

Key Points
Osmotic demyelination syndrome (ODS) can occur despite adherence to current hyponatremia correction guidelines, especially in patients with serum sodium <115 mEq/L.
Limit the rate of correction of serum sodium <8 mEq/L in any 24-hour period in these patients to minimize the risk of ODS.
Thiamine supplementation should be considered for any patient with hyponatremia whose dietary intake has been poor.
Keywords: acid/base and electrolyte disorders, central pontine myelinolysis, demyelinating diseases, hyponatremia, osmosis, osmotic demyelination syndrome, rate of correction
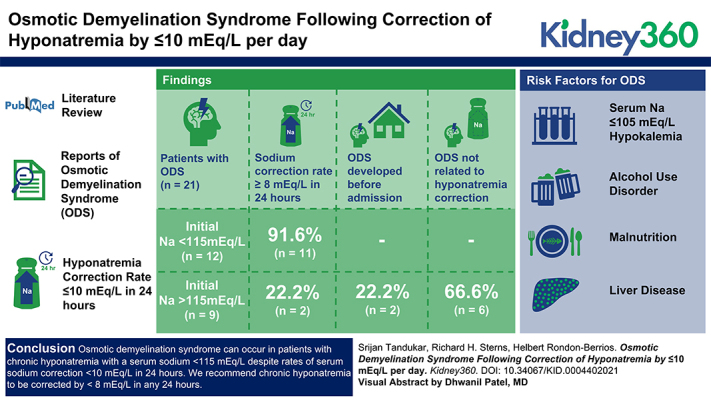
Visual Abstract
Abstract
Background
Overly rapid correction of chronic hyponatremia may lead to osmotic demyelination syndrome. European guidelines recommend a correction to ≤10 mEq/L in 24 hours to prevent this complication. However, osmotic demyelination syndrome may occur despite adherence to these guidelines.
Methods
We searched the literature for reports of osmotic demyelination syndrome with rates of correction of hyponatremia ≤10 mEq/L in 24 hours. The reports were reviewed to identify specific risk factors for this complication.
Results
We identified 19 publications with a total of 21 patients that were included in our analysis. The mean age was 52 years, of which 67% were male. All of the patients had community-acquired chronic hyponatremia. Twelve patients had an initial serum sodium <115 mEq/L, of which seven had an initial serum sodium ≤105 mEq/L. Other risk factors identified included alcohol use disorder (n=11), hypokalemia (n=5), liver disease (n=6), and malnutrition (n=11). The maximum rate of correction in patients with serum sodium <115 mEq/L was at least 8 mEq/L in all but one patient. In contrast, correction was <8 mEq/L in all but two patients with serum sodium ≥115 mEq/L. Among the latter group, osmotic demyelination syndrome developed before hospital admission or was unrelated to hyponatremia overcorrection. Four patients died (19%), five had full recovery (24%), and nine (42%) had varying degrees of residual neurologic deficits.
Conclusion
Osmotic demyelination syndrome can occur in patients with chronic hyponatremia with a serum sodium <115 mEq/L, despite rates of serum sodium correction ≤10 mEq/L in 24 hours. In patients with severe hyponatremia and high-risk features, especially those with serum sodium <115 mEq/L, we recommend limiting serum sodium correction to <8 mEq/L. Thiamine supplementation is advisable for any patient with hyponatremia whose dietary intake has been poor.
Introduction
To avoid osmotic demyelination syndrome (ODS) after treatment of chronic hyponatremia, a number of therapeutic limits have been proposed. The American Expert Panel Recommendations endorse a serum sodium (SNa) correction limit of 10–12 mEq/L in any 24-hour and 18 mEq/L in any 48-hour period for patients at average risk of ODS, and 8 mEq/L in any 24-hour period for patients at high risk of ODS (1), whereas the European Clinical Practice Guidelines recommend a daily limit of SNa correction of 10 mEq/L in the first 24 hours and 8 mEq/L during every 24 hours thereafter (2). In this study, we review reported cases of ODS that occurred after correction of hyponatremia by ≤10 mEq/L in the first 24 hours to identify the risk factors that predispose to ODS despite adherence to the European Clinical Practice Guidelines.
Materials and Methods
We searched PubMed using terms “osmotic demyelination syndrome,” “central pontine myelinolysis,” and “slow correction of hyponatremia” to identify English-language cases of patients with hyponatremia who developed ODS despite an SNa correction rate of ≤10 mEq/L in 24 hours. We then reviewed all studies and case reports cited by the articles identified in our PubMed search. We only included studies if the rate of correction had been documented to be ≤10 mEq/L in the first 24 hours of treatment followed by clinical or radiologic evidence of ODS. We excluded studies reporting the rate of correction in milliequivalents per liter per hour, or as an average value over several days, unless the daily change in SNa was explicitly stated. We also excluded studies reporting on multiple patients unless changes in SNa were provided for individual patients.
Whenever available, we recorded data on age, sex and race, comorbidities, and medications. We assumed hyponatremia to be chronic if it developed in the community and was present on admission or if it was hospital acquired with documentation of a normal SNa >48 hours before its treatment.
The initial SNa, serum potassium, creatinine, BUN levels along with other laboratory chemistries, serum and urine osmolalities, and urine electrolyte studies were recorded whenever available. The changes in SNa in the first 24- and 48-hour period, along with the absolute SNa values, were recorded. The approach to treatment of hyponatremia was reviewed. We recorded the administration of intravenous fluids—whether isotonic or hypertonic saline, the rate of infusion, volume infused, bolus versus continuous therapy, and with or without use of desmopressin. Other approaches to treatment—including fluid restriction, loop diuretics, salt tablets, urea, and vasopressin antagonists—were noted.
From the case histories, we recorded the nature of symptoms before treatment of hyponatremia, the day of onset of symptoms suggestive of ODS, the course of ODS symptoms, magnetic resonance imaging (MRI) findings, and the presence or absence of residual sequelae or a fatal outcome.
The presence of risk factors for ODS—such as SNa ≤105 mEq/L, alcohol use disorder, hypokalemia, liver disease with or without cirrhosis, malnutrition, and history of liver transplantation—was noted for each patient. The presence of factors that would predispose to overly rapid correction of hyponatremia was recorded when available, such as treatment of low dietary solute intake, treatment of hypovolemia, treatment of cortisol deficiency, resolution of transient syndrome of inappropriate antidiuresis (SIAD), discontinuation of thiazides and other causative medications of SIAD, and use of vasopressin antagonists. Any high-risk features of brain herniation were recorded, such as self-induced water intoxication (e.g., psychogenic polydipsia, ecstasy use, and endurance exercise), acute postoperative state, and underlying intracranial pathology.
Results
Selection of Studies for Analysis
A total of 21 patients met inclusion criteria: 18 patients from 17 case reports, and three patients from two retrospective observational case series.
Patient Characteristics and Clinical Presentation
The mean age of the patients was 52 years (range, 26–74 years) and 67% were male. Only two reports mentioned the patient’s race or ethnicity (one White and one Hispanic). There was a wide variation in the patients’ presenting complaints. Confusion and weakness were the most common complaints.
Hyponatremia and Rates of Correction
All of the patients had community-acquired hyponatremia. The initial mean SNa was 111 mEq/L (range, 94–130 mEq/L). Initial serum potassium values were only reported in six patients, five of whom were hypokalemic (2.1–3.2 mEq/L). Initial serum and urine osmolalities were reported in only six patients and ranged from 197 to 238 mOsm/kg and 97–452 mOsm/kg, respectively. Urine sodium values were reported in four patients, and ranged from 3 to 53 mEq/L.
The rate of correction of hyponatremia at 24 and 48 hours ranged from 2 to 10 mEq/L and 5–18 mEq/L, respectively (Figure 1). Consistent with most reports of ODS, 12 of the patients had initial SNa <115 mEq/L (range, 94–112 mEq/L). The maximum rate of correction in this group was at least 8 mEq/L in all but one of these patients (Table 1). By contrast, correction was <8 mEq/L in all but two patients with less severe hyponatremia (range, 115–130 mEq/L; Table 2).
Figure 1.

Osmotic demyelination syndrome in reported cases despite changes in serum sodium (SNa) that adhere to current hyponatremia correction guidelines. Open squares represent patients with initial SNa <115 mEq/L. Closed triangles represent patients with initial SNa ≥115 mEq/L. The dashed lines represent therapeutic limits recommended by current hyponatremia guidelines (12 mEq/L in 24 hours and 18 mEq/L in 48 hours). The case from Koul et al. (29) is not represented in this figure because changes in SNa at 24 and 48 hours were not reported. Two patients with initial SNa <115 mEq/L (John et al. [8] and Malhotra et al. [35]) corrected their SNa by 8 mEq/L and 16 mEq/L at 24 and 48 hours, respectively.
Table 1.
Patients with initial serum sodium <115 mEq/L who experienced osmotic demyelination syndrome
| Reference | Age/ Sex | Initial SNa (mEq/L) | Factors Increasing Risk of ODS | ΔSNa at 24 h (mEq/L) | ΔSNa at 48 h (mEq/L) | Maximum ΔSNa (mEq/L per day) | ODS Symptoms (Onset) | Brain MRI | Final Outcome |
| Koul et al. (29) | 47/M | 94 | Hypokalemia | Not reported | Not reported | ≤8a | Locked in (day 3) |
CPM and EPM | Died of sepsis |
| de Souza et al. (30) | 50/M | 98 | None mentioned | 4 | 13 | 9 | Dysarthria, dysphagia, rigidity, normal reflexes, positive glabellar tap, grasp and palmomental reflexes, bradykinesia, parkinsonian gait (day 15) |
CPM and EPM | Mild gait dysfunction, urge incontinence |
| John et al. (8) | 65/F | 100 | None mentioned | 8 | 16 | 8 | Dysphasia, psychosis, dyskinesia (day 3) |
No lesions | Resolution |
| Leens et al. (31) | 54/F | 100 | Alcohol use disorder; hypokalemia | 10 | 18 | 10 | Tetraplegic, pseudobulbar palsy, seizure, strabism (day 5) |
CPM and EPM | Near complete recovery |
| Al-Shaibany et al. (32) | 48/F | 101 | Alcohol use disorder; liver disease | 10 | 16 | 10 | Leg weakness, scanning speech (day 12) |
CPM | Improved at discharge |
| Silbert et al. (33) | 51/F | 101 | None mentioned | 10 | 14 | 10 | Behavioral changes, dysarthria (day 16) |
CPM and EPM | Partial resolution with residual pseudobulbar dysarthria |
| Reijnders et al. (34) | 57/M | 105 | Alcohol use disorder; malnutrition | 7 | 17 | 10 | Dysphagia, ataxia, spasticity (day 11) |
CPM | Near complete recovery |
| Malhotra et al. (35) | 48/M | 107 | Alcohol use disorder; liver disease; malnutrition | 8 | 16 | 8 | Ataxia, dysarthria, weakness, spastic quadriparesis (day 14) |
CPM | Ataxia improved |
| Dellabarca et al. (36) | 63/M | 109 | Alcohol use disorder; malnutrition | 10 | 17 | 10 | Dysarthria, dysphagia, bilateral cogwheel rigidity (day 10) |
CPM and EPM | Death due to respiratory failure |
| Yamada et al. (19) | 61/F | 109 | None mentioned | 5 | 10 | 5 | Spastic tetraparesis, dysarthria, nystagmus, disturbed consciousness (day 5) |
CPM | Complete resolution |
| Macmillan et al. (4) | 70–74/M | 111 | Alcohol use disorder; liver disease; malnutrition; hypokalemia |
10 | 13 | 10 | Symptoms not described by authors (day 4) |
Areas of lesions not specified | Unknown residual deficits |
| Dewitt et al. (37) | 37/F | 112 | Liver disease | 9 | 17 | 9 | Decerebrate posturing, disorientation, hyperreflexia, impaired horizontal eye movements (day 4) |
CPM | Improved with mild spasticity of lower extremities after 8 months |
SNa, serum sodium concentration; ODS, osmotic demyelination syndrome; ΔSNa, change in SNa; MRI, magnetic resonance imaging; M, male; CPM, central pontine myelinolysis; EPM, extrapontine myelinolysis; F, female.
Correction did not exceed 8 mEq/L on any day, precise values not provided; treated with 3% saline until SNa reached 120 mEq/L.
Table 2.
Patients with initial serum sodium ≥115 mEq/L who experienced osmotic demyelination syndrome
| Reference | Age/Sex | Initial SNa (mEq/L) | Factors Increasing Risk of ODS | ΔSNa at 24 h (mEq/L) | ΔSNa at 48 h (mEq/L) | Maximum ΔSNa (mEq/L per day) | ODS Symptoms (Onset) | Brain MRI | Final Outcome |
| Hu et al. (23) | 30/M | 115 | High ammonia levels | 5 | 9 | 5 | Dysarthria, dysphagia, exaggerated deep tendon reflexes (day 2) |
EPM | No improvement |
| Pradhan et al. (17) | 55/M | 115 | Malnutrition | 3 | 7 | 4 | Spastic quadriparesis, brisk deep tendon reflexes, positive Babinski in right (day 8) |
CPM | No improvement, patient death |
| Macmillan et al. (4) | 60–64/M | 115 | Alcohol use disorder; liver disease; malnutrition | 7 | 6 | 7 | Symptoms not described by authors (day 12) |
Areas of lesions not specified | Unknown residual deficits |
| Pradhan et al. (17) | 39/M | 118 | Malnutrition | 4 | 8 | 4 | Dysarthria, dysphagia, rigidity, impaired horizontal eye movement, decorticate posturing, brisk deep tendon reflexes, positive Babinski bilaterally (day 6) |
CPM and EPM | Incomplete recovery. Able to feed himself, speak few words, walk with support. Cogwheel rigidity and parkinsonian gait persisted |
| Zunga et al. (16) | 65/F | 121 | Hypokalemia | 6 | 12 | 6 | Altered sensorium, stuporous (on admission) |
EPM | Full recovery |
| Orakzai et al. (38) | 52/M | 122 | Alcohol use disorder; malnutrition | 8 | 16 | 8 | Nonverbal, decreased right hand grip strength, paralysis of other extremities, Babinski positive bilaterally, pupillary responses sluggish (day 5) |
CPM | Complete resolution |
| Jahan et al. (39) | 55/M | 123 | Alcohol use disorder; malnutrition | 5 | 5 | 5 | Encephalopathic, dysarthria, dysphagia, ophthalmoplegia, brisk deep tendon reflexes in all extremities (day 7) |
CPM and EPM | Dysarthria, dysphagia, neurologic exam improved “significantly” |
| Davenport et al. (21) | 63/M | 127 | Alcohol use disorder; hypokalemia; malnutrition | 9 | 15 | 10 (SNa 161 on day 7) |
Obtundation, comatose (day ?) |
CPM | Death |
| Shah et al. (3) | 26/M | 130 | Alcohol use disorder; liver disease; malnutrition |
2 | 6 | 4 | Never developed ODS symptoms | CPM (MRI done to evaluate lymphoma) | Remained asymptomatic |
SNa, serum sodium concentration; ODS, osmotic demyelination syndrome; ΔSNa, change in SNa; MRI, magnetic resonance imaging; M, male; EPM, extrapontine myelinolysis; CPM, central pontine myelinolysis; F, female.
Treatment Approach to Hyponatremia
Hypertonic saline was given to four patients (3% saline in three patients, 2.5% saline in one patient). Normal saline was given in 11 patients. Other treatment modalities—such as cessation of thiazide diuretics, fluid restriction, and oral salt tablets—were used in various combinations (Table 3 and 4).
Table 3.
Etiology and treatment of hyponatremia for patients with initial serum sodium <115 mEq/L who experienced osmotic demyelination syndrome
| Reference | Etiology of Hyponatremia | Treatment of Hyponatremia |
| Koul et al. (29) | Not mentioned by authors, presumably from a vasopressin-mediated process with reduced effective arterial blood volume | 3% saline till SNa reached 120, then NS given |
| de Souza et al. (30) | Vomiting from colchicine administration | Not mentioned by authors |
| John et al. (8) | SIAD secondary to small cell bronchogenic carcinoma of lung | Saline infusion (not specified as 3%), discontinuation of thiazide diuretic |
| Leens et al. (31) | Psychogenic polydipsia, thiazide diuretic use | Water restriction, NS with potassium added |
| Al-Shaibany et al. (32) | Watery diarrhea and vomiting | 3% saline at 150 ml bolus, then NS 50–100 ml/h |
| Silbert et al. (33) | Psychogenic polydipsia | Hypertonic saline 2.5% and fluid restriction |
| Reijnders et al. (34) | Thiazide diuretic use and poor intake | Discontinuation of thiazide diuretic, isotonic saline infusion |
| Malhotra et al. (35) | Excess fluid and low solute intake | Fluid restriction |
| Dellabarca et al. (36) | Not mentioned by authors, presumably from low solute intake | Intravenous saline infusion (not specified as 3%), water restriction |
| Yamada et al. (19) | Excess vasopressin stimulated by vomiting | Continuous infusion of 3% saline at 25 ml/h |
| Macmillan et al. (4) | Not mentioned by authors | Desmopressin |
| Dewitt et al. (37) | Not mentioned by authors | Not mentioned |
SNa, serum sodium; NS, 0.9% sodium chloride; SIAD, syndrome of inappropriate antidiuresis.
Table 4.
Etiology and treatment of hyponatremia for patients with initial serum sodium ≥115 mEq/L who experienced osmotic demyelination syndrome
| Reference | Etiology of Hyponatremia | Treatment of Hyponatremia |
| Hu et al. (23) | Not mentioned by authors | NS at 1 L over 24 h |
| Pradhan et al. (17) | Not mentioned by authors, presumably from low solute intake | NS and salt tablets |
| Macmillan et al. (4) | Not mentioned by authors | Not mentioned by the authors |
| Pradhan et al. (17) | Salt restriction, persistent vomiting | NS, salt tablets, discontinuation of thiazide diuretic and enalapril |
| Zunga et al. (16) | Not mentioned by authors, presumably from viral gastroenteritis | Only mentions “supportive treatment” without specifics |
| Orakzai et al. (38) | Not mentioned by authors | No specific therapy given |
| Jahan et al. (39) | Not mentioned by authors, presumably from nonbloody diarrhea | Free water restriction and “gentle” intravenous fluid repletion with NS |
| Davenport et al. (21) | SIAD secondary to pneumonia | NS |
| Shah et al. (3) | Not mentioned by authors | Salt tablets and NS |
NS, 0.9% sodium chloride; SIAD, syndrome of inappropriate antidiuresis.
ODS and Its Clinical Course
ODS occurred 2–16 days after correction of hyponatremia. One patient was described as asymptomatic (3) and, in two patients, no description of ODS features was provided (4). Eighteen patients presented with classic features of ODS: spasticity, hyperreflexia, quadriplegia, mutism, dysarthria, gait abnormalities, obtundation, and coma. However, one patient was asymptomatic and clinical features were not described in two patients. Psychotic features were reported in three patients. Respiratory failure requiring intubation was seen in two patients and another patient with hematemesis was intubated for airway protection. In all but one patient, the clinical diagnosis of ODS was confirmed by brain MRI findings of demyelination (nine with pontine findings, two with extrapontine, seven with pontine and extrapontine, and two unstated). Four patients (19%) died after the diagnosis of ODS. Complete resolution of ODS symptoms was seen in five patients (24%), with nine patients (42%) showing varying degrees of residual deficits (Tables 1 and 2).
Risk Factors for ODS
Among the risk factors for ODS, an SNa ≤105 mEq/L was reported in seven patients, hypokalemia in five, malnutrition in 11, alcohol use disorder in 11, and liver disease in six (Figure 2). None of the patients had a liver transplant. Factors predisposing to overly rapid correction of hyponatremia included low dietary solute intake in five patients, hypovolemia in seven, and discontinuation of diuretics in three.
Figure 2.

Risk factors for osmotic demyelination syndrome in reported cases based on initial serum sodium.
Discussion
The appropriate rates of correction of SNa in hyponatremia to prevent neurologic complications have been an area of debate for decades. The European Clinical Practice Guidelines state that the SNa should be limited to ≤10 mEq/L in the initial 24 hours and ≤8 mEq/L per day in the subsequent day (2). However, our literature review identified many cases of well-documented ODS in which the rate of correction of SNa was reported to adhere to these guidelines (Tables 1 and 2).
We divided our patients, somewhat arbitrarily, into two groups: 12 patients with SNa <115 mEq/L (Table 1), and nine patients with SNa ≥115 mEq/L (Table 2). We believe that findings in the group with SNa <115 mEq/L (range, 94–112 mEq/L) are most relevant to clinicians attempting to set an appropriate correction limit. In a literature review, Spasovski et al. (2) found that 96% (52 of 54) of patients with ODS had an initial SNa <120 mEq/L, and 85% (46 of 54) had an SNa < 115 mEq/L. Among hospitalized patients with hyponatremia, an SNa <115 mEq/L is less than half as common as an SNa between 115 and 119 mEq/L, and about one ninth as common as an SNa between 120 and 124 mEq/L (5). Therefore, because about 85% of patients with ODS have an SNa <115 mEq/L, the risk of ODS in a patient with an SNa <115 mEq/L is more than ten-fold greater than the risk in a patient with a higher SNa.
The 12 patients with SNa <115 mEq/L had a median SNa of 103 mEq/L, and all but one had risk factors that made them more susceptible to injury caused by a rapid increase in SNa (SNa ≤105 mEq/L, alcohol use disorder, hypokalemia, liver disease, and malnutrition). Characteristics of this group were similar to those identified in a nationwide study of 83 patients diagnosed with ODS in Sweden; in that study, most patients (87%) were hyponatremic (all chronic) with a median SNa of 104 (interquartile range, 99.5–110.5) mEq/L, 69% of the patients were alcoholic, and 68% were hypokalemic (6). Of note, Swedish national guidelines set a correction limit of SNa to ≤8 mEq/L in 24 hours, and all but six of the 83 patients (7%) exceeded this rate of correction. The cases we identified represent a small percentage of the cases published in the literature, but they confirm that ODS can occur when severe, chronic hyponatremia is corrected by ≤10 mEq/L or even by 8 mEq/L per 24 hours.
The susceptibility to ODS associated with extremely low SNa can be explained by the brain’s adaptation to hyponatremia. The fall in effective plasma tonicity that accompanies hyponatremia leads to the movement of water into brain cells, primarily astrocytes, which may lead to cerebral edema when hyponatremia is acute and severe (7). The dreaded complications of cerebral edema include seizures, obtundation, coma, brain herniation, and death. When hyponatremia develops more gradually, over ≥48 hours, the brain adapts such that the brain cells restore its normal volume by loss of potassium within the first 3 hours, and then organic osmolytes, including myo-inositol, glutamine, glutamate, taurine, glycine, aspartate, and creatine (8,9). When SNa falls <105 mEq/L, survival is not possible without the adaptive loss of organic osmolytes. Overly rapid correction of chronic hyponatremia is known to cause ODS with its attendant neurologic manifestations, which may be irreversible. Astrocytes play a critical role in maintaining the production, function, and maintenance of myelin-producing oligodendrocytes. They regulate neuroglial communication, neuronal excitability, and neurotransmission. The inability of the astrocytes to maintain intracellular volume homeostasis to changing extracellular osmolality is a key factor in the pathogenesis of ODS (9).
Correction of chronic hyponatremia subjects the astrocytes to hyperosmotic stress as they try to compensate for a rising extracellular osmolality by movement of osmolytes, mainly myo-inositol and glutamine/glutamate, back into the cell in an effort to prevent excessive cell shrinkage, injury, and apoptosis. When this osmoregulatory function of the astrocytes is disrupted, it leads to degeneration of both the myelin and oligodendrocytes, resulting in the clinicopathologic syndrome of ODS. These intracellular responses require phosphate, often in the form of ATP, along with other substrates, including amino acids, glucose, potassium, and magnesium (10). Patients who are malnourished and have a history of alcohol use disorder are often chronically deficient in these substrates and, therefore, may not be able to mount a compensatory response adequate to prevent glial cell shrinkage, apoptosis, and osmotic demyelination. Moreover, Häussinger et al. (11), using proton magnetic resonance spectroscopy, demonstrated marked depletion of brain myo-inositol stores in both cirrhosis with hepatic encephalopathy and in chronic hyponatremia.
Potassium ions act as effective osmoles like sodium ions. A hypokalemic state may jeopardize the early protective phase of the glial cells to hyperosmotic stress by the inability to rapidly move potassium ions into the cells to prevent intercompartmental osmolality difference (12). In addition, hypokalemia prevents insulin release, which is vital for glucose uptake by glial cells and generation of ATP that drives the cellular response to hyperosmotic stress (10). Correction of hypokalemia may also lead to overly rapid correction of hyponatremia and ODS independent of other factors (13). The severity of hyponatremia is directly proportional to the risk of ODS, because lower levels of SNa are more likely to be associated with overly rapid correction of hyponatremia and consequent ODS. The risk appears highest with SNa ≤105 mmol/L (14,15).
Nine of the reported patients that we identified had SNa ≥115 mEq/L (115–130 mEq/L), including five with an SNa >120 mEq/L, a level that is seldom associated with ODS (Table 2). At least two of these patients may have developed central pontine myelinolysis (CPM) before admission to the authors’ hospitals (3,16). One had been given intravenous fluids for gastroenteritis at a peripheral health center for 2 days and was transferred to the author’s hospital for altered consciousness (16). On admission, despite modest hyponatremia (120 mEq/L) the patient was stuporous and had bilateral Babinski signs. Another, with an SNa of 130 mEq/L on admission, was found to have asymptomatic CPM on a staging MRI for a newly diagnosed aggressive lymphoma (3). In addition to an unusually high SNa, six of the nine patients developed ODS, despite extremely slow correction (no more than 4–6 mEq/L in 24 hours). Two of these patients were reported by the same author and both had other unusual features (17): both had severe, persistent vomiting, with poor dietary intake and untreated hyponatremia for over a month, and both developed ODS despite azotemia (BUN, 78 and 128 mg/dl, respectively; creatinine, 3.3 and 7 mg/dl, respectively), which is thought to protect against ODS from rapid correction of hyponatremia (18). These two patients and a similar atypical patient with ESKD (19) and an SNa of 109 mEq/L (Table 1) who developed spastic quadriparesis and nystagmus after correction by only 5 mEq/L per day may have had thiamine deficiency, which has been associated with CPM and extrapontine myelinolysis (EPM) in patients who were not known to be hyponatremic (20). Another patient developed ODS after initially presenting with an SNa of 127 mEq/L (21); although the rate of correction never exceeded 10 mEq/L, he became severely hypernatremic, to a level of 161 mEq/L on the seventh day. ODS can be caused by hypernatremia even in the absence of hyponatremia (22); brain adaptations, even to mild hyponatremia, would be expected to increase the susceptibility to injury from hypernatremia. Two of the patients with SNa ≥115 mEq/L had hyperammonemia, one from cavernous transformation of the portal vein (23), and one from end stage liver disease (4). Patients with coexisting liver cirrhosis and chronic hyponatremia have swelling of the brain cells due to the combined effect of hyperammonemia and hypotonic hyponatremia (24). This is partly attributed to an increased level of intracellular glutamine in response to hyperammonemia with a compensatory loss of intracellular myo-inositol (11). Myo-inositol may serve as an antioxidant and has been shown to protect cells against osmotic stress in rats (25). In addition, patients with severe liver disease and hyperammonemia may develop lesions of CPM and EPM, even in the absence of hyponatremia (26).
Our study has important limitations. First, we relied on data available in individual case reports that are likely to include patients with unusual findings. Second, several case reports of ODS that could have potentially affected the outcome of the study were excluded if they did not explicitly report the SNa corrections rates per 24 hours. Third, there is likely a publication bias. Fourth, the study is underpowered to draw any firm conclusions.
In conclusion, published reports of ODS despite slow correction of hyponatremia are more convincing for patients with SNa <115 mEq/L (Table 1) than for patients with higher SNa (Table 2). The latter cases include patients who may have already had CPM or EPM at the time of their admission and patients with uncommon conditions that by themselves can result in CPM or EPM, even in the absence of hyponatremia; in such cases, it is difficult to ascribe neurologic complications to the very slow rise in SNa. Despite the limitations described, we believe that our findings can be useful in guiding treatment of patients with severe hyponatremia, especially those with SNa <115 mEq/L. In patients with conditions making them more susceptible to ODS (such as SNa ≤105 mEq/L, alcohol use disorder, hypokalemia, liver disease, malnutrition), well-documented ODS with a typical clinical course may complicate correction of chronic hyponatremia despite adherence to both the European Clinical Practice Guidelines (≤10 mEq/L per 24 hours) and to the recommendations of the American Expert Panel (≤8 mEq/L per 24 hours) (1,2). Therefore, it may be prudent to take a more conservative approach to hyponatremia correction than has been recommended by current guidelines in this high-risk patient population, as suggested by some experts (14,27,28). In such patients, we recommend limiting the rate of correction to less than (not equal to) 8 mEq/L in any 24 hour period to minimize the risk of ODS. Thiamine deficiency is commonly present in patients with hyponatremia and it constitutes an etiology for ODS independent of hyponatremia (20). Thiamine supplementation, a low-risk intervention, is advisable for any patient with hyponatremia whose dietary intake has been poor.
Disclosures
R.H. Sterns reports serving as a section editor for UpToDate. S. Tandukar reports having other interests in/relationships with the American Society of Nephrology and the American Society of Transplantation. The remaining author has nothing to disclose.
Funding
H. Rondon-Berrios is funded by a National Institute of Diabetes and Digestive and Kidney Diseases exploratory/developmental research grant R21DK122023.
Author Contributions
H. Rondon-Berrios and R.H. Sterns conceptualized the study, were responsible for formal analysis, and reviewed and edited the manuscript; H. Rondon-Berrios and S. Tandukar wrote the original draft and were responsible for data curation; and S. Tandukar was responsible for investigation.
References
- 1.Verbalis JG, Goldsmith SR, Greenberg A, Korzelius C, Schrier RW, Sterns RH, Thompson CJ: Diagnosis, evaluation, and treatment of hyponatremia: Expert panel recommendations. Am J Med 126[Suppl 1]: S1–S42, 2013. 10.1016/j.amjmed.2013.07.006 [DOI] [PubMed] [Google Scholar]
- 2.Spasovski G, Vanholder R, Allolio B, Annane D, Ball S, Bichet D, Decaux G, Fenske W, Hoorn EJ, Ichai C, Joannidis M, Soupart A, Zietse R, Haller M, van der Veer S, Van Biesen W, Nagler E; Hyponatraemia Guideline Development Group: Clinical practice guideline on diagnosis and treatment of hyponatraemia. Nephrol Dial Transplant 29: i1–i39, 2014. 10.1093/ndt/gfu040 [DOI] [PubMed] [Google Scholar]
- 3.Shah SO, Wang A, Mudambi L, Ghuznavi N, Fekete R: Asymptomatic central pontine myelinolysis: A case report. Case Rep Neurol 4: 167–172, 2012. 10.1159/000345225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacMillan TE, Cavalcanti RB: Outcomes in severe hyponatremia treated with and without desmopressin. Am J Med 131: 317.e1–317.e10, 2018. 10.1016/j.amjmed.2017.09.048 [DOI] [PubMed] [Google Scholar]
- 5.Chawla A, Sterns RH, Nigwekar SU, Cappuccio JD: Mortality and serum sodium: Do patients die from or with hyponatremia? Clin J Am Soc Nephrol 6: 960–965, 2011. 10.2215/CJN.10101110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aegisdottir H, Cooray C, Wirdefeldt K, Piehl F, Sveinsson O: Incidence of osmotic demyelination syndrome in Sweden: A nationwide study. Acta Neurol Scand 140: 342–349, 2019. 10.1111/ane.13150 [DOI] [PubMed] [Google Scholar]
- 7.King JD, Rosner MH: Osmotic demyelination syndrome. Am J Med Sci 339: 561–567, 2010. 10.1097/MAJ.0b013e3181d3cd78 [DOI] [PubMed] [Google Scholar]
- 8.John V, Evans P, Kalhan A: Delayed dyskinesia and prolonged psychosis in a patient presenting with profound hyponatraemia. Endocrinol Diabetes Metab Case Rep 2017: 16-0147, 2017. 10.1530/EDM-16-0147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicaise C, Marneffe C, Bouchat J, Gilloteaux J: Osmotic demyelination: From an oligodendrocyte to an astrocyte perspective. Int J Mol Sci 20: 1124, 2019. 10.3390/ijms20051124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pham PM, Pham PA, Pham SV, Pham PT, Pham PT, Pham PC: Correction of hyponatremia and osmotic demyelinating syndrome: have we neglected to think intracellularly? Clin Exp Nephrol 19: 489–495, 2015. 10.1007/s10157-014-1021-y [DOI] [PubMed] [Google Scholar]
- 11.Häussinger D, Laubenberger J, vom Dahl S, Ernst T, Bayer S, Langer M, Gerok W, Hennig J: Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 107: 1475–1480, 1994. 10.1016/0016-5085(94)90552-5 [DOI] [PubMed] [Google Scholar]
- 12.Lohr JW: Osmotic demyelination syndrome following correction of hyponatremia: Association with hypokalemia. Am J Med 96: 408–413, 1994. 10.1016/0002-9343(94)90166-X [DOI] [PubMed] [Google Scholar]
- 13.Berl T, Rastegar A: A patient with severe hyponatremia and hypokalemia: Osmotic demyelination following potassium repletion. Am J Kidney Dis 55: 742–748, 2010. 10.1053/j.ajkd.2009.12.024 [DOI] [PubMed] [Google Scholar]
- 14.Sterns RH: Treatment of severe hyponatremia. Clin J Am Soc Nephrol 13: 641–649, 2018. 10.2215/CJN.10440917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tandukar S, Rondon-Berrios H: Treatment of severe symptomatic hyponatremia. Physiol Rep 7: e14265, 2019. 10.14814/phy2.14265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zunga PM, Farooq O, Dar MI, Dar IH, Rashid S, Rather AQ, Basu JA, Ashraf M, Bhat JA: Extra pontine osmotic demyelination syndrome. Ann Neurosci 22: 51–53, 2015. 10.5214/ans.0972.7531.220212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pradhan S, Jha R, Singh MN, Gupta S, Phadke RV, Kher V: Central pontine myelinolysis following ‘slow’ correction of hyponatremia. Clin Neurol Neurosurg 97: 340–343, 1995. 10.1016/0303-8467(95)00060-W [DOI] [PubMed] [Google Scholar]
- 18.Soupart A, Penninckx R, Stenuit A, Decaux G: Azotemia (48 h) decreases the risk of brain damage in rats after correction of chronic hyponatremia. Brain Res 852: 167–172, 2000. 10.1016/S0006-8993(99)02259-3 [DOI] [PubMed] [Google Scholar]
- 19.Yamada H, Takano K, Ayuzawa N, Seki G, Fujita T: Relowering of serum Na for osmotic demyelinating syndrome. Case Rep Neurol Med 2012: 704639, 2012. 10.1155/2012/704639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergin PS, Harvey P: Wernicke’s encephalopathy and central pontine myelinolysis associated with hyperemesis gravidarum. BMJ 305: 517–518, 1992. 10.1136/bmj.305.6852.517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davenport C, Liew A, Vic Lau P, Smith D, Thompson CJ, Kearns G, Agha A: Central pontine myelinolysis secondary to hypokalaemic nephrogenic diabetes insipidus. Ann Clin Biochem 47: 86–89, 2010. 10.1258/acb.2009.009094 [DOI] [PubMed] [Google Scholar]
- 22.Pena-Polanco JE, Rondon-Berrios H: Quiz page November 2016: Kidney and neurologic complications in the treatment of a patient with hepatic encephalopathy. Am J Kidney Dis 68: A15–A17, 2016. 10.1053/j.ajkd.2016.07.020 [DOI] [PubMed] [Google Scholar]
- 23.Hu Q, Zhang C, Liu J, Xu F, Zhu S: Extrapontine myelinolysis in a case of portal cavernous transformation following slow correction of chronic hyponatremia. Neurol Sci 34: 1831–1833, 2013. 10.1007/s10072-013-1311-2 [DOI] [PubMed] [Google Scholar]
- 24.Córdoba J, García-Martinez R, Simón-Talero M: Hyponatremic and hepatic encephalopathies: similarities, differences and coexistence. Metab Brain Dis 25: 73–80, 2010. 10.1007/s11011-010-9172-3 [DOI] [PubMed] [Google Scholar]
- 25.Silver SM, Schroeder BM, Sterns RH, Rojiani AM: Myoinositol administration improves survival and reduces myelinolysis after rapid correction of chronic hyponatremia in rats. J Neuropathol Exp Neurol 65: 37–44, 2006. 10.1097/01.jnen.0000195938.02292.39 [DOI] [PubMed] [Google Scholar]
- 26.Chang Y, An DH, Xing Y, Qi X: Central pontine and extrapontine myelinolysis associated with acute hepatic dysfunction. Neurol Sci 33: 673–676, 2012. 10.1007/s10072-011-0838-3 [DOI] [PubMed] [Google Scholar]
- 27.Adrogué HJ, Madias NE: The challenge of hyponatremia. J Am Soc Nephrol 23: 1140–1148, 2012. 10.1681/ASN.2012020128 [DOI] [PubMed] [Google Scholar]
- 28.Oh MS, Kim HJ, Carroll HJ: Recommendations for treatment of symptomatic hyponatremia. Nephron 70: 143–150, 1995. 10.1159/000188576 [DOI] [PubMed] [Google Scholar]
- 29.Koul PA, Khan UH, Jan RA, Shah S, Qadri AB, Wani B, Ashraf M, Ahmad F, Bazaz SR: Osmotic demyelination syndrome following slow correction of hyponatremia: Possible role of hypokalemia. Indian J Crit Care Med 17: 231–233, 2013. 10.4103/0972-5229.118433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Souza A, Desai PK: More often striatal myelinolysis than pontine? A consecutive series of patients with osmotic demyelination syndrome. Neurol Res 34: 262–271, 2012. 10.1179/1743132812Y.0000000009 [DOI] [PubMed] [Google Scholar]
- 31.Leens C, Mukendi R, Forêt F, Hacourt A, Devuyst O, Colin IM: Central and extrapontine myelinolysis in a patient in spite of a careful correction of hyponatremia. Clin Nephrol 55: 248–253, 2001 [PubMed] [Google Scholar]
- 32.Al-Shaibany A, Shabahat S, Shahid M: A case of central pontine myelinolysis due to severe acute prolonged hyponatremia in our county hospital. IJMA 3: 1316–1321, 2021 [Google Scholar]
- 33.Silbert PL, Knezevic WV, Peake HI, Khangure M: Behavioural changes due to pontine and extrapontine myelinolysis. Med J Aust 157: 487–488, 1992. 10.5694/j.1326-5377.1992.tb137316.x [DOI] [PubMed] [Google Scholar]
- 34.Reijnders TDY, Janssen WMT, Niamut SML, Kramer AB: Role of risk factors in developing osmotic demyelination syndrome during correction of hyponatremia: A case study. Cureus 12: e6547, 2020. 10.7759/cureus.6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malhotra K, Ortega L: Central pontine myelinolysis with meticulous correction of hyponatraemia in chronic alcoholics. BMJ Case Rep 2013: bcr2013009970, 2013. 10.1136/bcr-2013-009970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dellabarca C, Servilla KS, Hart B, Murata GH, Tzamaloukas AH: Osmotic myelinolysis following chronic hyponatremia corrected at an overall rate consistent with current recommendations. Int Urol Nephrol 37: 171–173, 2005. 10.1007/s11255-004-4770-9 [DOI] [PubMed] [Google Scholar]
- 37.DeWitt LD, Buonanno FS, Kistler JP, Zeffiro T, DeLaPaz RL, Brady TJ, Rosen BR, Pykett IL: Central pontine myelinolysis: Demonstration by nuclear magnetic resonance. Neurology 34: 570–576, 1984. 10.1212/WNL.34.5.570 [DOI] [PubMed] [Google Scholar]
- 38.Orakzai RH, Orakzai SH, Hasley PB: Treating hyponatremia: How slow is safe? Central pontine myelinolysis despite appropriate correction of hyponatremia. Eur J Intern Med 19: e29–e31, 2008. 10.1016/j.ejim.2007.08.009 [DOI] [PubMed] [Google Scholar]
- 39.Jahan M, Sharma S, Rehmani R: Osmotic demyelination syndrome despite appropriate hyponatremia correction. Cureus 12: e8209, 2020. 10.7759/cureus.8209 [DOI] [PMC free article] [PubMed] [Google Scholar]