

Abstract
Background:
Chronic arsenic exposure via drinking water is associated with an increased risk of developing cancer and noncancer chronic diseases. Pre-mRNAs are often subject to alternative splicing, generating mRNA isoforms encoding functionally distinct protein isoforms. The resulting imbalance in isoform species can result in pathogenic changes in critical signaling pathways. Alternative splicing as a mechanism of arsenic-induced toxicity and carcinogenicity is understudied.
Objective:
This study aimed to accurately profile differential alternative splicing events in human keratinocytes induced by chronic arsenic exposure that might play a role in carcinogenesis.
Methods:
Independent quadruplicate cultures of immortalized human keratinocytes (HaCaT) were maintained continuously for 28 wk with 0 or sodium arsenite. RNA-sequencing (RNA-Seq) was performed with poly(A) RNA isolated from cells harvested at 7, 19, and 28 wk with subsequent replicate multivariate analysis of transcript splicing (rMATS) analysis to detect and quantify differential alternative splicing events. Reverse transcriptase-polymerase chain reaction (RT-PCR) for selected alternative splicing events was performed to validate RNA-Seq predictions. Functional enrichment was performed by gene ontology (GO) analysis of the differential alternative splicing event data set at each time point.
Results:
At least 600 differential alternative splicing events were detected at each time point tested, comprising all the five main types of alternative splicing and occurring in both open reading frames (ORFs) and untranslated regions (UTRs). Based on functional relevance ELK4, SHC1, and XRRA1 were selected for validation of predicted alternative splicing events at 7 wk by RT-PCR. Densitometric analysis of RT-PCR data corroborated the rMATS predicted alternative splicing for all three events. Protein expression validation of the selected alternative splicing events was challenging given that very few isoform-specific antibodies are available. GO analysis demonstrated that the enriched terms in differential alternatively spliced mRNAs changed dynamically with the time of exposure. Notably, RNA metabolism and splicing regulation pathways were enriched at the 7-wk time point, when the greatest number of differentially alternatively spliced mRNAs are detected. Our preliminary proteomic analysis demonstrated that the expression of the canonical isoforms of the splice regulators DDX42, RMB25, and SRRM2 were induced upon chronic arsenic exposure, corroborating the splicing predictions.
Discussion:
These results using cultures of HaCaT cells suggest that arsenic exposure disrupted an alternative splice factor network and induced time-dependent genome-wide differential alternative splicing that likely contributed to the changing proteomic landscape in arsenic-induced carcinogenesis. However, significant challenges remain in corroborating alternative splicing data at the proteomic level. https://doi.org/10.1289/EHP9676
Introduction
Chronic exposure to arsenic is a well-documented environmental health crisis, endangering over individuals, the vast majority in Asia (Podgorski and Berg 2020). Such exposure causes several cancers (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans 2012; NTP 2016) and increases the risk of development of several noncancer health outcomes (Ghosh et al. 2007; Navas-Acien et al. 2008; Tsuji et al. 2014). Skin cancers, including cutaneous squamous cell carcinoma (cSCC), basal cell carcinoma, and Bowen’s disease are the most common malignant outcomes of chronic arsenic exposure (Banerjee et al. 2011). cSCC is metastatic and often fatal, with high incidence of recurrence and low survival rates (Waldman and Schmults 2019).
Elucidating the molecular etiology of arsenic-induced cancers, especially skin cancers, has been of special interest for more than two decades. The immortalized but nonmalignant human keratinocyte cell line, HaCaT, is an important tool used to study arsenic-induced skin carcinogenesis. HaCaT cells exposed to arsenite for 28 wk acquired an aggressive cSCC phenotype (Pi et al. 2008; Sun et al. 2009). This exposure is toxicologically relevant given that it is consistent with the blood arsenic levels of chronically arsenic-exposed populations in China (Pi et al. 2000) and Mexico (Gonsebatt et al. 1994, 1997). Using this cell line model, early cellular changes were detectable by 7 wk of exposure, and transformation-related changes started at 19 wk and continued through 28 wk, when transformation was often complete as demonstrated by epithelial-to-mesenchymal transition (Banerjee et al. 2021; Pi et al. 2008; Sun et al. 2009). This model thus provides an excellent opportunity to follow the longitudinal events involved in the process of arsenic-induced skin carcinogenesis.
Although several different mechanisms and biological pathways have been proposed to be involved in the process of arsenic-induced skin carcinogenesis, it is not clear how they interact to influence carcinogenesis (Hunt et al. 2014). Recent technological advances and studies of several cancers across different experimental models have clearly indicated that such dysregulated signaling cascades engage in extensive molecular crosstalk and are regulated in a temporally dynamic manner (Boja and Rodriguez 2014). It has also been demonstrated that chronic arsenic exposure has led to changes in the proteomic landscape in exposed individuals (Bailey et al. 2014; De Loma et al. 2020), target tissues (Lantz et al. 2007), and in vitro models (Mir et al. 2017; Udensi et al. 2014; Zhou et al. 2017). So, how arsenic causes changes in the proteomic landscape that is regulated in a dynamic manner with time, starting from exposure initiation through to the development of cancer, remains an unanswered question. Multiple mechanisms can give rise to changes in the proteome, including differential transcription and translation (Hoogendijk et al. 2019; Ishikawa 2021; Iwasaki and Ingolia 2017; Vogel and Marcotte 2012), alternative splicing (Liu et al. 2017), and differential protein degradation (Grandi and Bantscheff 2019). Several of these mechanisms have been studied regarding arsenic toxicity (Al-Eryani et al. 2018a; Berglund et al. 2009; Chen et al. 2020b; Riedmann et al. 2015; Tam and Wang 2020), but the role of differential alternative splicing remains poorly understood. One previous study in the arsenic-exposed Ad12-SV40 2B cell line model of arsenic-induced lung carcinogenesis demonstrated 104 differential alternative splicing events (Riedmann et al. 2015). A recent review discussed the effects of arsenic-induced DNA methylation on alternative splicing (Saintilnord and Fondufe-Mittendorf 2021). These observations added to the hypothesis that differential alternative splicing could be playing a critical role in chronic arsenic exposure-induced cSCC.
Alternative splicing and its dysregulation have been recently recognized as a potent mechanism in carcinogenesis through modulation of gene expression by posttranscriptional generation of mRNA and proteome diversity (Kim et al. 2018). Differential alternative splicing generates several mRNA isoforms from the same gene by variable combinations of the available exons (Keren et al. 2010). Alternative splicing can add or remove amino acids, introduce a termination codon, or change the reading frame. It might affect gene expression by removing or inserting regulatory elements controlling translation, mRNA stability, or localization (Faustino and Cooper 2003). In the last few years, alternative splicing has emerged as a central player in the intricate and nuanced gene expression regulation in health and disease by tightly fine-tuning the relative protein isoform expression (Kim et al. 2018) and generating regulatory micropeptides and regulatory mRNA molecules (Alvarez-Dominguez et al. 2017). Alternative splicing is regulated by a complex interplay of trans-acting factors and associated proteins that promote or inhibit spliceosome formation (Han et al. 2017; Ule and Blencowe 2019). Chromatin- and transcription-associated components can also impact the splicing process (Braunschweig et al. 2013). Recent studies have shown signaling proteins are capable of catalyzing posttranslational modifications of splicing proteins through different mechanisms, such as by altering the synthesis or degradation (Heyd and Lynch 2011), function or localization (Heyd et al. 2008; Yang and Carstens 2017) of key splicing regulators. In this case, the downstream alternative splicing events occurred often in response to cellular stimuli (Han et al. 2017; Heyd and Lynch 2011). Experimental evidence strongly supports alternative splicing as a key underlying mechanism in oncogenesis (El Marabti and Younis 2018; Song et al. 2018; Urbanski et al. 2018).
Thus, it is evident that differential alternative splicing could be playing an important role in chronic arsenic exposure-induced cSCC. However, no experimental data exist to determine if this is truly the case. In the present work, we investigated the dynamic changes in alternative splicing over time, at multiple time points spanning the course of arsenic exposure (7, 19, and 28 wk) in the human keratinocyte cell line HaCaT. We further performed gene ontology (GO) analysis to explore the biological pathways and networks that are affected by such differential alternative splicing events at each time point. Thus, this study aimed to understand how individual differential alternative splicing events together could modulate development of arsenic-induced cSCC at different stages.
Methods
Chemicals
Sodium arsenite (; Chemical Abstracts Service Registry No. 7784-0698) was obtained from Thermo Fisher Scientific Inc. Single-thaw aliquots of were prepared in ultrapure DNase/RNase-free distilled water (Thermo Fisher Scientific Inc.) and were thawed immediately before use. Minimum Essential Media (MEM) alpha modification media, trypsin, ethylenediaminetetraacetic acid (EDTA), penicillin/streptomycin, and l-glutamine were obtained from Thermo Fisher Scientific Inc. Fetal bovine serum (FBS; characterized) was obtained from Hyclone.
Cell Culture
This study employed a well-characterized HaCaT cell line model for chronic arsenic exposure-induced cSCC (Banerjee et al. 2021; Pi et al. 2008). The HaCaT cell line was developed by spontaneous immortalization of human keratinocytes cultured continuously with low at 38.5°C (Boukamp et al. 1988; Colombo et al. 2017). These cells lack the SV40 T-antigen or human papillomavirus genomic sequences (Boukamp et al. 1988) but do contain two point mutations in the TP53 gene (Henseleit et al. 1997). However, arsenic exposure inhibits the functions of TP53 (Ganapathy et al. 2016; Shen et al. 2008), thus alleviating concerns about a nonfunctional TP53 in this cell line with respect to arsenic-induced cSCC. The HaCaT cells were the kind gift of Dr. Tai Hao Quan, University of Michigan. Briefly, cells were cultured in MEM alpha modification media (Cat. #12561056; Thermo Fisher Scientific Inc.) supplemented with 10% FBS, penicillin, streptomycin, and l-glutamine. The MEM media contains , which has previously been demonstrated to not affect the basal phenotype (Colombo et al. 2017). Expression of keratin 14, keratin 10, or involucrin 14 in cells maintained at confluence was similar in high () or low () media, especially when the cells were grown for 14 d or more (Colombo et al. 2017). Quadruplicate biological replicates were generated by maintaining multiple cultures of cells (4 with 0 and 4 with ) independently for 28 wk. Cells were passaged twice a week (at confluency), and 1 million cells were plated per dish every time and kept in a 37°C tissue culture incubator with 5% carbon dioxide. Cells were harvested at 7,- 19-, and 28-wk time points for isolation of RNA and whole cell lysates. Human liver cancer cells (HepG2) cells were the kind gift of Dr. Kyung Hong, University of Louisville. These cells were cultured as described previously (Barker et al. 2006). Cells were grown for one passage and whole cell lysates were harvested for immunoblot analysis.
Authentication of the HaCaT Cell Line
Authentication of the HaCaT cell line was performed through short tandem repeat (STR) mapping, outsourced to a commercial third-party vendor (Labcorp). Genomic DNA was isolated from each HaCaT cell culture following previously established protocol with minor modifications (Miller et al. 1988). Briefly, after lysis, proteins were precipitated by adding one-fourth volume saturated sodium chloride (NaCl; ) solution followed by centrifugation (, 30 min at room temperature) and collection of the DNA-containing supernatant. The genomic DNA was precipitated by adding two volumes ice-cold 96% ethanol, collected with a Pasteur pipette hook, rinsed in 70% ethanol, and allowed to air dry (3–5 min). The air-dried precipitate of genomic DNA was resuspended in Tris-EDTA (TE) buffer [ Tris-hydrochloride (Tris-HCl), EDTA disodium salt (), pH 8.0] and incubated with RNAse A (37°C; 3 h) to remove traces of contaminating RNA. Purified genomic DNA was then precipitated overnight at following the addition of one-fifth volume acidified ammonium acetate and 2–2.5 volumes ice-cold 96% ethanol. Finally, the precipitated purified genomic DNA was collected by Pasteur pipette hook, air dried, and resuspended in TE buffer. Purified genomic DNA was isolated at the beginning and the end of the experiment and sent to Labcorp’s Cell Line Testing Division. STR mapping was performed employing polymerase chain reaction (PCR) and capillary electrophoresis on a 3130xl genetic analyzer (Applied Biosystems). The 13 core Combined DNA Index System STR loci plus Pentameric repeat (PENTA) E and PENTA D, and the sex-determining locus, amelogenin, were analyzed using the commercially available PowerPlex 16HS amplification kit (Promega Corporation; mouse marker included) and GeneMapper ID software (version 3.2.1; Applied Biosystems). Appropriate positive and negative controls were used concurrently throughout the analysis. Authentication of each cell line and absence of mouse DNA was confirmed by entering the STR DNA profile of each tested culture into the Cellosaurus database (Table S1). Authentication is defined as having a percentage match with the reference STR profile at when using the American National Standards Institute/American Type Culture Collection guidelines (ASN-0002-2011).
RNA Isolation and Complementary DNA Generation
Total RNA was purified using the mirVana RNA Isolation Kit (ThermoFisher Scientific Inc.), as recommended by the manufacturer. Briefly, cells were lysed, and organic extraction was performed using phenol-chloroform. After column washes, the RNA was eluted in of preheated (95°C) elution buffer and stored at . The isolated RNA was checked for integrity using the Agilent Bioanalyzer 2100 system and quantified using a Qubit fluorometric assay (Thermo Fisher Scientific), following the manufacturer’s instructions. All samples used had an RNA integrity number of . Complementary DNA was generated from total RNA using the SuperScript IV First Strand Synthesis System (Thermo Fisher Scientific Inc.), as per the manufacturer’s instructions. Briefly, of total RNA was reverse transcribed into cDNA using primers, and the input RNA was subsequently degraded by incubation with E. coli RNaseH (supplied with the SuperScript IV First Strand Synthesis System kit) at 37°C for 20 min, as per the manufacturer’s instruction.
RNA-Sequencing and Alternative Splicing Analysis
mRNA library preparation, cluster generation, and sequencing were performed at the University of Louisville Center for Genetics and Molecular Medicine DNA Facility Core, as described previously (Banerjee et al. 2021). The libraries were prepared from total RNA, using the Illumina Truseq Stranded mRNA kit, according to the manufacturer’s protocol. Absence of adapter dimers and consistent library size ( for mRNA) was confirmed using the Agilent Bioanalyzer 2100. The library concentration and sequencing behavior was assessed in relation to a standardized spike-in of PhiX Control v3 Library using a Nano MiSeq sequencing flow cell (Illumina). All pooled samples were run simultaneously on the Illumina NextSeq500 instrument on four flow cells, using paired-end sequencing with the sequencing kit 500 high-output (version 2; 300 cycles). The RNA-sequencing (RNA-Seq) data are available at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database under accession no. GSE107054. Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was used in the first step for both read quality check and adapter removal, and the reads were mapped to the human genome (i.e., hg38) using the Spliced Transcripts Alignment to a Reference aligner (Dobin et al. 2013). Subsequently, replicate multivariate analysis of transcript splicing (rMATS) (Shen et al. 2014) was used to identify differential alternative splicing events corresponding to all five major types of alternative splicing patterns (Figure 1) [i.e., skipped exon (SE), mutually exclusive exons (MXE), alternative 3′ splice site (A3′SS), alternative 5′ splice site (A5′SS), and retained intron (RI)], between arsenic-exposed and unexposed samples. For each alternative splicing event, both the reads mapped to the splice junctions and the reads mapped to the exon body were used as the input for rMATS. Each comparison was made to identify differentially spliced events with an associated change in exon usage () of these events. To compute -values and false discovery rates (FDRs) of splicing events with cutoff, rMATS was run using the -c 0.0001 parameter. Differential alternative splicing events were detected with an FDR of and . The difference in the proportion of the two isoforms of the transcript was expressed as the change in mean percentage spliced-inform included (mean ) (Shen et al. 2014).
Figure 1.

Most common types of alternative splicing. Exons are shown in rectangles, and introns as lines. The products of the splicing process are shown on the right. Black and dotted lines indicate differential splicing outcomes. Adapted with permission from Dlamini et al. (2017). Note: A3′SS, alternative 3′ splice site; A5′SS, alternative 5′ splice site; MXE, mutually exclusive exon; RI, retained intron; SE, skipped exon.
Determination of Differentially Expressed Alternative Splicing Events in Open Reading Frames and Untranslated Regions
To determine the alternative splicing events that could potentially have an effect on the polypeptide sequence, we used the Galaxy web platform (https://genome.ucsc.edu). Alternative splicing events and untranslated region (UTR) intervals were uploaded into the platform, and the public server at usegalaxy.org was used to remove intervals based on overlaps between the two files. The remaining alternative splicing events were categorized as occurring within the open reading frame (ORF).
Shortlisting of Differentially Expressed SE Events at the 7-Wk Time Point
To identify predicted differential alternative splicing events for Reverse Transcriptase (RT)-PCR validation, we focused on SE events from the 7-wk arsenic-exposed data set. Differential SE events were further shortlisted on the basis of , FDR , and at least 10 fragments per kilobase of transcript per million fragments mapped reads for both the included and skipped isoforms combined in at least one of the control or arsenic-exposed samples cumulatively. The shortlisted events were further filtered based on the cancer relevance of the host gene to identify three predicted differential alternative splicing events for validation of RNA-Seq data through RT-PCR.
RT-PCR and Densitometric Analyses for Selected Alternative Splicing Events
To co-amplify both the predicted isoforms of each shortlisted gene, primers were designed on the flanking exons, as described previously (Cho et al. 2015; Leparc and Mitra 2007) using Primer3 (version 0.4.0) (Untergasser et al. 2012) and synthesized by Integrated DNA Technologies. Primer sequences are detailed in Table S2. The primers so designed were capable of co-amplifying both the spliced-in as well as the spliced-out isoforms in each case in a single amplification reaction. Information about the predicted isoforms, and mean values are provided in Table S3. Amplification reactions were carried out using of cDNA from each sample (4 each exposed to 0 or ). All PCR amplifications were carried out on a Veriti 96-Well Thermal Cycler (Applied Biosystems). The cycling conditions for each amplification reaction are provided in Table S2. The amplification products were resolved by electrophoresis in 6% polyacrylamide gels (Acrylamide/Bis Solution, 19:1; BIO-RAD), stained with RedSafe dye (Bulldog Bio). Image acquisition was done using FOTO/Analyst FX (Thermo Fisher Scientific Inc.) and quantified using Image J software (Schneider et al. 2012). Raw data from densitometric analyses were divided by the isoform size to determine the number of events for skipped/included isoforms. The value of for each sample was calculated according to the following formula:
Cloning and Sequencing of the Isoforms
To confirm that the observed RT-PCR bands were specific to the genes E26 transformation-specific Transcription Factor ELK4 (ELK4), X-ray Radiation Resistance Associated 1 (XRRA1), and Sarcoma Homology 2 Domain Containing (SHC) Adaptor Protein 1 (SHC1), respectively, and the isoforms of interest, cloning and sequencing were performed. RT-PCR bands corresponding to predicted included (ELK4 and XRRA1), SE isoforms (ELK4, SHC1, and XRRA1), and one other abundant band (182 bp band for ELK4 and 304 bp band for SHC1) were gel eluted and purified (Figure S1) employing NucleoSpin Gel and PCR Clean-Up kit (TaKaRa Bio USA, Inc.), following the manufacturer’s protocol. A two-step nested PCR was performed for downstream cloning of the predicted included SHC1 isoform (352 bp) given its low abundance. The first round of amplification was performed using the SHC1 primers listed in Table S2, which produced a 352-bp band that was subsequently used as the template for the second round of amplification using internal primers [forward: AGACTCCATGAGGCCCTGACGGAGT (3′–5′), and reverse: TTGACTGGAGGACCTCCACACAACCCATGTACT (3′–5′)] corresponding to exon junctions in the predicted SHC1-included isoform to generate a 265-bp fragment. The cycling conditions for the first round of PCR reaction are detailed in Table S2. The two-step PCR conditions for the second round comprised of 95°C for 5 min, followed by 30 cycles of 95°C for 15 s and 68°C for 30 s, plus a final extension at 72°C for 5 min.
RT-PCR products were purified and cloned using either the PCR Cloning Kit (NEB) or the TA Cloning Kit (Thermo Fisher Scientific Inc.) and transformed into competent E. coli DH10B or strains, respectively, following the manufacturer’s instructions. Plasmid DNA was isolated from transformed colonies using Miniprep kit (Qiagen), and each sample was subjected to PCR amplification using NEB- and Thermo Fisher–provided primers (for the PCR and TA cloning kits, respectively) against the flanking sequences in the vector backbone (primer sequences for the cloning kits and cycling conditions are listed in Table S2). Following confirmation of clone insertion into the plasmid by PCR, two clones of each PCR product were subjected to Sanger sequencing using the same manufacturer-provided sequencing primers and the BigDye Terminator (version 3.1) Cycle Sequencing Kit (Thermo Fisher Scientific Inc.) and on an ABI 3130xl Genetic Analyzer, equipped with a capillary array. Sequence analysis was performed using the Chromas software (version 2.6.5) (Technelysium Pty Ltd.). The sequences were searched via Nucleotide BLAST against the Homo sapiens (Taxid: 9606) genome on the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome).
Immunoblotting
Cells were lysed with a cell lysis buffer containing Tris-HCl, pH 7.4; EDTA; 0.1% sodium dodecyl sulfate (SDS); phenylmethylsulfonyl fluoride (PMSF); sodium vanadate; and protease inhibitor cocktail (Thermo Fisher Scientific). After sonication, cell lysates were centrifuged at at 4°C for 15 min. Protein concentrations were determined using the Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Proteins were separated by electrophoresis in SDS-polyacrylamide (Acrylamide/Bis Solution, 29:1; BIO-RAD) gels of an appropriate percentage (Table S4). They were then electroblotted onto polyvinylidene fluoride membranes (Thermo Fisher Scientific). To verify equal loading and efficient transfer, membranes were stained with Coomassie Brilliant Blue R250 (Thermo Fisher Scientific) (Goldman et al. 2016). Membranes were then blocked in 5% milk in Tris buffer saline plus Tween 20 (TBST: Tris-HCl, pH 7.4; NaCl; 0.1% Tween 20) at room temperature for 1 h. Blots were incubated with the requisite antibodies overnight at 4°C. A list of the antibodies used in the study and the dilutions used thereof are detailed in Table S4. Blots were incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibody, as detailed in Table S4 (in 1% skim milk in TBST) and incubated with an enhanced chemiluminescent substrate (Thermo Fisher Scientific) or with the SuperSignal West Atto Ultimate Sensitivity substrate (Thermo Fisher Scientific) afterward. Images were acquired using FOTO/Analyst FX. Densitometric analyses were done using ImageJ software. If necessary, blots were stripped by incubation at 37°C for 30 min at with Restore PLUS western blot stripping buffer (Thermo Fisher Scientific Inc.).
GO Analysis for Alternatively Spliced Genes
Functional enrichment was performed by GO analysis of the differential alternative splicing event data set at each time point (7, 19, and 28 wk) using the web-based g:Profiler tool (https://biit.cs.ut.ee/gprofiler/) (Raudvere et al. 2019). For the analysis, we selected GO terms biological process (GO:BP), molecular function (GO:MF), biological pathways Kyoto Encyclopedia of Genes and Genomes (GO:KEGG), and REACTOME (GO:REAC). The significant threshold was calculated employing the set counts and sizes threshold method for multiple testing because it has been demonstrated to provide better accuracy compared with other commonly used multiple testing methods (Reimand et al. 2007). This approach helped us to surpass well-known issues with the assumption of independence for multiple testing with the other methods that are violated during GO analysis (Reimand et al. 2007). The analysis provided output in terms of enriched functional pathways categorized by GO terms (GO:BP, GO:MF, GO:KEGG, and GO:REAC) along with a list of genes from our input data set belonging to each pathway, as well as -values adjusted for multiple testing. The adjusted -values for each of the enriched pathways was further transformed into a positive number [] to better visualize the data.
Proteomic Analysis
Proteomic experiments were conducted as previously described (Zheng et al. 2020). Briefly, tandem-mass tagging (TMT) approaches were used to determine relative changes in protein abundance of canonical isoform of selected splicing-related genes (Figure S3, Excel Table S2) as a function of arsenic exposure. Cell lysates used for immunoblot studies were also used for proteomic studies. Individual lysate samples ( protein) in were mixed with an equal volume of triethylammonium bicarbonate, pH 7.55, and was taken for digestion with an S-Trap Micro Spin column (Protifi, LLC), which alleviated the need for concentration/desalting on the C18 column. dithiothreitol was used to adjust the sample to a final concentration of and then heated to 95°C for 10 min. Samples were cooled to room temperature, adjusted to iodoacetamide (IAA) using IAA in liquid chromatography–mass spectrometry (LC-MS)–grade water, and incubated at room temperature for 30 min in the dark. MS-grade trypsin (Pierce) was added to a 1:25 ratio with the sample (:) and incubated per the S-trap protocol at 47°C for 1 h. Tryptic digests were resuspended in LC-MS–grade water and the absorbance at of a aliquot was measured on a Nanodrop 2000 (blanked to LC-MS–grade water). Two readings were taken on each sample aliquot, and the average value was used to compute peptide concentration based on the extinction coefficient provided by the Nanodrop software, Version 1.6 (Thermo Fisher). Aliquots () were derivatized with individual aliquots of TMT 10plex (Thermo Fisher) reagents and subsequently admixed prior to the first dimensional separation using high-pH reversed-phase chromatography. First dimensional peptide separations were conducted on a U3000SD ultra-high-performance liquid chromatography (uHPLC) system (ThermoFisher Scientific) including an LPG-3400SD pump, WPS-3000SL Analytical Autosampler, TCC-3000SD Column Thermostat, VWD-3100 Detector with an stainless steel analytical flow cell, and an AFC-3000 fraction collector with a PEEK low-flow drop former. All separations were achieved using a Waters VanGuard cartridge and BEH XBridge C18 column with a freshly prepared binary gradient of solvent system including buffer A [] and buffer B () using LC-MS–grade water (ThermoFisher; #W6-4), (ThermoFisher; #A955-4), and HPLC-grade (Fluka; #17843). The flow rate throughout the separation was , and the total program time was 95 min. The columns were equilibrated for 5 min with 100% A. The concentrated and desalted samples () were dissolved in 5% , pH 10, loaded onto the Waters BEH column, and eluted using a step gradient profile as follows: 0% B from 0 to 1 min, 0% to 10% B over 4 min, 10% to 40% B over 60 min, 40% to 98% B over 10 min, 98% B for 5 min, 98% to 0% B over 0.1 min, and 0% B for 9.9 min. Sample elution was followed by absorbance at . Fractions were collected in Eppendorf tubes. Fraction 1 included the 5-min load volume. Fractions 2–19 were collected in 1.25-min intervals such that tube 2 contained fractions 2, 20, and 38 (each concatenated fraction equals the admixture of , , and fractions). Fraction 20 collected the last 6.25 min of eluate ( of eluate). Sample tubes were transferred to a Thermo Electron speed-vacuum for drying. All dried samples were stored at until one dimension liquid chromatography-MS analysis was performed. The dried samples were resuspended in 2% /0.1% formic acid, and LC-MS data were collected in a top-10 data-dependent analysis using the Orbitrap Elite-ETD system with a mass resolution of 60,000 (for first dimension mass spectrometry (MS1) and MS2). Differential protein expression for the canonical isoforms of the genes of interest was estimated using ProteomeDiscoverer 2.2.0.388 using MASCOT Server (version 2.5.1) (Matrix Science) and SequestHT (ThermoFisher Scientific Inc.).
Statistical Analyses
For longitudinal comparisons (event distribution/location of events in a gene), homogeneity analyses were carried out. Densitometric data for RT-PCR were analyzed by one-tailed Mann-Whitney test, whereas those for immunoblots were analyzed using two-tailed -tests. was considered significant for all tests. All statistical analyses were performed using GraphPad Prism (version 9.0.1; GraphPad). All the graphs were generated with GraphPad Prism, whereas the Venn diagram was generated using Venn diagram plotter (https://omics.pnl.gov/software/venn-diagram-plotter). Pie charts were generated using Microsoft-Excel.
Results
Approach
In this study, we first determined the differential alternative splicing events in HaCaT cells exposed to arsenite for 7, 19, and 28 wk compared to passage-matched unexposed controls. Next, we validated some of the alternative splicing predictions employing RT-PCR. Finally, we performed GO analysis to gain insight into the biological pathways that might be modulated by arsenic-induced differential splicing events.
RNA-Seq and rMATS Analysis of HaCaT Cells Exposed to for up to 28 Wk
RNA-Seq followed by rMATS analyses demonstrated that several hundred differential alternative splicing events occurred at each of the three time points tested (Table 1). Interestingly, even the time point that had the lowest number of differential AS events had 626 events (i.e., the 19-wk time point), whereas the 7-wk time point had the most differential alternative splicing events (1,779). These differential events encompassed 1,267, 506, and 726 genes at the 7-, 19-, and 28-wk time points, respectively (Table 1). Further, at each time point there was a fairly constant proportion of genes with multiple differential alternative splicing events (25%, 24%, and 28% at the 7-, 19-, and 28-wk time points, respectively; Table 1). Most of the genes were represented uniquely at each time point, but there was some overlap (not necessarily the same events), as shown in Figure 2A.
Table 1.
Differential alternative splicing events upon chronic arsenic exposure at different time points.
| Event type | 7 wk | 19 wk | 28 wk |
|---|---|---|---|
| Total events () | 1,779 | 626 | 927 |
| Total unique genes [ (%)] | 1,267 (100) | 506 (100) | 726 (100) |
| Genes with multiple events [ (%)] | 312 (24.6) | 120 (23.7) | 201 (27.7) |
| Genes with a single event [ (%)] | 955 (75.4) | 386 (76.3) | 525 (72.3) |
Figure 2.

Evaluation of differential alternative splicing events in HaCaT cells chronically exposed to sodium arsenite. (A) Venn diagram depicting the number of unique genes with differential alternative splicing event(s) at each time point and their overlap at different time points. The overlap in genes with differential splicing event does not necessarily mean the splicing event occurring is also the same. (B) Relative proportion of different alternative splicing subtypes [i.e., skipped exon (SE), mutually exclusive exons (MXE), alternative 3′ splice site (A3′SS), alternative 5′ splice site (A5′SS), and retained intron (RI)] at each time point. The number of differential alternative splicing events stratified by subtypes at different time points are detailed in Table S5. (C) Relative proportion of differential alternative splicing events occurring in the open reading frame (ORF) or the untranslated region (UTR) at each time point. The number and percentage of differential alternative splicing events in the ORFs and UTRs at different time points are detailed in Table 2. (D) Relative proportion of genes with differential alternative splicing events in the ORF only, UTR only, or both at each time point tested. The number and percentage of differential alternative splicing events in the ORF and UTRs at different time points are detailed in Table S6.
Alternative splicing pattern analysis by rMATS showed that chronic arsenic exposure induced all five different types tested (SE, MXE, A3′SS, A5′SS, and RI) at each time point (Figure 2B; Table S5) in HaCaT cells. Importantly, the relative proportions of each subtype were variable across the different time points. There was an inverse relationship between the proportions of SE and MXE events (Figure 2B; Table S5). SE was the most common alternative splicing type at 7 wk; however, by 28 wk, MXE was the predominant type (Figure 2B). Homogeneity analysis demonstrated that the alternative splicing event distribution patterns were significantly different at each time point ().
Categorization of Each Splicing Event Using the Galaxy Platform
We wanted to determine if the predicted differential alternative splicing events were likely to give rise to different protein isoforms or had a potential regulatory impact. Intersection of our differential alternative splicing data set with the Galaxy data set allowed us to categorize each event based on whether they occurred in the ORF or the UTR of a gene. Apart from A3′SS and A5′SS at the 7-wk time point, events were distributed between the ORF and the UTRs (Table 2). Analysis of the event localization data showed that the relative distribution of events in the ORF and the UTR at the three time points were significantly different (homogeneity ; ; Figure 2C, Table 2). Although most of the genes had alternative splicing events either only in the UTR or only in the ORF, there was a small fraction of genes at each time point with events in both UTRs and ORFs (Figure 2D; Table S6). In addition, there was a clear trend of decrease in the percentage of genes with events in both UTRs and ORFs at 19 and 28 wk compared with 7 wk (Figure 2D). Homogeneity analyses demonstrated that the alternative splicing event localization patterns were significantly different at each time point ().
Table 2.
Differential alternative splicing events [ (%)] in the ORF and UTRs at different time points.
| Category | 7 wk | 19 wk | 28 wk | |||
|---|---|---|---|---|---|---|
| ORF | UTR | ORF | UTR | ORF | UTR | |
| SE | 707 (61.5) | 442 (38.5) | 168 (54.0) | 143 (46.0) | 185 (53.6) | 160 (46.7) |
| MXE | 79 (58.1) | 57 (41.9) | 135 (67.8) | 64 (32.2) | 264 (69.8) | 114 (30.2) |
| A5′SS | 99 (100.0) | 0 (0.0) | 20 (46.5) | 23 (53.5) | 19 (44.2) | 24 (55.8) |
| A3′SS | 149 (100.0) | 0 (0.0) | 18 (66.7) | 9 (33.3) | 41 (73.2) | 15 (26.8) |
| RI | 132 (53.7) | 114 (46.3) | 24 (52.2) | 22 (47.8) | 45 (42.9) | 60 (57.1) |
| Total | 1,166 (65.5) | 613 (34.5) | 365 (58.3) | 261 (41.7) | 554 (59.8) | 373 (40.2) |
Note: The event numbers and percentages presented in this table correspond to Figure 2C. A3′SS, alternative 3′ splice site; A5′SS, alternative 5′ splice site; MXE, mutually exclusive exon; ORF, open reading frame; RI, retained intron; SE, skipped exon; UTR, untranslated region.
Selection of Predicted SE Events at the 7-Wk Time Point for RT-PCR Validation
We wanted to validate some of the bioinformatically predicted differential alternative splicing events by RT-PCR to demonstrate the reliability of the rMATS predictions. We decided to concentrate on the differential SE events at the 7-wk time point, the most numerous events across all alternative splicing classes and time points (Table 1). To shortlist the SE events with the maximum probability of detection, we filtered the data set based on criteria outlined in the “Shortlisting of Differentially Expressed SE Events at the 7-Wk Time Point” subsection of the “Methods” section. Of the possible 1,149 differential SE events, 42 met the filtering criteria. Isoforms in selected SE events were identified by comparison of the included and SEs with the Ensembl database release 82 (Howe et al. 2021). Interestingly, several of these alternative splicing events did not correspond to any known isoform annotated in the Ensembl database. Of the 42 shortlisted SE events, both the included and SE isoforms were annotated for only 7 (16.7%) of these predicted alternative splicing events, whereas 19 (45.2%) had 1 annotated isoform, and 16 (38.1%) had none. We wanted to explore whether we could validate each of these scenarios with RT-PCR and chose three SE events in the genes XRRA1, SHC1, and ELK4. XRRA1 had both included/skipped isoforms annotated (XRRA1-202/201; Ensembl database), and SHC1 had only the skipped isoform annotated (Isoform X12; NCBI Reference Sequence: XM_011509898.2), whereas none of the isoforms were annotated for ELK4 (Table S3).
Validation of Selected Alternative Splicing Predictions Using RT-PCR
Our RT-PCR data demonstrated that for each of these three predicted alternative splicing events, we were able to co-amplify the included/skipped isoforms (Figure 3). Statistical analysis of the densitometric data demonstrated that value was higher for XRRA1, but lower for both SHC1 and ELK4, in 7-wk arsenic-treated HaCaT cells compared with control cells, as predicted by rMATS analysis of the RNA-Seq data (Figure 3A; Table S7).
Figure 3.
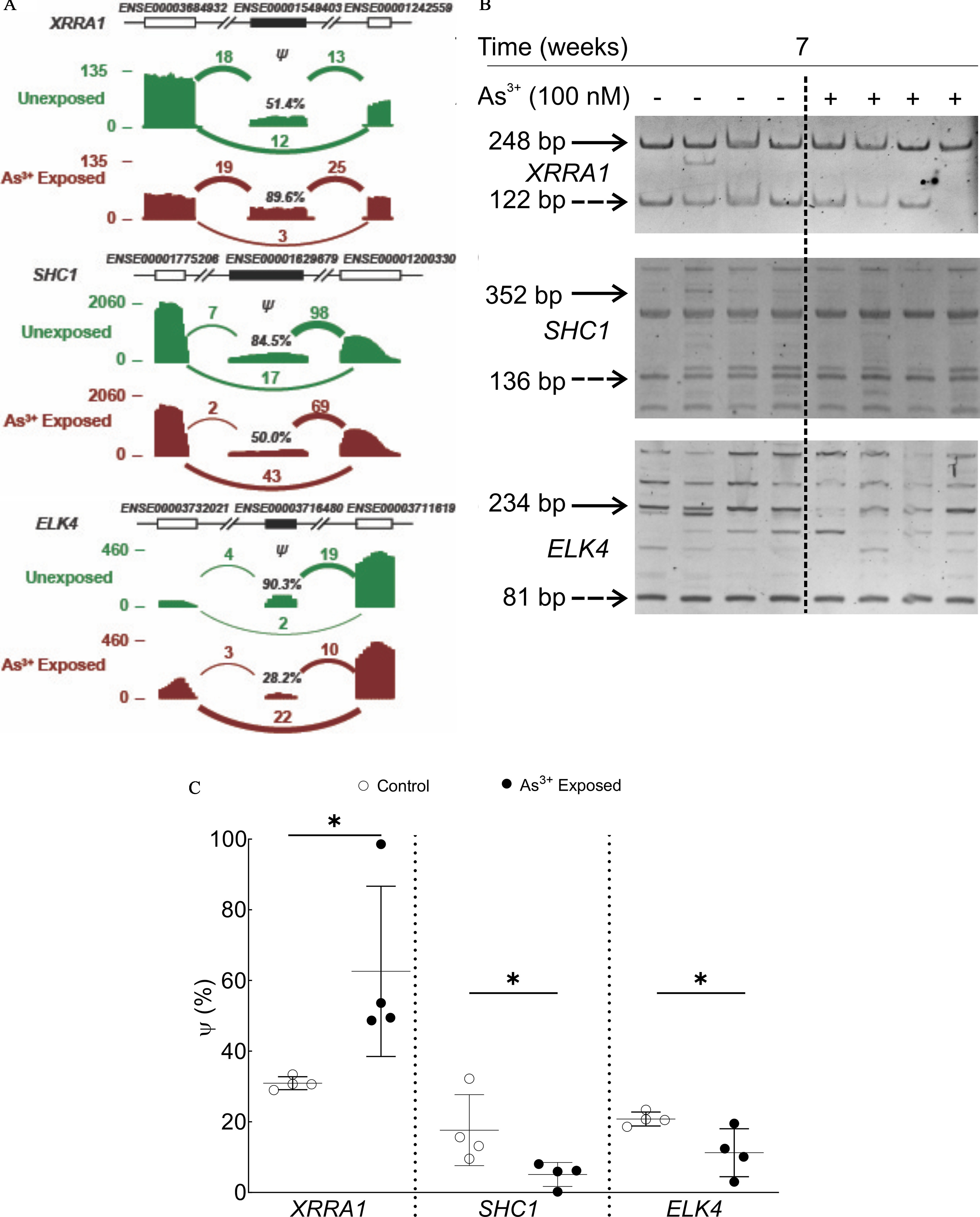
Validation of the differential splicing predictions by RT-PCR. (A) Read density plots indicating the RNA-Seq read counts and estimated exon inclusion levels for XRRA1, SHC1, and ELK4 in arsenic-exposed (red, lower in each pair) and unexposed (green, upper in each pair) HaCaT cells at 7 wk. Arcs represent splice junctions with the number of reads mapped to the junction indicated by the thickness of the arc. (B) RT-PCR analysis of RNA-Seq predicted isoform expression in control and 7-wk arsenic-exposed HaCaT cells for XRRA1, SHC1, and ELK4. The solid arrows indicate the predicted included isoform, whereas the dashed arrows indicate the skipped isoform for each event. (C) Densitometric analysis of XRRA1, SHC1, and ELK4 RT-PCR. Raw data from densitometric analyses were divided by the isoform size in each case to determine the number of events for skipped/included isoforms. The value of for each sample was calculated according to the following formula: . All predicted isoforms identified by RNA-Seq and validated by RT-PCR were cloned and sequenced to confirm identity. *, by one-tailed Mann-Whitney test. The numerical values for the densitometric analysis () for each differential splicing event are presented in Table S7. Note: RT-PCR, real-time polymerase chain reaction; RNA-Seq, RNA-sequencing; SD, standard deviation.
Sequencing of Predicted and Other Bands from RT-PCR Experiments
Given that we detected more bands in our RT-PCR experiments than predicted by the RNA-Seq and rMATS analysis for SHC1 and ELK4, we sequenced the cDNA fragments in some of these bands to ensure they were other isoforms of the same genes and not an outcome of nonspecific primer binding and amplification elsewhere in the genome. The included/skipped bands for each gene along with at least one more major isoform (for ELK4 and SHC1) were cloned and sequenced. Sequencing data corroborated the rMATS prediction for the included/skipped isoform pair for all three genes (Table S8). All the additional bands sequenced were found to be flanked by the predicted flanking exons as predicted by rMATS analysis. Novel SHC1 and ELK4 isoforms (93 bp and 182 bp, respectively) were discovered. For SHC1, the new isoform contained an A5′SS on exon 1 and a novel exon covering the intron sequence (GenBank accession no. MZ161200), whereas the ELK4 novel isoform contained exons 5, 8, and 9 (GenBank accession no. MZ161201). The details of all the annotated isoforms identified and those sequenced are provided in Figure S1.
Immunoblots for Isoforms of the Shortlisted Genes
Once we had validated the rMATS predictions by RT-PCR, we sought to validate the RNA predictions at the protein level. Furthermore, we also wanted to determine whether we could detect any quantitative changes in the canonical protein isoforms for the other genes for which one or both the predicted isoforms were unannotated. We detected the three major annotated isoforms of SHC1 at 66, 52, and 46 kDa, and there was no difference in the expression of any of these isoforms upon 7-wk arsenic exposure (Figure S2A,B,E). However, none of these isoforms corresponded to the predicted alternatively spliced isoforms, and we did not see any other bands in the blot suggestive of other isoforms (at least that could be detected with this antibody). We did not detect any difference in the levels of the ELK4 canonical isoform at 45 kDa, and no other band indicative of the predicted novel isoforms was observed (Figure S2C–E). For XRRA1, only two antibodies are commercially available. One of the antibodies (ab102681; immunogen: aa 1-50) was supposed to recognize the canonical isoform (XRRA1-202; 89.864 kDa), whereas the other one (ab235784; immunogen: aa 1-400) was expected to recognize both the isoforms. Unfortunately, both the antibodies showed multiple bands between 50 and 250 kDa both with our HaCaT cell lysates as well as the manufacturer-suggested HepG2-positive control lysates (Figure S2F). Consequently, we could not distinguish whether these bands represented possible splice isoforms or were random nonspecific bands.
Analysis of Enriched GO Terms of Alternatively Spliced Genes from HaCaT Cells Exposed to for up to 28 Wk
GO analysis identified 95, 2, and 21 functional enrichment terms at the 7-, 19-, and 28-wk time points, respectively (Figure 4A–C; Excel Table S1). Longitudinal comparison of the enrichment terms demonstrated that the enriched terms were mostly unique at each time point, with relatively few overlaps (Figure 4D). One enrichment term (protein binding) was common at all three time points.
Figure 4.
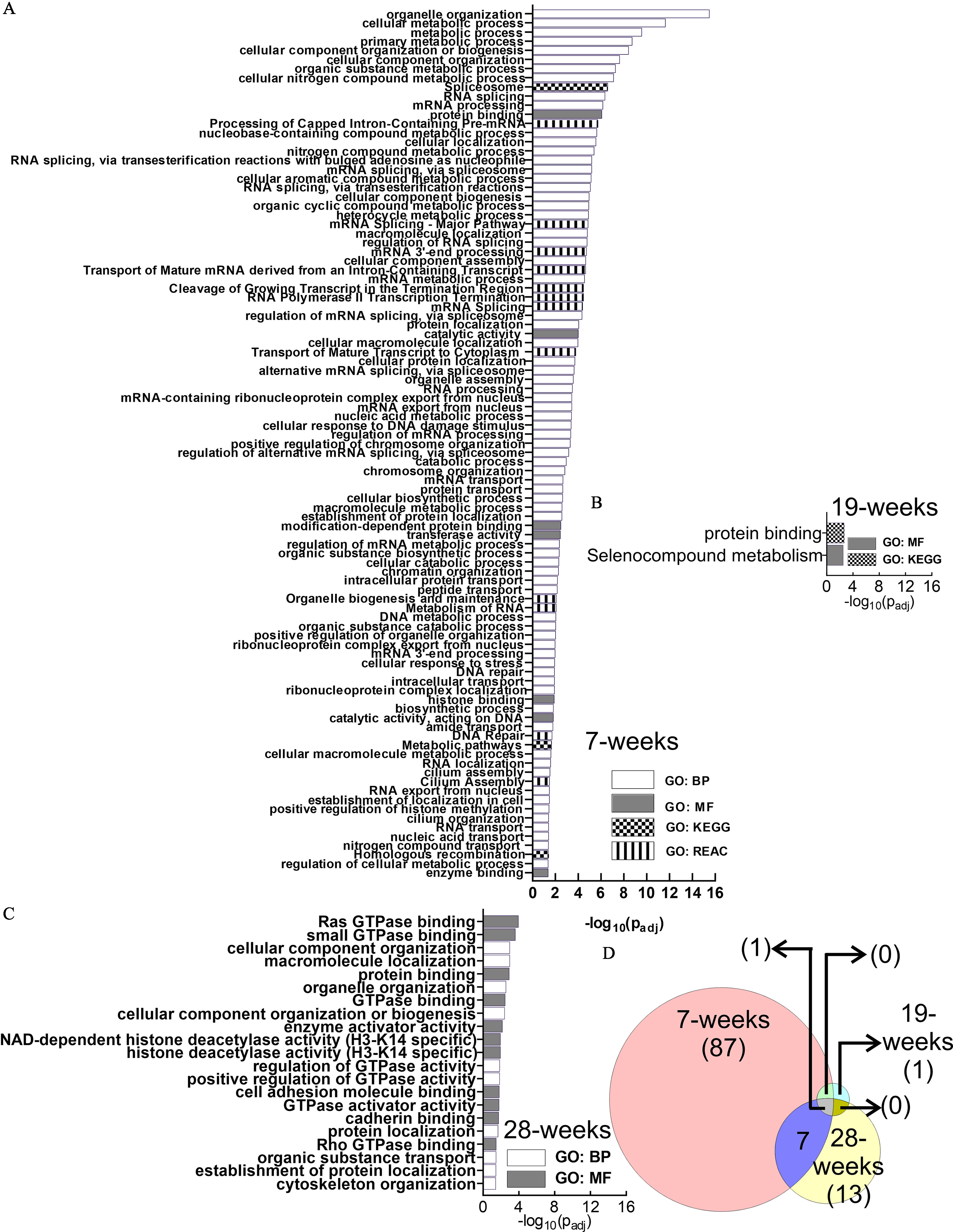
GO Functional enrichment analysis of the differential alternative splicing events in HaCaT cells induced by arsenic exposure. (A) 7 wk, (B) 19 wk, and (C) 28 wk. GO terms depicted in (A–C) are presented in the same order as in Excel Table S1 [in ascending order of ]. (D) Venn diagram describing the number of enriched pathways at each time point and their overlap at different time points. Note: BP, biological process; GO, gene ontology; GTPase, guanosine triphosphatase; KEGG, Kyoto Encyclopedia of Genes and Genomes; MF, molecular function; REAC, Reactome.
RNA Metabolism and Splicing Regulation Pathways in HaCaT Cells Exposed to for 7 Wk
Examination of the 7-wk enriched pathways demonstrated that several of them were related to RNA metabolism (Figure 5A), including splicing (Figure 5B). In fact, almost one-third of the enriched terms (31/95; 32.6%) were related to RNA metabolism, whereas about one-third of all RNA metabolism-related enriched GO terms (11/31; 35.5%) belonged to splicing only (Figure 5C). Thus, splicing and splicing regulatory processes contributed to of all predicted enriched pathways at the 7-wk time point.
Figure 5.

Examination of the 7-wk enriched pathways. (A) Enriched RNA metabolism-related pathways. (B) Splicing-related pathways are included among the RNA metabolism pathways. (C) Fractions of enriched terms related to RNA metabolism and splicing: 32.6% of the enriched terms are related to RNA metabolism, whereas 35.5% of all RNA metabolism-related terms belong to splicing.
Splicing–Proteome Relationship at the 7-Wk Time Point
To investigate the relationship between differential splicing prediction and protein expression, proteomic analysis was performed on the 7-wk data sets. We specifically concentrated on the genes and events that were represented in the enriched splicing GO terms (Figure 5B). We further applied multiple filters on the 62 genes (and differential events thereof) that were uniquely represented in one or more of these GO terms to provide us with optimal opportunities for proteomic detection and validation (Figure S3A). We chose genes that were represented in at least 6 of the 11 enriched GO terms (Figure S3B), had differential SE or RI events, and for which both transcript isoforms were annotated in the Ensembl database. Furthermore, we shortlisted events that had at least five counts for included and skipped isoforms in each sample by rMATS to have a probabilistic chance of being detectable at the protein level. Finally, we decided to focus only on those events for which one isoform was likely to give rise to the canonical protein isoform for the gene, whereas the other would undergo nonsense-mediated decay and would not be translated to a protein isoform (Figure S3A). Thus, our strategy was to investigate the possible changes in the expression of the canonical isoform for each of these selected genes (17 genes, corresponding to 27 differential splicing events; Excel Table S2) upon chronic arsenic exposure for 7 wk.
The expected transcript and protein isoforms, as well as the expected and experimental outcomes for the protein expression data, are presented in Excel Table S2. Of the 17 shortlisted genes (corresponding to 27 events), expression of two genes could not be detected (U2AF1L4; Uniprot ID: Q8WU68-1 and DDX5; Uniprot ID: P17844-1). Protein expression patterns for three genes (corresponding to four events) corroborated the rMATS predictions. Chronic arsenic exposure induced the protein expression for canonical isoforms of DDX42 (Uniprot ID: Q86XP3-1; encoded by Transcript: DDX42-202), RBM25 (Uniprot ID: P49756-1; encoded by Transcript: RBM25-201), and SRRM2 (Uniprot ID: Q9UQ35-1; encoded by Transcript: SRRM2-201) as predicted based on rMATS derived mean values (Excel Table S2).
Discussion
Mechanisms of arsenic exposure-induced carcinogenesis, especially skin carcinogenesis, have been widely studied. Several mechanisms have been hypothesized with differing levels of experimental support as well as controversy (Hunt et al. 2014). Arsenite did not induce point mutations in a Chinese hamster ovary cell line (Li and Rossman 1989), but it is widely accepted to be clastogenic in nature (Roy Burman et al. 2021). Considerable evidence also exists for other molecular mechanisms, including, for example, inhibition of DNA damage repair, modulation of cellular signaling cascades, and interaction with protein sulfhydryls, which have been previously covered in excellent reviews (Hunt et al. 2014; Muenyi et al. 2015; Rossman 2003; States 2015; Tam et al. 2020; Tam and Wang 2020).
New research has suggested that the transcriptome, epitranscriptome, and proteome are modulated in arsenic-induced carcinogenesis (Eckstein et al. 2017a; Riedmann et al. 2015; Saintilnord and Fondufe-Mittendorf 2021). Such changes were often associated with carcinogenesis-related processes (Martinez et al. 2011; Minatel et al. 2018; Mir et al. 2017). Although mechanisms including dysregulation of micro RNA (miRNA) expression, altered global DNA methylation, and histone modification patterns have been postulated to be partially responsible (Cardoso et al. 2018; Seow et al. 2014), significant knowledge gaps still exist (Cardoso et al. 2018; States et al. 2011). The present work is the first study to our knowledge to explore the role of genome-wide differential alternative splicing in a model of chronic arsenic exposure-induced skin carcinogenesis. The present work provides evidence of global differential alternative splicing events as a potent force in nuanced regulation of gene expression during the process of chronic arsenic exposure-induced cSCC.
Our data clearly demonstrate the scope and extent of genome-wide differential alternative splicing events induced by chronic exposure to environmentally relevant level of arsenic across different time points in immortalized human keratinocytes (HaCaT). At the 19-wk time point, arsenic-exposed HaCaT cells had the smallest number of differential alternative splicing events (626), which was still times more than that reported in arsenic-transformed BEAS-2B cells (Riedmann et al. 2015). This quantitative difference could reflect a combination of factors, including differences in cell line (skin vs. lung), arsenic exposure ( vs. ), exposure time (up to 28 wk vs. 16 wk), or the experimental platform used (RNA-Seq vs. microarray hybridization).
Our study included in-depth coverage of the transcriptome ( read depth), which enabled a thorough examination of the differential alternative splicing events at multiple time points that represented the process of cancer development. This approach contrasts with most other studies that examined either cancer tissues (Sanyal et al. 2020) or transformed cell lines (Eckstein et al. 2017b) when malignancy had already developed. Our unique longitudinal approach corresponds to the three different stages of transformation, evidenced by changes in the growth curve pattern and epithelial-to-mesenchymal transition: 7 wk (early transformation changes), 19 wk (beginning of transformation), and 28 wk (transformed cells) (Banerjee et al. 2021). This approach overcame the challenge of delineating which changes could potentially contribute to carcinogenesis and which were outcomes of transformation. We demonstrated that arsenic-induced differential splicing events in HaCaT cells changed in a temporally dynamic manner, as exemplified by very few overlaps in genes harboring these events (Figure 2A) and overrepresented pathways (Figure 4D) across multiple time points, in agreement with similar observations at the miRNA- and mRNA-expression levels, as recently reported (Banerjee et al. 2021). Furthermore, the use of four independent biological quadruplicates for each experimental condition at each time point, as opposed to technical replicates, made our data set less vulnerable to predicting possible stochastic events as false positives or negatives. We acknowledge that stochastic events often set cells into a cancerous trajectory followed by clonal selection for cells harboring such events (Gagliardi et al. 2020; Li et al. 2019; Marongiu and Laconi 2020). However, drawing empirical inference about causal stochastic events requires single-cell RNA-Seq analysis (Marinov et al. 2014), which was beyond the scope of the present work.
Both the localization and the nature of splicing events in our data set varied with time (Figure 2C,D, Table 2). The proportion of MXE events increased over time, whereas that of SE events decreased consistently between 7 and 28 wk (Figure 2B; Table S5). Reports in the literature have suggested that MXE events translate into protein isoforms more often than other classes of splice variants (Floor and Doudna 2016; Hatje et al. 2017; Weatheritt et al. 2016) and result in disease-associated mutations (Hatje et al. 2017; Huang et al. 2018). Thus, a higher proportion of MXE at 28 wk could potentially lead to alternative protein isoforms and proteome diversity (Hatje et al. 2017), possibly contributing to carcinogenesis. Collectively, our data suggest that the global alternative splicing landscape in HaCaT cells changed in several different ways upon chronic arsenic exposure and was regulated temporally.
The use of next-generation sequencing technologies coupled with bioinformatic tools has revolutionized the way we study and understand genome function; however, the results still are predictive in nature and must be cross-validated by other techniques. We chose three differential splicing events in three genes based on their role in carcinogenesis (Terada 2019; Wang et al. 2017; Zhu et al. 2020). XRRA1 plays a role in repair response against radiation-induced DNA damage and has been implicated in the etiology of colorectal cancer (Wang et al. 2017); SHC1 is a central regulator of tyrosine kinase signaling essential to signal transduction resulting in cell proliferation, differentiation, migration, metabolism and programmed cell death (Ahn et al. 2017); and ELK4 is an important molecule in the regulation of c-fos transcriptional expression (Janknecht et al. 1995). c-Fos dysregulation had been implicated in cell proliferation, differentiation, and oncogenic progression (Velazquez et al. 2015). Our RT-PCR based assays corroborated three selected predicted SE events at the 7-wk time point and provided evidence of the reliability of the differential alternative splicing analyses. Interestingly, for two of the three events (ELK4 and SHC1), several additional PCR bands were found to be co-amplified along with the predicted included/skipped isoforms (Figure 3; Figure S1). When we compared the sizes of these other amplification products with the isoform information available in the Ensembl database, many of them corresponded with other annotated isoform(s) for these genes. This finding is not surprising given that other exon(s) are often present in between the flanking exons predicted by the rMATS for the predicted SE events. Consequently, it was possible to have several isoforms (besides the predicted included/skipped pair) with a wide variety of exon/intron combination if they were present between the predicted flanking exons. This feature also explained the occurrence of the amplification bands in our RT-PCR experiments that did not correspond to any annotated isoform but, rather, corresponded to isoforms that could arise from unique permutations of one or more additional exons located between the flanking exons. Given our experimental design that depended on primers designed to bind to the flanking exons, any combination of exons that lay in between these flanking exons could potentially be co-amplified along with the predicted included/skipped isoforms. The sequencing analysis of additional bands in ELK4 and SHC1 elucidated this point clearly.
Next, we investigated the possible functional or regulatory impacts of the differential splicing events. At each time point, the differential splicing events were distributed between ORFs and UTRs. The changes in the ORFs can lead to distinct protein isoforms but can also influence protein stability, degradation through nonsense-mediated decay, and protein localization by altering signal sequences and sites of posttranslational modifications (Mockenhaupt and Makeyev 2015). 5′UTRs contain important cis-regulatory elements and sequences with the topological structure essential for binding trans-regulatory elements to activate or repress translation of the ORF downstream (Araujo et al. 2012; Mockenhaupt and Makeyev 2015). 3′UTRs include miRNA seed sequences, polyadenylation sites, and mRNA localization signals (Mockenhaupt and Makeyev 2015). Recent data have suggested that alternative polyadenylation through differential 3′UTR splicing can induce isoform-specific mRNA degradation under conditions of acute arsenic exposure-induced cellular stress (Zheng et al. 2018).
Given that chronic arsenic exposure induced hundreds and even thousands of differential alternative splicing events, the question persisted as to their functional implications. Clearly, the effects of differential splicing were more nuanced and complicated than the simple increase or decrease in the levels of mRNA or protein isoforms. An increase in an exon usage (especially, the poison cassette exon) could lead to a decrease in protein levels by introducing a premature termination codon and target the transcript to nonsense-mediated decay (Hamid and Makeyev 2014; Lareau et al. 2007). Conversely, an increased skipping of such exons would lead to an increase in the translation of canonical isoform, instead of producing a distinct protein isoform (Lareau et al. 2007). Differential alternative splicing could change the relative proportion of isoforms without necessarily altering the total mRNA expressed. Furthermore, differential alternative splicing events could simply alter the localization of the same protein isoform without altering its steady-state amount (Ciolli Mattioli et al. 2019). Issues such as these made it difficult to draw a direct linear relationship between differential alternative splicing events with the final transcriptome/proteome expression.
Several other factors also need to be considered. First, rMATS produced a readout consisting of the alternatively spliced exon along with the two flanking exons. This could lead to a scenario where several transcript isoforms of a gene might have that particular combination corresponding to either included or skipped or both forms. Secondly, a considerable proportion of the isoforms predicted by rMATS from this data set are currently unannotated in any database. We also discovered a completely novel exon in SHC1 between the flanking exons, which further increased the possible number of isoforms detected by RT-PCR. In a cellular context, there could be many more such novel exons across the genome that are yet to be identified. This added another layer of complexity to interpreting the alternative splicing data and their possible functional implications. In addition, several genes had multiple differential alternative splicing events occurring at the same time point. However, the nature of the rMATS output made it impossible to deduce whether these different events were independent mRNA isoforms or were occurring simultaneously in the same mRNA isoform. These issues are inherent with the RNA-Seq platform where the read lengths are typically short. One potential solution could be the application of long-read sequencing technologies, but these methods currently have issues of high error rate and low throughput, limiting their usefulness for the analysis of global alternative splicing (Weirather et al. 2017).
Considering the well-documented pitfalls of all these approaches, we decided to perform a GO analysis to identify and target pathways that are enriched in genes undergoing differential alternative splicing (Liu and Rabadan 2021). Longitudinal overlap data from our GO analysis demonstrated that the pathways enriched in transcripts with differential alternative splicing events changed dynamically with the time of exposure (Figure 4; Excel Table S1). At 7 wk, RNA metabolism pathways (including mRNA processing, transport, and splicing) were found to be abundant (Figure 5; Excel Table S1). Both mRNA processing and transport are intricately connected with protein expression and proteomic profile, and their dysregulation is widely accepted to be significant in carcinogenesis (Borden 2020; Goodall and Wickramasinghe 2021; Zhang et al. 2021b). Dysregulation of splicing pathways, splice regulators, and networks in cancers have also been well documented (Bessa et al. 2020; Ding et al. 2020; Kim et al. 2018; Roy Burman et al. 2021; Sciarrillo et al. 2020). In addition, GO analysis in these alternatively spliced genes indicated RNA splicing as an enriched term. At the 19-wk time point, GO analysis predicted the selenoprotein metabolism pathway to be enriched. The association of selenoproteins in metastasis and tumorigenesis is well studied (Marciel and Hoffmann 2017; Short and Williams 2017), and, importantly, arsenic and selenium also have a mutually protective effect (Zwolak and Zaporowska 2012; Zwolak 2020). At 28-wk, guanosine triphosphatase (GTPase) binding and regulation of GTPase activity were represented multiple times (Figure 4; Excel Table S1). The role of GTPases in cancer has been studied widely and confirmed by independent studies (Aspenström 2018; Boudhraa et al. 2020; Kazanietz and Caloca 2017; Vega and Ridley 2008). Thus, our GO data were in agreement with the accepted mechanisms of carcinogenesis through different stages of tumorigenesis (Aspenström 2018; Kim et al. 2018; Ouyang et al. 2021; Zheng et al. 2021).
Although individual splicing changes might be small, they are coordinated in a genome-wide manner and are capable of generating significant biological effects through combination (Kelemen et al. 2013). In agreement, our GO analysis suggested that alternative splicing was dynamically integrated with other layers of gene regulation, such as altered miRNA and mRNA expression, upon chronic arsenic exposure (Al-Eryani et al. 2018a, 2018b; Banerjee et al. 2021). The results from the GO analysis validated our approach in identifying important target pathways that might be regulated through differential alternative splicing upon chronic arsenic exposure rather than attempting to functionally characterize the possible effects of individual differential splicing events.
Application of both antibody- or MS-based techniques can identify and quantitate differentially expressed isoforms. However, after analyzing publications regarding alternative splicing, Kelemen et al. (2013) indicated that the functions of most alternative exons are unknown and that most changes caused by alternative splicing were subtle and often hard to detect. Very few isoform-specific antibodies are available, and the coverage of the proteome is extremely low. Results from our immunoblot experiments made this point very clear (Figure S2). In any case, antibody-based techniques are not suitable for high-throughput proteome-wide validation of differential alternative splicing events. High throughput is an advantage of proteomic platforms, but differential posttranslational modifications in differentially spliced isoforms could still be a complicated, confounding interpretation. Furthermore, there are obvious issues with low-abundance isoforms and labeling efficiency (Wasinger et al. 2013).
Our preliminary proteomic analysis also provided experimental support for the hypothesis that splicing regulation itself was dysregulated upon chronic arsenic exposure for 7-wk. We demonstrated that the expression of the canonical isoform for three well-known broad-spectrum splice regulators (DDX42, RMB25, and SRRM2) were induced in the whole-cell lysates, corroborating splicing predictions (Table Excel Table S2). Each of these three molecules have known roles in pathogenesis of multiple cancer types (Cai et al. 2017; Chen et al. 2020a; Ge et al. 2019; Hińcza et al. 2019). This finding was quite remarkable given that almost all these splice regulators should be enriched in the nuclear fraction. This also perhaps partly explained why we did not see differential expression of some of the shortlisted splicing-related genes in the whole cell lysates upon chronic arsenic exposure. In the future, it would be important to investigate whether chronic arsenic exposure modulates the expression of these molecules in the nuclear fraction.
A few more caveats also need to be taken into account while interpreting our preliminary proteomic results. First, phosphorylation plays a key role in modulating the activity, localization, and splice site choice of splice factors (Long et al. 2019; Misteli 1999; Naro and Sette 2013), as well as in carcinogenesis (Naro and Sette 2013). It is possible that the functional effect of a relatively small (and statistically nonsignificant) change in protein expression (for example, 7% induction in SRRM1 canonical isoform Q8IYB3-1; Excel Table S2) could have been amplified by the alterations in the phosphorylation leading to important downstream effects. In fact, most of these splice factors could be differentially phosphorylated, impacting their function and their mass (Aubol et al. 2003). Such alternative phosphorylation makes it difficult for MS methods to identify them as the same molecule (Wu et al. 2018). In addition, it is possible that, in some cases, the change in mean value was driven largely by changes in the isoform that underwent nonsense-mediated decay. In such instances, the protein coding isoform was relatively unlikely to show any considerable change. For example, the included MAGOHB-208 isoform underwent a 56% reduction in rMATS counts upon chronic arsenic exposure, which in large part drove the mean value (0.099) rather than a large change in the counts of the excluded MAGOHB-201 isoform that codes for the canonical protein isoform (Uniprot ID: Q96A72; Excel Table S2). Furthermore, in some cases, there could be an imperfect binding between miRNA and mRNA, leading to suppression of the target protein translation without affecting the mRNA expression. This targeting by miRNAs could also be driving the discordance between some of the splicing predictions and proteomic data. For example, miR-744 was induced upon chronic arsenic exposure for 7-wk (Banerjee et al. 2021). This miRNA targets HNRNPC-202, which encodes for the canonical protein isoform P07910-1 (Zhang et al. 2021a). The canonical isoform was predicted to be induced by rMATS analysis (mean : 0.104; Excel Table S2), but the canonical protein isoform expression did not differ (Excel Table S2). All these observations and caveats strengthened our stance that proteomic validation of alternative splicing events is still in its infancy and faces multiple technological challenges. New bioinformatic as well as technological pipelines need to be developed to surpass these obstacles.
Given the widespread nature of differential splicing in our data set, it was important to consider how chronic arsenic exposure mediated the genome-wide cascade of alternative splicing. This modulation could be achieved through dysregulation of broad-spectrum splice factor function through zinc displacement (Banerjee et al. 2020) or by differential splicing of other splice regulators such as serine- and arginine-rich splice factor proteins, as in our 7-wk data set (Excel Table S2). Such arsenic-mediated functional abrogation of a few upstream splice regulators of several other splice regulators could affect the downstream splicing of a large number of targets simultaneously. Future studies could explore the role of dysregulated splicing regulation in arsenic-mediated cSCC.
In conclusion, we demonstrated that chronic arsenic exposure temporally modulated genome-wide differential alternative splicing events in HaCaT cells in terms of splicing type, genes, and biological processes involved and in the location of the events in the transcripts. This modulation of alternative splicing could lead to changes in the proteomic landscape with respect to steady-state expression, localization, and posttranslational modifications. Another important question is whether some or all these differential splicing events are reversible upon removal of exposure and, if so, until which time point. Although empirical investigation of that issue was beyond the scope of the present work, existing literature has provided some clues. In a previous study on BEAS-2B cell lines exposed chronically to arsenic, removal of arsenic exposure was shown to reverse some of the differential alternative splicing events compared with continuous arsenic exposure condition (Riedmann et al. 2015). Based on this observation, we hypothesize that at least some of the differential alternative splicing events in our data sets are likely to be reversed upon removal of arsenic, at least at the early time points. Ultimately, functional characterization of the effects of such large numbers of differential alternative splicing events occurring simultaneously with respect to proteome changes remains a challenge. Identifying target pathways and their functions rather than focusing on individual events will aid in understanding the effects influenced by differential alternative splicing at a genome-wide scale.
Supplementary Material
Acknowledgments
We thank E. Hudson, Center for Genetics and Molecular Medicine DNA Facility Core, University of Louisville for mRNA library preparation and RNA-sequencing (RNA-Seq) data generation. We also thank S. Waigel and A. Wise-Mitchell, Genomics Facility, University of Louisville, for assistance with RNA quality assessment.
This work was supported by the National Institutes of Health (NIH)/National Institute of Environmental Health Sciences (R21ES023627, R21ES030334, R01ES027778, and P30ES030283); KY INBRE Investigator Development Awards from the NIH/National Institute of General Medical Sciences (NIGMS; P20GM103436 and P20GM113226), and by the NIGMS Academic Research Enhancement Awards (R15GM126446).
RNA-Seq data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/) under the accession no. GSE107054.
References
- Ahn R, Sabourin V, Bolt AM, Hébert S, Totten S, De Jay N, et al. 2017. The Shc1 adaptor simultaneously balances Stat1 and Stat3 activity to promote breast cancer immune suppression. Nat Commun 8:14638, PMID: , 10.1038/ncomms14638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Eryani L, Jenkins SF, States VA, Pan J, Malone JC, Rai SN, et al. 2018a. miRNA expression profiles of premalignant and malignant arsenic-induced skin lesions. PLoS One 13(8):e0202579, PMID: , 10.1371/journal.pone.0202579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Eryani L, Waigel S, Tyagi A, Peremarti J, Jenkins SF, Damodaran C, et al. 2018b. Differentially expressed mRNA targets of differentially expressed miRNAs predict changes in the TP53 axis and carcinogenesis related pathways in human keratinocytes chronically exposed to arsenic. Toxicol Sci 162(2):645–654, PMID: , 10.1093/toxsci/kfx292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dominguez JR, Zhang X, Hu W. 2017. Widespread and dynamic translational control of red blood cell development. Blood 129(5):619–629, PMID: , 10.1182/blood-2016-09-741835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo PR, Yoon K, Ko D, Smith AD, Qiao M, Suresh U, et al. 2012. Before it gets started: regulating translation at the 5′ UTR. Comp Funct Genomics 2012:475731, PMID: , 10.1155/2012/475731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspenström P. 2018. Activated rho GTPases in cancer—the beginning of a new paradigm. Int J Mol Sci 19(12):3949, PMID: , 10.3390/ijms19123949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubol BE, Chakrabarti S, Ngo J, Shaffer J, Nolen B, Fu XD, et al. 2003. Processive phosphorylation of alternative splicing factor/splicing factor 2. Proc Natl Acad Sci USA 100(22):12601–12606, PMID: , 10.1073/pnas.1635129100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey KA, Laine J, Rager JE, Sebastian E, Olshan A, Smeester L, et al. 2014. Prenatal arsenic exposure and shifts in the newborn proteome: interindividual differences in tumor necrosis factor (TNF)-responsive signaling. Toxicol Sci 139(2):328–337, PMID: , 10.1093/toxsci/kfu053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee M, Bhattacharjee P, Giri AK. 2011. Arsenic-induced cancers: a review with special reference to gene, environment and their interaction. Genes Environ 33(4):128–140, 10.3123/jemsge.33.128. [DOI] [Google Scholar]
- Banerjee M, Ferragut Cardoso A, Al-Eryani L, Pan J, Kalbfleisch TS, Srivastava S, et al. 2021. Dynamic alteration in miRNA and mRNA expression profiles at different stages of chronic arsenic exposure-induced carcinogenesis in a human cell culture model of skin cancer. Arch Toxicol 95(7):2351–2365, PMID: , 10.1007/s00204-021-03084-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee M, Ferragut Cardoso AP, Lykoudi A, Wilkey DW, Pan J, Watson WH, et al. 2020. Arsenite exposure displaces zinc from ZRANB2 leading to altered splicing. Chem Res Toxicol 33(6):1403–1417, PMID: , 10.1021/acs.chemrestox.9b00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DF, Husain A, Neale JR, Martini BD, Zhang X, Doll MA, et al. 2006. Functional properties of an alternative, tissue-specific promoter for human arylamine N-acetyltransferase 1. Pharmacogenet Genomics 16(7):515–525, PMID: , 10.1097/01.fpc.0000215066.29342.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund SR, Santana AR, Li D, Rice RH, Rocke DM, Goldberg Z. 2009. Proteomic analysis of low dose arsenic and ionizing radiation exposure on keratinocytes. Proteomics 9(7):1925–1938, PMID: , 10.1002/pmic.200800118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessa C, Matos P, Jordan P, Gonçalves V. 2020. Alternative splicing: expanding the landscape of cancer biomarkers and therapeutics. Int J Mol Sci 21(23):9032, PMID: , 10.3390/ijms21239032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boja ES, Rodriguez H. 2014. Proteogenomic convergence for understanding cancer pathways and networks. Clin Proteomics 11(1):22, PMID: , 10.1186/1559-0275-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden KLB. 2020. The nuclear pore complex and mRNA export in cancer. Cancers (Basel) 13(1):42, PMID: , 10.3390/cancers13010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudhraa Z, Carmona E, Provencher D, Mes-Masson AM. 2020. Ran GTPase: a key player in tumor progression and metastasis. Front Cell Dev Biol 8:345, PMID: , 10.3389/fcell.2020.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106(3):761–771, PMID: , 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. 2013. Dynamic integration of splicing within gene regulatory pathways. Cell 152(6):1252–1269, PMID: , 10.1016/j.cell.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Chen ZX, Rane G, Satendra Singh S, Choo Z, Wang C, et al. 2017. Wanted DEAD/H or alive: helicases winding up in cancers. J Natl Cancer Inst 109(6):djw278, PMID: , 10.1093/jnci/djw278. [DOI] [PubMed] [Google Scholar]
- Cardoso APF, Al-Eryani L, States JC. 2018. Arsenic-induced carcinogenesis: the impact of miRNA dysregulation. Toxicol Sci 165(2):284–290, PMID: , 10.1093/toxsci/kfy128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lv L, Zhan Z, Wang X, You Z, Luo X, et al. 2020a. Silencing of long noncoding RNA SRRM2-AS exerts suppressive effects on angiogenesis in nasopharyngeal carcinoma via activating MYLK-mediated cGMP-PKG signaling pathway. J Cell Physiol 235(11):7757–7768, PMID: , 10.1002/jcp.29382. [DOI] [PubMed] [Google Scholar]
- Chen YT, Ou Yang WT, Juang HH, Chen CL, Chen HW, Tsui KH, et al. 2020b. Proteomic characterization of arsenic and cadmium exposure in bladder cells. Rapid Commun Mass Spectrom 34(suppl 1):e8578, PMID: , 10.1002/rcm.8578. [DOI] [PubMed] [Google Scholar]
- Cho S, Moon H, Loh TJ, Jang HN, Liu Y, Zhou J, et al. 2015. Splicing inhibition of U2AF65 leads to alternative exon skipping. Proc Natl Acad Sci USA 112(32):9926–9931, PMID: , 10.1073/pnas.1500639112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciolli Mattioli C, Rom A, Franke V, Imami K, Arrey G, Terne M, et al. 2019. Alternative 3′ UTRs direct localization of functionally diverse protein isoforms in neuronal compartments. Nucleic Acids Res 47(5):2560–2573, PMID: , 10.1093/nar/gky1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo I, Sangiovanni E, Maggio R, Mattozzi C, Zava S, Corbett Y, et al. 2017. HaCaT cells as a reliable in vitro differentiation model to dissect the inflammatory/repair response of human keratinocytes. Mediators Inflamm 2017:7435621, PMID: , 10.1155/2017/7435621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Loma J, Gliga AR, Levi M, Ascui F, Gardon J, Tirado N, et al. 2020. Arsenic exposure and cancer-related proteins in urine of indigenous Bolivian women. Front Public Health 8:605123, PMID: , 10.3389/fpubh.2020.605123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Feng G, Yang M. 2020. Prognostic role of alternative splicing events in head and neck squamous cell carcinoma. Cancer Cell Int 20:168, PMID: , 10.1186/s12935-020-01249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlamini Z, Mokoena F, Hull R. 2017. Abnormalities in alternative splicing in diabetes: therapeutic targets. J Mol Endocrinol 59(2):R93–R107, PMID: , 10.1530/JME-17-0049. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21, PMID: , 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein M, Eleazer R, Rea M, Fondufe-Mittendorf Y. 2017a. Epigenomic reprogramming in inorganic arsenic-mediated gene expression patterns during carcinogenesis. Rev Environ Health 32(1–2):93–103, PMID: , 10.1515/reveh-2016-0025. [DOI] [PubMed] [Google Scholar]
- Eckstein M, Rea M, Fondufe-Mittendorf YN. 2017b. Transient and permanent changes in DNA methylation patterns in inorganic arsenic-mediated epithelial-to-mesenchymal transition. Toxicol Appl Pharmacol 331:6–17, PMID: , 10.1016/j.taap.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Marabti E, Younis I. 2018. The cancer spliceome: reprograming of alternative splicing in cancer. Front Mol Biosci 5:80, PMID: , 10.3389/fmolb.2018.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA. 2003. Pre-mRNA splicing and human disease. Genes Dev 17(4):419–437, PMID: , 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- Floor SN, Doudna JA. 2016. Tunable protein synthesis by transcript isoforms in human cells. Elife 5:e10921, PMID: , 10.7554/eLife.10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi A, Dugué PA, Nøst TH, Southey MC, Buchanan DD, Schmidt DF, et al. 2020. Stochastic epigenetic mutations are associated with risk of breast cancer, lung cancer, and mature B-cell neoplasms. Cancer Epidemiol Biomarkers Prev 29(10):2026–2037, PMID: , 10.1158/1055-9965.EPI-20-0451. [DOI] [PubMed] [Google Scholar]
- Ganapathy S, Li P, Fagman J, Yu T, Lafontant J, Zhang G, et al. 2016. Low doses of arsenic, via perturbing p53, promotes tumorigenesis. Toxicol Appl Pharmacol 306:98–104, PMID: , 10.1016/j.taap.2016.07.009. [DOI] [PubMed] [Google Scholar]
- Ge Y, Schuster MB, Pundhir S, Rapin N, Bagger FO, Sidiropoulos N, et al. 2019. The splicing factor RBM25 controls MYC activity in acute myeloid leukemia. Nat Commun 10(1):172, PMID: , 10.1038/s41467-018-08076-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Banerjee M, De Chaudhuri S, Chowdhury R, Das JK, Mukherjee A, et al. 2007. Comparison of health effects between individuals with and without skin lesions in the population exposed to arsenic through drinking water in West Bengal, India. J Expo Sci Environ Epidemiol 17(3):215–223, PMID: , 10.1038/sj.jes.7500510. [DOI] [PubMed] [Google Scholar]
- Goldman A, Harper S, Speicher DW. 2016. Detection of proteins on blot membranes. Curr Protoc Protein Sci 86:10.18.1–10.18.11, PMID: , 10.1002/cpps.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsebatt ME, Vega L, Montero R, Garcia-Vargas G, Del Razo LM, Albores A, et al. 1994. Lymphocyte replicating ability in individuals exposed to arsenic via drinking water. Mutat Res 313(2–3):293–299, PMID: , 10.1016/0165-1161(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Gonsebatt ME, Vega L, Salazar AM, Montero R, Guzmán P, Blas J, et al. 1997. Cytogenetic effects in human exposure to arsenic. Mutat Res 386(3):219–228, PMID: , 10.1016/S1383-5742(97)00009-4. [DOI] [PubMed] [Google Scholar]
- Goodall GJ, Wickramasinghe VO. 2021. RNA in cancer. Nat Rev Cancer 21(1):22–36, PMID: , 10.1038/s41568-020-00306-0. [DOI] [PubMed] [Google Scholar]
- Grandi P, Bantscheff M. 2019. Advanced proteomics approaches to unravel protein homeostasis. Drug Discov Today Technol 31:99–108, PMID: , 10.1016/j.ddtec.2019.02.001. [DOI] [PubMed] [Google Scholar]
- Hamid FM, Makeyev EV. 2014. Emerging functions of alternative splicing coupled with nonsense-mediated decay. Biochem Soc Trans 42(4):1168–1173, PMID: , 10.1042/BST20140066. [DOI] [PubMed] [Google Scholar]
- Han H, Braunschweig U, Gonatopoulos-Pournatzis T, Weatheritt RJ, Hirsch CL, Ha KCH, et al. 2017. Multilayered control of alternative splicing regulatory networks by transcription factors. Mol Cell 65(3):539–553.e7, PMID: , 10.1016/j.molcel.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Hatje K, Rahman RU, Vidal RO, Simm D, Hammesfahr B, Bansal V, et al. 2017. The landscape of human mutually exclusive splicing. Mol Syst Biol 13(12):959, PMID: , 10.15252/msb.20177728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henseleit U, Zhang J, Wanner R, Haase I, Kolde G, Rosenbach T. 1997. Role of p53 in UVB-induced apoptosis in human HaCaT keratinocytes. J Invest Dermatol 109(6):722–727, PMID: , 10.1111/1523-1747.ep12340708. [DOI] [PubMed] [Google Scholar]
- Heyd F, Carmo-Fonseca M, Möröy T. 2008. Differential isoform expression and interaction with the P32 regulatory protein controls the subcellular localization of the splicing factor U2AF26. J Biol Chem 283(28):19636–19645, PMID: , 10.1074/jbc.M801014200. [DOI] [PubMed] [Google Scholar]
- Heyd F, Lynch KW. 2011. DEGRADE, MOVE, REGROUP: signaling control of splicing proteins. Trends Biochem Sci 36(8):397–404, PMID: , 10.1016/j.tibs.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hińcza K, Kowalik A, Kowalska A. 2019. Current knowledge of germline genetic risk factors for the development of non-medullary thyroid cancer. Genes (Basel) 10(7):482, PMID: , 10.3390/genes10070482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogendijk AJ, Pourfarzad F, Aarts CEM, Tool ATJ, Hiemstra IH, Grassi L, et al. 2019. Dynamic transcriptome-proteome correlation networks reveal human myeloid differentiation and neutrophil-specific programming. Cell Rep 29(8):2505–2519.e4, PMID: , 10.1016/j.celrep.2019.10.082. [DOI] [PubMed] [Google Scholar]
- Howe KL, Achuthan P, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, et al. 2021. Ensembl 2021. Nucleic Acids Res 49(D1):D884–D891, PMID: , 10.1093/nar/gkaa942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZG, He RQ, Mo ZN. 2018. Prognostic value and potential function of splicing events in prostate adenocarcinoma. Int J Oncol 53(6):2473–2487, PMID: , 10.3892/ijo.2018.4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt KM, Srivastava RK, Elmets CA, Athar M. 2014. The mechanistic basis of arsenicosis: pathogenesis of skin cancer. Cancer Lett 354(2):211–219, PMID: , 10.1016/j.canlet.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. 2012. Arsenic, metals, fibres, and dusts. IARC Monogr Eval Carcinog Risks Hum 100(pt C):11–465, PMID: . [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K. 2021. Multilayered regulation of proteome stoichiometry. Curr Genet 67(6):883–890, PMID: , 10.1007/s00294-021-01205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Ingolia NT. 2017. The growing toolbox for protein synthesis studies. Trends Biochem Sci 42(8):612–624, PMID: , 10.1016/j.tibs.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R, Cahill MA, Nordheim A. 1995. Signal integration at the c-fos promoter. Carcinogenesis 16(3):443–450, PMID: , 10.1093/carcin/16.3.443. [DOI] [PubMed] [Google Scholar]
- Kazanietz MG, Caloca MJ. 2017. The Rac GTPase in cancer: from old concepts to new paradigms. Cancer Res 77(20):5445–5451, PMID: , 10.1158/0008-5472.CAN-17-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, et al. 2013. Function of alternative splicing. Gene 514(1):1–30, PMID: , 10.1016/j.gene.2012.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G. 2010. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11(5):345–355, PMID: , 10.1038/nrg2776. [DOI] [PubMed] [Google Scholar]
- Kim HK, Pham MHC, Ko KS, Rhee BD, Han J. 2018. Alternative splicing isoforms in health and disease. Pflugers Arch 470(7):995–1016, PMID: , 10.1007/s00424-018-2136-x. [DOI] [PubMed] [Google Scholar]
- Lantz RC, Lynch BJ, Boitano S, Poplin GS, Littau S, Tsaprailis G, et al. 2007. Pulmonary biomarkers based on alterations in protein expression after exposure to arsenic. Environ Health Perspect 115(4):586–591, PMID: , 10.1289/ehp.9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. 2007. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446(7138):926–929, PMID: , 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- Leparc GG, Mitra RD. 2007. A sensitive procedure to detect alternatively spliced mRNA in pooled-tissue samples. Nucleic Acids Res 35(21):e146, PMID: , 10.1093/nar/gkm989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JH, Rossman TG. 1989. Mechanism of comutagenesis of sodium arsenite with N-methyl-N-nitrosourea. Biol Trace Elem Res 21:373–381, PMID: , 10.1007/BF02917278. [DOI] [PubMed] [Google Scholar]
- Li L, Tian T, Zhang X. 2019. Stochastic modelling of multistage carcinogenesis and progression of human lung cancer. J Theor Biol 479:81–89, PMID: , 10.1016/j.jtbi.2019.07.006. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gonzàlez-Porta M, Santos S, Brazma A, Marioni JC, Aebersold R, et al. 2017. Impact of alternative splicing on the human proteome. Cell Rep 20(5):1229–1241, PMID: , 10.1016/j.celrep.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Rabadan R. 2021. Computing the role of alternative splicing in cancer. Trends Cancer 7(4):347–358, PMID: , 10.1016/j.trecan.2020.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y, Sou WH, Yung KWY, Liu H, Wan SWC, Li Q, et al. 2019. Distinct mechanisms govern the phosphorylation of different SR protein splicing factors. J Biol Chem 294(4):1312–1327, PMID: , 10.1074/jbc.RA118.003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciel MP, Hoffmann PR. 2017. Selenoproteins and metastasis. Adv Cancer Res 136:85–108, PMID: , 10.1016/bs.acr.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, et al. 2014. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res 24(3):496–510, PMID: , 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marongiu F, Laconi E. 2020. Cell competition in liver carcinogenesis. World J Hepatol 12(8):475–484, PMID: , 10.4254/wjh.v12.i8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez VD, Becker-Santos DD, Vucic EA, Lam S, Lam WL. 2011. Induction of human squamous cell-type carcinomas by arsenic. J Skin Cancer 2011:454157, PMID: , 10.1155/2011/454157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16(3):1215, PMID: , 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minatel BC, Sage AP, Anderson C, Hubaux R, Marshall EA, Lam WL, et al. 2018. Environmental arsenic exposure: from genetic susceptibility to pathogenesis. Environ Int 112:183–197, PMID: , 10.1016/j.envint.2017.12.017. [DOI] [PubMed] [Google Scholar]
- Mir SA, Pinto SM, Paul S, Raja R, Nanjappa V, Syed N, et al. 2017. SILAC-based quantitative proteomic analysis reveals widespread molecular alterations in human skin keratinocytes upon chronic arsenic exposure. Proteomics 17(6):1600257, PMID: , 10.1002/pmic.201600257. [DOI] [PubMed] [Google Scholar]
- Misteli T. 1999. RNA splicing: what has phosphorylation got to do with it? Curr Biol 9(6):R198–R200, PMID: , 10.1016/S0960-9822(99)80128-6. [DOI] [PubMed] [Google Scholar]
- Mockenhaupt S, Makeyev EV. 2015. Non-coding functions of alternative pre-mRNA splicing in development. Semin Cell Dev Biol 47–48:32–39, PMID: , 10.1016/j.semcdb.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenyi CS, Ljungman M, States JC. 2015. Arsenic disruption of DNA damage responses—potential role in carcinogenesis and chemotherapy. Biomolecules 5(4):2184–2193, PMID: , 10.3390/biom5042184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naro C, Sette C. 2013. Phosphorylation-mediated regulation of alternative splicing in cancer. Int J Cell Biol 2013:151839, PMID: , 10.1155/2013/151839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas-Acien A, Silbergeld EK, Pastor-Barriuso R, Guallar E. 2008. Arsenic exposure and prevalence of type 2 diabetes in US adults. JAMA 300(7):814–822, PMID: , 10.1001/jama.300.7.814. [DOI] [PubMed] [Google Scholar]
- NTP (National Toxicology Program). 2016. Report on Carcinogens, Fourteenth Edition. Research Triangle Park, NC: U.S. Department of Health and Human Services, Public Health Service. [Google Scholar]
- Ouyang Y, Xia K, Yang X, Zhang S, Wang L, Ren S, et al. 2021. Alternative splicing acts as an independent prognosticator in ovarian carcinoma. Sci Rep 11(1):10413, PMID: , 10.1038/s41598-021-89778-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Diwan BA, Sun Y, Liu J, Qu W, He Y, et al. 2008. Arsenic-induced malignant transformation of human keratinocytes: involvement of Nrf2. Free Radic Biol Med 45(5):651–658, PMID: , 10.1016/j.freeradbiomed.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Kumagai Y, Sun G, Yamauchi H, Yoshida T, Iso H, et al. 2000. Decreased serum concentrations of nitric oxide metabolites among Chinese in an endemic area of chronic arsenic poisoning in Inner Mongolia. Free Radic Biol Med 28(7):1137–1142, PMID: , 10.1016/S0891-5849(00)00209-4. [DOI] [PubMed] [Google Scholar]
- Podgorski J, Berg M. 2020. Global threat of arsenic in groundwater. Science 368(6493):845–850, PMID: , 10.1126/science.aba1510. [DOI] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. 2019. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47(W1):W191–W198, PMID: , 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Kull M, Peterson H, Hansen J, Vilo J. 2007. g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 35(suppl 2):W193–W200, PMID: , 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmann C, Ma Y, Melikishvili M, Godfrey SG, Zhang Z, Chen KC, et al. 2015. Inorganic arsenic-induced cellular transformation is coupled with genome wide changes in chromatin structure, transcriptome and splicing patterns. BMC Genomics 16(1):212, PMID: , 10.1186/s12864-015-1295-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman TG. 2003. Mechanism of arsenic carcinogenesis: an integrated approach. Mutat Res 533(1–2):37–65, PMID: , 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Roy Burman D, Das S, Das C, Bhattacharya R. 2021. Alternative splicing modulates cancer aggressiveness: role in EMT/metastasis and chemoresistance. Mol Biol Rep 48(1):897–914, PMID: , 10.1007/s11033-020-06094-y. [DOI] [PubMed] [Google Scholar]
- Saintilnord WN, Fondufe-Mittendorf Y. 2021. Arsenic-induced epigenetic changes in cancer development. Semin Cancer Biol 76:195–205, PMID: , 10.1016/j.semcancer.2021.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal T, Paul M, Bhattacharjee S, Bhattacharjee P. 2020. Epigenetic alteration of mitochondrial biogenesis regulatory genes in arsenic exposed individuals (with and without skin lesions) and in skin cancer tissues: a case control study. Chemosphere 258:127305, PMID: , 10.1016/j.chemosphere.2020.127305. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675, PMID: , 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarrillo R, Wojtuszkiewicz A, Assaraf YG, Jansen G, Kaspers GJL, Giovannetti E, et al. 2020. The role of alternative splicing in cancer: from oncogenesis to drug resistance. Drug Resist Updat 53:100728, PMID: , 10.1016/j.drup.2020.100728. [DOI] [PubMed] [Google Scholar]
- Seow WJ, Kile ML, Baccarelli AA, Pan WC, Byun HM, Mostofa G, et al. 2014. Epigenome-wide DNA methylation changes with development of arsenic-induced skin lesions in Bangladesh: a case–control follow-up study. Environ Mol Mutagen 55(6):449–456, PMID: , 10.1002/em.21860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Lee J, Weinfeld M, Le XC. 2008. Attenuation of DNA damage-induced p53 expression by arsenic: a possible mechanism for arsenic co-carcinogenesis. Mol Carcinog 47(7):508–518, PMID: , 10.1002/mc.20406. [DOI] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, et al. 2014. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci USA 111(51):E5593–E5601, PMID: , 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SP, Williams CS. 2017. Selenoproteins in tumorigenesis and cancer progression. Adv Cancer Res 136:49–83, PMID: , 10.1016/bs.acr.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Zeng Z, Wei H, Wang Z. 2018. Alternative splicing in cancers: from aberrant regulation to new therapeutics. Semin Cell Dev Biol 75:13–22, PMID: , 10.1016/j.semcdb.2017.09.018. [DOI] [PubMed] [Google Scholar]
- States JC. 2015. Disruption of mitotic progression by arsenic. Biol Trace Elem Res 166(1):34–40, PMID: , 10.1007/s12011-015-0306-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- States JC, Barchowsky A, Cartwright IL, Reichard JF, Futscher BW, Lantz RC. 2011. Arsenic toxicology: translating between experimental models and human pathology. Environ Health Perspect 119(10):1356–1363, PMID: , 10.1289/ehp.1103441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Pi J, Wang X, Tokar EJ, Liu J, Waalkes MP. 2009. Aberrant cytokeratin expression during arsenic-induced acquired malignant phenotype in human HaCaT keratinocytes consistent with epidermal carcinogenesis. Toxicology 262(2):162–170, PMID: , 10.1016/j.tox.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam LM, Price NE, Wang Y. 2020. Molecular mechanisms of arsenic-induced disruption of DNA repair. Chem Res Toxicol 33(3):709–726, PMID: , 10.1021/acs.chemrestox.9b00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam LM, Wang Y. 2020. Arsenic exposure and compromised protein quality control. Chem Res Toxicol 33(7):1594–1604, PMID: , 10.1021/acs.chemrestox.0c00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada LS. 2019. Shc and the mechanotransduction of cellular anchorage and metastasis. Small GTPases 10(1):64–71, PMID: , 10.1080/21541248.2016.1273172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji JS, Perez V, Garry MR, Alexander DD. 2014. Association of low-level arsenic exposure in drinking water with cardiovascular disease: a systematic review and risk assessment. Toxicology 323:78–94, PMID: , 10.1016/j.tox.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Udensi UK, Tackett AJ, Byrum S, Avaritt NL, Sengupta D, Moreland LW, et al. 2014. Proteomics-based identification of differentially abundant proteins from human keratinocytes exposed to arsenic trioxide. J Proteomics Bioinform 7(7):166–178, PMID: , 10.4172/jpb.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Blencowe BJ. 2019. Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol Cell 76(2):329–345, PMID: , 10.1016/j.molcel.2019.09.017. [DOI] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. 2012. Primer3–new capabilities and interfaces. Nucleic Acids Res 40(15):e115, PMID: , 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanski LM, Leclair N, Anczuków O. 2018. Alternative-splicing defects in cancer: splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip Rev RNA 9(4):e1476, PMID: , 10.1002/wrna.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega FM, Ridley AJ. 2008. Rho GTPases in cancer cell biology. FEBS Lett 582(14):2093–2101, PMID: , 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Velazquez FN, Caputto BL, Boussin FD. 2015. c-Fos importance for brain development. Aging (Albany NY) 7(12):1028–1029, PMID: , 10.18632/aging.100862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. 2012. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13(4):227–232, PMID: , 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman A, Schmults C. 2019. Cutaneous squamous cell carcinoma. Hematol Oncol Clin North Am 33(1):1–12, PMID: , 10.1016/j.hoc.2018.08.001. [DOI] [PubMed] [Google Scholar]
- Wang W, Guo M, Xia X, Zhang C, Zeng Y, Wu S. 2017. XRRA1 targets ATM/CHK1/2-mediated DNA repair in colorectal cancer. BioMed Res Int 2017:5718968, PMID: , 10.1155/2017/5718968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasinger VC, Zeng M, Yau Y. 2013. Current status and advances in quantitative proteomic mass spectrometry. Int J Proteomics 2013:180605, PMID: , 10.1155/2013/180605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatheritt RJ, Sterne-Weiler T, Blencowe BJ. 2016. The ribosome-engaged landscape of alternative splicing. Nat Struct Mol Biol 23(12):1117–1123, PMID: , 10.1038/nsmb.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirather JL, de Cesare M, Wang Y, Piazza P, Sebastiano V, Wang XJ, et al. 2017. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res 6:100, PMID: , 10.12688/f1000research.10571.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Tiambeng TN, Cai W, Chen B, Lin Z, Gregorich ZR, et al. 2018. Impact of phosphorylation on the mass spectrometry quantification of intact phosphoproteins. Anal Chem 90(8):4935–4939, PMID: , 10.1021/acs.analchem.7b05246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Carstens RP. 2017. Alternative splicing regulates distinct subcellular localization of epithelial splicing regulatory protein 1 (Esrp1) isoforms. Sci Rep 7(1):3848, PMID: , 10.1038/s41598-017-03180-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liao K, Liu D. 2021a. MicroRNA-744-5p is downregulated in colorectal cancer and targets SEPT2 to suppress the malignant phenotype. Mol Med Rep 23(1):54, PMID: , 10.3892/mmr.2020.11692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu L, Qiu Q, Zhou Q, Ding J, Lu Y, et al. 2021b. Alternative polyadenylation: methods, mechanism, function, and role in cancer. J Exp Clin Cancer Res 40(1):51, PMID: , 10.1186/s13046-021-01852-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Wang R, Ding Q, Wang T, Xie B, Wei L, et al. 2018. Cellular stress alters 3′UTR landscape through alternative polyadenylation and isoform-specific degradation. Nat Commun 9(1):2268, PMID: , 10.1038/s41467-018-04730-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Feng L, Yin Y, Yu C, He X, Zhu J, et al. 2021. Comprehensive analysis of aberrant alternative splicing related to carcinogenesis and prognosis of papillary thyroid cancer. Aging (Albany NY) 13(19):23149–23168, PMID: , 10.18632/aging.203608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Merchant ML, Burke TJ, Ritzenthaler JD, Li M, Gaweda AE, et al. 2020. Redox states of protein cysteines in pathways of protein turnover and cytoskeleton dynamics are changed with aging and reversed by Slc7a11 restoration in mouse lung fibroblasts. Oxid Med Cell Longev 2020:2468986, PMID: , 10.1155/2020/2468986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Su J, Wu Z, Wang C, Zhong W, et al. 2017. Integration of microRNAome, proteomics and metabolomics to analyze arsenic-induced malignant cell transformation. Oncotarget 8(53):90879–90896, PMID: , 10.18632/oncotarget.18741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Song J, Guo Y, Huang Z, Chen X, Dang X, et al. 2020. LAMB3 promotes tumour progression through the AKT-FOXO3/4 axis and is transcriptionally regulated by the BRD2/acetylated ELK4 complex in colorectal cancer. Oncogene 39(24):4666–4680, PMID: , 10.1038/s41388-020-1321-5. [DOI] [PubMed] [Google Scholar]
- Zwolak I. 2020. The role of selenium in arsenic and cadmium toxicity: an updated review of scientific literature. Biol Trace Elem Res 193(1):44–63, PMID: , 10.1007/s12011-019-01691-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak I, Zaporowska H. 2012. Selenium interactions and toxicity: a review. Selenium interactions and toxicity. Cell Biol Toxicol 28(1):31–46, PMID: , 10.1007/s10565-011-9203-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.