholar]
- 70.Shimizu H, Fujita T: New short interfering RNA-based therapies for glomerulonephritis. Nat Rev Nephrol 7: 407–415, 2011 [DOI] [PubMed] [Google Scholar]
- 71.Coellar JD, Long J, Danesh FR: Long noncoding RNAs and their therapeutic promise in diabetic nephropathy. Nephron 145: 404–414, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roda A, Pellicciari R, Gioiello A, Neri F, Camborata C, Passeri D, De Franco F, Spinozzi S, Colliva C, Adorini L, Montagnani M, Aldini R: Semisynthetic bile acid FXR and TGR5 agonists: Physicochemical properties, pharmacokinetics, and metabolism in the rat. J Pharmacol Exp Ther 350: 56–68, 2014 [DOI] [PubMed] [Google Scholar]