

Abstract
Potential roles of euchromatic histone methyltransferase 2 (EHMT2 or G9a) in invasion and metastasis are not well understood in non-small cell lung cancer (NSCLC). Here we investigated the effect and underlying mechanisms of G9a, and therapeutic implications of targeting G9a in the invasion and metastasis of NSCLC. Overexpression of G9a significantly enhanced in vitro proliferation and invasion, while knockdown of G9a drastically suppressed in vivo growth and metastasis of A549 and H1299 NSCLC cells. Knockdown or inhibition of G9a significantly decreased the expression of FAK protein and activation of FAK pathway. Additionally, defactinib, a potent FAK inhibitor, partially abolished the G9a-enhanced invasion in these NSCLC cells. Furthermore, targeting G9a was found to suppress nuclear factor-kappa B (NF-κB) transcriptional activity in NSCLC cells through stabilizing NF-κB inhibitor alpha (IκBα), while an NF-κB inhibitor Parthenilide partially abolished the G9a-enhanced FAK activation, which suggests that G9a-enhanced invasion and activation of FAK is mediated by elevated NF-κB activity. Notably, a strong positive correlation between the immunohistochemical staining of G9a and phosphorylated FAK proteins was identified in H1299 xenografts and 159 cases of NSCLC tissues (R = 0.408).
Implications:
The findings of this study strongly demonstrate that G9a may promote invasion and metastasis of NSCLC cells by enhancing FAK signaling pathway via elevating NF-κB transcriptional activity, indicating potential significance and therapeutic implications of these pathways in the invasion and metastasis of NSCLCs that overexpress G9a protein.
Keywords: NSCLC, Histone methyltransferase G9a, Invasion, Metastasis, Focal adhesion kinase (FAK), Nuclear factor-kappa B (NF-kB), NF-κB inhibitor alpha (IκBα)
Introduction
Lung cancer is a major global health problem and is the leading cause of cancer-related mortality in both men and women (1). Although advances including lung cancer screening, targeted therapies, and immunotherapy have improved outcomes in patients with lung cancer (2), the overall 5-year survival of lung cancer patients is still just 5% for patients with advanced lung cancer (3). Therefore, novel therapeutic approaches for lung cancer are urgently needed.
G9a histone methyltransferase encoded by the euchromatic histone-lysine N-methyltransferase 2 (EHMT2) is responsible for catalyzing mono-methylation and di-methylation of Histone H3 Lysine 9 (H3K9me1 and H3K9me2), and plays an important role in regulating gene expression (4). G9a epigenetically blocks tumor suppressors and activates oncogenes leading to carcinogenesis and cancer cell growth. Previous studies have shown that G9a expression is elevated in many types of human cancers, including pancreatic, bladder and breast cancers (5–7). Overexpression of G9a is associated with enhanced proliferation and metastasis in many cancer types. However, the underlying mechanisms about how G9a participates in lung cancer metastasis are still poorly understood.
Tumor metastasis is the major cause of cancer-related death, yet precise mechanisms of metastasis remain incompletely understood (8). Focal Adhesion Kinase (FAK) has been shown to play a central role in metastasis. FAK is a non-receptor cytoplasmic tyrosine kinase which is transcriptionally regulated by P53 and NF-κB (9). It contributes to almost every aspect of tumor metastasis, as well as cancer cell adhesion, growth, migration and invasion (10,11). Enhanced phosphorylation of FAK at specific sites, especially at Y397, has been reported in a number of cancer types (12). Studies have shown that FAK is up-regulated in non-small cell lung cancers (NSCLC) (13) and is associated with an aggressive clinical course (14). In patients with KRAS mutation, FAK is a potential druggable target as it is a downstream effector of KRAS signaling (15). In KRAS-driven lung adenocarcinomas, FAK inhibitors showed potent antitumor effects in KRAS-G12V-INK4a/ARF-deficient lung cancers in mice (16). Preclinical data in KRAS-mutated lung cancers showed that inhibition of FAK resulted in sustained DNA damage by suppressing DNA repair mechanisms and enhancing radiation sensitivity (17). In addition, FAK is considered to be a potential therapeutic target to prevent tumor metastasis in a number of solid tumors (18).
Recently, we reported that G9a was frequently overexpressed in NSCLC tissues and that targeting G9a potently suppressed the growth of NSCLC cells, suggesting that G9a may be a therapeutic target for NSCLC (19). In this study, we extended our investigation into the role of G9a in NSCLC cellular migration, invasion, and metastasis. We studied potential mechanisms of G9a-mediated invasion and its impact on FAK signaling pathway in NSCLC. Additionally, we explored the effect of targeting G9a on invasive potential and the activation of FAK signaling pathway in NSCLC cells.
Materials and Methods
Cultured cell lines and treated inhibitors
Two human RAS mutated non-small cell lung adenocarcinoma cell lines were used in this study. Both cell lines were purchased from American Type Culture Collection (ATCC, USA). All of these cell lines were authenticated by Integrative Genomics Core by using short tandem repeat polymorphism analysis. H1299 (p53-Null, N-RAS mutation) was cultured in RPMI 1640 (ATCC, USA) medium and A549 (p53-Wild-type, K-RAS mutation) was cultured with DMEM/F12 (Corning, USA) medium. For all cell lines, cell authentic growth medium was supplemented with 10% FBS, 100 U/mL penicillin and 100 mg/mL streptomycin. Cell lines were tested Mycoplasma free using colorimetric mycoplasma detection assay with HEK-Blue™-2 cells. Cells used for experiments were within 20 passages from thawing. UNC0638, a selective inhibitor for G9a and GLP methyltransferase, was purchased from Cayman Chemical Company (USA) and used at a concentration of 2.5 μM and 5 μM. Defactinib hydrochloride, a FAK inhibitor which inhibits FAK phosphorylation at Tyr397, was purchased from MedChemExpress (USA) and used at a concentration of 1μM. Parthenilide, NF-κB inhibitor, was purchased from Abcam and used at a concentration of 20 μM.
Plasmids, Cell transfection, and qRT-PCR
G9a mRNA (isoform a) was cloned into pcDNA3.1 vector with Hind III and EcoR I (New England BioLabs, USA). Two independent G9a siRNAs: Cat No.10630318 (5’-GGCAUCUCAGGAGGAUGCCAAUGAA-3’) and Cat No. 10620319 (5’-UUCAUUGGCAUCCUCCUGAGAUGCC-3’) purchased from Thermo Fisher Scientific Corporation (Carlsbad, USA) were used to silence G9a expression by using RNAi maxi reagent (Invitrogen, USA) as describe previously (20). A FAK siRNA (Cat#: sc-29310) was purchased from Santa Cruz Company. Cells were transfected with G9a-pcDNA3.1 plasmid and pcDNA3.1 plasmid as control by using Lipofectamine 3000 reagent (Invitrogen, USA) according to the manufacturer’s protocol. Total RNAs from cells were extracted with RNeasy Mini Kit (Cat#: 74104) and cDNA was synthesized from 1.0 μg RNA with QuantiTech@ Reverse Transcription Kit (Cat#: 205313) which were purchased from Qiagen Company. EMHT2 (Forward: GGACATCATCACTCATGCGGAAA, Reverse: GCAAAACCATGTCCAAACCAGG) PTK2 (Forward: GCAATGGAGCGAGTATTAAAGGTC, Reverse: TGGCCACATGCTTTACTTTGTGAC) and GAPDH (Forward: GGGAAGGTGAAGGTCGGAGTC, Reverse: GAGTTAAAAGCAGCCCTGGTGAC) NFKBIA (Forward: GCCATCATCCATGAAGAAAAG, Reverse: GGAGTCTGCAGGTTCTT) primers were used in the qRT-PCR assay to quantitatively measure the mRNA expression. Power SYBR@ Green PCR Master Mix was purchased from Life Technology LTD (Cat #: 4367695). Data was presented as ratio to control to show the relative expression of target, normalized to internal control and relative to the calibration sample.
Cell proliferation, Migration and Invasion Assays
For cell proliferation, cells were seeded as 1×105 per well in 6 well plates. After 72 hours, cells were harvested and counted with Cello-meter Spectrum (Nexcellom Bioscience, USA). For migration assay, after transfected with siRNA, plasmids or treated with inhibitors for 48 hours, cells were detached from tissue culture plate by using 0.25% Trypsin-EDTA solution, and pelleted by centrifugation. The cells were resuspended in serum free culture medium and seeded as 5×105 per well in a 6-well plate. After 24 hours, cells were scratched with the tip of 20ul pipette and washed twice with PBS. Cells were then cultured with serum free medium and treated with inhibitors. Images were captured immediately after scratching (0 hour), and then at 24h for H1299 and 36h for A549. Five locations were imaged for each well. Migration distances were measured by image J software. Three measurements were performed for each image and average migratory distance was determined. Cell invasion assay was performed using 8 μm Matrigel invasion chambers (Corning, USA). Cells were detached after being transfected or treated with inhibitors for 48 hours as previously described (21). Then 2.5×104 cells were seeded on top of the insert with serum-free medium. Lower chambers were filled with growth medium with 10% FBS. The culture medium was continuously supplemented with respective inhibitor in the inhibitors treated group. After 22 hours, media containing the remaining cells that did not migrate from the top of the membrane was carefully removed. Cells were fixed by placing the insert in 500 μl 10% formalin for 15 min, air-drying for 10 min at room temperature, and staining with 1% crystal violet solution for 10 minutes. The fixed inserts were then washed with distilled water. Trans-wells were imaged by microscope and cells were counted using Qu path software (22). Data were analyzed and presented as the ratio of the average of total number in treated group divided by the average of total number in the paired control group.
Western Blot Analysis
Total cellular protein samples were extracted with SDS lysis buffer and heated in 95 °C for 7 minutes. Proteins were separated by electrophoresis and transferred onto Polyvinylidenefluoride (PVDF) membranes (Thermo Fisher, USA). Membrane was blocked with 5% nonfat milk in PBST for 40 min in room temperature and blotted with primary and secondary antibodies, respectively. A primary antibody against β-actin (1:3000) was purchased from Santa Cruz Biotechnology. FAK (1:1000), pFAK (Tyr397) (1:1000), IκBα (1:1500), H3K9-me2 (1: 2000) antibodies were purchased from Cell Signaling Technology. Antibody against G9a (1:1000) was purchased from GeneTex Inc (USA).
Luciferase Reporter Assay of NF-κB
The pSI-Check2-hRluc-NFkB-firefly reporter plasmid was a gift from Qing Deng (Addgene plasmid # 106979; http://n2t.net/addgene:106979; RRID: Addgene_106979) (23). For each well, 50 ng NF-κB reporter plasmids were transfected into cells at a concentration of 3×103 per well for H1299 and 5×103 per well for A549 in 96-well plates using lipofectamine 3000 reagent, with either siRNAs or the overexpression plasmids. For inhibitor experiments, cells were treated with UNC0638 12 hours after transfection with the NF-κB plasmid. At 72 hours post transfection, cells were washed with PBS and lysed with 40 μl 1× passive lysis buffer. Firefly and renilla luciferase activity were detected by using Dual-Luciferase® Reporter Assay System (Promega, USA). For each group, five replicate experiments were performed. Firefly and renilla luciferase activities were determined and calculated as described previously (24).
In vivo Xenograft and Metastasis Model
All animal protocols were approved by the institutional animal care and use committee (IACUC, 16005) of City of Hope and performed in the animal facility at City of Hope in accordance with federal, local, and institutional guidelines. H1299 cell line was transfected with G9a-shRNA-GFP plasmid and transfected cells were selected with puromycin. NOD/SCID/IL2R gamma null mice (NSG, 24–27 g, 8–10 weeks of age, Female: Male = 1: 1) from Jackson Labs (Bar Harbor, USA) were used for xenograft experiment. A suspension of 5×106 tumor cells (H1299 shRNA-control and H1299 G9a-KD) in 0.1 ml RPMI 1640 was injected into the subcutaneous dorsa of mice at the proximal midline (25). About 3 weeks after injection, mice were euthanized by CO2 inhalation, and tumors were excised and fixed for IHC staining. To investigate the impact of G9 knockdown on in vivo metastasis formation, a suspension of 3×106 GFP-expressing parental H1299 cells transfected with a scramble control shRNA (shCtrl) and G9a-attenuated H1299 cells (G9a-KD) in which G9a is stably knocked down by a G9a specific shRNA in 0.1 ml PBS was injected into the tail vein of mice, respectively. About 4 weeks after injection, mice were euthanized by CO2 inhalation; livers and lungs were collected and frozen or fixed immediately. The metastatic nodules of each cancer cell in the frozen liver and lung tissue slides were analyzed and imaged by GFP imaging using a fluorescence microscope (Nikon).
Immunohistochemistry Analysis
This study was reviewed and approved by the Institutional Review Board (IRB#: 13240) of City of Hope National Medical Center. All subjects gave written informed consent. A total of 250 patients with lung adenocarcinoma and squamous carcinoma who underwent surgical resection for curative intent between 2002 and 2014 without preoperative chemotherapy or radiation therapy were included, as previously reported (19). Formaldehyde-fixed paraffin embedded (FFPE) tumor samples were sectioned and stained for G9a, H3K9me2 and pFAK (Tyr397) at the pathology core laboratory, as previously described (26). Antibody against pFAK (Tyr397) (1:2000) was purchased from Invitrogen (Cat#: 700255). Antibody against G9a (1:300) was purchased from GeneTex (Cat#: 129153). Antibody against H3K9me2 (1:200) was purchased from Abcam (Cat#: 5327). G9a and pFAK (Tyr397) IHC staining was scored according to different expression positive percentage and intensity as 0 negative, + minimal, ++ moderate and +++ strong.
Data Analysis
All experiments were performed in duplicates or triplicates and repeated at least two times. For group-group statistics analysis, data were analyzed for variation and significance using Student’s T test. All data are shown as mean ± SD. Statistical significance was set at P < 0.05. Pearson’s correlation coefficient was used to measure correlation of G9a and pFAK (Tyr397) gene expression.
Results
G9a regulates PTK2 gene expression in NSCLC cells
Through RNA-Seq analysis (see GSE113493 on NCBI GEO website) and gene set enrichment analysis, we found that knockdown of G9a significantly suppressed gene sets of cell motility and cell adhesion signaling pathways of NSCLC cells, which are critical for cancer invasion and metastasis (19); notably, PTK2 that encodes FAK protein is among these significantly downregulated genes in the gene sets (Figure 1A), suggesting that PTK2 may represent an important G9a target. To further validate the finding and explore the association between G9a and FAK expression, we first knocked down G9a in H1299 and A549 NSCLC cells by two independent siRNAs. As shown in Figure 1B, in both A549 and H1299 cell lines, upon knockdown of G9a, the level of H3K9me2 was decreased, and FAK protein was also dramatically decreased in both H1299 and A549 cell lines. qRT-PCR analysis showed that G9a mRNA expression level was significantly silenced in both H1299 and A549 cell lines transfected with specific G9a siRNAs (Figure 1C, P < 0.01). Simultaneously, mRNA expression of PTK2 gene was down regulated in these two cells (Figure 1D). These data indicate that FAK expression is associated with G9a expression in NSCLC.
Figure 1. G9a regulates PTK2 gene expression in NSCLC cells.

A. RNAseq data of mRNA levels of EMHT2 (G9a) gene and PTK2 (FAK) gene in G9a-attenuated A549 and H1299 cells by gene-specific siRNAs. B. Western blot analysis shows H3K9me2 and FAK was decreased upon silence of G9a in A549 and H1299 cells. qRT-PCR analysis of mRNAs. C. EMHT2 gene and D. PTK2 gene in G9a-attenuated A549 and H1299 cells. Data is presented as percentages related to the control cells transfected with scramble siRNAs, and is for averages ± standard division based on data of three independent experiments (** P < 0.01, *** P < 0.001, compared to control cells).
Knockdown and inhibition of G9a suppresses cell migratory and invasive potential of NSCLC cells
To investigate the potential roles of G9a in cell migration and invasion of NSCLCs, cancer cells were first transfected with two different G9a-specific siRNAs for in vitro migration and invasion assays. We observed that cell proliferation was also significantly suppressed (P < 0.01) upon G9a knockdown in these two lung cancer cells (Figure S1A & B). As shown in Figure 2A, compared to the control group, cell migration potential was significantly suppressed upon knockdown of G9a in both H1299 (upper panel) and A549 (lower panel) cell lines. A significantly decreased migration distance (P < 0.001) was observed in both H1299 and A549 cells upon G9a knockdown (Figure 2B).
Figure 2. Knockdown and inhibition of G9a suppresses cell migratory and invasive potential of NSCLC cells.

A. Representative images for scratch assays of A549 and H1299 cells transfected with control siRNA (Ctrl siRNA) and G9 siRNA 1 and 2 (G9a si1/2) (scale bar: 400 μm). B. Quantitative analysis of migration distances in G9a-attenuated NSCLC cells. C. Representative images for Matrigel trans-well invasion assays of A549 and H1299 cells transfected with control siRNA (Ctrl siRNA) and G9 siRNA 1 and 2 (G9a si1/2), (scale bar: 2000 μm). D. Quantitative analysis of invasive cells in G9a-attenuated NSCLC cells. E. Western blots shows H3K9me2 was decreased by G9a inhibitor UNC0638 in of A549 and H1299 cells. F. Quantitative analysis of decreased cell migration (left panel) and invasion (right panel) in A549 and H1299 cells treated with UNC0638. Data is normalized to the untreated control cells, and is presented as percentages related to the control cells (* P < 0.05, ** P < 0.01, *** P < 0.001, compared to control cells).
Invasion assay was carried out in G9a-attenuated cells. As shown in Figure 2C, cells transfected with G9a siRNA showed lower invasive potential compared to cell transfected with the control siRNA in both H1299 (upper panel) and A549 (lower panel) cells. Statistics analysis showed that compared to the controls, the invasive potential of the G9a-attenuated groups decreased significantly (P < 0.05) in the two NSCLC cells (Figure 2D).
To examine if pharmacological inhibition on G9a activity will also suppress the invasive potential of lung cancer, UNC0638, a selected G9a methyltransferase inhibitor (27), was used to suppress the G9a activity. As shown in Figure 2E, a drastically decrease of H3K9me2 protein was found in cancer cells treated with UNC0638, indicating the methyltransferase activity of G9a was inhibited significantly. Similar to the data of G9a knockdown experiment, cell migration in these two cells was also suppressed by UNC0638 treatment (Figure S2A). Compared to the control group, a significant decrease was observed in UNC0638 treated cells (Figure 2F, left panel) in these two cells (P < 0.05). Besides, the number of invasive cells in A549 and H1299 cell lines was also significantly decreased by UNC0638 treatment (Figure S2B). Quantitative analysis of invasive data was presented in Figure 2F, right panel (P < 0.05). Therefore, above data suggests G9a plays an important role in migratory and invasive potential of NSCLC cell lines.
Inhibition of G9a suppresses the activation of FAK signal pathway in NCSLC
Considering the critical role of FAK in cancer migration and invasion, we hypothesize that G9a may regulate cell invasion and migration through FAK signal pathway. To investigate this underlying mechanism, total protein was extracted from cells either transfected with G9a siRNA or treated with UNC0638. Western blot analysis showed that, compared to control siRNA group, FAK and phosphorylation of FAK (Figure 3A) were drastically decreased in both A549 and H1299 cells upon G9a knockdown. Similarly, after being treated with UNC0638 for 72 hours, total FAK and phospho-FAK (pFAK at Tyr397, an autophosphorylation site on the activated FAK that is used as an indicator for FAK activation) proteins were decreased in the both two cell lines (Figure 3B). In contrast, overexpression of G9a significantly elevated the levels of FAK and pFAK (Tyr397) (Figure 3C). These data indicated that G9a positively regulates the expression of PTK2 gene and activation of FAK signal pathway in NSCLC cells.
Figure 3. Inhibition of G9a suppresses the activation of FAK signal pathway in NCSLC.

A. Western blot analysis of G9a, total FAK and pFAK proteins in G9a-attenuated NSCLC cells. B. Western blots analysis of decreased G9a, H3K9me2, total FAK and pFAK proteins in the cells treated with UNC0638. C. Western blot analysis of G9a, total FAK and pFAK proteins in A549 and H1299 cells, and the relative protein levels in the cancer cells overexpressing G9a were presented as the normalized folds (under each band) to the control cells transfected with empty plasmid (set as 1).
Overexpression of G9a enhances cell migratory and invasive potential of NCSLC cells
To further validate the role of G9a in migration, invasion and activation of FAK signal pathway of NSCLC cells, we inserted G9a gene into the pcDNA 3.1 vector and examined if G9a overexpression will enhance the invasion and activation of FAK signaling pathway. Consistently, compared to control cells, we observed overexpression of G9a significantly increased lung cancer cell proliferation (Figure S3 A&B, P < 0.05). Meanwhile, migration of NSCLC cells (Figure 4A) was significantly increased with overexpressed G9a protein (P < 0.001, Figure 4B & C). Furthermore, compared to control group, the invasion of NSCLC cells was also increased significantly with overexpression of G9a protein (Figure 4D & E, P < 0.05). These data further demonstrated a key role of G9a in cell invasion, migration as well as activation of FAK signal pathway in NSCLC.
Figure 4. Overexpression of G9a enhances cell migratory and invasive potential of NCSLC cells.

A. Western blot analysis of overexpressed G9a in H1299 and A549 cells. B. Representative images for the scratch assays of H1299 cells overexpressing G9a and A549 cells overexpressing G9a (at 0h and 24h for H1299 / 36h for A549). C. Quantitative analysis of migration distances in the cells overexpressing G9a. D. Representative images for Matrigel transwell invasion assays of G9a-overexpressed A549 and H1299 cells. E. Quantitative analysis for invasive cells in G9a-overexpressed NSCLC cell lines. Data are presented as percentage of the control cells (* P < 0.05, ** P < 0.01. *** P < 0.001; compared to the control cells).
G9a regulates cell migration and invasion through FAK signal pathway
To investigate whether G9a promotes cell invasion and migration directly or indirectly through activating FAK signal pathway, a rescue experiment was designed and performed. In this experiment we investigated if knockdown of FAK by a FAK-specific siRNA can suppress G9a-enhanced FAK activation and cell invasion in NSCLC cells. As shown in Figure 5A, overexpression of G9a enhanced the phosphorylation of FAK, while knockdown of FAK significantly attenuated or even completely abolished the elevated phosphorylated FAK. While cell migration (Figure 5B & C) and invasion (Figure 5D & E) increased after overexpression of G9a, similar to the change in phosphorylated FAK, the enhanced migration and invasion was reversed by knockdown of FAK siRNA in these two cell lines. In addition, the FAK inhibitor also suppressed G9a-enhanced FAK activation and cellular invasion in these lung cancer cells (Figure S4). Therefore, the above data suggests that the increased cell invasion by overexpression of G9a was partially abolished by FAK siRNA as well as FAK inhibitor, indicating that G9a promotes cell invasion and migration via activating of FAK signal pathway.
Figure 5. FAK knockdown attenuates the enhanced cell migration and invasion by G9a overexpression.

A. Western blot analysis showing that activation of FAK signal pathway was attenuated by knockdown of FAK by siRNA in G9a-overexpressed NSCLC cells. B. Representative images of scratch assays. 0h and 24h/ 36h for the controls (pcDNA 3.1) and G9a-overexpressed (G9a-pcDNA 3.1) H1299 / A549 upon knockdown of FAK by its specific siRNA, respectively. C. Quantitative analysis of migration distances in the control and G9a-overexpressed A549 and H1299 cells after FAK is attenuated by its specific siRNA. D. Representative images for Matrigel transwell invasion assays of the controls (pcDNA 3.1) and G9a-overexpressed (G9a-pcDNA 3.1) H1299 and A549 transfected with control and FAK siRNA. E. Quantitative analysis for invasive cells in the controls and G9a-overexpressed A549 and H1299 cancer cells upon knockdown of FAK. Data are presented as percentages of the control cells transfected with the empty plasmid pcDNA 3.1 (* P < 0.05, ** P < 0.01, *** P < 0.001, NS represents for no significance; compared to the controls cells).
G9a activates FAK signal pathway by elevating NF-kB transcriptional activity
Studies have already demonstrated that FAK was transcriptionally regulated by P53 and NF-κB transcription factors (9). We observed the regulation of FAK expression by G9a in both p53-wildtype A549 and p53-null H1299 cells. Considering that, we hypothesized that G9a might activate FAK expression through NF-κB signaling pathway. Therefore, we investigated the effect of knockdown and overexpression of G9a on transcriptional activity of the NF-κB-controlled luciferase reporter. Dual-luciferase assays showed that silencing of G9a significantly suppressed NF-κB luciferase activity (Figure 6A, P < 0.05), while overexpression of G9a significantly increased NF-κB activity (Figure 6B, P < 0.01) in these two NSCLC cell lines. UNC0638 also significantly suppressed NF-κB luciferase activity in both the cell lines (Figure 6C, P < 0.05). Interestingly, the level of IκBα protein, an inhibitor of NF-κB signaling pathway, was found to be upregulated by knockdown of G9a and downregulated by overexpression of G9a (Figure 6D), indicating that suppression of G9a on IκBα expression may contribute to the over-activation of NF-κB signaling pathway.
Figure 6. G9a activates FAK signal pathway by elevating NF-kB transcriptional activity.

A. Knockdown of G9a suppressed NF-κB luciferase reporter activity in H1299 and A549 cells (Ctrl siR for control siRNA, G9a si1/2 for G9a siRNA1/2; * P < 0.05, ** P < 0.01, compared to the control siRNA). B. Increased NF-κB luciferase reporter activity in G9a-overexpressed (G9+) A549 and H1299 cells; (** P < 0.01, compared to the control cells transfected with pcDNA 3.1). C. NF-κB luciferase reporter activity was suppressed in these cancer cells treated with UNC0638 (* P < 0.05, ** P < 0.01, compared to the control cells treated with DMSO). D. Western blot analysis of up-regulated IκBα (upper panel) in G9a-attenuated cancer cells and decreased IκBα (lower panel) in G9-overexpressed cancer cells. E. Western blots shows NF-κB inhibitor Parthenilide partially abolished the elevated pFAK (Tyr397) by overexpression of G9a. F. qRT-PCR analysis of EMHT2 and NFKBIA mRNAs in G9a-attenuated (left panel) and G9a-overexpressed (right panel) H1299 and A549 cells. G. Western blot analysis of G9a and IκBα proteins in the controls and G9a-attenuated H1299 cells treated with Cycloheximide (CHX). Cells were first transfected with either control siRNA or G9a siRNA, at 48h post-transfection, cells were then treated with 25 ug/ml CHX. Data are presented as percentages of the control cells (* P < 0.05, ** P < 0.01, *** P < 0.001, NS represents for no significance; compared to the controls cells).
To further investigate whether G9a activates FAK signal pathway through NF-κB, we treated the G9a-overexpressed H1299 cells with a NF-κB inhibitor, Parthenilide. Western blot analysis showed that NF-κB inhibitor could partially abolish the elevated phosphorylation of FAK (Tyr397) that was caused by overexpression of G9a (Figure 6E).
To further address how G9a regulates IκBα protein. qRT-PCR was used to measure the mRNA expression level of NFKBIA gene which encodes IκBα protein. Expression of NFKBIA was not significantly modulated by either knockdown of G9a (Figure 6F, left panel) nor overexpression of G9a in H1299 cells (Figure 6F, right panel), indicating the regulation may be at the post-transcription level. Next, we investigated if G9a impacts the stability of IκBα. Cycloheximide was used to suppress protein synthesis to check the stability of IκBα protein. As shown in Figure 6G, knockdown of G9a stabilized IκBα protein, leading to inhibition of NF-κB signaling pathway. Taken together, these data indicate that G9a knockdown inhibits FAK signal pathway partially through inhibiting NF-κB signal pathway via stabilizing IκBα protein, although the exact mechanisms for IκBα stabilized by G9a remains to be further explored.
The impact of knockdown of G9a on in vivo growth, metastasis, FAK activation and the correlation of G9a to pFAK proteins in NSCLC tissues
We first investigated the correlation between G9a expression with in vivo growth and FAK activation in xenografts using stable G9a-KD H1299 cells. As shown in Figure 7A, the tumor sizes of G9a-KD group were significantly smaller than the control group. Compared to the xenograft tissues of the controls, the IHC intensities of G9a and H3K9me2 were strongly decreased in G9a-attenuated xenograft tissues, and consistent with the in vitro data, pFAK (Tyr397) levels were down-regulated dramatically in G9a-attenuated xenograft tissues (Figure 7B).
Figure 7. The impact of knockdown of G9a on in vivo growth, metastasis, FAK activation and the correlation of G9a to pFAK proteins in NSCLC tissues.
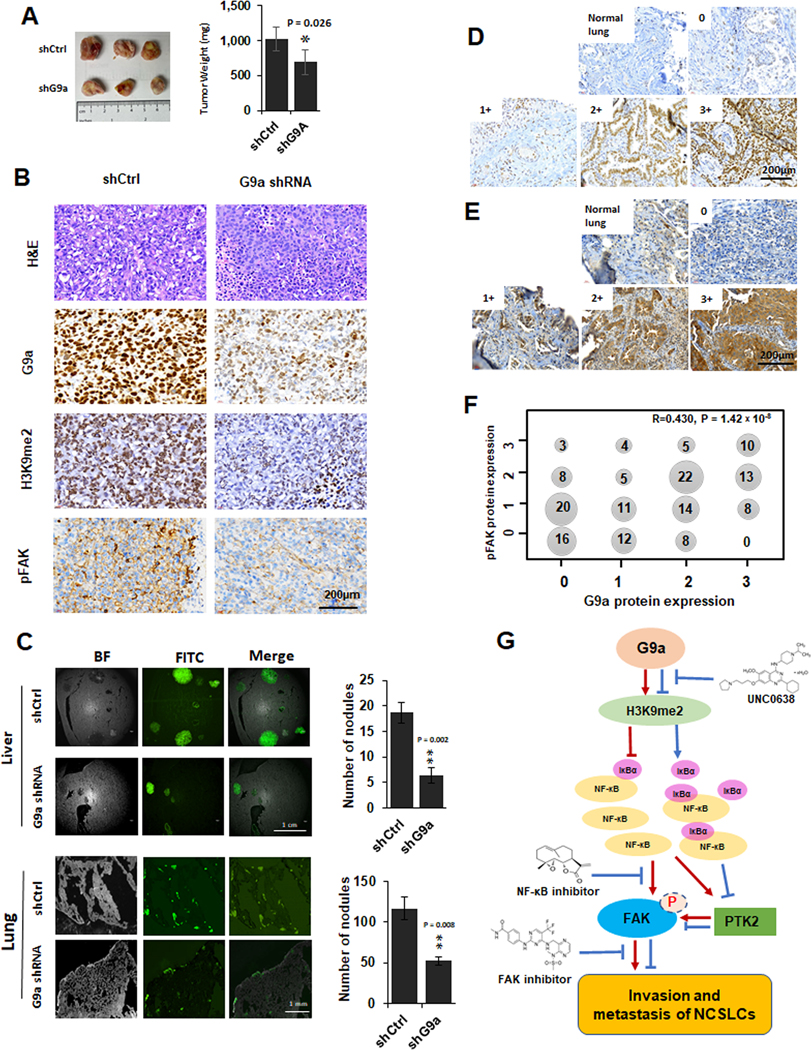
A. The images (left panel) and weighs (right panel) of xenografts generated by the control cells and G9a-attenuated H1299 (compared to the control xenografts, * P < 0.05. B. Representative images for H&E and G9a, H3K9me2 and pFAK IHC staining in the xenograft tissues of the control and G9a-attenuated H1299 cancer cells (scale bar: 200 μm). C. left panel, Representative GFP images for liver metastasis (the upper of left panel) and lung metastasis (the lower of left panel) formed by the GFP-labeled control and G9a-attenuated H1299 cells. Right panel, Statistical analysis of the metastasized cells and the number of nodules in liver (the upper of right panel) and lung (the right of lower panel) (** P < 0.01, compared to the control cells). D&E. Representative images for G9a (E) proteins and pFAK (F) proteins scored as 0, 1+, 2+, 3+ in NSCLC tissues (Scale bar: 200 μm). F. Cross distribution and correlation of G9a IHC scores to pFAK IHC scores in 159 NSCLC tissues. X axis is for G9a IHC scores, Y axis is for pFAK scores. Number in dot is the sample size with various G9a and pFAK. R is the Spearman’s correlation coefficient for G9a and pFAK. G. Schematic diagram of the potential mechanisms for G9a-enhanced metastasis, modulated FAK and NF-κB signaling pathways, and related therapeutic implications in NSCLC.
To further examine the potential role of G9a in metastasis formation, parental and G9a-attenuated H1299 cells were injected into the tail veins of mice to establish in vivo metastasis. We observed that, at about 4 weeks of post-injection, H1299 cells developed visible metastasis in both liver (Figure 7C, the left of upper panel) and lung (Figure 7C, the left of lower panel). Metastatic tumors were more numerous and larger in livers than lungs. Compared with control H1299 cells, G9a-KD H1299 formed fewer and smaller nodules in both liver tissues (Figure 7C, the right of upper panel, Figure S5A) and lung tissues (Figure 7C, the right of lower panel, Figure S5b). Similarly, pFAK (Tyr397) levels were also dramatically decreased in metastasis of G9a-attenuated H1299 cells (data not shown), suggesting the activation of FAK signaling pathway was suppressed upon G9a knockdown.
Furthermore, we analyzed the correlation between G9a and FAK expression after IHC staining of pFAK (Tyr397). IHC analysis demonstrated nuclear G9a staining (Figure 7D), and quantitative analysis of G9a on IHC slides was conducted as previously reported (19). IHC analysis showed cytoplasmic pFAK (Tyr397) staining in tumors and staining was absent in adjacent normal cells. pFAK (Tyr397) IHC staining in the same tissue arrays were quantitatively scored using the same scoring method, and representative images of IHC scoring are shown in Figure 7E. Pearson correlation analysis was used to examine the correlation between G9a and pFAK (Tyr397) IHC staining in these tumor tissues, and the analysis revealed that pFAK (Tyr397) staining was significantly correlated with G9a staining (Figure 7F; R = 0.408, P < 0.001).
These data indicated that overexpression of G9a may enhance activation of FAK signaling pathway and then promote invasion and metastasis in NSCLC.
Discussion
This study demonstrated for the first time that G9a may promote invasion and metastasis of NSCLC by activating FAK signaling pathway. Our data suggest that in NSCLC, G9a promotes the degradation of IκBα protein, and then may activate NF-κB signaling pathway, which then promotes FAK expression, enhances cancer invasion and metastasis. Given the important role of both G9a and FAK in cancer cell growth and metastasis, this study underscores the potential role of G9a inhibition as a therapeutic target for lung cancer.
Functionally, G9a is responsible for mono or di-methylation of H3K9 and its activity leads to silencing of a number of tumor suppressor genes including CDH1, DUSP5 and SPRY4, etc. (28). Overexpression of G9a is closely associated with tumorigenesis and metastasis in many cancers. Recently, G9a has been demonstrated to promote tumor metastasis by upregulating ITGB3 in gastric cancer (29). It is also responsible for liver cancer development and metastasis by silencing tumor suppressor RARRES2 (30). In lung cancer, we previously reported that G9a promotes tumor growth via enhancing Wnt signaling pathway and HP1α and silencing APC2 expression (19). A previous study demonstrated that G9a promotes lung cancer metastasis via silencing Ep-CAM (31) and CASP1 (32). G9a inhibition significantly suppressed the migration and invasion enhanced by overexpression of Snail2 in lung cancer cells (33). In contrast, a recent study demonstrated that depletion of G9a during tumorigenesis enriches tumors in tumor-propagating cells and accelerates disease progression metastasis (34). Recently, we also found that G9a is involved in lung cancer stemness through epigenetic mechanisms of maintaining DNA methylation of multiple lung cancer stem cell genes and their expression (35). Consistent with findings of many other studies, we also found in this study that G9a promotes cancer invasion and metastasis through a novel mechanism, and targeting G9a suppressed invasion and metastasis in lung cancer. Studies revealed that G9a inhibitor UNC0638 reduced growth and metastasis of breast cancer and significantly suppressed epithelial-mesenchymal transition-mediated cellular migration and invasion (36). Another G9a methyltransferase activity inhibitor, UNC0646, was shown to suppress hepatocellular tumor growth but activate cellular apoptosis and metastasis (37).
One of novel findings of the study is that FAK signaling pathway may represent a downstream target of G9a. FAK plays a pivotal role in transducing signals from the plasma membrane to the nucleus, and phosphorylation of FAK promotes cancer cell growth, invasion, and angiogenesis (38). Studies have demonstrated that high level of pFAK (Tyr397) is associated with aggressive behavior of lung cancer, including rapid growth, frequent metastasis and poor overall survival (39). Several phase I and phase II clinical trials are currently undergoing for FAK inhibitors in cancers (40). And we further showed that FAK inhibition can significantly attenuated G9a-enhanced invasion. Considering the advanced improvements on FAK inhibitor, FAK inhibition may represent a potential therapy for lung cancer patients with overexpressed G9a.
Furthermore, this study has demonstrated for the first time that G9a may regulate FAK through NF-κB signaling pathway. In the study, we have found that G9a activates NF-κB signaling pathway in lung cancer. NF-κB activity not only promotes tumor cells proliferation, suppresses apoptosis, and attracts angiogenesis, but it also induces epithelial-mesenchymal transition, which facilitates distant metastasis (41). In lung cancer cells expressing oncogenic K-Ras, NF-κB signaling pathway has been shown to be activated in a K-Ras-dependent manner, and represent a potential therapeutic target for K-Ras-induced lung cancer (42). In many cancers, the primary regulation of the NF-κB pathway is through the association of NF-κB complexes with IκB proteins. IκBα is a major inhibitor of NF-κB signaling pathway that forms a complex with NF-kappaB subunits p50 and p65, with the degradation of IκBα, P50 and P65 translocate to the nucleus to regulate downstream gene expression (41). For example, a previous study demonstrated that Tetraspanin 15 (TSPAN15) promoted esophageal cancer metastasis via activating NF-κB through promoting the degradation of IκBα (43). Previous study reported that inhibition of G9a epigenetically increases activation of NF-κB signaling pathway in breast cancer (44). In this study, we revealed that the elevated expression of G9a promoted degradation of IκBα protein, and then activate NF-κB. NF-κB signaling pathway has previously been shown to be associated with FAK activation and increased cell proliferation in lung cancer (45). Therefore, although the exact underlying and association between NF-κB and FAK signaling pathways should be further explored, our data have indicated that the NF-κB signaling pathway may positively contribute to the activation of FAK signaling pathway and cancer metastasis. We have also demonstrated that NF-κB inhibition suppress G9a-enhanced invasion, indicating it may also represent a potential druggable target for lung cancer overexpressing G9a protein.
In our previous study, we found G9a activated Wnt signaling pathway. Wnt signaling usually negatively regulates NF-κB signaling pathway in liver, colon and breast cancer (46–48). But such correlation was not found in head and neck cancer (49), suggesting a context-specific mechanism for the interaction between two signaling pathways. In lung cancer, a study revealed that overexpression of ING4 gene induced growth inhibition of A549 cells by inactivation of Wnt signaling pathway partially through suppressing NF-κB signaling pathway (50), indicating a potential positive interaction among the two pathways. In our studies, we have found that G9a may enhance activation of Wnt and NF-κB signaling pathways in NSCLC cells. And we have also shown overactivation Wnt signaling pathway does not affect NF-κB signaling pathway in NSCLC cells (Figure S6).
In summary, we have revealed a novel mechanism by which G9a mediates invasion and metastasis in NSCLC. We have demonstrated that the role of G9a in lung cancer invasion and metastasis is mediated in part by enhanced activation of FAK that appears to be mediated by NF-κB signaling pathway (Figure 7G), suggesting a potential positive interaction between G9a, NF-κB pathway, and FAK pathway in NSCLC cells.
Supplementary Material
Acknowledgements
We acknowledge the generous support of the Baum Family Foundation in support of this laboratory. The authors thank Michael Lewallen at pathology core for IHC analysis and Michael Nelson at the light microscope core for trans-well imaging and cell counting.
Financial support: Research reported in this publication is supported by the V Foundation (DR), the Doris Duke Charitable Foundation (DR) and the National Cancer Institute of the National Institutes of Health (NIH P30CA33572) using several core facilities.
List of abbreviations
- G9a
Euchromatic histone-lysine N-methyltransferase 2 (EHMT2)
- FAK
focal adhesion kinase
- NF-κB
Nuclear Factor kappa-light-chain-enhancer of activated B cells
- NSCLC
non-small cell lung cancer
- H3K9me2
di-methylation at the 9th lysine residue of the histone H3 protein
- Ep-CAM
Epithelial cell adhesion molecule
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- CDH1
cadherin 1
- DUSP5
Dual-specificity phosphatase 5
- SPRY4
sprout RTK Signaling Antagonist 4
- RARRES2
Retinoic Acid Receptor Responder 2
- ITGB3
Integrin beta-3
- KRAS
Kirsten rat sarcoma 2 viral oncogene homolog
- ATCC
American Type Culture Collection
- GLP
G9a-like protein
- pFAK
phosphorylation of FAK
- IκBα
inhibitor of NF-κB
- IKK
IκB kinase
- HP1α
heterochromatin protein 1α
- APC2
adenomatous polyposis coli protein 2
- SATB2
special AT-rich sequence-binding protein 2
Footnotes
Supplementary Data
Figure S1–S6 was shown in supplementary material.
The authors declare no potential conflicts of interest.
References:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature 2018;553(7689):446–54 doi nature25183 [pii] 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 3.Heron M, Anderson RN. Changes in the Leading Cause of Death: Recent Patterns in Heart Disease and Cancer Mortality. NCHS Data Brief 2016(254):1–8. [PubMed] [Google Scholar]
- 4.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev 2011;25(8):781–8 doi 25/8/781 [pii] 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urrutia G, Salmonson A, Toro-Zapata J, de Assuncao TM, Mathison A, Dusetti N, et al. Combined Targeting of G9a and Checkpoint Kinase 1 Synergistically Inhibits Pancreatic Cancer Cell Growth by Replication Fork Collapse. Mol Cancer Res 2019. doi 1541–7786.MCR-19–0490 [pii] 10.1158/1541-7786.MCR-19-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Segovia C, San Jose-Eneriz E, Munera-Maravilla E, Martinez-Fernandez M, Garate L, Miranda E, et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat Med 2019;25(7):1073–81 doi 10.1038/s41591-019-0499-y10.1038/s41591-019-0499-y [pii]. [DOI] [PubMed] [Google Scholar]
- 7.Wang YF, Zhang J, Su Y, Shen YY, Jiang DX, Hou YY, et al. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat Commun 2017;8(1):274 doi 10.1038/s41467-017-00350-910.1038/s41467-017-00350-9 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011;331(6024):1559–64 doi 331/6024/1559 [pii] 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 9.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta 2004;1678(2–3):111–25 doi 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 2014;14(9):598–610 doi nrc3792 [pii] 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen M, Jiang YZ, Wei Y, Ell B, Sheng X, Esposito M, et al. Tinagl1 Suppresses Triple-Negative Breast Cancer Progression and Metastasis by Simultaneously Inhibiting Integrin/FAK and EGFR Signaling. Cancer Cell 2019;35(1):64–80 e7 doi 10.1016/j.ccell.2018.11.016. [DOI] [PubMed] [Google Scholar]
- 12.Aronsohn MS, Brown HM, Hauptman G, Kornberg LJ. Expression of focal adhesion kinase and phosphorylated focal adhesion kinase in squamous cell carcinoma of the larynx. Laryngoscope 2003;113(11):1944–8 doi 10.1097/00005537-200311000-00017. [DOI] [PubMed] [Google Scholar]
- 13.Carelli S, Zadra G, Vaira V, Falleni M, Bottiglieri L, Nosotti M, et al. Up-regulation of focal adhesion kinase in non-small cell lung cancer. Lung Cancer 2006;53(3):263–71 doi 10.1016/j.lungcan.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Aboubakar Nana F, Lecocq M, Ladjemi MZ, Detry B, Dupasquier S, Feron O, et al. Therapeutic Potential of Focal Adhesion Kinase Inhibition in Small Cell Lung Cancer. Mol Cancer Ther 2019;18(1):17–27 doi 10.1158/1535-7163.MCT-18-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matikas A, Mistriotis D, Georgoulias V, Kotsakis A. Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity. Crit Rev Oncol Hematol 2017;110:1–12 doi 10.1016/j.critrevonc.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Konstantinidou G, Ramadori G, Torti F, Kangasniemi K, Ramirez RE, Cai Y, et al. RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov 2013;3(4):444–57 doi 10.1158/2159-8290.CD-12-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang KJ, Constanzo JD, Venkateswaran N, Melegari M, Ilcheva M, Morales JC, et al. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clin Cancer Res 2016;22(23):5851–63 doi 10.1158/1078-0432.CCR-15-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu HJ, Hao M, Yeo SK, Guan JL. FAK signaling in cancer-associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene 2020;39(12):2539–49 doi 10.1038/s41388-020-1162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang K, Wang J, Yang L, Yuan YC, Tong TR, Wu J, et al. Targeting histone methyltransferase G9a inhibits growth and Wnt signaling pathway by epigenetically regulating HP1alpha and APC2 gene expression in non-small cell lung cancer. Mol Cancer 2018;17(1):153 doi 10.1186/s12943-018-0896-810.1186/s12943-018-0896-8 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang K, Gao H, Wu X, Wang J, Zhou W, Sun G, et al. Frequent overexpression of HMGA2 in human atypical teratoid/rhabdoid tumor and its correlation with let-7a3/let-7b miRNA. Clin Cancer Res 2014;20(5):1179–89 doi 10.1158/1078-0432.CCR-13-1452. [DOI] [PubMed] [Google Scholar]
- 21.Zhang K, Wang J, Wang J, Luh F, Liu X, Yang L, et al. LKB1 deficiency promotes proliferation and invasion of glioblastoma through activation of mTOR and focal adhesion kinase signaling pathways. American journal of cancer research 2019;9(8):1650–63. [PMC free article] [PubMed] [Google Scholar]
- 22.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Scientific reports 2017;7(1):16878 doi 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou W, Pal AS, Hsu AY, Gurol T, Zhu X, Wirbisky-Hershberger SE, et al. MicroRNA-223 Suppresses the Canonical NF-kappaB Pathway in Basal Keratinocytes to Dampen Neutrophilic Inflammation. Cell Rep 2018;22(7):1810–23 doi S2211–1247(18)30107–4 [pii] 10.1016/j.celrep.2018.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang K, Lu J, Mori T, Smith-Powell L, Synold TW, Chen S, et al. Baicalin increases VEGF expression and angiogenesis by activating the ERR{alpha}/PGC-1{alpha} pathway. Cardiovascular research 2011;89(2):426–35 doi 10.1093/cvr/cvq296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang K, Yang L, Wang J, Sun T, Guo Y, Nelson R, et al. Ubiquitin-specific protease 22 is critical to in vivo angiogenesis, growth and metastasis of non-small cell lung cancer. Cell Commun Signal 2019;17(1):167 doi 10.1186/s12964-019-0480-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang K, Keymeulen S, Nelson R, Tong TR, Yuan YC, Yun X, et al. Overexpression of Flap Endonuclease 1 Correlates with Enhanced Proliferation and Poor Prognosis of Non-Small-Cell Lung Cancer. The American journal of pathology 2018;188(1):242–51 doi 10.1016/j.ajpath.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nature chemical biology 2011;7(8):566–74 doi 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wozniak RJ, Klimecki WT, Lau SS, Feinstein Y, Futscher BW. 5-Aza-2’-deoxycytidine-mediated reductions in G9a histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene 2007;26(1):77–90 doi 10.1038/sj.onc.1209763. [DOI] [PubMed] [Google Scholar]
- 29.Hu L, Zang MD, Wang HX, Zhang BG, Wang ZQ, Fan ZY, et al. G9A promotes gastric cancer metastasis by upregulating ITGB3 in a SET domain-independent manner. Cell Death Dis 2018;9(3):278 doi 10.1038/s41419-018-0322-610.1038/s41419-018-0322-6 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei L, Chiu DK, Tsang FH, Law CT, Cheng CL, Au SL, et al. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J Hepatol 2017;67(4):758–69 doi 10.1016/j.jhep.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Chen MW, Hua KT, Kao HJ, Chi CC, Wei LH, Johansson G, et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer research 2010;70(20):7830–40 doi 10.1158/0008-5472.CAN-10-0833. [DOI] [PubMed] [Google Scholar]
- 32.Huang T, Zhang P, Li W, Zhao T, Zhang Z, Chen S, et al. G9A promotes tumor cell growth and invasion by silencing CASP1 in non-small-cell lung cancer cells. Cell Death Dis 2017;8(4):e2726 doi 10.1038/cddis.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Y, Zheng Y, Dai M, Wu J, Yu B, Zhang H, et al. Snail2 induced E-cadherin suppression and metastasis in lung carcinoma facilitated by G9a and HDACs. Cell Adh Migr 2019;13(1):285–92 doi 10.1080/19336918.2019.1638689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rowbotham SP, Li F, Dost AFM, Louie SM, Marsh BP, Pessina P, et al. H3K9 methyltransferases and demethylases control lung tumor-propagating cells and lung cancer progression. Nat Commun 2018;9(1):4559 doi 10.1038/s41467-018-07077-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pangeni RP, Yang L, Zhang K, Wang J, Li W, Guo C, et al. G9a regulates tumorigenicity and stemness through genome-wide DNA methylation reprogramming in non-small cell lung cancer. Clin Epigenetics 2020;12(1):88 doi 10.1186/s13148-020-00879-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu XR, Zhou LH, Hu JX, Liu LM, Wan HP, Zhang XQ. UNC0638, a G9a inhibitor, suppresses epithelialmesenchymal transitionmediated cellular migration and invasion in triple negative breast cancer. Mol Med Rep 2018;17(2):2239–44 doi 10.3892/mmr.2017.8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu M, Toh TB, Hooi L, Lim JJ, Zhang X, Chow EK. Nanodiamond-Mediated Delivery of a G9a Inhibitor for Hepatocellular Carcinoma Therapy. ACS Appl Mater Interfaces 2019;11(49):45427–41 doi 10.1021/acsami.9b16323. [DOI] [PubMed] [Google Scholar]
- 38.Thanapprapasr K, Nartthanarung A, Thanapprapasr D, Jinawath A. pFAK-Y397 overexpression as both a prognostic and a predictive biomarker for patients with metastatic osteosarcoma. PLoS One 2017;12(8):e0182989 doi 10.1371/journal.pone.0182989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aboubakar Nana F, Hoton D, Ambroise J, Lecocq M, Vanderputten M, Sibille Y, et al. Increased Expression and Activation of FAK in Small-Cell Lung Cancer Compared to Non-Small-Cell Lung Cancer. Cancers (Basel) 2019;11(10) doi 10.3390/cancers11101526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohanty A, Pharaon RR, Nam A, Salgia S, Kulkarni P, Massarelli E. FAK-targeted and combination therapies for the treatment of cancer: an overview of phase I and II clinical trials. Expert Opin Investig Drugs 2020:1–11 doi 10.1080/13543784.2020.1740680. [DOI] [PubMed] [Google Scholar]
- 41.Xia Y, Shen S, Verma IM. NF-kappaB, an active player in human cancers. Cancer Immunol Res 2014;2(9):823–30 doi 10.1158/2326-6066.CIR-14-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basseres DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer research 2010;70(9):3537–46 doi 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B, Zhang Z, Li L, Qin YR, Liu H, Jiang C, et al. TSPAN15 interacts with BTRC to promote oesophageal squamous cell carcinoma metastasis via activating NF-kappaB signaling. Nat Commun 2018;9(1):1423 doi 10.1038/s41467-018-03716-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park SE, Yi HJ, Suh N, Park YY, Koh JY, Jeong SY, et al. Inhibition of EHMT2/G9a epigenetically increases the transcription of Beclin-1 via an increase in ROS and activation of NF-kappaB. Oncotarget 2016;7(26):39796–808 doi 10.18632/oncotarget.9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia Y, Yeddula N, Leblanc M, Ke E, Zhang Y, Oldfield E, et al. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat Cell Biol 2012;14(3):257–65 doi 10.1038/ncb2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, et al. beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell 2002;2(4):323–34 doi 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- 47.Du Q, Zhang X, Cardinal J, Cao Z, Guo Z, Shao L, et al. Wnt/beta-catenin signaling regulates cytokine-induced human inducible nitric oxide synthase expression by inhibiting nuclear factor-kappaB activation in cancer cells. Cancer Res 2009;69(9):3764–71 doi 10.1158/0008-5472.CAN-09-0014. [DOI] [PubMed] [Google Scholar]
- 48.Moreau M, Mourah S, Dosquet C. beta-Catenin and NF-kappaB cooperate to regulate the uPA/uPAR system in cancer cells. Int J Cancer 2011;128(6):1280–92 doi 10.1002/ijc.25455. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez-Pinilla M, Rodriguez-Peralto JL, Hitt R, Sanchez JJ, Sanchez-Verde L, Alameda F, et al. beta-Catenin, Nf-kappaB and FAS protein expression are independent events in head and neck cancer: study of their association with clinical parameters. Cancer Lett 2005;230(1):141–8 doi 10.1016/j.canlet.2004.12.045. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Cai L, Liang M, Wang Y, Yang J, Zhao Y. ING4 induces cell growth inhibition in human lung adenocarcinoma A549 cells by means of Wnt-1/beta-catenin signaling pathway. Anat Rec (Hoboken) 2008;291(5):593–600 doi 10.1002/ar.20685. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.