

Abstract
Background
Despite improved surgical approaches for chronic limb‐threatening ischemia (CLTI), amputation rates remain high and contributing tissue‐level factors remain unknown. The purpose of this study was twofold: (1) to identify differences between the healthy adult and CLTI limb muscle proteome, and (2) to identify differences in the limb muscle proteome of CLTI patients prior to surgical intervention or at the time of amputation.
Methods and results
Gastrocnemius muscle was collected from non‐ischemic controls (n = 19) and either pre‐interventional surgery (n = 10) or at amputation outcome (n = 29) CLTI patients. All samples were subjected to isobaric tandem‐mass‐tag‐assisted proteomics. The mitochondrion was the primary classification of downregulated proteins (> 70%) in CLTI limb muscles and paralleled robust functional mitochondrial impairment. Upregulated proteins (> 38%) were largely from the extracellular matrix. Across the two independent sites, 39 proteins were downregulated and 12 upregulated uniformly. Pre‐interventional CLTI muscles revealed a robust upregulation of mitochondrial proteins but modest functional impairments in fatty acid oxidation as compared with controls. Comparison of pre‐intervention and amputation CLTI limb muscles revealed mitochondrial proteome and functional deficits similar to that between amputation and non‐ischemic controls. Interestingly, these observed changes occurred despite 62% of the amputation CLTI patients having undergone a prior surgical intervention.
Conclusions
The CLTI proteome supports failing mitochondria as a phenotype that is unique to amputation outcomes. The signature of pre‐intervention CLTI muscle reveals stable mitochondrial protein abundance that is insufficient to uniformly prevent functional impairments. Taken together, these findings support the need for future longitudinal investigations aimed to determine whether mitochondrial failure is causally involved in amputation outcomes from CLTI.
Keywords: metabolism, peripheral artery disease, surgery, vascular disease
Proteomic analyses of CLTI muscle specimens identify a distinct limb muscle proteome signature characterized by a severe mitochondriopathy in amputation‐stage specimens, which was discovered to differ from non‐PAD controls and CLTI pre‐surgical intervention limb muscles. Coincident with the proteome signature, mitochondrial function was severely impaired in amputation‐stage CLTI specimens. Compared to non‐PAD controls, interventional‐stage CLTI specimens displayed an upregulation of the mitochondrial proteome; however, this did not afford any improvement in mitochondrial oxidative phosphorylation.

Abbreviations
- ABI
ankle‐brachial index
- CLTI
chronic limb threatening ischemia
- MALE
major adverse limb event
- OXPHOS
oxidative phosphorylation
- PAD
peripheral artery disease
1. INTRODUCTION
The most severe manifestation of peripheral artery disease (PAD) is chronic limb‐threatening ischemia (CLTI). While this occurs in only 5–10% of patients, it carries substantially greater risk for major limb amputation, cardiovascular events, and mortality. 1 , 2 CLTI patients often present with more complex patterns of disease and although endovascular recanalization or revascularization (bypass) surgical procedures have improved technically, resolution of symptoms occurs in only 25% of CLTI patients. 3 In fact, major adverse limb events remain unacceptably high, 4 , 5 highlighting the dire need to better understand CLTI pathobiology. Recent work using wide‐ranging “omics” and screening technologies have revealed tissue characteristics unique to the CLTI presentation (compared to non‐PAD or mild claudicants), including the transcriptome, 6 , 7 , 8 cytokine profile, 9 metabolome, 10 , 11 mitochondrial function, 6 , 12 , 13 and lipid profiles 11 of the limb muscle. Each of these studies has provided new insight into the biological underpinnings of the affected CLTI limb, a necessity to develop new ideas to improve lower limb tissue level perfusion and/or alleviate the severe myopathy that persists in these patients. The proteome of the CLTI peripheral limb muscle has not been described but is a likely functional outcome to the previously described changes. Given that mRNA changes do not necessarily result in differences in abundance, analysis of the CLTI proteome would provide a detailed biological portrait to drive functional therapeutic target design for CLTI. The purpose of this study was twofold: (1) to identify differences between the non‐ischemic control and CLTI limb muscle proteome, and (2) to identify differences in the limb muscle proteome of CLTI patients prior to surgical intervention and at the time of amputation. Non‐PAD control and CLTI patients were recruited across two independent vascular surgery practices and limb muscle tissues collected for isobaric tandem‐mass‐tag (TMT) assisted quantitative proteomics.
HIGHLIGHTS
Proteomic analysis of CLTI muscle specimens identifies a distinct limb muscle proteome signature characterized by a severe mitochondriopathy in amputation‐stage specimens, which was discovered to differ from non‐PAD controls and CLTI presurgical intervention limb muscles. Coincident with the proteome signature, mitochondrial function was severely impaired in amputation‐stage CLTI specimens.
Compared to non‐PAD controls, interventional‐stage CLTI specimens displayed an upregulation of the mitochondrial proteome; however this did not afford any improvement in mitochondrial oxidative phosphorylation.
Regardless of CLTI treatment stage, the largest class of upregulated proteins were related to the extracellular matrix.
2. MATERIALS AND METHODS
2.1. Study populations and specimen collection
Gastrocnemius muscle specimens were collected from 26 older adult non‐PAD controls (Con) and 39 CLTI patients across two vascular clinics (University of Florida and East Carolina University). Five presurgery patients underwent bypass interventions and five underwent endovascular procedures. Muscle specimens were collected within the confines of the operating rooms (CLTI patients) or via percutaneous muscle biopsy using sterile procedures previously described. 6 , 14 A portion of the muscle was quickly trimmed of fat/connective tissue and snap‐frozen in liquid nitrogen for proteomic analysis. Another portion of the fresh muscle specimen was processed immediately for mitochondrial function assessment as described in detail previously. 6 Non‐PAD control participants enrolled either had an ankle‐brachial index (ABI) greater than or equal to 1.0 (University of Florida) or based on diagnosis‐ and symptom‐free by self‐report (East Carolina University). This study was approved by the institutional review boards at the University of Florida, Malcom Randall VA Medical Center, and East Carolina University. All study procedures were carried out according to the Declaration of Helsinki and participants were fully informed about the research and informed consent was obtained.
2.2. TMT‐labeled proteomics analysis of skeletal muscle
University of Florida: Frozen muscle samples (∼10 mg) from each population (n = 10 control, n = 10 CLTI presurgery, and n = 10 CLTI amputation) were homogenized in ice‐cold CHAPS lysis buffer (150 mM KCl, 50 mM HEPES, 0.1% CHAPS, 1× cOmplete ULTRA mini EDTA‐free protease inhibitor tablet, pH 7.4) using a glass Teflon homogenizer (Wheaton). To release protein bound to DNA, the lysates were further processed with syringe lysis (lysate passaging 10 times through a 23‐gauge needle attached to a 1 ml syringe). Following centrifugation (16,000 × g) for 10 min at 4°C, the supernatant was collected and the protein concentration of each sample was determined using BCA protein assay kit (ThermoFisher Scientific Cat. no. SL256970). To achieve reduction and alkylation of cysteine residues, 5 μl of 200 mM Bond‐Breaker TCEP solution (ThermoFisher Scientific cat. no.77720) was added to 200 μg protein sample and incubated at 55°C for 1 h. This step was followed by addition of 5 μl of 375 mM iodoacetamide addition and incubation at room temperature for 30 min during which the samples were protected from light. The samples, thereafter, underwent chloroform/methanol/water precipitation and brief decanting (∼2 min) using a vacuum centrifuge. Following the precipitation, the pellets were re‐suspended with 100 μl of TEAB lysis buffer (ThermoFisher Scientific cat. no. 90114), 5 μl of trypsin was added, and incubated overnight at 37°C for protein digestion. Subsequently, the peptide samples were labeled using 11‐plex tandem mass tag (TMT)‐based isobaric stable isotope label reagents following the manufacturer's instructions (ThermoFisher Scientific cat. no. A34808). In brief, each TMT tag vial was dissolved with 41 μl of anhydrous acetonitrile (ThermoFisher Scientific ca. no. 51101) and the TMT label reagent was added to each peptide sample followed by 1 h incubation protected from light at room temperature with gentle shaking. After quenching (∼15 min) the reaction with 8 μl of 5% hydroxylamine (ThermoFisher Scientific ca. no. 90115), labeled samples were submitted to the UF ICBR Proteomics Core facility where the samples were combined and purified using C18 spin columns for mass spectroscopy quantification. Each 11‐plex TMT kit involved analysis of a pooled control sample that was used to account for batch variability and normalization across runs.
East Carolina University: Frozen muscle samples (∼10 mg) from each population (n = 16 control and n = 19 amputation) were combined into 9 grouped Control (7 samples contained 2 patients each) and 11 grouped amputation aliquots (8 samples contained 2 patients each) and lysed in ice‐cold 8 M Urea Lysis Buffer (8 M urea in 40 mM Tris, pH 8.0, 30 mM NaCl, 1 mM CaCl2, 1x cOmplete ULTRA mini EDTA‐free protease inhibitor tablet), as described previously. 15 Pooling muscle specimens were performed to maximize patient specimen use across the available resources. Each individual TMT 10‐plex kit involved analysis of a pooled control sample that was used to account for batch variability and normalization across runs. Homogenates were subjected to three freeze‐thaw cycles and further disrupted by sonication with a probe sonicator in three 5 second bursts (Q Sonica #CL‐188; amplitude of 30). Centrifugation was performed at 10,000 × g for 10 min at 4°C to pellet insoluble material. Protein concentration was determined by BCA, and equal amounts of protein (100 μg) were subjected to reduction and alkylation (5 mM DTT at 32°C for 30 min, 15 mM iodoacetamide for 30 min in the dark). Unreacted iodoacetamide was quenched by the addition of DTT up to 15 mM. Digestion was first performed with Lys C (ThermoFisher Scientific, cat. no. 90307; 1:100 w:w; 1 μg enzyme per 100 μg protein) for 4 h at 32°C. Following dilution to 1.5 M urea with 40 mM Tris (pH 8.0), 30 mM NaCl, 1 mM CaCl2, the samples were trypsin digested overnight (Promega; cat. no. V5113; 50:1 w/w, protein:enzyme) at 32°C. Samples were acidified to 0.5% TFA and centrifuged at 4000 × g for 10 min at 4°C. Supernatant containing soluble peptides was desalted on a 50 mg tC18 SEP‐PAK solid‐phase extraction column (Waters, cat. no. WAT054955) and eluted (500 μL 25% acetonitrile/0.1% TFA and 2 × 500 μL 50% acetonitrile/0.1% TFA). Eluate was frozen and lyophilized. TMT labeling was performed as previously described. 15 The 20 samples from skeletal muscle were re‐suspended in 100 μL of 200 mM triethylammonium bicarbonate (TEAB), mixed with a unique 10‐plex Tandem Mass Tag (TMT10) reagent (0.8 mg re‐suspended in 50 μL100% acetonitrile) from 2× TMT10 kits, and shaken for 4 h at room temperature (ThermoFisher Scientific). Following quenching with 0.8 μL 50% hydroxylamine, all samples from each kit were combined into respective tubes, frozen, and lyophilized. Multiplexed samples were re‐suspended in ∼1 mL of 0.5% TFA and again subjected to solid‐phase extraction, but with a 100 mg tC18 SEP‐PAK SPE column (Waters, cat. no. WAT023590). The multiplexed peptide samples were subjected to high pH reversed‐phase fractionation according to the manufacturer's instructions (ThermoFisher Scientific, cat. no. 84868). Following elution, fractions (8 total fractions per kit) were frozen and lyophilized.
2.3. LC‐MS/MS for TMT proteomics
University of Florida: Labeled peptides were desalted with C18‐solid‐phase extraction and dissolved in strong cation exchange (SCX) solvent A (25% (v/v) acetonitrile, 10 mM ammonium formate, and 0.1% (v/v) formic acid, pH 2.8). The peptides were fractionated using an Agilent HPLC 1260 with a polysulfoethyl A column (2.1 × 100 mm, 5 μm, 300 Å; PolyLC, Columbia, MD, USA). Peptides were eluted with a linear gradient of 0–20% solvent B (25% (v/v) acetonitrile and 500 mM ammonium formate, pH 6.8) over 50 min followed by ramping up to 100% solvent B in 5 min. The absorbance at 280 nm was monitored and a total of 16 fractions were collected. The fractions were lyophilized, desalted, and resuspended in LC solvent A (0.1% formic acid in 99.9% water (v/v)). A hybrid quadrupole Orbitrap (Q Exactive Plus) MS system (Thermo Fisher Scientific, Bremen, Germany) was used with high energy collision dissociation (HCD) in each MS and MS/MS cycle. The MS system was interfaced with an automated Easy‐nLC 1000 system (Thermo Fisher Scientific, Bremen, Germany). Each sample fraction was loaded onto an Acclaim Pepmap 100 pre‐column (20 mm × 75 μm; 3 μm‐C18) and separated on a PepMap RSLC analytical column (250 mm × 75 μm; 2 μm‐C18) at a flow rate at 350 nl/min during a linear gradient from solvent A (0.1% formic acid (v/v)) to 30% solvent B (0.1% formic acid (v/v) and 99.9% acetonitrile (v/v)) for 95 min, to 98% solvent B for 15 min, and hold 98% solvent B for additional 30 min. Full MS scans were acquired in the Orbitrap mass analyzer over m/z 400–2000 range with resolution 70,000 at 200 m/z. The top ten most intense peaks with charge state ≥ 3 were fragmented in the HCD collision cell normalized collision energy of 28%, (the isolation window was 0.7 m/z). The maximum ion injection times for the survey scan and the MS/MS scans were 250 ms, respectively and the ion target values were set to 3e6 and 1e6, respectively. Selected sequenced ions were dynamically excluded for 60 s.
East Carolina University: nLC‐MS/MS was performed as described previously. 15 Peptide fractions were suspended in 0.1% formic acid (0.25 μg/μL) following peptide quantification (ThermoFisher Scientific). All samples were subjected to nanoLC‐MS/MS analysis using an UltiMate 3000 RSLCnano system (ThermoFisher Scientific) coupled to a Q Exactive PlusHybrid Quadrupole‐Orbitrap mass spectrometer (ThermoFisher Scientific) via nanoelectrospray ionization source. For each injection of 4 μL (1 μg), the sample was first trapped on an Acclaim PepMap 100 20 mm × 0.075 mm trapping column (ThermoFisher Scientific) 5 μl/min at 98/2 v/v water/acetonitrile with 0.1% formic acid, after which the analytical separation was performed over a 90‐min gradient (flow rate of 300 nanoliters/min) of 3–30% acetonitrile using a 2 μm EASY‐Spray PepMap RSLC C18 75 μm × 250 mm column (ThermoFisher Scientific). Column temperature was 55°C. MS1 was performed at 70,000 resolution, AGC target of 3 × 106 ions and a maximum IT of 60 ms (scan range 300–1750 m/z). MS2 spectra were collected by data‐dependent acquisition (DDA) of the top 20 most abundant precursor ions with a charge greater than 1 per MS1 scan, with dynamic exclusion enabled for 45 s. Precursor ions filtered with a 0.7 m/z isolation window and fragmented with a normalized collision energy of 30. MS2 scans were performed at 35,000 resolution, AGC target of 1 × 105 ions, and a maximum IT of 60 ms.
2.4. Data analysis for proteomics
University of Florida: The raw MS/MS data files were processed by a thorough database searching approach considering biological modification and amino acid substitution against Uniprot Homo sapiens [9606] database (downloaded on May 1, 2020; 188,558 entries) using the Proteome Discoverer v2.4 (Thermo Fisher Scientific), with the SEQUEST algorithm. 16 The following parameters were used for all the searching: peptide tolerance at 10 ppm, tandem MS tolerance at ± 0.02 Da, peptide charges of 2+ to 5+, trypsin as the enzyme, allowing one missed cleavage, TMT label (229.163 Da on peptide N‐term and K) and carbamidomethyl (C) as fixed modifications, oxidation (15.995 Da on M) as a variable modification. The false discovery rate (FDR) was calculated using Percolator algorithm in the Proteome Discoverer workflow based on the search results against a decoy database and was set at 1% FDR. For protein quantification, only MS/MS spectra that were unique to a particular protein and where the sum of the signal‐to‐noise ratios for all the peak pairs > 9 were used for quantification. M2 reporter (TMT) intensities were summed together for each TMT channel, each channel's sum was divided by the average of all channels’ sums, resulting in channel‐specific loading control normalization factors to correct for any deviation from equal protein input in the 11‐plex experiments. Reporter intensities were divided by the loading control normalization factors for each respective TMT channel. All loading control‐normalized reporter intensities were converted to log2 space and the average value from the combined samples was subtracted from each sample specific measurement to normalize the relative measurements to the mean. For each comparison, protein abundances were analyzed for group average, standard deviation, two‐tailed Student's t‐test (equal variance), and a Benjamini‐Hochberg 17 adjusted P‐value. All raw proteomics data are available online using accession numbers PXD021849 (Proteome Xchange 18 ) or JPST000852 (jPOST Repository 19 ). Gene ontology (GO) analyses were performed using PANTHER GO Enrichment analysis 20 , 21 , 22 using P < 0.05.
East Carolina University: Proteome Discoverer 2.2 (PDv2.2) was used for raw data analysis, with default search parameters including oxidation (15.995 Da on M) as a variable modification and carbamidomethyl (57.021 Da on C) and TMT (229.163 Da on peptide N‐term and K) as fixed modifications, and two missed cleavages (full trypsin specificity). Data were searched against Uniprot Homo sapiens database. PSMs were filtered to a 1% FDR and then grouped into unique peptides while maintaining a 1% FDR. Peptides were grouped into proteins using the rules of strict parsimony and proteins were filtered to 1% FDR using the Protein FDR Validator node of PD2.2. MS2 reporter ion intensities for all PSMs having co‐isolation interference below 0.5 (50% of the ion current in the isolation window) and an average S/N > 10 for reporter ions were summed together at the peptide and protein level. Imputation was performed via low abundance resampling. The protein group tab in the PDv2.2 results was exported as tab delimited.txt. files, and analyzed as previously described. 15 M2 reporter (TMT) intensities were summed together for each TMT channel, each channel's sum was divided by the average of all channels’ sums, resulting in channel‐specific loading control normalization factors to correct for any deviation from equal protein input in the 10‐plex experiments. Reporter intensities were divided by the loading control normalization factors for each respective TMT channel. All loading control‐normalized reporter intensities were converted to log2 space and the average value from the combined samples was subtracted from each sample‐specific measurement to normalize the relative measurements to the mean. For statistical comparison of Con to CTLI, condition average, standard deviation, P‐value (P, two‐tailed Student's t‐test, assuming equal variance), and adjusted P‐value (P adjusted, Benjamini Hochberg FDR correction) were calculated. 17 For protein‐level quantification, only Master Proteins—or the most statistically significant protein representing a group of parsimonious proteins containing common peptides identified at 1% FDR—were used for quantitative comparison. All raw data are available online using accession number PXD025810 for Proteome Xchange 18 and accession number JPST001157 for jPOST Repository. 19 Gene ontology (GO) analyses were performed using PANTHER GO Enrichment analysis 20 , 21 , 22 using P < 0.05.
2.5. Preparation of permeabilized muscle fibers
A portion of the muscle biopsy specimen was dissected and immediately placed in ice‐cold buffer X (50 mM K‐MES, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 20 mM taurine, 5.7 mM ATP, 14.3 mM phosphocreatine, and 6.56 mM MgCl2‐6H2O, pH 7.1) for preparation of permeabilized fiber bundles. 6 , 14 , 23 Fiber bundles were mechanically separated along their longitudinal axis using needle‐tipped forceps under a dissecting scope, and subsequently permeabilized with saponin (30 μg/ml) for 30 min at 4°C on a nutating mixer, and then washed in cold buffer Z (105 mM K‐MES, 30 mM KCl, 1 mM EGTA, 10 mM K2HPO4, 5 mM MgCl2‐6H2O, 0.5 mg/ml BSA, pH 7.1) for 15 min until analysis. Prior to loading fiber bundles for experimentation, they were gently blotted on a kim wipe for exactly 5 s and a wet weight was obtained using a Mettler Toledo MX5 microbalance.
2.6. Mitochondrial respiration measurements
High‐resolution O2 consumption measurements 6 , 24 were conducted at 37˚C in buffer Z (in mmol/l) (105 K‐MES, 30 KCl, 1 EGTA, 10 K2HPO4, 5 MgCl26H2O, 0.5 mg/ml BSA, pH 7.1), supplemented with creatine monohydrate (5 mM), using the OROBOROS O2K Oxygraph. To assess mitochondrial function in physiologically relevant conditions, this study utilized a novel creatine‐kinase clamp system to set the level of cellular energy demand to which the fiber bundles were exposed. 25 , 26 , 27 , 28 First, bundles were energized with either carbohydrate (5 mM pyruvate and 2.5 mM malate) or fatty acid (0.2 mM octanoylcarnitine and 2.5 mM malate) and measurements of state 2 oxygen consumption were collected. Next, the creatine kinase clamp was added including 20 U/ml creatine kinase, 5 mM ATP, and 1 mM phosphocreatine (PCr) to mimic a near‐maximal exercise condition. Subsequent additions of PCr were added step‐wise to bring the cellular energy demand down to resting conditions. The slope of the relationship between cellular energy demand (∆GATP) and oxygen consumption (JO2) was calculated. The rate of respiration was expressed as pmol/sec/mg fiber dry weight. All respiration measurements were conducted at 37°C and a working range [O2] of ∼350 to 200 μM.
2.7. Statistical analysis
This was cross‐sectional study in which participants were enrolled and analyses performed based on the available resources. Due to the exploratory nature, a priori sample size calculations were not performed. Data are presented as mean ± SD. Normality of all data was assessed using the Shapiro‐Wilk test. Data that were found to not be normally distributed were analyzed using a Kruskal‐Wallis test. Comparisons across the three groups were done using a one‐way ANOVA with Tukey's post hoc multiple comparisons when pairwise comparisons were appropriate. Chi‐Square analysis was used to determine differences in population proportions for relevant clinical characteristics. All statistical analysis was performed in GraphPad Prism (Version 8.0) or using Vassar Stats (http://vassarstats.net). For proteomics analysis, a Benjamini‐Hochberg 17 adjusted P < 0.1 was considered significant with a false discovery rate (FDR) of 0.05. In all other cases, P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Participant characteristics
In this study, limb muscle specimens were collected from non‐ischemic control participants and CLTI patients from two vascular clinics. All sample collection, protein digestion, and labeling, and mass spectrometry analysis were performed independently at each institution. Table 1 displays the physical and clinical characteristics of the 65 study participants across both sites. The physical characteristics for CLTI and non‐ischemic control groups were relatively well‐matched in age, although the control group had less comorbid conditions and less medication usage. Gastrocnemius muscle specimens from CLTI patients were harvested from the operating room immediately prior to a surgical intervention (n = 10, 50% endovascular and 50% revascularization) or amputation (n = 29). CLTI patient comorbidities across both sites included hypertension (90%), hyperlipidemia (82%), diabetes (69%), coronary artery disease (54%), chronic obstructive pulmonary disease (23%), and renal disease (33%). The majority (74%) of CLTI patients were either former or active smokers. Of the CLTI patients whose tissue was collected at the time of amputation, 62% had previously undergone a surgical intervention.
TABLE 1.
Patient characteristics
| University of Florida | East Carolina University | ||||||
|---|---|---|---|---|---|---|---|
| CLTI | CLTI | ||||||
| Characteristic |
Control (N = 10) |
Presurgery (N = 10) |
Amputation (N = 10) |
P‐value (Χ2 or ANOVA) |
Control (N = 16) |
Amputation (N = 19) |
P value (Χ2 or t‐test) |
| Mean age (SD) ‐ yr | 73.9 (7.8) | 64.5 (9.4) | 69.5 (6.2) | 0.043 † | 62.4 (8.4) | 64.6 (10.7) | 0.195 ‡ |
| Female sex – no. (%) | 4 (40) | 0 (0) | 1 (10) | 0.044 | 9 (56) | 6 (32) | 0.269 |
| Overweight/Obese (BMI ≥ 25) – no. (%) | 9 (90) | 7 (70) | 8 (80) | 0.535 | 12 (75) | 12 (63) | 0.476 |
| Ankle‐brachial index (ABI) – (SD) | 1.1 (0.1) | 0.57 (0.27) § | 0.54 (0.35) § | 0.017 † | ND | 0.13 (0.2) § | ND |
| Rutherford Classification – no. (%) | |||||||
| 0 | 10 (100) | 0 (0) | 0 (0) | <0.001 | 16 (100) | 0 (0) | <0.001 |
| 3 | 0 (0) | 2 (20) | 0 (0) | 0.093 | 0 (0) | 0 (0) | ND |
| 4 | 0 (0) | 4 (40) | 4 (40) | 0.624 | 0 (0) | 2 (11) | <0.001 |
| 5 | 0 (0) | 4 (40) | 4 (40) | 0.646 | 0 (0) | 10 (52) | <0.001 |
| 6 | 0 (0) | 0 (0) | 2(20) | 0.454 | 0 (0) | 7 (37) | <0.001 |
| Hemoglobin A1C (SD) | ND | 7.4 (1.1) | 7.7 (1.2) | 0.505 ‡ | ND | 5.7 (0.8) | ND |
| Medical history – no. (%) | |||||||
| Diabetes mellitus type I or II | 4 (40) | 6 (60) | 9 (90) | 0.065 | 0 (0) | 12 (63) | 0.002 |
| Hypertension | 7 (70) | 10 (100) | 10 (100) | 0.536 | 7 (21) | 15 (79) | 0.223 |
| Hyperlipidemia | 4 (40) | 10 (100) | 10 (100) | 0.006 | 1 (6) | 12 (63) | 0.014 |
| Coronary artery disease | 1 (10) | 6 (60) | 9 (90) | 0.001 | 0 (0) | 6 (32) | 0.039 |
| Renal disease | 0 (0) | 1 (10) | 3 (30) | 0.133 | 0 (0) | 9 (47) | 0.009 |
| Chronic obstructive pulmonary disease | 1 (10) | 4 (40) | 3 (30) | 0.303 | 0 (0) | 2 (19) | 0.315 |
| Tobacco use – no. (%) | 4 (40) | 7 (70) | 9 (90) | 0.058 | 0 (0) | 13 (68) | 0.002 |
| Former smoker | 3 (30) | 4 (40) | 7 (70) | 0.175 | 0 (0) | 6 (32) | 0.039 |
| Current smoker | 1 (10) | 3 (30) | 2 (20) | 0.535 | 0 (0) | 7 (36) | 0.024 |
| Medication used – no. (%) | |||||||
| Aspirin | 4 (40) | 8 (80) | 9 (90) | 0.035 | 1 (6) | 11 (58) | 0.019 |
| Statin | 4 (40) | 10 (100) | 10 (100) | <0.001 | 2 (13) | 11 (58) | 0.052 |
| ACE inhibitor | 5 (50) | 5 (50) | 6 (60) | 0.874 | 2 (13) | 6 (32) | 0.254 |
| Cilostazol | 0 (0) | 3 (30) | 4 (40) | 0.089 | 0 (0) | 0 (0) | ND |
| Metformin | 4 (40) | 3 (30) | 4 (40) | 0.865 | 0 (0) | 1 (5) | 0.920 |
| Previous vascular intervention – no. (%) | 0 (0) | 0 (0) | 5 (50) | 0.003 | 0 (0) | 13 (68) | ND |
ANOVA was performed. Chi‐squared (X2) analysis was performed to determine differences in population proportions.
t‐test (two‐tailed) was performed. SD, standard deviation.
Three amputation and two pre‐surgery CLTI patients at UF, and five CLTI patients at ECU had non‐compressible vessels precluding ABI measurement.
3.2. The CLTI limb muscle proteome includes unique alterations to the mitochondrion and extracellular matrix
To get a comprehensive and unbiased view of the proteomic landscape in the limb muscles of CLTI patients, muscle specimens from CLTI patients undergoing amputation and non‐ischemic controls without PAD were collected in two independent vascular clinics in separate geographic locations, individually prepared, and independently analyzed (Figure 1A). Analysis of tissues from patients of the first site (University of Florida) yielded 1130 identified proteins with at least two unique peptides quantified across all 30 muscle samples. At the second site (East Carolina University), a total of 1871 proteins were detected across all 35 patient samples. Volcano plots showing differential protein abundances in CLTI specimens versus controls are shown for each independent clinic in Figure 1B. Gene ontology and protein class analysis of differentially expressed proteins indicated that more than 70% of the downregulated proteins were from the mitochondrion (Figure 1C,E). Venn diagram analysis of data (Figure 1D) identified 39 significantly downregulated proteins across both independent clinical sites (Table 2). The largest proportion (>38%) of CLTI specific upregulated proteins within each independent site population were related to the extracellular matrix/space. Venn diagram analysis revealed 12 proteins significantly upregulated at both vascular clinics (Table 3).
FIGURE 1.
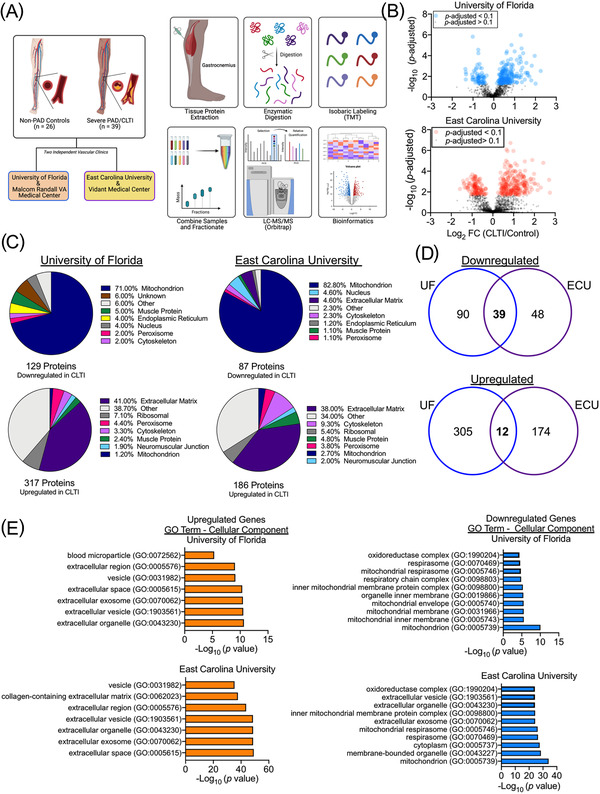
Proteomic Signature of Amputated CLTI limbs. (A) Graphical description of sample collection, processing, and bioinformatics analysis. (B) Volcano plots of proteins identified at both independent vascular clinics. (C) Pie charts depicting the proportions of differentially expressed proteins (upregulated and downregulated) by cellular organelle/component at both sites. (D) Venn diagrams showing similarities and differences in differentially expressed proteins in CLTI specimens across the two clinical sites. (E) Gene ontology analysis of differentially expressed proteins in CLTI demonstrate near identical proteome signatures across clinics
TABLE 2.
Downregulated proteins in CLTI amputation specimens identified at both sites
| University of Florida | East Carolina University | ||||
|---|---|---|---|---|---|
| Gene ID | Gene description | Fold change (CLTI/Control) | P‐value | Fold change (CLTI/Control) | P‐value |
| ACAA2 | 3‐Ketoacyl‐CoA thiolase | 0.64 | 0.00474 | 0.54 | 0.00273 |
| ACADS | Short‐chain‐specific acyl‐CoA dehydrogenase | 0.61 | 0.00000 | 0.43 | 0.00384 |
| ACAT1 | Acetyl‐CoA acetyltransferase | 0.56 | 0.00006 | 0.48 | 0.00035 |
| ACO2 | Aconitate hydratase | 0.73 | 0.00720 | 0.54 | 0.00260 |
| AK3 | GTP:AMP phosphotransferase AK3 | 0.55 | 0.00011 | 0.42 | 0.00224 |
| ALDH5A1 | Succinate‐semialdehyde dehydrogenase | 0.62 | 0.00006 | 0.38 | 0.00172 |
| APOO | MICOS complex subunit MIC26 | 0.62 | 0.00147 | 0.55 | 0.00412 |
| CKMT2 | creatine kinase, mitochondrial 2 | 0.50 | 0.00000 | 0.45 | 0.00145 |
| COQ9 | Ubiquinone biosynthesis protein | 0.59 | 0.00078 | 0.51 | 0.00240 |
| COX4I1 | Cytochrome c oxidase subunit 4 isoform 1 | 0.72 | 0.00615 | 0.56 | 0.00030 |
| COX5A | Cytochrome c oxidase subunit Va | 0.82 | 0.02496 | 0.55 | 0.00035 |
| COX7C | Cytochrome c oxidase subunit 7C | 0.69 | 0.00104 | 0.57 | 0.00185 |
| CRAT | Carnitine O‐acetyltransferase | 0.63 | 0.00014 | 0.70 | 0.00246 |
| CS | Citrate synthase | 0.72 | 0.00493 | 0.58 | 0.00718 |
| CYCS | Cytochrome c | 0.82 | 0.03651 | 0.37 | 0.00085 |
| DECR1 | 2,4‐dienoyl‐CoA reductase | 0.75 | 0.03406 | 0.66 | 0.00052 |
| DLST | Dihydrolipoyllysine‐residue succinyltransferase component of 2‐oxoglutarate dehydrogenase complex | 0.76 | 0.01305 | 0.49 | 0.00653 |
| ECHS1 | Enoyl Coenzyme A hydratase, short chain, 1 | 0.70 | 0.00207 | 0.65 | 0.00100 |
| FHL3 | Four and a half LIM domains protein 3 | 0.80 | 0.02060 | 0.58 | 0.00687 |
| GATD3B | Glutamine amidotransferase‐like class 1 domain‐containing protein 3B | 0.53 | 0.00006 | 0.64 | 0.00061 |
| GOT1 | Aspartate aminotransferase | 0.58 | 0.00014 | 0.55 | 0.00053 |
| HSDL2 | Hydroxysteroid dehydrogenase‐like protein 2 | 0.67 | 0.00024 | 0.56 | 0.00175 |
| IDH2 | Isocitrate dehydrogenase 2 | 0.68 | 0.00179 | 0.56 | 0.00032 |
| MDH2 | Malate dehydrogenase 2 | 0.61 | 0.00289 | 0.61 | 0.00393 |
| NDUFA10 | NADH dehydrogenase (Ubiquinone) 1 alpha subcomplex, 10 | 0.52 | 0.00047 | 0.39 | 0.00137 |
| NDUFC2 | NADH dehydrogenase [ubiquinone] 1 subunit C2 | 0.59 | 0.00058 | 0.63 | 0.00132 |
| NDUFS1 | NADH‐ubiquinone oxidoreductase 75 kDa subunit | 0.74 | 0.00557 | 0.59 | 0.00091 |
| NDUFS6 | NADH dehydrogenase [ubiquinone] iron‐sulfur protein 6 | 0.65 | 0.00038 | 0.55 | 0.00299 |
| NDUFV1 | NADH dehydrogenase [ubiquinone] flavoprotein 1 | 0.68 | 0.00146 | 0.50 | 0.00062 |
| NIPSNAP2 | Protein NipSnap homolog 2 | 0.62 | 0.00855 | 0.68 | 0.00182 |
| OGDH | 2‐oxoglutarate dehydrogenase | 0.74 | 0.00803 | 0.63 | 0.00681 |
| PDHA1 | Pyruvate dehydrogenase E1 component subunit alpha | 0.76 | 0.02951 | 0.55 | 0.00341 |
| SAMM50 | Sorting and assembly machinery component 50 homolog | 0.66 | 0.00070 | 0.53 | 0.00158 |
| SLC25A11 | Mitochondrial 2‐oxoglutarate/malate carrier protein | 0.65 | 0.00622 | 0.59 | 0.00334 |
| SLC25A4 | ADP/ATP translocase 1 | 0.71 | 0.00079 | 0.56 | 0.00162 |
| SUCLA2 | Succinate–CoA ligase [ADP‐forming] subunit beta | 0.62 | 0.00004 | 0.33 | 0.00509 |
| UQCRB | Cytochrome b‐c1 complex subunit 7 | 0.69 | 0.00130 | 0.54 | 0.00341 |
| UQCRC1 | Cytochrome b‐c1 complex subunit 1 | 0.68 | 0.00134 | 0.59 | 0.00666 |
| UQCRC2 | Cytochrome b‐c1 complex subunit 2 | 0.62 | 0.00044 | 0.66 | 0.00440 |
TABLE 3.
Upregulated proteins in CLTI amputation specimens identified at both sites
| University of Florida | East Carolina University | ||||
|---|---|---|---|---|---|
| Gene ID | Gene description | Fold change (CLTI/Control) | P‐value | Fold change (CLTI/Control) | P‐value |
| ADH1B | All‐trans‐retinol dehydrogenase [NAD(+)] ADH1B | 2.2 | 0.00023 | 3.9 | 0.00002 |
| AKR1C1 | Aldo‐keto reductase family 1 member C1 | 1.8 | 0.00620 | 5.4 | 0.02791 |
| AMBP | Protein AMBP | 2.2 | 0.00974 | 4.5 | <0.00001 |
| CFL1 | Cofilin‐1 | 1.3 | 0.00388 | 7.7 | <0.00001 |
| DDX39B | Isoform 2 of Spliceosome RNA helicase DDX39B | 1.4 | 0.01493 | 5.7 | <0.00001 |
| F13A1 | Coagulation factor XIII A chain | 2.6 | 0.00048 | 18.7 | <0.00001 |
| FTL | Ferritin light chain | 3.4 | 0.00001 | 18.4 | <0.00001 |
| MYH10 | Isoform 4 of Myosin‐10 | 1.5 | 0.01277 | 32.6 | <0.00001 |
| MYO1C | Unconventional myosin‐Ic | 1.5 | 0.00650 | 24.3 | <0.00001 |
| PA2G4 | Proliferation‐associated protein 2G4 | 1.4 | 0.00163 | 4.8 | <0.00001 |
| PPIA | Peptidyl‐prolyl cis‐trans isomerase | 1.9 | 0.00002 | 7.6 | <0.00001 |
| TNPO1 | Transportin‐1 | 1.4 | 0.02530 | 3.3 | <0.00001 |
3.3. Mitochondrial failure is the primary proteome signature of CLTI amputation
To understand the consequences of the downregulated mitochondrial proteome in CLTI, we analyzed mitochondrial oxidative phosphorylation (OXPHOS) using high‐resolution respirometry in permeabilized myofibers preparations. Using these methods, we previously demonstrated a uniform mitochondrial functional signature amongst CLTI amputees which was distinguishable from non‐PAD controls and mild claudicating PAD patients. 6 Here, we specifically analyzed OXPHOS function in permeabilized myofibers using a novel creatine‐kinase clamp protocol. This protocol more closely mimics a patient's physiologic stress‐test by analyzing mitochondrial oxygen consumption across a range of energy demands, similar to the conditions occurring when resting and transitioning to exercise (Figure 2A). Notably, OXPHOS performance in CLTI specimens using either carbohydrate or lipid fuel sources was substantially lower compared to non‐ischemic controls (Figure 2B). Affiliated heat maps (organized by their individual roles in mitochondrial OXPHOS) of the mitochondrial proteome demonstrate clearly divergent proteomic signatures between CLTI and controls (Figure 2C).
FIGURE 2.
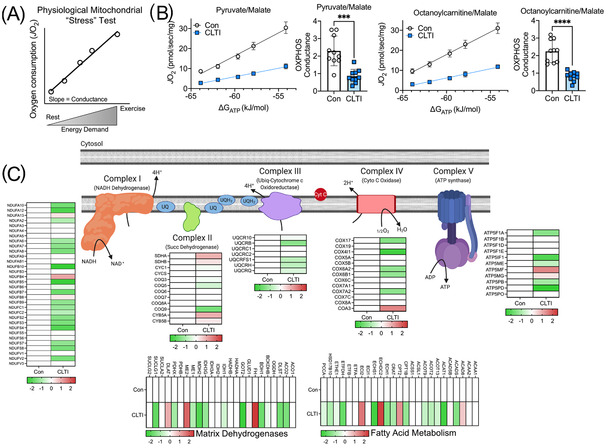
Proteomic changes impart a functional deficit in mitochondria in non‐salvageable CLTI. (A) Graphical depiction of novel mitochondrial “stress” test that mimics physiological levels of energy demand. (B) Quantitative analysis of muscle mitochondrial function supported by carbohydrate (pyruvate/malate) and fatty acid (octanoylcarnitine/malate) demonstrate substantial impairment in mitochondrial function in CLTI (n = 10/group). (C) Proteome differences (Log2 fold change) for non‐PAD control and CLTI amputation specimens according to their role in mitochondrial energy transduction. ***P < 0.001, ****P < 0.0001 using two‐tailed t‐test
3.4. The CLTI limb muscle proteome prior to surgical intervention differs from non‐PAD control limbs
The rates of successful limb salvage and resolution of CLTI symptoms resulting from endovascular or revascularization procedures are relatively low despite improvements in surgical techniques. For this experiment, we sought to examine whether differences in the CLTI limb muscle proteome immediately prior to surgical intervention would uniquely define this patient population and perhaps help identify new temporal clinical predictors of major adverse limb events. Compared to non‐ischemic control samples, those collected from presurgical CLTI limbs possessed 708 differentially abundant proteins (688 upregulated, and 20 downregulated) (Figure 3A,B). Consistent with CLTI amputation specimens, the largest proportion (26%) of upregulated proteins were related to the extracellular matrix (Figure 3C). However, in stark contrast to amputation specimens, the pre‐surgical tissues displayed a robust upregulation of mitochondrial proteins (Figure 3C). In fact, gene ontology analysis indicated the most significant pathways upregulated were related to mitochondrial metabolism (Figure 3D). Heat maps of quantified proteins involved in mitochondrial energy production are shown in Figure 3E. Interestingly, the compensatory upregulation of mitochondrial protein expression did not result in improved OXPHOS function. Functional analysis of mitochondrial OXPHOS indicated that mitochondrial function in CLTI patients before surgery was normal when fuel by carbohydrates (pyruvate/malate) but modestly (∼25%) impaired when fueled by lipid substrates (Figure 3F).
FIGURE 3.
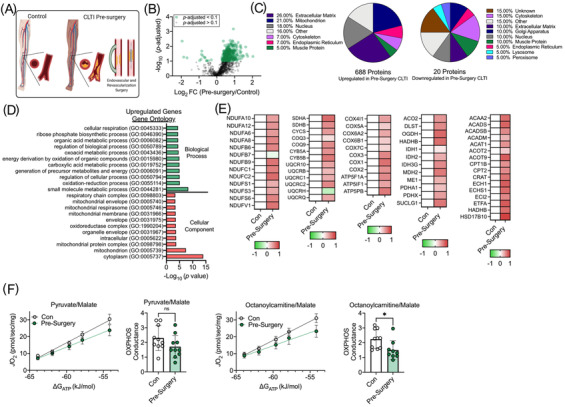
Mitochondrial signature is not present in CLTI prior to surgical intervention. (A) Proteome comparison and (B) Volcano plot between non‐PAD controls and CLTI patients prior to surgical intervention. (C) Pie charts depicting the proportions of differentially expressed proteins (upregulated and downregulated) by cellular organelle/component. (D) Gene ontology identifies upregulation of mitochondrial proteins in CLTI specimens prior to surgical intervention. (E) Heat maps of select mitochondrial protein abundances (Log2 fold change). (F) Functional analysis of muscle mitochondria identified a modest impairment in fatty acid oxidation but no difference in carbohydrate oxidation (n = 9‐10/group). *P < 0.05 using two‐tailed t‐test
3.5. The mitochondrial proteome differentiates presurgery and amputation CLTI patient limb muscles
We next directly compared the proteomic signatures of muscles from CLTI patients collected before surgical intervention to those acquired at the time of amputation (Figure 4A). Three hundred fourty‐five proteins were differentially abundant between these populations, with the majority (283 out of 345, 82%) downregulated (Figure 4B,C). Further evaluation of downregulated proteins indicated that 53% were mitochondrial‐related (Figure 4C) which was paralleled in gene ontology analysis (Figure 4D). Heat maps of proteins involved in mitochondrial energy production are shown in Figure 4E. Physiologically, mitochondrial function was significantly decreased in CLTI amputation muscles compared with those analyzed prior to surgery (Figure 4F). Our results indicate sweeping downregulation of the mitochondrial proteome and failure of mitochondrial oxidative phosphorylation defines amputation‐stage muscle specimens in CLTI.
FIGURE 4.
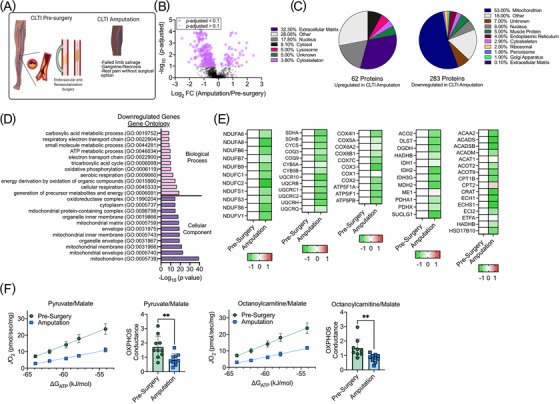
Proteomic signature distinguishes major adverse limb events from presurgery CLTI. (A) Proteome comparison and (B) Volcano plot between CLTI amputations and CLTI patients prior to surgical intervention. (C) Pie charts depicting the proportions of differentially expressed proteins (upregulated and downregulated) by cellular organelle/component. (D) Gene ontology identifies downregulated proteins CLTI amputations compared to CLTI specimens prior to surgical intervention. (E) Heat maps of select mitochondrial protein abundances (Log2 fold change). (F) Functional analysis of muscle mitochondria identified a severe impairment in mitochondrial oxidative phosphorylation in CLTI amputations that is not present prior to surgical intervention (n = 9‐10/group). **P < 0.01 using two‐tailed t‐test
4. DISCUSSION
The CLTI presentation of PAD is characterized by significant morbidity, mortality, and substantial health care costs. 1 , 2 , 29 It is most often coupled with considerable comorbid conditions contributing to poor outcomes. These conditions include diabetes, renal disease, and heart disease. Revascularization or interventions focused on restorative blood flow are the cornerstone of therapy for CLTI. 4 , 30 Resolution of CLTI symptoms remains low despite endovascular and open surgical techniques and technologies improving significantly over the past two decades. 31 A limited understanding of tissue‐level characteristics in these patients has undoubtedly led to blunted therapeutic design during this period. The results of this study reveal a distinct proteome signature at the time of major adverse limb events (i.e. amputations) in CLTI that is distinct from both non‐PAD control limbs and CLTI limbs prior to surgical intervention. Bioinformatics revealed biological themes and cellular components unique to CLTI amputations including decreased mitochondrial, oxidative phosphorylation, and cellular respiration. The largest class of upregulated proteins in limb muscle at amputation were related to extracellular matrix remodeling. Importantly, bioinformatics produced near‐identical gene ontology across tissues that were independently obtained, processed, and analyzed from two vascular clinics.
Skeletal muscle function is a strong predictor for morbidity and mortality in PAD patients, regardless of symptomatic presentation. 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 Walking performance and mitochondrial health specifically are strong predictors of disease severity in PAD patients. 6 , 40 , 41 , 42 Combined with our previous transcriptomics work, 6 the proteomic data here supports a uniform downregulation of mitochondrial related genes and proteins in the CLTI limb muscle at the time of amputation. We previously established the unique functional deficits of CLTI patient limb muscle mitochondria at this disease stage. 6 In an attempt to fully understand the functional impact of downregulated mitochondrial proteins identified herein we employed a different strategy, using a novel creatine‐kinase energy clamp system to assess mitochondrial function. Using this specific methodology, the myofibers are exposed to increasing cellular energy demand (mimicking a range contractile activity) akin to a stress test. The relationship between mitochondrial oxygen consumption and energy demands facilitates a calculation of conductance through the oxidative metabolic system (termed OXPHOS conductance herein). 25 , 43 Myofibers prepared from amputation limbs exhibited low OXPHOS conductance, fully demonstrating that the muscle mitochondria are incapable of adequately responding to changes of energy demand by increasing oxidative phosphorylation. This would lead to increased reliance on non‐oxidative metabolic pathways, especially during increased levels of energy demand, such as those required for normal activities of daily living. This is undoubtedly a problem, as we have also demonstrated decreases in the limb muscle transcriptome related to glycolytic flux in CLTI patients. 44 Additionally, functional glycolytic flux is blunted in primary muscle myotubes from these patients in vitro. This wholesale failure of energy charge in the limb muscles of CLTI patients at amputation may provide bioenergetic insight into the exercise‐, and general physical activity‐ intolerance that plagues this clinical presentation.
Interestingly, we report that catastrophic deficits in oxidative metabolism and the mitochondrial proteome discovered at amputation do not uniformly characterize CLTI limbs prior to surgical intervention. Although there were some similarities with increased protein abundances between these populations related to extracellular matrix remodeling, the second‐largest class of upregulated proteins (21%) in CLTI presurgery specimens were related to mitochondrial health. Increased mitochondrial proteins in the CLTI patients prior to surgical intervention, in stark contrast to the widespread downregulation that occurs at the time of amputation, would seem to indicate that these tissues are still capable of mounting some sort of biological/bioenergetic response to the chronic ischemic insult. Of particular relevance to this idea, however, is that 62% of the CLTI amputation patients enrolled in this study across both clinical sites had previously undergone a surgical intervention of some kind to restore limb muscle blood flow. A sufficient mitochondrial proteomic signature in these patients at the time of intervention might logically lead one to assume that the limb tissues would be efficiently capable of utilizing oxidative metabolism to regenerate or restore muscle function after surgery. Unfortunately, this does not appear to be the case. Our data actually suggest that limb pathology in CLTI does not necessarily involve a progressive decline in mitochondrial health as a result of reduced blood flow to the limb muscles, but rather the wholesale collapse of mitochondrial health in non‐salvageable limbs. Frustratingly, in CLTI patients this seems to occur at high frequency regardless of the restoration of blood flow and independent of vessel patency after intervention. This also seems to occur in these patients despite relatively stable total mitochondrial numbers according to established biomarkers including citrate synthase activity, mtDNA/nDNA, and cardiolipin content. 6 Collectively, these data paint a clear picture of the non‐salvageable limb with a microenvironment populated by fragile limb muscle mitochondria.
Development of fibrosis has been previously reported in muscle biopsy specimens from PAD patients. 45 , 46 Uniformity in expanded extracellular matrix protein abundance across the CLTI population supports the histological characterization of the limb muscles from these patients as pro‐fibrotic. 7 , 47 The expansion of non‐contractile tissues in response to limb ischemia is not limited to humans, as this is also a hallmark of inbred strains of mice that do not regenerate and recover limb muscle perfusion or function after hindlimb ischemia. 48 , 49 , 50 , 51 Fibrosis, in general, is a common occurrence in muscles that fail to adequately respond to a degenerative insult. Remodeling of the extracellular matrix has important functional roles to facilitate cell proliferation, migration, differentiation, and survival 52 that are processes crucial for both angiogenesis and muscle regeneration. Additionally, emerging evidence has begun to link aberrant mitochondrial function to organ fibrosis 53 suggesting that the observed proteome changes could be mechanistically linked histopathology in CLTI limb muscle. Currently, the functional impact of pathologic remodeling of extracellular compartment in the ischemic limb is unknown but would likely contribute to poor muscle contractile performance and activity intolerance in PAD/CLTI patients. 34 , 54 To this end, it is noteworthy to consider that numerous approved drugs exert their effects by modulating extracellular matrix remodeling. 52 Whether these classes of drugs can improve limb function in CLTI has not been investigated to date but provides an interesting therapeutic avenue.
5. STUDY LIMITATIONS
There were some limitations to the current study. First, this work was cross‐sectional in nature, therefore no causal inferences should be made regarding the role of proteome and mitochondrial functions changes as tissue‐level mechanisms leading to limb amputation. Future studies to rigorously establish causality in limb muscle mitochondrial function in amputation outcomes would require several things, including: (1) establishment of mitochondrial dysfunction as a primary determinant of amputation/pathologic outcomes (clinically/pre‐clinically, respectively), (2) determination of sufficiency for limb muscle mitochondrial function in the prevention of the CLTI phenotype/presentation (preclinically/clinically, respectively), and/or (3) repeated limb muscle biopsies in the same patient cohorts coupled with proteomic and mitochondrial analyses. Second, this exploratory proteomics analysis was performed on a relatively small sample size (65 patients) and thus should be considered provisionary in nature. Despite the relatively small sample size, confidence in the identified proteome changes is enhanced by the fact that two independent patient cohorts from separate sites and independently‐conducted proteomic analyses identified common cellular components that were altered in CLTI. However, differences in the methods used for tissue lysis/digestion, TMT‐labeling, and mass spectrometry analysis across sites may have diminished the ability to identify more common protein targets across sites. Fourth, there may be modest differences in the clinical severity of presurgery and amputation CLTI patients, indicated by the Rutherford classification but not hemodynamic outcomes (ABI), which could have impacted the proteome signature.
6. CONCLUSIONS
Amputation outcomes in CLTI are defined by a failing cellular microenvironment that may occur despite technically successful surgical intervention. The underlying mechanisms contributing to the unacceptably high amputation rates in this patient population are still somewhat unclear, however this work contributes critical information to a recently expanding book on these patients. 6 , 7 , 8 , 10 , 11 , 44 , 47 , 55 This study, in particular, builds on transcriptomics data to highlight a deficit in limb muscle mitochondrial function and the corresponding mitochondrial proteome that defines the non‐salvageable ischemic limb. Although the exact clinical application of these results is unknown presently, several applications can be envisioned. First, the observation that amputation tissues display a mitochondrial deficit that is distinguishable from CLTI tissues prior to surgical intervention suggests that future studies involving longitudinal analysis of mitochondrial health both prior to‐ and following surgical intervention are needed to determine if mitochondrial health can identify patients with the greatest risk for amputation. While not as technically in depth as myofiber isolation and direct substrate‐driven OXPHOS interrogation, mitochondrial analyses can be more easily performed using non‐invasive technologies. 56 , 57 Given that a muscle mitochondrial deficit also distinguishes pathology in CLTI‐susceptible mice, 58 and mitochondrial‐targeted therapy improves outcomes in mice with ischemic mitochondriopathy, 59 it is reasonable to hypothesize that therapeutic targeting mitochondrial health in CLTI may prove beneficial. This is an exciting avenue for innovative advancement in patients with few interventional options at present.
DISCLOSURES
The authors have no conflicts, financial or otherwise, to report.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Jin Koh and staff at the Interdisciplinary Center for Biotechnology Research at the University of Florida for assistance with LC‐MS/MS processing. This study was supported by National Institutes of Health (NIH) grants including P30AG028740 (Pilot funds awarded to TER), R01HL149704 (TER), R01HL150003 (JMM), R01HL157659 (JMM), R01DK119274 (SAB), R01HL148597 (STS), the American Heart Association ‐ 18CDA34110044 (TER) and Department of Defense grant W81XWH‐19‐1‐0213 (KF‐W).
Ryan TE, Kim K, Scali ST, et al. Interventional‐ and amputation‐stage muscle proteomes in the chronically threatened ischemic limb. Clin Transl Med. 2022;12:e658. 10.1002/ctm2.658
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available online using accession numbers PXD021849 and PXD025810 (Proteome Xchange) or JPST000852 and JPST001157 (jPOST Repository).
REFERENCES
- 1. Soga Y, Iida O, Takahara M, et al. Two‐year life expectancy in patients with critical limb ischemia. JACC Cardiovasc Interv. 2014;7:1444‐1449. [DOI] [PubMed] [Google Scholar]
- 2. Shishehbor MH, White CJ, Gray BH, et al. Critical limb ischemia an expert statement. Journal of the American College of Cardiology. 2016;68:2002‐2015. [DOI] [PubMed] [Google Scholar]
- 3. Anand SS, Caron F, Eikelboom JW, et al. Major adverse limb events and mortality in patients with peripheral artery disease. J Am Coll Cardiol. 2018;71:2306‐2315. [DOI] [PubMed] [Google Scholar]
- 4. Smith AD, Hawkins AT, Schaumeier MJ, de Vos MS, Conte MS, Nguyen LL. Predictors of major amputation despite patent bypass grafts. J Vasc Surg. 2016;63:1279‐1288. [DOI] [PubMed] [Google Scholar]
- 5. Vallabhaneni R, Kalbaugh CA, Kouri A, Farber MA, WA Marston. Current accepted hemodynamic criteria for critical limb ischemia do not accurately stratify patients at high risk for limb loss. J Vasc Surg. 2016;63:105‐112. [DOI] [PubMed] [Google Scholar]
- 6. Ryan TE, Yamaguchi DJ, Schmidt CA, et al. Extensive skeletal muscle cell mitochondriopathy distinguishes critical limb ischemia patients from claudicants. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cong GZ, Cui XD, Ferrari R, et al. Fibrosis distinguishes critical limb ischemia patients from claudicants in a transcriptomic and histologic analysis. J Clin Med. 2020;9:3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Terwilliger ZS, Ryan TE, Goldberg EJ, et al. Racial differences in the limb skeletal muscle transcriptional programs of patients with critical limb ischemia. Vasc Med. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jalkanen J, Hollmen M, Maksimow M, Jalkanen S, Hakovirta H. Serum cytokine levels differ according to major cardiovascular risk factors in patients with lower limb atherosclerosis. Cytokine. 2019;114:74‐80. [DOI] [PubMed] [Google Scholar]
- 10. Ismaeel A, Franco ME, Lavado R, et al. Altered metabolomic profile in patients with peripheral artery disease. J Clin Med. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khattri RB, Kim K, Thome T, et al. Unique metabolomic profile of skeletal muscle in chronic limb threatening ischemia. J Clin Med. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pipinos II, Sharov VG, Shepard AD, et al. Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J Vasc Surg. 2003;38:827‐832. [DOI] [PubMed] [Google Scholar]
- 13. Pipinos II, Judge AR, Zhu Z, et al. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic Biol Med. 2006;41:262‐269. [DOI] [PubMed] [Google Scholar]
- 14. Ryan TE, Brophy P, Lin CT, Hickner RC, Neufer PD. Assessment of in vivo skeletal muscle mitochondrial respiratory capacity in humans by near‐infrared spectroscopy: a comparison with in situ measurements. J Physiol. 2014;592:3231‐3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McLaughlin KL, Kew KA, McClung JM, Fisher‐Wellman KH. Subcellular proteomics combined with bioenergetic phenotyping reveals protein biomarkers of respiratory insufficiency in the setting of proofreading‐deficient mitochondrial polymerase. Sci Rep‐Uk. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eng JK, Mccormack AL, Yates JR. An approach to correlate tandem mass‐spectral data of peptides with amino‐acid‐sequences in a protein database. J Am Soc Mass Spectr. 1994;5:976‐989. [DOI] [PubMed] [Google Scholar]
- 17. Benjamini Y, Hochberg Y. Controlling the false discovery rate ‐ a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289‐300. [Google Scholar]
- 18. Deutsch EW, Csordas A, Sun Z, et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45:D1100‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Okuda S, Watanabe Y, Moriya Y, et al. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. 2017;45:D1107‐D1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mi HY, Muruganujan A, Ebert D, Huang XS, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO‐slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47:D419‐D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carbon S, Douglass E, Good BM, et al. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325‐D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perry CG, Kane DA, Lin CT, et al. Inhibiting myosin‐ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J. 2011;437:215‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fisher‐Wellman KH, Lin CT, Ryan TE, et al. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy‐consuming redox circuit. Biochem J. 2015;467:271‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fisher‐Wellman KH, Davidson MT, Narowski TM, Lin CT, Koves TR, Muoio DM. Mitochondrial diagnostics: a multiplexed assay platform for comprehensive assessment of mitochondrial energy fluxes. Cell Rep. 2018;24:3593‐3606. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thome T, Salyers ZR, Kumar RA, et al. Uremic metabolites impair skeletal muscle mitochondrial energetics through disruption of the electron transport system and matrix dehydrogenase activity. Am J Physiol‐Cell Ph. 2019;317:C701‐C713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thome T, Kumar RA, Burke SK, et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. Jci Insight. 2020;6(1):e139826. 10.1172/jci.insight.139826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fisher‐Wellman KH, Draper JA, Davidson MT, et al. Respiratory phenomics across multiple models of protein hyperacylation in cardiac mitochondria reveals a marginal impact on bioenergetics. Cell Rep. 2019;26:1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Agarwal S, Sud K, Shishehbor MH. Nationwide trends of hospital admission and outcomes among critical limb ischemia patients from 2003–2011. J Am Coll Cardiol. 2016;67:1901‐1913. [DOI] [PubMed] [Google Scholar]
- 30. Reinecke H, Unrath M, Freisinger E, et al. Peripheral arterial disease and critical limb ischaemia: still poor outcomes and lack of guideline adherence. Eur Heart J. 2015;36:932‐938. [DOI] [PubMed] [Google Scholar]
- 31. Norgren L, Hiatt WR, Dormandy JA, et al. Inter‐society consensus for the management of peripheral arterial disease (TASC II). J Vasc Surg. 2007;45(Suppl S):S5‐67. [DOI] [PubMed] [Google Scholar]
- 32. McDermott MM, Liu K, Ferrucci L, et al. Decline in functional performance predicts later increased mobility loss and mortality in peripheral arterial disease. J Am Coll Cardiol. 2011;57:962‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jain A, Liu K, Ferrucci L, et al. The Walking Impairment Questionnaire stair‐climbing score predicts mortality in men and women with peripheral arterial disease. J Vasc Surg. 2012;55:1662‐1673. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McDermott MM, Liu K, Tian L, et al. Calf muscle characteristics, strength measures, and mortality in peripheral arterial disease: a longitudinal study. J Am Coll Cardiol. 2012;59:1159‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jain A, Liu K, Ferrucci L, et al. Declining walking impairment questionnaire scores are associated with subsequent increased mortality in peripheral artery disease. J Am Coll Cardiol. 2013;61:1820‐1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leeper NJ, Myers J, Zhou M, et al. Exercise capacity is the strongest predictor of mortality in patients with peripheral arterial disease. J Vasc Surg. 2013;57:728‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thompson JR, Swanson SA, Haynatzki G, et al. Protein concentration and mitochondrial content in the gastrocnemius predicts mortality rates in patients with peripheral arterial disease. Annals of surgery. 2015;261:605‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsubara Y, Matsumoto T, Aoyagi Y, et al. Sarcopenia is a prognostic factor for overall survival in patients with critical limb ischemia. J Vasc Surg. 2015;61:945‐950. [DOI] [PubMed] [Google Scholar]
- 39. White SH, McDermott MM, Sufit RL, et al. Walking performance is positively correlated to calf muscle fiber size in peripheral artery disease subjects, but fibers show aberrant mitophagy: an observational study. J Transl Med. 2016;14:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. AlGhatrif M, Zane A, Oberdier M, et al. Lower mitochondrial energy production of the thigh muscles in patients with low‐normal ankle‐brachial index. J Am Heart Assoc. 2017;6:e006604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McDermott MM, Peterson CA, Sufit R, et al. Peripheral artery disease, calf skeletal muscle mitochondrial DNA copy number, and functional performance. Vasc Med. 2018. 1358863X18765667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gonzalez‐Freire M, Moore AZ, Peterson CA, et al. Associations of peripheral artery disease with calf skeletal muscle mitochondrial DNA heteroplasmy. J Am Heart Assoc. 2020;9:e015197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52:2793‐2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ryan TE, Schmidt CA, Tarpey MD, et al. PFKFB3‐mediated glycolysis rescues myopathic outcomes in the ischemic limb. JCI Insight. 2020;5:e139628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ha DM, Carpenter LC, Koutakis P, et al. Transforming growth factor‐beta 1 produced by vascular smooth muscle cells predicts fibrosis in the gastrocnemius of patients with peripheral artery disease. Journal of translational medicine. 2016;14:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kosmac K, Gonzalez‐Freire M, McDermott MM, et al. Correlations of calf muscle macrophage content with muscle properties and walking performance in peripheral artery disease. J Am Heart Assoc. 2020;9:e015929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mietus CJ, Lackner TJ, Karvelis PS, et al. Abnormal microvascular architecture, fibrosis, and pericyte characteristics in the calf muscle of peripheral artery disease patients with claudication and critical limb ischemia. J Clin Med. 2020;9:2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McClung JM, McCord TJ, Keum S, et al. Skeletal muscle‐specific genetic determinants contribute to the differential strain‐dependent effects of hindlimb ischemia in mice. Am J Pathol. 2012;180:2156‐2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McClung JM, McCord TJ, Southerland K, et al. Subacute limb ischemia induces skeletal muscle injury in genetically susceptible mice independent of vascular density. J Vasc Surg. 2016;64:1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McClung JM, McCord TJ, Ryan TE, et al. BAG3 (Bcl‐2‐associated athanogene‐3) coding variant in mice determines susceptibility to ischemic limb muscle myopathy by directing autophagy. Circulation. 2017;136:281‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schmidt CA, Amorese AJ, Ryan TE, et al. Strain‐dependent variation in acute ischemic muscle injury. Am J Pathol. 2018;188:1246‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61:198‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li XY, Zhang W, Cao QT, et al. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Singh N, Liu K, Tian L, et al. Leg strength predicts mortality in men but not in women with peripheral arterial disease. J Vasc Surg. 2010;52:624‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Azab S, Britz‐McKibbin P, Zamzam A, Abdin R, Qadura M. Serum metabolomic profile of critical limb‐threatening ischemia patients. J Vasc Surg. 2020;72:E214‐E214. [Google Scholar]
- 56. Pipinos II, Shepard AD, Anagnostopoulos PV, Katsamouris A, Boska MD. Phosphorus 31 nuclear magnetic resonance spectroscopy suggests a mitochondrial defect in claudicating skeletal muscle. J Vasc Surg. 2000;31:944‐952. [DOI] [PubMed] [Google Scholar]
- 57. West AM, Anderson JD, Epstein FH, et al. Percutaneous intervention in peripheral artery disease improves calf muscle phosphocreatine recovery kinetics: a pilot study. Vasc Med. 2012;17:3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schmidt CA, Ryan TE, Lin CT, et al. Diminished force production and mitochondrial respiratory deficits are strain‐dependent myopathies of subacute limb ischemia. J Vasc Surg. 2017;65:1504‐1514. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ryan TE, Schmidt CA, Alleman RJ, et al. Mitochondrial therapy improves limb perfusion and myopathy following hindlimb ischemia. J Mol Cell Cardiol. 2016;97:191‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available online using accession numbers PXD021849 and PXD025810 (Proteome Xchange) or JPST000852 and JPST001157 (jPOST Repository).