

Abstract
Aims
Cardiac electrical activity is extraordinarily robust. However, when it goes wrong it can have fatal consequences. Electrical activity in the heart is controlled by the carefully orchestrated activity of more than a dozen different ion conductances. While there is considerable variability in cardiac ion channel expression levels between individuals, studies in rodents have indicated that there are modules of ion channels whose expression co-vary. The aim of this study was to investigate whether meta-analytic co-expression analysis of large-scale gene expression datasets could identify modules of co-expressed cardiac ion channel genes in human hearts that are of functional importance.
Methods and results
Meta-analysis of 3653 public human RNA-seq datasets identified a strong correlation between expression of CACNA1C (L-type calcium current, ICaL) and KCNH2 (rapid delayed rectifier K+ current, IKr), which was also observed in human adult heart tissue samples. In silico modelling suggested that co-expression of CACNA1C and KCNH2 would limit the variability in action potential duration seen with variations in expression of ion channel genes and reduce susceptibility to early afterdepolarizations, a surrogate marker for proarrhythmia. We also found that levels of KCNH2 and CACNA1C expression are correlated in human-induced pluripotent stem cell-derived cardiac myocytes and the levels of CACNA1C and KCNH2 expression were inversely correlated with the magnitude of changes in repolarization duration following inhibition of IKr.
Conclusion
Meta-analytic approaches of multiple independent human gene expression datasets can be used to identify gene modules that are important for regulating heart function. Specifically, we have verified that there is co-expression of CACNA1C and KCNH2 ion channel genes in human heart tissue, and in silico analyses suggest that CACNA1C–KCNH2 co-expression increases the robustness of cardiac electrical activity.
Keywords: Calcium channels, Potassium channels, Gene expression, Computational modelling, Cardiac arrhythmia
Graphical Abstract

1. Introduction
Robust electrical signalling in the heart is critical for co-ordinating the efficient pumping of blood around the body. Failure of cardiac electrical signalling, even for just a few minutes, can have fatal consequences, with sudden cardiac death accounting for up to 10% of deaths in our community.1 While we now have therapies for reducing the incidence of proarrhythmic triggers (e.g. beta-blockers2) and devices for terminating potentially fatal arrhythmias when they occur (e.g. implantable cardiac defibrillators3), predicting in advance who is more or less susceptible to sudden cardiac death and so who would most benefit from these therapies has proved to be extraordinarily difficult.4
The action potential (AP) of excitable cells, such as cardiac myocytes and neurons, reflects the orchestrated activity of at least a dozen distinct ion channels and electrogenic transporters. In such complex systems, with so many different molecular players, and hence degrees of freedom, it is not surprising that there are multiple combinations of different ion channel expression levels, that can repolarize the cell equally well.5,6 This has led to a paradigm shift in computational cardiology that relies not on generating idealized models based on mean data but rather development of populations of models that account for the observed variability in molecular inputs.7–9 A corollary of this concept is that while there are multiple possible ‘good-enough solutions’ that satisfy baseline requirements, not all combinations of ion channel expression will prove equally robust when faced with pathological insults.6
There is considerable heterogeneity of ion channel expression both within and between hearts.6 This heterogeneity, however, is not completely random. Rather there are reported examples of pairs or small modules of genes whose level of expression co-vary. For example, Bányász et al.10 and Rees et al.11 demonstrated that the expression of ICaL is correlated with the sum of the major repolarizing ion currents in guinea-pig and mouse, respectively. Furthermore, Rees and colleagues showed that in mice, this module of gene expression was important for reducing the variability in the magnitude of the calcium transient. To date, there have been no similar studies in human heart tissue. Furthermore, the human heart expresses a different complement of repolarization channels compared to rodent hearts.12 So even if this pattern was conserved, it is likely that different potassium channels would be involved in the human heart.
Outside the cardiovascular field, there is a considerable literature looking at the identification of co-expression networks among large-scale gene expression datasets.13,14 Such networks have been shown to encode functional information,15 with co-expression reflecting co-regulation and co-functionality.11,16 Furthermore, meta-analytical methods have been developed, which aggregate large numbers of individual networks across multiple independent experiments to average away noise and remove bias that may be inherent in any one experimental approach thereby reinforcing those correlations that reflect real signals.17,18
The aim of this study was to test the hypothesis that meta-analytic co-expression analysis of large-scale gene expression datasets would identify modules of co-expressed cardiac ion channel genes that are of functional importance for the human heart. This approach identified multiple pairs of co-expressed genes. To investigate whether pairs of co-expressed genes identified could be functionally significant, we focused on one co-expressed pair: CACNA1C (encodes the L-type calcium current, ICaL) and KCNH2 [encodes the rapid component of the delayed rectifier potassium current (IKr) which is often referred to as the human ether a-go-go related gene]. Using in silico approaches to generate populations of models of cardiac electrical activity with and without constrained patterns of co-expressed genes, we show that co-variation of ICaL and IKr reduced the emergence of proarrhythmic early afterdepolarizations (EADs) and this protection persisted in the face of highly variable expression of other ion channels. Our modelling data also suggested that hearts with high expression of ICaL and IKr are likely to be more susceptible to EADs when exposed to drugs that block IKr, the underlying basis of drug-induced long QT syndrome,19 whereas low expression of ICaL and IKr is associated with the greatest prolongation of repolarization duration without generation of EADs. This later prediction was verified in human cardiac myocytes derived from induced pluripotent stem cells. Together, our data, confirm that meta-analytic approaches of large-scale gene expression datasets are a valuable means for identification of gene modules that are important for regulating heart function.
2. Methods
A detailed version of the materials and methods section appears in the Supplementary material online that accompanies this article.
2.1 Analysis of public RNA-seq datasets
An aggregate co-expression gene network was built from public data, similar to that described previously.18 Briefly, 75 human RNA-seq expression experiments (listed in Supplementary material online, Table S1) that passed quality control and had a least 10 samples (3653 samples in total) were downloaded from the Gemma database.20 (see Supplementary material online for more details). A co-expression network was generated for each experiment by calculating Spearman’s correlation coefficients between every gene pair and then ranking these values.18 An aggregate gene co-expression network was then generated by averaging across all the individual networks, and re-ranking the final network. This final aggregate network was then used to determine the co-expression ranking between genes that encode for the set of ion channels and calcium handling proteins that determine the shape and duration of the human ventricular AP, the so-called rhythmonome gene subset (Supplementary material online, Table S2).
2.2 GTEx dataset
Gene expression data from v8 of the GTEx atlas21 were downloaded from their webportal (https://gtexportal.org/home/). The dataset is human post-mortem RNA-seq across 31 tissues from 948 individuals, and included 429 atria samples and 432 samples from the left ventricle. Given that there may be significant mRNA degradation during the time between death and harvesting human heart samples, we repeated our analysis using only those samples with RNA integrity (RIN) scores >7.0; which corresponded to 472 heart samples (237 atria, 235 left ventricle).
2.3 Human-induced pluripotent stem cell-derived cardiac myocytes
Human iPSC lines, generated from healthy patients by Stanford Cardiovascular Institute Biobank, as previously described,22 were a generous donation from Joseph Wu (Standford Cardiovascular Institute). HiPSC colonies were maintained on Matrigel® (Corning) coated plates in chemically defined medium (mTeSR1™, StemCell technologies), and passaged using Dispase (StemCell technologies). HiPSC were differentiated using protocols as described previously (Perry 2020).23 The CMs were seeded on a Matrigel-coated 96-well plate (Greiner Bio-One) and maintained in CM maintenance medium for 10–15 days before they were harvested for mRNA expression analysis. Total RNA was obtained from 40 000 hiPSC-CMs lysed using QIAzol Lysis reagent (Qiagen). RNA was purified using miRNeasy® Mini Kits (Qiagen) and analysed using Agilent Bioanalyzer pico-chip. RNA samples with RIN values >7.5 were hybridized with probes designed to detect 35 known rhythmonome genes using nCounter (NanoString Technologies), which was performed at the Ramaciotti Centre for Genomics (UNSW). For quantitative comparisons of different mRNA species across different runs, we normalized mRNA levels, relative to GAPDH and HPRT1, to account for any variation in mRNA loading levels.
Extracellular field potentials were recorded from hiPSC-CM monolayers using a Maestro-APEX multi-electrode array (MEA) system (Axion Biosystems, Atlanta, GA, USA) as previously described.24 Field potential durations (FPD) were measured from the peak depolarization spike to the peak of the repolarization wave. After 30 min of pacing equilibration, electrograms were recorded for 5 min in control conditions, 5 min in the presence of cisapride (200 nM), and for 5 min in the presence of cisparide (200nM)+nifedipine (50 nM). Measurements were averaged over the last 10 beats of each recording period. Twelve wells were plated for each line and all wells from which signals were recorded (n = 7–12) were included in the analysis. After the MEA recordings were completed, cells were harvested for mRNA analysis, as described above.
2.4 Statistical analysis
Summary data are shown as box and whiskers (median, box: 25–75%, extremities: 5–95%). Correlation coefficients for linear regressions were calculated in Prism (GraphPad Software, San Diego, CA) and statistical significance for whether the slope of the line was significantly different to zero was determined using the F-test.
2.5 Computer modelling
Human cardiac APs were simulated using a modified version of the endocardial configuration of the O’Hara-Rudy (ORD11) model,25 with key conductances modified so that the model could reproduce the clinical phenotypes of long QT syndrome Types 1, 2, and 3 and maintain normal calcium transients as described by Mann et al.26 and Krogh-Madsen et al.27 The code was adapted to run in the Brain Dynamics Toolbox for Matlab.28 To incorporate population variability in ion channel expression levels the maximum conductance for each current was multiplied by conductance scalar (Gx), that was drawn from a random log-normal distribution,29 with unit mean and variance that was systematically manipulated from 0.05 to 0.5. All models were paced at 1 Hz with a stimulus of −40 mV and duration of 1 ms and allowed to equilibrate for 300 beats. We then analysed the next four beats (to allow for the possibility of development of alternans) after the equilibration stage. Individual beats were classified as ectopic if they had secondary peaks that were separated by >100 ms. The set of four beats were further classified as alternans if the profile of any of the APs deviated from each other by >1 mV at any time point. In a second set of simulations, we repeated the same method as above except that the random multipliers applied to both ICaL and IKr were identical. This case, we denote co-expression, whereas the former case, we denote independent expression.
3. Results
3.1 Meta-analysis of cardiac ion channel gene expression
The shape and duration of APs in cardiac myocytes are determined by the orchestrated activity of voltage-gated sodium, calcium, and potassium channels, as well as a series of electrogenic transporters that regulate intracellular ion concentrations. These channels, transporters, and related intracellular calcium handling proteins are encoded by a few dozen genes, sometimes referred to as the rhythmonome30 (a full list is provided in Supplementary material online, Table S2).
To determine whether there were any co-expression patterns among the rhythmonome genes in human samples, we first undertook an untargeted screen for possible expression correlation patterns in publicly available RNA-seq datasets obtained from a diverse range of independent experiments (Figure 1). Ranked correlation coefficients from an aggregate co-expression network that contain data from 3653 samples are illustrated in the heatmap in Figure 1A. High-ranked correlations indicate similarity of transcriptional profiles between the genes. A clustering analysis, as shown by the dendrogram in Figure 1A, groups genes according to their correlation similarities, as defined by their aggregated ranked correlations (see colour code in Figure 1A). There is a large cluster of yellow/green squares in the bottom left corner indicating that there are significant levels of correlation among many of the genes. Within this cluster, one can discern two sub-clusters. The cluster of yellow-green squares corresponding to 13 genes in the bottom left corner (red-dashed box in Figure 1A) encode for proteins that regulate calcium fluxes as well as the transient outward K+ current (KCND3), which helps to maintain the plateau potential at a level that maximizes calcium influx.31 A second sub-cluster, in the upper right quadrant of the main cluster, encompasses 10 genes (black-dashed box in Figure 1A), including KCNH2, SCN5A, KCNJ12, and KCNIP2, that encode for ion channel proteins important for regulating AP duration (APD). It should be noted that there are many pairs of genes within this larger cluster that have highly ranked correlations but are separated into different sub-clusters (see below).
Figure 1.

(A) Sub-network of co-expression ranking across 35 genes encoding for cardiac ion channels or calcium homeostasis proteins. Spearman’s correlation coefficients are colour coded from blue: low-ranked correlations to yellow: high-ranked correlations. The dendrogram illustrates the modules of genes with high levels of similarity in their transcriptional profiles. Red- and black-dashed boxes highlight sub-networks of correlated genes. (B) Connectivity of genes to KCNH2 (encodes for IKr) and CACNA1C (encodes for ICaL). The list of 75 RNA-seq experiments interrogated is summarized in Supplementary material online, Table S2.
Overall, the connectivity within the set of rhythmonome genes is high in comparison to their connectivity to all other genes in the co-expression network (node degree analysis, P ∼ 5.7e-14), with a central gene being CACNA1C, the gene encoding the alpha subunit of the L-type calcium channel (Supplementary material online, Figure S1). Although CACNA1C clusters in the group of calcium handling genes in the bottom left quadrant of the main cluster in Figure 1A, it also shows high correlation with the cluster of ion channel genes in the top right quadrant of Figure 1A. Similarly, KCNH2, which encodes for IKr, is included in the ion channel cluster, however the strongest individual connection to KCNH2 is with CACNA1C (see Figure 1B).
There is a considerable evidence in the literature to suggest that gene pairs or modules identified by meta-analytic co-expression analysis encode functional information.11,15,16 To test whether this also applies to the human cardiac rhythmonome genes, we chose to focus our validation studies on the CACNA1C–KCNH2 gene pair. First, it was the strongest correlation observed for KCNH2. Second, CANCA1C-encoded calcium channels and KCNH2-encoded potassium channels are known to be important contributors to repolarization in the human heart.32 Third, previous studies in rodents have shown that there is co-expression of ICaL and repolarization currents,10,11 suggesting that this may be a relationship conserved across species. And finally, there are highly specific pharmacological tools to enable specific dissection of the functional role of these channels.33
3.2 Co-expression of CACNA1C and KCNH2 in human heart tissue
The public RNA-seq datasets included in our analyses spanned a wide range of tissue types and different experiments. This was a deliberate strategy to minimize the confounder effects that may be seen in any one type of experimental approach. Unfortunately, there are too few human heart samples to undertake specific co-expression meta-analysis just on heart samples. So, to investigate whether these co-expression patterns were observed in human heart, we looked at the GTEx dataset of post-mortem tissue samples.21 While we acknowledge there are limitations associated with the GTEx dataset, for example, the majority of samples is from diseased hearts and there is a significant variation in the time between death and tissue collection, it is clear that there is a correlation between the expression of CACNA1C and KCNH2 (as well as between SCN5A and KCNH2) (Figure 2). Furthermore, this correlation was much stronger in the ventricular samples than in the atrial samples. It is also worth noting that in other tissues, collected as part of the GTEx dataset, that show robust expression of both KCNH2 and CACNA1C, the correlation between expression of KCNH2 and CACNA1C was also observed (e.g. oesophagus and colon; see Supplementary material online, Figure S3).
Figure 2.

Expression levels of (A) CACNA1C and (B) SCN5A vs. expression levels of KCNH2 in the GTEx dataset. Expression levels are expressed as log(transcripts per million+1). Data points for samples with a RNA integrity number (RIN) >7.0 are shown in black (237 atria, 235 left ventricle samples) and all other sample data points are shown in grey (192 atria, 197 left ventricle samples). The left panel shows all heart samples, the middle panel shows atrial appendage samples, and the right panel shows left ventricular samples. Correlation coefficients shown in black and grey at the bottom right of each panel correspond to the full dataset (grey) and the samples with a high RIN (black). Correlation coefficients for linear fits to the data are shown in each panel. It is notable that correlations are stronger for the ventricular samples than for atrial samples.
3.3 In silico analysis of impact of co-expression of CACNA1C and KCNH2 on cardiac electrical activity
Given the correlation between CACNA1C and KCNH2 expression was most apparent in ventricular tissue, and these two genes encode for the most important currents (ICaL, inward current and IKr, outward current) controlling repolarization in the human ventricular AP under resting conditions,29 we next investigated how the co-expression of these two genes would influence ventricular repolarization. Initially, we used an in silico approach based on the modified O-Hara-Rudy model of the human ventricular myocyte with the baseline conductance scalars modified so that the model could reproduce the clinical phenotypes of long QT syndrome Types 1, 2, and 3 and maintain normal calcium transients as previously described26,27 (Supplementary material online, Figure S4).
First, we simulated a population of 1000 human cardiac APs where the ion conductances for each cell were scaled randomly using a log-normal distribution, that is, meaning the probability of the conductance being doubled was the same as that of it being halved (see Supplementary material online, Figure S5 for details of generation of scalars). The population of models generated when all conductance scalars were varied independently (Supplementary material online, Movie S1) are denoted independent in Figure 3. We then repeated the simulation but forced the conductance scaling factors for IKr and ICaL to be identical to each other, while the scaling factors for all other conductances were allowed to vary independently, denoted co-expression in Figure 3 (and see Supplementary material online, Movie S2).
Figure 3.

APs for independent (blue) and co-expression of GKr-GCaL (red) for (A) low scalar variability (σ2=0.05) and (B) high scalar variability (σ2=0.25). Note the presence of EADs in six of the APs in the independent group with σ2=0.25. The last four beats for each of the populations of 1000 APs are shown in the movies in Supplementary material online. (C) Histograms of log(APD90/mean APD90) distributions for co-expressed (red) or independent (blue) GKr-GCaL simulations with σ2=0.05, 0.1, 0.15, 0.2, 0.25. The numbers in parentheses above the 0.2 and 0.25 groups indicate the number of EADs in each independent scalars group. APs and incidence of EADs for an extended range of variances are shown in Supplementary material online, Figure S6.
The range of APD90 values for both independent and co-expression cell populations becomes broader as the level of variability is increased (Figure 3A–C). Unsurprisingly, the spread of APD90 values in the cells with identical GCaL–GKr scalars is always narrower than in the cell populations with independent GCaL–GKr scalar values. For example, in the case of Figure 3B, the variance of the APD90 values was 0.026 for the co-expression dataset but 0.046 for the independent dataset. Another notable feature of the data in Figure 3 is that EADs begin to appear in the cell population with independent scalars when the scalar variability, σ2, exceeds 0.20 (in addition, see Supplementary material online, Figure S6). The number of cells with an EAD is indicated in parentheses above each distribution in Figure 3C.
3.4 Impact of co-expression of CACNA1C and KCNH2 on response to drug block of IKr
We next investigated whether coupling of the conductance scalars for GKr and GCaL influenced the generation of EADs in response to a pathological stimulus. Specifically, we investigated how cells respond to drug block of IKr which is the predominant cause of drug-induced long QT syndrome.19 Populations of APs obtained for independent and co-expression populations with IKr block of 0%, 50%, and 80% are illustrated in Figure 4. As the extent of IKr block is increased (from A to C), the proportion of APs producing EADs increases. It is also clear that at lower levels of IKr block, EADs were more frequent when GCaL and GKr scalars were modulated independently (see Figure 4B and inset to Figure 4D). However, the proportion of simulations developing EADs in both the independent and co-expression populations becomes similar when the extent of IKr block exceeds 80% (Figure 4D).
Figure 4.

APs for co-expressed GKr-GCaL (red) and independent (blue) GKr-GCaL populations (n = 1000), with scalar variance σ2=0.25, for (A) control, (B) 50% IKr block, and (C) 80% IKr block. (D) Plot of % of AP simulations with EADs vs. % IKr block (co-expression, red and independent, blue). The inset in panel (D) shows an expanded view of the % EADs at small levels of IKr block.
It is well established that the extent of repolarization prolongation seen in patients prescribed drugs that block IKr is highly variable and only a subset of patients exposed to drugs that block IKr,34 or with a mutation causing 50% loss of IKr function,32 will develop life-threatening arrhythmias. This is consistent with the prediction made by our simulated drug block experiments shown in Figure 4. We therefore asked whether the data from the co-expression datasets could tell us anything about what factors might predispose to lesser or greater extent of AP prolongation and/or the development of EADs in the presence of a drug that blocks IKr. Analysis of the subset of scalars within the co-expression dataset that produced the 50 longest APDs without EADs, compared to the subset of scalars that resulted in APs with EADs, is illustrated in Figure 5A and B, respectively. Notably, the longest APs without EADs had low GCaL scalars (and hence low GKr scalars before addition of drug block). Conversely, the APs that developed EADs had higher GCaL and GKr scalars. In Figure 5C, we have plotted the APD90 values for cells in the highest (red) and lowest (blue) quartiles of GCal–GKr scalars. As expected, the low GCaL group showed longer APD90 values on an average compared to the high GCaL group (see the continuous lines in Figure 5C). Furthermore, for the 70% IKr block scenario, 44% of the high GCaL group have developed EADs whereas only 7% of the low GCaL group have developed EADs (compare red and blue bars at 70% drug block in Figure 5C). Thus, higher GCaL is associated with a greater risk of developing EADs in response to moderate levels of IKr block. A similar pattern of results was observed when GCaL and GKr were allowed to vary independently, except that in this scenario the difference between the high GCaL and low GCaL groups was even more dramatic at lower levels of IKr block (Supplementary material online, Figure S7). A corollary of our prediction that patients with high GCaL are more susceptible to EADs when exposed to a drug that blocks IKr, is that co-administration of a drug that blocks ICaL would reduce the incidence of EADs A drug that inhibited ICaL by 20% caused a modest decrease in the percentage of cells with EADs and reduction of ICaL by 50% had a correspondingly larger effect (Supplementary material online, Figure S8).
Figure 5.

Scalars (mean±SD) for the population of cells with GKr-GCaL co-expression. (A) 50 cells with the longest APD90 values and no EAD with increasing % IKr block. (B) cells with EADs with increasing % IKr block. The GCaL and GKr scalars are highlighted in yellow. (C) APD90 values (left axis) for the highest (red) and lowest (blue) quartile of cells according to baseline GCaL scalar. The continuous lines show the mean value and shaded area shows ±1 SD. The percentage of EADs in each quartile is shown as columns (see axis on right side of graph). The corresponding plots for the population of cells with independent GKr-GCaL scalars are show in Supplementary material online, Figure S7.
3.5 Validation of in silico predictions in human cardiac myocytes derived from induced pluripotent stem cells
So far, we have identified modules of co-expressed genes in RNA-seq datasets, demonstrated using in silico modelling of the human ventricular AP that co-expression of CACNA1C and KCNH2 would produce more robust electrical signalling, and shown that the extent of prolongation of repolarization seen following IKr drug block should be inversely proportional to the level of CACNA1C/KCNH2 expression. We next wished to investigate whether this could be verified in heart tissue, given that KCNH2-encoded IKr is the major repolarization current in human heart but not in rodents.12 Due to the difficulties in obtaining adult human heart cells, we chose to use human-induced pluripotent stem cell-derived cardiac myocyte cell lines to determine whether the predictions from our in silico prediction holds up in human heart cells. First, we extracted mRNA from human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs)35 obtained from patients with no known heart disease. All samples contained high levels of cardiac marker genes (MYL7, GATA4, and NKX2.5) (Figure 6A). The levels of expression of most rhythmonome genes showed variations between the samples that spanned approximately an order of magnitude. As these cell lines represent an embryonic phenotype, they have low levels of expression of a number of ion channel genes, including, for example, SCN5A, KCND3, and KCNJ2. However, they do express robust levels of KCNH2 and CACNA1C. Most importantly, there was also a strong correlation between the level of expression of KCNH2 and CACNA1C (Figure 6B, r2=0.68).
Figure 6.
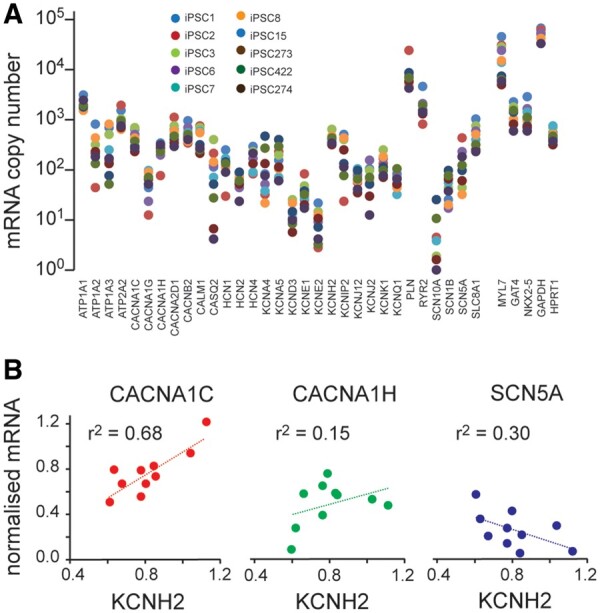
(A) Plot of all mRNAs (log axis) for 10 hiPSC-CM lines. Note that there are high levels of expression of cardiac markers (MYL7 as well as NKX2-5 and GATA4) in all cell lines. In addition, there is less variability in the levels for the housekeeping genes (GAPDH and HPRT1) compared to that seen for the ion channels and calcium handling proteins. The mRNA data for panel (A) are available in the GEO database under accession number GSE146667. (B) Correlation plots for normalized mRNA expression KCNH2 vs. genes that encode the major depolarizing currents (CACNA1C, CACNA1H, and SCN5A). To account for any differences in mRNA loading between runs, levels of expression have been normalized to GAPDH. Normalizing the data to HPRT1 or to the geometric mean of HPRT1 and GAPDH gave very similar results (Supplementary material online, Figure S9). Correlation coefficients for linear fits to the data are shown in each panel.
We next measured the duration of repolarization from extracellular field potentials (FPD) recorded from monolayers of hiPSC-derived cardiac myocytes paced at 1 Hz and exposed to drugs that specifically block IKr, cisapride,33 and ICaL, nifedipine.33 Cisapride (200 nM) caused a prolongation of repolarization that varied between 17.9 ± 7.4 ms (mean±SD, n = 12) for the least sensitive cell line to 98 ± 41 ms (mean±SD, n = 11) for the most sensitive cell line (Figure 7A). There was also a significant inverse correlation between the extent of FPD prolongation and the [KCNH2–mRNA] measured for each cell line (Figure 7B). Furthermore, the extent of FPD prolongation caused by cisapride in each cell line was almost exactly reversed by Nifedipine (50 nM) (Figure 7A).
Figure 7.

(A) Example extracellular electrograms recorded from two separate hiPSC-CM lines under control (black) conditions, after 5 min perfusion with cisparide (200 nM, red) followed by 5 min perfusion with cisapride (200 nM)+nifedipine (50 nM, blue). In cell line 15-c1, there was a 130 ms prolongation in field potential duration (FPD) which almost completely reversed on addition of nifedipine. In cell line 273-c1, there was a much smaller prolongation of 17 ms with addition of cisapride that again reversed on the addition of Nifedipine. (B) Summary of ΔFPD for six cell lines plotted against the level of KCNH2 mRNA expression (normalized to GAPDH expression). Data shown as box and whiskers (median, box: 25–75%, extremities: 5–95%, mean: filled circle, n = 7–12 replicates for each cell line). The line of best fit is shown as a solid line with 95% confidence intervals as dashed lines; the slope of the line is significantly different to zero (F-test, P = 0.0010). The micro-electrode array data for panels (A) and (B) are available in the DRYAD repository at doi: https://doi.org/10.5061/dryad.612jm640k. The mRNA data for panels (B) and (C) are available in the GEO database under accession number GSE146667.
4. Discussion
A better understanding of the molecular and cellular level control of cardiac electrical activity could pave the way for improved detection of patients at risk of cardiac arrhythmias. Here, we report that the levels of KCNH2 and CACNA1C mRNA expression, in generic tissue RNA-seq datasets (Figure 1) and the GTEx Atlas of heart tissue samples (Figure 2), vary considerably from person to person but that the levels of KCNH2 and CACNA1C expression are tightly correlated. Our in silico population modelling of the human ventricular AP shows that co-expression of IKr (encoded by KCNH2) and ICaL (encoded by CACNA1C), in the context of random variations in the densities of all cardiac ion channels, results in a narrower range of APD90 values at baseline (Figure 3) and helps to protect against the development of EADs (Figures 3 and 4). Our modelling approach also suggests that individuals with higher ICaL levels will be more susceptible to EADs when exposed to drugs that block IKr (Figure 5), whereas the subset of individuals with lower levels of ICaL will develop more prolongation of repolarization without developing EADs. Importantly, independent data obtained from human iPSC-derived cardiac myocytes have validated key predictions from our in silico analyses (Figure 7).
Over the last few years, numerous groups have shown that there is considerable heterogeneity of ion channel expression among excitable cells, including neurons5 and cardiac myocytes.6,10,36 Furthermore, since the pioneering work of Marder and colleagues, the presence of modules of co-expressed ion channel genes has also been well appreciated.14 These previous studies, however, relied on patch clamp analysis of isolated cells10,14 and qPCR analysis14 or single-molecule fluorescence in situ hybridization36 of individual ion channel mRNAs, which are highly laborious techniques and so have been restricted to only a few important ion channel genes. The advent of high-throughput transcriptomic analyses has greatly facilitated the identification of conserved networks of co-expressed gene modules among the entire set of expressed genes.18 By applying meta-analytic approaches to a large number of independent datasets one can more readily discern genuine co-expression signals from noise, as well as explore smaller gene modules.18 In our analysis of a subset of rhythmonome genes within a large library of public RNA-seq datasets, we identified multiple correlations within the subset of genes that predominantly affects calcium handling, the subset that predominantly affects membrane potential as well as between these two subsets of genes (see Figure 1). The nodes within each of these clusters that are most closely connected within the rhythmonome relative to all other genes are KCNH2 and CACNA1C (Supplementary material online, Figure S1). This is analogous to a network of network,37 where the KCNH2–CACNA1C link provides an interconnection of the two sub-networks. Independent evidence to corroborate an important link between calcium handling and regulation of cardiac AP duration comes from the large genome-wide association studies of QT interval duration which identified SNPs in a number of calcium handling genes as well as KCNH2 as being important determinants of QT interval in the population.38
Due to the large number of associations, we were testing for in our network analyses, it is possible that some would occur by chance. It was therefore critical that we investigated whether any of the identified correlations could be verified in human heart tissue and whether they were of functional importance. That we were able to confirm the presence of important co-expression modules in an independent dataset, that is, GTEx21 heart tissue samples (see Figure 2 and Supplementary material online, Figure S2) as well as in hiPSC-CM (see Figure 6) provides important corroborative evidence to support the conclusion that co-expression patterns are likely to be real and have physiological relevance. The patterns of co-expressed genes we observed also show some important similarities to previous studies in rodents. Bányász et al.10 and Rees et al.11 have both demonstrated that the expression of ICaL is correlated with the sum of the major repolarizing ion currents in guinea-pig and mouse, respectively. However, the molecular players involved in cardiac electrical activity in rodents are quite distinct to humans.12 In humans, the most important determinant of repolarization duration at resting heart rates in the absence of adrenergic stimulation is IKr,29 whereas in guinea-pig IKs and IKr play equally important roles10 and in mice the fast component of the transient outward current (Ito,f) and the ultra-rapid delayed rectifier (IKur) are the major repolarization currents.11 Our study, however, is the first to demonstrate the importance of co-expressed genes important for repolarization in human heart tissue.
Identifying modules of co-expressed genes are the first step in seeking to understand the logic of complex systems.6 The next challenge is understanding how such modules impact function in health and disease. Recently, Rees et al.,11 have demonstrated that modules of co-expressed depolarization and repolarization currents can help to ensure normal amplitude calcium transients in murine heart cells, a critical determinant of overall heart function. We have extended these studies to show that, co-expression of KCNH2 (repolarization) and CACNA1C (depolarization) channels not only limits the extent of variability in duration of the ventricular AP (Figure 3) it also helps to protect the heart from early after depolarizations when they are exposed to drugs that block IKr (Figure 5).
It is worth noting that IKr is close to fully activated during the cardiac AP at resting heart rates.39 Conversely, ICaL, is only partially activated under resting conditions and Ca2+ fluxes through CACNA1C channels can increase markedly under conditions of adrenergic stress or exercise.39 An unopposed increase in ICaL would likely be proarrhythmic. However, adrenergic stress also results in a marked increase in K+ flux through the slow delayed rectifier K+ channels encoded for by KCNQ1 and KCNE1.39 Our meta-analysis data (Figure 1) do not indicate a close correlation between KCNQ1/KCNE1 and CACNA1C, suggesting that these channels are not co-regulated at a transcriptional level. However, given that acute responses to adrenergic stress are mediated by post-translational modifications, it is likely co-regulation of ICaL and IKs occurs at a post-transcriptional level. Indeed, there is considerable evidence for co-regulation of cardiac ion channels occurring at post-transcriptional levels.36,40 For example, recent work from Robertson and colleagues demonstrated co-translation of SCN5A and KCNH2 channels in human iPSC-derived cardiac myocytes.36 There is also evidence to suggest that different ion channels traffic together to reach similar destinations at the cell surface, for example, the co-assembly of INa, Ito, and IK1 at intercalated discs40–42 has an important influence on intercellular conduction. Thus, any complete cardiac electrophysiology modelling approach will have to include not just co-regulation at the level of expression but also co-regulation occurring at the post-transcriptional level.
Our in silico studies, incorporating co-expression of KCNH2 and CACNA1C, have provided two important insights into the nature of inter-individual risk for developing arrhythmias in response to drugs that block IKr, the major cause of drug-induced cardiac arrhythmias.19 First, cells with low GCaL (and hence low GKr at baseline) exhibited the greatest prolongation of AP duration when exposed to IKr drug block (Figures 5 and 7). Second, cells with high GCaL (and hence high GKr at baseline) showed greater propensity for development of EADs at moderate levels of IKr drug block (Figure 5). This observation also provides a plausible explanation for the well-described clinical observation that prolongation of the QT interval is an imperfect surrogate marker for proarrhythmic risk.43 A corollary of the observation that a high GCaL increases the susceptibility to EADs in response to drug block of IKr is that the co-administration of an ICaL blocker should reduce the risk of EADs (Supplementary material online, Figure S8). This is consistent with the observation that the administration of magnesium, which is a mild calcium channel blocker, is helpful in the acute management of patients with drug-induced torsades de pointes,19 and conversely that hypomagnesaemia, which would stimulate ICaL, can exacerbate torsades de pointes.44 It is also consistent with the observation that drugs that block ICaL and IKr (e.g. verapamil) are not associated with drug-induced arrhythmias45 and that verapamil prevented the development of torsades de pointes in rabbit hearts exposed to an IKr blocker.46 However, given that calcium channel blockers are contra-indicated in some ventricular arrhythmias,47 and the likelihood that patients who have drug-induced LQTS may have other underlying cardiac conditions,19 one should be cautious about prescribing calcium channel blockers. Conversely, it would be reasonable to consider using calcium channel blockers to treat patients with LQTS type 2 (i.e. patients with an isolated loss of IKr function) who continue to have cardiac events despite treatment with ß-blockers.48
4.1 Limitations
It is much more problematic to study gene expression in adult human samples, as opposed to laboratory animals, as there is necessarily a much longer time interval between death and obtaining tissue samples to process for mRNA expression. For this reason, in addition to analysing the full GTEx dataset, we also restricted our analysis to only those samples where the RIN value was >7, which resulted in exclusion of ∼half of the samples. Similarly, it is difficult to obtain human heart samples for functional studies. For this reason, we used human iPSC studies. It should be noted, however, that some ion channels important for function in adult cardiac myocytes are not expressed at significant levels in immature cardiac myocytes49 and so iPSC-CM may not be the ideal system for looking at ion channel modules other than KCNH2 and CACNA1C. A further potential limitation of using iPSC-derived cardiac myocytes is the possibility that differences in maturity between wells and between cell lines could contribute to the differences we observed in our functional studies. This does not detract from the observation that wells that contained cells with higher levels of expression of CACNA1C and KCNH2 showed less prolongation of FPD in response to IKr drug block, however, we acknowledge that the inter-individual differences we observed in immature cardiac myocytes may be larger than the differences one might see in adult myocytes.
4.2 Conclusion
Co-expression of CACNA1C and KCNH2 ion channel genes in the heart enhances the robustness of cardiac electrical activity. Furthermore, we show that the subset of the population who have above average levels of calcium channel expression are most at risk from arrhythmias caused by drug block of the rapid delayed rectifier potassium current. We also suggest that the combination of technologies we have used should be applicable to identifying modules of co-expressed genes important in other fundamental physiological processes as well as investigating how the different modules of co-expressed genes interact to co-ordinate whole-of-cell function. Similar approaches should also be applicable to investigating the transcription factors and/or networks that control the co-expression of rhythmonome gene modules.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
This work was supported by the Victor Chang Cardiac Research Institute Innovation Centre, funded by the NSW Government. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this article were obtained from the GTEx Portal on 16 February 2020.
Conflict of interest: none declared.
Contributor Information
Sara Ballouz, Garvan-Weizmann Centre for Cellular Genomics, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst NSW 2010, Australia; University of New South Wales, Sydney, Kensington, NSW 2052, Australia; Stanley Institute for Cognitive Genomics, Cold Spring Harbor Laboratory, Cold Spring Harbor, One Bungtown Road, NY 11724, USA.
Melissa M Mangala, Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool Street, Darlinghurst, New South Wales 2010, Australia.
Matthew D Perry, University of New South Wales, Sydney, Kensington, NSW 2052, Australia; Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool Street, Darlinghurst, New South Wales 2010, Australia.
Stewart Heitmann, Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool Street, Darlinghurst, New South Wales 2010, Australia.
Jesse A Gillis, Stanley Institute for Cognitive Genomics, Cold Spring Harbor Laboratory, Cold Spring Harbor, One Bungtown Road, NY 11724, USA.
Adam P Hill, University of New South Wales, Sydney, Kensington, NSW 2052, Australia; Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool Street, Darlinghurst, New South Wales 2010, Australia.
Jamie I Vandenberg, University of New South Wales, Sydney, Kensington, NSW 2052, Australia; Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool Street, Darlinghurst, New South Wales 2010, Australia.
Funding
This work was supported by grants from the National Health and Medical Research Council (NHMRC) (App1116948 to J.I.V., App1074386 to J.I.V., and App1164518 to A.P.H.) and the National Institutes of Health (NIH) (R01LM012736 to J.A.G. and R01MH113005 to J.A.G.).
Data availability
Raw data for MEA recordings have been deposited at DRYAD database: doi: https://doi.org/10.5061/dryad.612jm640k. Details of the Nanostring codes and the raw data for mRNA expression levels have been deposited at National Center for Biotechnology Information Gene Expression Omnibus database under accession GSE146667. Code for the analysis of the public RNA Seq datasets is available at https://github.com/sarbal/hERG-cal and for the AP modelling is available at https://git.victorchang.edu.au/projects/CC/repos/ordcoreg.
Translational perspective
Here, we show, using meta-analysis of multiple independent human gene expression datasets, that there is co-expression of KCNH2–CACNA1C in human heart tissue which was then confirmed in human cardiac myocytes derived from induced pluripotent stem cells. Both in silico and functional studies show that the co-expression of CACNA1C and KCNH2 increases the robustness of cardiac electrical signalling. Our data also suggest that those patients who express higher levels of CACNA1C and KCNH2 are likely to be more susceptible to arrhythmias when exposed to drugs that block IKr, the major cause of drug-induced cardiac arrhythmias.
References
- 1. Zipes DP, Wellens HJ. Sudden cardiac death. Circulation 1998;98:2334–2351. [DOI] [PubMed] [Google Scholar]
- 2. Goyal V, Jassal DS, Dhalla NS. Pathophysiology and prevention of sudden cardiac death. Can J Physiol Pharmacol 2016;94:237–244. [DOI] [PubMed] [Google Scholar]
- 3. Moss AJ. MADIT‐I and MADIT‐II. J Cardiovasc Electrophysiol 2003;14(Suppl. 9):S96–S98. [DOI] [PubMed] [Google Scholar]
- 4. Behr E, Ensam B. New approaches to predicting the risk of sudden death. Clin Med 2016;16:283–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marder E, Goaillard J-M. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci 2006;7:563–574. [DOI] [PubMed] [Google Scholar]
- 6. Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y, Lusis AJ. “Good enough solutions” and the genetics of complex diseases. Circ Res 2012;111:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prinz AA, Billimoria CP, Marder E. Alternative to hand-tuning conductance-based models: construction and analysis of databases of model neurons. J Neurophysiol 2003;90:3998–4015. [DOI] [PubMed] [Google Scholar]
- 8. Britton OJ, Bueno-Orovio A, Van Ammel K, Lu HR, Towart R, Gallacher DJ, Rodriguez B. Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc Natl Acad Sci U S A 2013;110:E2098–E2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sarkar AX, Sobie EA. Quantification of repolarization reserve to understand inter-patient variability in the response to pro-arrhythmic drugs: a computational analysis. Heart Rhythm 2011;8:1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bányász T, Horváth B, Jian Z, Izu LT, Chen-Izu Y. Sequential dissection of multiple ionic currents in single cardiac myocytes under action potential-clamp. J Mol Cell Cardiol 2011;50:578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rees CM, Yang J-H, Santolini M, Lusis AJ, Weiss JN, Karma A. The Ca2+ transient as a feedback sensor controlling cardiomyocyte ionic conductances in mouse populations. eLife 2018;7:4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang CL-H. Murine electrophysiological models of cardiac arrhythmogenesis. Physiol Rev 2017;97:283–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O’Leary T, Sutton AC, Marder E. Computational models in the age of large datasets. Curr Opin Neurobiol 2015;32:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schulz DJ, Goaillard J-M, Marder EE. Quantitative expression profiling of identified neurons reveals cell-specific constraints on highly variable levels of gene expression. Proc Natl Acad Sci U S A 2007;104:13187–13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 1998;95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaiteri C, Ding Y, French B, Tseng GC, Sibille E. Beyond modules and hubs: the potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav 2014;13:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P. Coexpression analysis of human genes across many microarray data sets. Genome Res 2004;14:1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ballouz S, Verleyen W, Gillis J. Guidance for RNA-seq co-expression network construction and analysis: safety in numbers. Bioinformatics 2015;31:2123–2130. [DOI] [PubMed] [Google Scholar]
- 19. Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med 2004;350:1013–1022. [DOI] [PubMed] [Google Scholar]
- 20. Zoubarev A, Hamer KM, Keshav KD, McCarthy EL, Santos JRC, Van Rossum T, McDonald C, Hall A, Wan X, Lim R, Gillis J, Pavlidis P. Gemma: a resource for the reuse, sharing and meta-analysis of expression profiling data. Bioinformatics 2012;28:2272–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. The GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burridge PW, Diecke S, Matsa E, Sharma A, Wu H, Wu JC. Modeling cardiovascular diseases with patient-specific human pluripotent stem cell-derived cardiomyocytes. Methods Mol Biol 2016;1353:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, Plowright AT, Needham EJ, Wang QD, Gregorevic P, Xin M, Thomas WG, Parton RG, Nielsen LK, Launikonis BS, James DE, Elliott DA, Porrello ER, Hudson JE. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci U S A 2017;114:E8372–E8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perry MD, Ng CA, Mangala MM, Ng TYM, Hines AD, Liang W, Xu MJO, Hill AP, Vandenberg JI. Pharmacological activation of IKr in models of long QT Type 2 risks overcorrection of repolarization. Cardiovasc Res 2020;116:1434–1445. [DOI] [PubMed] [Google Scholar]
- 25. O’Hara T, Virág L, Varró A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 2011;7:e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mann SA, Imtiaz M, Winbo A, Rydberg A, Perry MD, Couderc JP, Polonsky B, McNitt S, Zareba W, Hill AP, Vandenberg JI. Convergence of models of human ventricular myocyte electrophysiology after global optimization to recapitulate clinical long QT phenotypes. J Mol Cell Cardiol 2016;100:25–34. [DOI] [PubMed] [Google Scholar]
- 27. Krogh-Madsen T, Jacobson AF, Ortega FA, Christini DJ. Global optimization of ventricular myocyte model to multi-variable objective improves predictions of drug-induced torsades de pointes. Front Physiol 2017;8:e1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heitmann S, Aburn MJ, Breakspear M. The brain dynamics toolbox for Matlab. Neurocomputing 2018;315:82–88. [Google Scholar]
- 29. Sadrieh A, Mann SA, Subbiah RN, Domanski L, Taylor JA, Vandenberg JI, Hill AP. Quantifying the origins of population variability in cardiac electrical activity through sensitivity analysis of the electrocardiogram. J Physiol 2013;591:4207–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bush WS, Crawford DC, Alexander C, George AL Jr, Roden DM, Ritchie MD. Genetic variation in the rhythmonome: ethnic variation and haplotype structure in candidate genes for arrhythmias. Pharmacogenomics 2009;10:1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sah R, Ramirez RJ, Backx PH. Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate. Circ Res 2002;90:165–173. [DOI] [PubMed] [Google Scholar]
- 32. Shimizu W, Moss AJ, Wilde AAM, Towbin JA, Ackerman MJ, January CT, Tester DJ, Zareba W, Robinson JL, Qi M, Vincent GM, Kaufman ES, Hofman N, Noda T, Kamakura S, Miyamoto Y, Shah S, Amin V, Goldenberg I, Andrews ML, McNitt S. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol 2009;54:2052–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harris K, Aylott M, Cui Y, Louttit JB, McMahon NC, Sridhar A. Comparison of electrophysiological data from human-induced pluripotent stem cell-derived cardiomyocytes to functional preclinical safety assays. Toxicol Sci 2013;134:412–426. [DOI] [PubMed] [Google Scholar]
- 34. Pedersen HS, Elming H, Seibæk M, Burchardt H, Brendorp B, Torp-Pedersen C, Køber L. Risk factors and predictors of Torsade de Pointes ventricular tachycardia in patients with left ventricular systolic dysfunction receiving dofetilide. Am J Cardiol 2007;100:876–880. [DOI] [PubMed] [Google Scholar]
- 35. Sayed N, Liu C, Wu JC. Translation of human-induced pluripotent stem cells. J Am Coll Cardiol 2016;67:2161–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eichel CA, Ríos-Pérez EB, Liu F, Jameson MB, Jones DK, Knickelbine JJ, Robertson GA. A microtranslatome coordinately regulates sodium and potassium currents in the human heart. Elife 2019;8:e52654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Min B, Zheng M. Correlated network of networks enhances robustness against catastrophic failures. PLoS One 2018;13:e0195539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lundby A, Rossin EJ, Steffensen AB, Acha MR, Newton-Cheh C, Pfeufer A, Lynch SNQT Interval International GWAS Consortium (QT-IGC)Olesen SP, Brunak S, Ellinor PT, Jukema JW, Trompet S, Ford I, Macfarlane PW, Krijthe BP, Hofman A, Uitterlinden AG, Stricker BH, Nathoe HM, Spiering W, Daly MJ, Asselbergs FW, van der Harst P, Milan DJ, de Bakker PI, Lage K, Olsen JV. Annotation of loci from genome-wide association studies using tissue-specific quantitative interaction proteomics. Nat Methods 2014;11:868–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 2005;85:1205–1253. [DOI] [PubMed] [Google Scholar]
- 40. Ponce-Balbuena D, Guerrero-Serna G, Valdivia CR, Caballero R, Diez-Guerra FJ, Jiménez-Vázquez EN, Ramírez RJ, Monteiro da Rocha A, Herron TJ, Campbell KF, Willis BC, Alvarado FJ, Zarzoso M, Kaur K, Pérez-Hernández M, Matamoros M, Valdivia HH, Delpón E, Jalife J. Cardiac Kir2.1 and NaV1.5 channels traffic together to the sarcolemma to control excitability. Circ Res 2018;122:1501–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR, Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 2012;109:E2134–E2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deschênes I, Armoundas AA, Jones SP, Tomaselli GF. Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J Mol Cell Cardiol 2008;45:336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J 2014;167:292–300. [DOI] [PubMed] [Google Scholar]
- 44. Roden DM, Iansmith DH. Effects of low potassium or magnesium concentrations on isolated cardiac tissue. Am J Med 1987;82:18–23. [DOI] [PubMed] [Google Scholar]
- 45. Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT, Wallis R, Camm AJ, Hammond TG. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 2003;58:32–45. [DOI] [PubMed] [Google Scholar]
- 46. Farkas AS, Makra P, Csík N, Orosz S, Shattock MJ, Fülöp F, Forster T, Csanády M, Papp JG, Varró A, Farkas A. The role of the Na+/Ca2+ exchanger, INa and ICaL in the genesis of dofetilide-induced torsades de pointes in isolated, AV-blocked rabbit hearts. Br J Pharmacol 2009;156:920–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Akhtar M, Tchou P, Jazayeri M. Use of calcium channel entry blockers in the treatment of cardiac arrhythmias. Circ 1989;80:IV31–IV39. [PubMed] [Google Scholar]
- 48. Komiya N, Tanaka K, Doi Y, Fukae S, Nakao K, Isomoto S, Seto S, Yano K. A patient with LQTS in whom verapamil administration and permanent pacemaker implantation were useful for preventing torsade de pointes. Pacing Clin Electrophysiol 2004;27:123–124. [DOI] [PubMed] [Google Scholar]
- 49. Robertson C, Tran DD, George SC. Concise review: maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem Cells 2013;31:829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data for MEA recordings have been deposited at DRYAD database: doi: https://doi.org/10.5061/dryad.612jm640k. Details of the Nanostring codes and the raw data for mRNA expression levels have been deposited at National Center for Biotechnology Information Gene Expression Omnibus database under accession GSE146667. Code for the analysis of the public RNA Seq datasets is available at https://github.com/sarbal/hERG-cal and for the AP modelling is available at https://git.victorchang.edu.au/projects/CC/repos/ordcoreg.