

Abstract
Primary age-related tauopathy (PART) is a neurodegenerative pathology with features distinct from but also overlapping with Alzheimer disease (AD). While both exhibit Alzheimer-type temporal lobe neurofibrillary degeneration alongside amnestic cognitive impairment, PART develops independently of amyloid-β (Aβ) plaques. The pathogenesis of PART is not known, but evidence suggests an association with genes that promote tau pathology and others that protect from Aβ toxicity. Here, we performed a genetic association study in an autopsy cohort of individuals with PART (n = 647) using Braak neurofibrillary tangle stage as a quantitative trait. We found some significant associations with candidate loci associated with AD (SLC24A4, MS4A6A, HS3ST1) and progressive supranuclear palsy (MAPT and EIF2AK3). Genome-wide association analysis revealed a novel significant association with a single nucleotide polymorphism on chromosome 4 (rs56405341) in a locus containing three genes, including JADE1 which was significantly upregulated in tangle-bearing neurons by single-soma RNA-seq. Immunohistochemical studies using antisera targeting JADE1 protein revealed localization within tau aggregates in autopsy brains with four microtubule-binding domain repeats (4R) isoforms and mixed 3R/4R, but not with 3R exclusively. Co-immunoprecipitation in post-mortem human PART brain tissue revealed a specific binding of JADE1 protein to four repeat tau lacking N-terminal inserts (0N4R). Finally, knockdown of the Drosophila JADE1 homolog rhinoceros (rno) enhanced tau-induced toxicity and apoptosis in vivo in a humanized 0N4R mutant tau knock-in model, as quantified by rough eye phenotype and terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) in the fly brain. Together, these findings indicate that PART has a genetic architecture that partially overlaps with AD and other tauopathies and suggests a novel role for JADE1 as a modifier of neurofibrillary degeneration.
Introduction
Primary age-related tauopathy (PART) is nearly ubiquitously observed to varying degrees of severity in the brains of aged individuals. PART develops in the absence Aβ plaques yet manifests as neurofibrillary tangles (NFT) composed of abnormal aggregates of the microtubule-associated protein tau that are morphologically, biochemically and ultrastructurally identical to those in early to moderate-stage Alzheimer Disease (AD) [23]. A neuropathological diagnosis of PART does not imply cognitive impairment; individuals with PART can be normal, mildly cognitively impaired (MCI) or demented [40, 101]. Most individuals with PART remain cognitively normal; however, those clinically affected usually develop amnestic cognitive changes [7, 10, 40, 69]. Given the similarities between PART and AD, PART as a diagnostic construct would have greater value if it were shown to arise independently [16, 28]. The observation that some individuals of advanced age at autopsy show substantial NFT pathology without evidence of amyloid deposition provides strong evidence that it does. However, the argument can be made that PART might be a component of the AD spectrum, and Aβ pathology might have eventually developed in such individuals had they lived longer [28, 53]. Thus, the question remains to what extent patients with a neuropathological diagnosis of PART have divergent or overlapping risk factors with AD and other dementias [8, 48].
Much of the mechanistic knowledge surrounding tauopathy stems from genetic studies [95]. Autosomal dominant mutations in the microtubule-associated protein tau gene (MAPT) in coding regions can interfere with microtubule binding or promote transition to toxic forms [38]. Mutations in splice sites disrupt alternative mRNA splicing of the tau mRNA transcript, modifying the ratio of three repeat (3R) and four repeat (4R) tau isoforms leading to downstream pathological sequelae [38], while alternative splicing of the N-terminal exons may also play a role in modulating toxic tau specifically through regulation of microtubule stabilization [21, 26, 72]. However, MAPT mutations are rare. In contrast, PART occurs sporadically and nearly ubiquitously with advanced age in individuals without MAPT mutations. Thus, studying PART may reveal common genetic drivers of tauopathy and their association with abnormalities in tau proteostasis and isoform expression. The MAPT 17q21.31 locus is defined by a large ~ 900 kb inversion consisting of two distinct common haplotypes, H1 and H2. The more frequent H1 has been associated with increased risk for PART and several other sporadic tauopathies including APOE ε4-negative AD, progressive supranuclear palsy, corticobasal degeneration, as well as Parkinson disease which is not classically considered a tauopathy [18, 35, 46, 49, 68, 83]. Research focusing on the genetics of PART has consistently failed to show an association with the APOE ε4 allele, the strongest risk locus in AD [43, 65, 69, 83]. This is not surprising, given the strong association of APOE ε4 with early deposition of amyloid beta (Aβ) peptide. Outside of PART, one of the largest AD GWAS has identified 21 risk loci [52]. However, APOE, the strongest AD risk locus may be confounding associations independently related to tauopathy [46]. Another study confirmed APOE ε4 was not associated with PART but did find associations in PART with AD alleles in the BIN1, PTK2B, and CR1 loci [61]. Taken together, these data suggest an unexplored genetic risk driving tauopathy that might be revealed by conducting association studies in PART.
The neuropathological overlap between PART and AD allows for a unique opportunity to study mechanisms of tau-mediated AD-type neurodegeneration independently of Aβ. However, PART cannot be reliably diagnosed pre-mortem because of non-specific symptomology and accompanying age-related comorbid dementing pathologies. Therefore, discovering unique drivers is challenging antemortem [43, 45, 70, 71, 80, 85]. Assembling an autopsy cohort large enough to assess its genetics on a genome-wide scale has not yet been attempted. In collaboration with 21 domestic and international brain biorepositories, we performed the first GWAS in PART, using tau pathology as a quantitative trait, and compared our findings to known tauopathy risk loci. We then examined expression of candidate genes in our strongest risk locus (chromosome 4q28.2) using bulk RNA sequencing, single cell RNA-sequencing, and immunohistochemistry. We identified gene for apoptosis and differentiation in epithelia (JADE1) as a potential candidate gene. Finally, we validated our findings biochemically in human postmortem brain and identified a JADE1-tau protein interaction. Studies in Drosophila suggest that JADE1 plays a neuroprotective role in tauopathy. The work presented here not only further informs the genetics of PART, but also suggests a novel role for JADE1 in tauopathy.
Materials and methods
Cohort
Fresh-frozen brain tissue was obtained from the contributing centers (Supplementary Table 1, online resource). All tissue was used in accordance with the relevant guidelines and regulations of the respective institutions. Inclusion criteria were individuals with normal cognition, mild cognitive impairment (any type) and dementia. Cognitive status was determined either premortem or postmortem by a clinical chart review, mini-mental score, or clinical dementia rating [33, 64]. Neuropathological assessments were performed at the respective centers using standardized criteria including Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuritic plaque assessment and Braak neurofibrillary tangle staging [15, 62]. Neuropathological inclusion criteria were Braak tangle stage of 0–IV and neuritic plaque severity CERAD score of 0 [15, 62]. In addition, formalin fixed paraffin-embedded tissue sections were obtained and reevaluated by the study investigators to confirm the lack of Aβ and degree of PART tau pathology as described by Walker et al. [97]. Clinical exclusion criteria were motor neuron disease, parkinsonism, and frontotemporal dementia. Neuropathological exclusion criteria were other degenerative diseases associated with NFTs (i.e., AD, progressive supranuclear palsy [PSP], corticobasal degeneration [CBD], chronic traumatic encephalopathy [CTE], frontotemporal lobar degeneration-tau [FTLD-tau], Pick disease (PiD), Guam amyotrophic lateral sclerosis/parkinsonism–dementia, subacute sclerosing panencephalitis, globular glial tauopathy). Individuals with aging-related tau astrogliopathy (ARTAG) were not excluded [51]. In addition to PART cases, neuropathologically confirmed cases of AD, PSP, CTE, CBD, argyrophilic grain disease (AGD) and PiD (n = 3 for each, Supplemental Table 6) were examined for convergent or divergent staining patterns.
Genotyping
High-throughput isolation of DNA was performed using the MagMAX DNA Multi-Sample Ultra 2.0 Kit on a King-Fisher Flex robotic system (Thermofisher, Waltham, MA). Fresh frozen cortical brain tissue (20–40 mg) was placed into a deep-well plate and treated with 480 μl of Proteinase K mix (Proteinase K, phosphate buffered saline [pH 7.4], Binding Enhancer) and incubated overnight at 65 °C at 800 rpm on a shaking plate. Genomic DNA was isolated and purified using magnetic particles. DNA quality control was performed using a nanodrop spectrophotometer (concentration > 50 ng/μl, 260/280 ratio 1.7–2.2). Genotyping was performed using single nucleotide polymorphism (SNP) microarrays (Infinium Global Screening Array v2.4. or Infinium OmniExpress-24; Illumina, San Diego, CA). Raw genotype files were converted to PLINK-compatible files using GenomeStudio software (Illumina, San Diego, CA).
Genetic analysis
PLINK v1.9 was used to perform quality control [78]. SNP exclusion criteria included minor allele frequency < 1%, genotyping call-rate filter less then 95%, and Hardy–Weinberg threshold of 1 × 10−6 [3]. Individuals with discordant sex, non-European ancestry, genotyping failure of > 5%, or relatedness of > 0.1 were excluded. A principal component analysis (PCA) was performed to identify population sub-structure using EIGENSTRAT v6.1.4 and the 1000 genomes reference panel [20, 76]. Samples were excluded if they were five standard deviations away from the European population cluster (Supplementary Fig. 1, online resource). PCA was performed again on the genomic data with non-Europeans excluded to generate new PCs, of which the first four were used as covariates in the regression model. All data was imputed on the University of Michigan server using mini-mac3 and HRC reference panel [24, 60]. Imputed variants with MAF < 0.01 and a dosage R2 < 0.7 were excluded. A quantitative trait association test was run on 647 PART cases vs. Gaussian-normalized Braak stage using conditional linear regression and age, sex, principal component 1–4 and SNP chip array (batch) as covariates. The analysis was run separately on each genotyping array and a meta-analysis was performed using METAL (2011 release) [99]. Regional genome-wide association plots were created with LocusZoom [77] (GRCh37) and other plots were created using R v4.0 and ggplot2 v3.3.5.
Expression quantitative trait loci (eQTL) analysis
For eQTL analysis, publicly available Religious Orders Study and Rush Memory and Aging Project (ROS-MAP) whole genome sequencing (WGS) and bulk RNA sequencing data were obtained from the Synapse portal (syn3219045 and syn8456629, respectively) and analyzed using techniques described elsewhere [25, 88]. Briefly, RNA was analyzed from the dorsolateral prefrontal cortex of 452 postmortem samples and a linear regression was performed stratified by the lead SNP’s genotype and JADE1 mRNA expression. Statistics were performed using R v4.0 and plotted in ggplot2 v3.3.5.
Single-cell mRNA profiling in tangle-containing neurons
Identification of differentially expressed genes in single neuronal somata with and without NFTs was performed by analyzing a transcriptomic data set of isolated neurons from Brodmann area 9 post-mortem human brain from individuals with AD (Braak stage VI) previously reported by Otero-Garcia et al. [73]. This data set consists of single neuronal soma transcriptomes obtained by mechanical dissociation of frozen tissue, followed by fluorescence-activated cell sorting (FACS) using p-tau (AT8) and MAP2 antisera to isolate single NFT-positive and NFT-negative neurons, mRNA capture using 10 × Chromium v2 and sequenced with Illumina Novaseq 6000. Briefly, the data was normalized using the global-scaling function “LogNormalize” which normalizes the gene expression measurements for each cell by the total expression, multiplies this by a scale factor (10,000), and log-transforms the result (natural log). For dimensionality reduction and clustering, Seurat v2.4 MultiCCA was used with the top 4200 highly variable genes, selecting the first 26 canonical correlation vectors aligned by sample as grouping variable. For visualization, Seurat v2.4 FindClusters function was used with the following parameters: 1:22 PCs; 1.0 resolution; 100 random start positions and 10 iterations per random start; and 30 k for the k-nearest neighbor algorithm). Gene cluster-specific markers were identified by comparing the gene expression levels for each specific cluster to all other cells using the Wilcoxon rank-sum test. Genes detected in ≥ 25% of cells with positive log fold changes > 0.25 and adjusted p values < 0.05 were included. Clusters containing cells with nonspecific identities were removed from further analysis. Differential gene expression (DGE) between neurons with and without NFTs for each cell-type cluster was assessed using the MAST generalized linear model, selecting an adjusted p value of < 0.05 as cutoff [31].
Immunohistochemistry
Formalin-fixed paraffin-embedded (FFPE) tissue sections (5 μm) on charged slides were baked at 70 °C and immunohistochemistry (IHC) was performed on a Ventana Benchmark XT automatic stainer (Rouche, Tucson, AZ). Antigen retrieval was done using citric acid buffer (CC1) for 1 h followed by primary antibody incubation for approximately 40 min. A secondary antibody, 3,3’-diaminobenzidine (DAB) was then applied. For slides that were doubled-labeled, DAB and alkaline phosphatase were used for visualization. Slides were stained with antibodies to JADE1 (1:100, Proteintech, Rosemont, IL) and hyperphosphorylated tau (p-tau, AT8, 1:1000, Invitrogen, Waltham, MA). To ensure specificity of the JADE1 antisera, a peptide competition was performed using a blocking peptide. The JADE1 antibody was combined with a fivefold (by weight) excess of blocking peptide (Proteintech, Rosemont, IL) in 500 μl of PBS and incubated overnight at 4 °C. Whole slide images (WSI) were visualized and scanned using an Aperio CS2 digital slide scanner (Leica Biosystems, Wetzlar Germany).
Biochemical analysis
Western blots were performed using homogenized fresh-frozen brain tissue. Samples were placed in a micro-tube homogenizer (SP Bel-Art, Wayne, NJ) in 10 volumes (wt/vol) of ice-cold Pierce RIPA buffer (Thermo Fisher Scientific, Waltham, MA) containing Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific), and incubated on ice for 30 min. For each sample, 20 μg of proteins were boiled in 1×Laemmli sample buffer (Bio-Rad, Hercules, CA) for 5 min, run on 10% Criterion TGX Precast Gels (Bio-Rad, Hercules, CA), blotted to nitrocellulose membranes, and stained with JADE1 antisera (1:2000). Horseradish peroxidase-labeled secondary anti-rabbit antisera (1:20,000; Vector Labs, Burlingame, CA) was detected by SuperSignal West Pico PLUS Chemiluminescent Substrate or Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific). To quantify and standardize protein levels, GAPDH was detected with GAPDH antisera (6C5, 1:20,000; Abcam, Cambridge, MA) and total protein was detected with Amido Black (Sigma-Aldrich, St. Louis, MO) as previously described [2]. Chemiluminescence was measured in a ChemiDoc Imaging System (Bio-Rad), and relative optical densities were determined using AlphaEaseFC software, version 4.0.1 (Alpha Innotech, San Jose, CA), normalized to GAPDH and total protein loaded.
Co-immunoprecipitation (IP) assays were performed using fresh-frozen brain tissue homogenized in a glass-Teflon homogenizer at 500 rpm in 10 volumes (wt/vol) of ice-cold lysis buffer containing 50 mM Tris, pH 7.8, 0.5% NP40, 150 mM NaCl, 1 mM EDTA, and Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Samples were incubated on ice for 30 min, and centrifuged at 1000×g for 10 min. The supernatant was collected and used for immunoprecipitation. In a microcentrifuge tube, 70 ul of supernatant, Lysis buffer (930 μl) and 2 μg of either JADE1 antisera or anti-0N tau antisera (EPR21726, Abcam, Cambridge, MA) were combined and incubated overnight at 4 °C. Antisera-negative and IgG isotype controls, either normal rabbit IgG (PeproTech, Rocky Hill, NJ) or Mouse IgG1 kappa, (clone: P3.6.2.8.1, Thermo Fisher Scientific, Waltham, MA), were carried out. 20 ul of Pierce Protein A/G Agarose beads (Thermo Fisher Scientific, Waltham, MA) was added to each reaction, and the mixture was incubated for 1 h at 4 °C. Agarose beads were pelleted at 1000 × g for 5 min at 4 °C, supernatant was removed, 1 ml of ice-cold lysis buffer was added, and pellet was washed by inverting tube several times. Beads were washed 4 times, each time repeating the centrifugation step above. After the final wash, pelleted beads were resuspended in 40 μl of 1×Laemmli sample buffer (Bio-Rad, Hercules, CA) and boiled for 5 min. The samples were then centrifuged to pellet the agarose beads followed by SDS-PAGE analysis of the supernatant. Fifteen μl of samples for JADE1 detection and 5 μl for tau with tau isoform ladders (rPeptide, Watkinsville, GA) were run on 10% PROTEAN TGX Precast Gels (Bio-Rad, Hercules, CA), blotted to nitrocellulose membranes, and stained with JADE1 antisera (1:2000), total tau antisera (HT7, 1:3000; Thermo Fisher Scientific), three microtubule repeat domain tau antisera (3R, 8E6/C11, 1:2000; MilliporeSigma, St. Louis, MO), four microtubule binding domain repeat tau antisera (4R, 1:2000; Cosmo Bio, Carlsbad, CA), pThr231 tau antisera (RZ3, 1:200; a gift from Dr. Peter Davies), pSer396/pSer404 tau antisera (PHF1, 1:500; a gift from Dr. Peter Davies), pSer202 tau antisera (CP13, 1:500; a gift from Dr. Peter Davies), pSer202/pThr205 tau antisera (AT8, 1:1000; Thermo Fisher Scientific), pSer214 tau antisera (S214; 44-742G, 1:1000; Thermo Fisher Scientific, Waltham, MA), or pSer409 tau antisera (PG5, 1:200; a gift from Dr. Peter Davies). Horseradish peroxidase-labeled conformation-sensitive secondary anti-mouse IgG for IP or anti-rabbit VeriBlot for IP Detection antibody (both at 1:20,000; Abcam, Cambridge, MA) was detected by SuperSignal West Femto Maximum Sensitivity substrate (Thermo Fisher Scientific).
A dephosphorylation assay was performed using fresh-frozen brain tissue homogenized with a glass-Teflon homogenizer at 500 rpm in 10 volumes (wt/vol) of ice-cold lysis buffer containing 50 mM Tris, pH 7.8, 0.5% NP40, 150 mM NaCl, and Halt protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA). Samples were incubated on ice for 30 min, centrifuged at 1000 ×g for 10 min, and supernatant was collected. Reaction mixtures (51 μl) consisted of 39 μl of supernatant, 1 μl of protease inhibitor cocktail, 5 μl of 10X NEBuffer for protein metallophosphatases, 5 μl of 10 mM MnCl2, 1 μl of lambda protein phosphatase (New England BioLabs, Ipswich, MA). Each mixture was incubated at 30 °C for either 1, 2, 3, or 4 h. Additional 1 μl of lambda protein phosphatase and 1 μl of protease inhibitor cocktail were added in each mixture every hour.
Proximity ligation assay
5 μm-thick FFPE hippocampal sections were mounted on charged slides and stained using a Duolink kit (MilliporeSigma, St. Louis, MO). Sections were deparaffinized and incubated in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) at 95 °C for 20 min, washed in running water, incubated in 0.2% Tween 20 in PBS at room temperature for 20 min, and washed in PBS 3 times for 5 min. Blocking assays were performed using Duolink In Situ Red Starter Kit Mouse/Rabbit (MilliporeSigma, St. Louis, MO) according to the manufacture’s protocol with JADE1 antisera (1:20) and 0N tau antisera (1:500; Bio-Legend, San Diego, CA). Two control assays were also performed, one with JADE1 antisera only, and the other with anti-0N tau antisera. All samples were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Sections were imaged using an Axioview fluorescent microscope (Carl Zeiss, Oberkochen, Germany) and processed using Zen Blue software v3.4 (Carl Zeiss).
In vivo Drosophila model
Drosophila stocks, crosses, and aging were performed at 25 °C for the duration of the experiment and an equal number of male and female flies were used. The GAL4-UAS expression system and the pan-neuronal elav-GAL4 driver were used to control transgenic gene expression. Analyses were run on four fly groups (Bloomington stock rnoRNAi line number 57774): elav-GAL4 driver in the heterozygous state (control, elav-GAL4/ +), elav-Gal4 positive plus rnoRNAi positive group (control + rnoRNAi, elav-GAL4/ +; UAS-rnoRNAi/ +), elav-Gal4 positive transgenic human UAS-tauR406W tau group (0N4R Tau, elav-GAL4/ +; UAS-TauR406W), elav-Gal4 positive human transgenic UAS-tauR406W tau plus rnoRNAi positive group (0N4R Tau + rnoRNAi, elav-GAL4/ +; UAS-TauR406W/UAS-rnoRNAi). An additional rnoRNAi line was tested but did not produce progeny when crossed to tau transgenic Drosophila, as often occurs with strong enhancers (Bloomington stock rnoRNAi line number 62880). Flies were aged to 10 d, at which point terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay was performed in the brain (n = 6 per genotype) and a blinded assessment of the fly eye phenotype was performed (n = 16 per genotype). TUNEL was performed on 4 μm FFPE fly heads. Quantification of TUNEL-positive nuclei was performed throughout the entire brain using DAB-based detection and bright field microscopy and representative images were taken in the fly cortex. Fly eye phenotype scoring was performed blindly using light microscopy. The blinded rater semi-quantitively evaluated the eye for four distinct qualities including roughness, size, shape and conical shape on a 1–5 scale (five being the most severe phenotype) and an average summary score was calculated. Representative fly eye images were taken using a scanning electron microscope (JEOL, Peabody, MA) using an accelerating voltage of 20 kV.
Statistical analysis
For GWAS, statistical analysis was performed in PLINK and our genome-wide significance value was < 5 × 10−8, which is Bonferroni-corrected for all the independent common SNPs across the human genome. Genome-wide suggestive significant value was set at < 5 × 10−6. All other statistical analyses were performed in R v4.0. For non-normally distributed data a Wilcox test was used to test for significance, and an ANOVA was used for normally distributed data.
Results
We assembled a cohort of aged individuals to enable our genetic study of primary age-related tauopathy (PART) in which all were neuropathologically confirmed to be devoid of neuritic plaques. Brain tissue samples were obtained from 21 biorepositories (n = 857). While each center had performed a comprehensive neurodegeneration workup, we restained and reassessed these cases as a component of our ongoing histological studies [97]. Because some subjects upon restaining were found to have a mild neuritic plaque burden (CERAD 0), the final number of subjects in the study was reduced to 647. Neurofibrillary tangles (NFTs) were assessed using the Braak staging system, ranging from 0 to IV with all stages well represented, as is consistent with PART (Table 1). It should be noted that many Braak stage 0 subjects were observed to have rare tau pathology when assessed using immunohistochemistry which is compatible with that stage. The average age of death was 83 years, and the number of male and females was approximately equal. Braak stage II was the most frequent (n = 189), with relatively equal number of donors with Braak stages III (n = 152) and I (n = 142), and the fewest with Braak stages 0 (n = 71) and IV (n = 93). Braak staging performed at each center was not disproportionally skewed (Supplementary Fig. 2a, online resource). Sixty-six percent of the donors were cognitively normal. However, amongst Braak stages I–IV, there was an equal number of cognitively impaired subjects across each stage. We evaluated the association of age at death with Braak stage with respect to cognitive status and found a positive correlation that does not significantly change when cognitive status is accounted for in the model (Supplementary Fig. 2b, online resource). Comorbid cerebrovascular disease, which is common in this cohort, is likely responsible for the variation in cognitive impairment [39]. In summary, our cohort consists of older individuals, with a range of dementia severity, restricted regional tau distribution and varying severity of burden, demonstrating the diversity of both the clinical and neuropathological features of the condition.
Table 1.
Cohort data
| Braak stage | n | Average age of death in years (range) | Male (%) | Female (%) | Normal (%) | MCI (%) | Dementia (%)* |
|---|---|---|---|---|---|---|---|
| 0 | 71 | 71.2 (51–104) | 48 (7.5) | 23 (3.5) | 58 (8.9) | 7 (1.0) | 6 (0.9) |
| I | 142 | 78.1 (53–108) | 88 (13.6) | 54 (8.3) | 99 (15.3) | 17 (2.6) | 22 (3.4) |
| II | 189 | 84.1 (51–107) | 98 (15.1) | 91 (14.0) | 126 (19.4) | 40 (6.1) | 21 (3.2) |
| III | 152 | 88.8 (68–105) | 59 (9.1) | 93 (14.8) | 96 (14.8) | 29 (4.4) | 26 (4.0) |
| IV | 93 | 91.2 (67–106) | 29 (4.4) | 64 (9.8) | 50 (7.7) | 22 (3.4) | 21 (3.2) |
| Total (647) | 647 | 83.5 (51–108) | 322 (49.7) | 325 (50.2) | 429 (66.3) | 115 (17.7) | 96 (14.8) |
7 samples did not have cognative status available, MCI mild cognitive impairment
Using this cohort, we ran a quantitative trait association analysis across the entire genome to characterize genetic risk for PART. Our model adjusted for age, sex, genotyping platform, and principal components 1–4 and produced an inflation λ of 1.04 (Supplementary Fig. 3a, online resource). Using Braak stage as a quantitative trait revealed suggestive signals in 14 loci and a nominally genome-wide significant signal on chromosome 4q28.2 (Fig. 1a, rs56405341; linear regression β = 0.35, standard error = 0.06, p = 4.82 × 10−8, Table 2, Supplementary Table 3, online resource). The genome-wide significant variant (rs56405341) has a minor allele frequency of 0.27. Assessment of this region indicated this SNP is not within the footprint of any specific gene, but is near C4orf33, SCLT1 and JADE1 (Fig. 1b), and in high linkage disequilibrium with 22 other SNPs (r2 > 0.8, Supplementary table 4, online resource). Further examination of the homozygous and heterozygous alleles using strip chart showed a significant relationship between higher Braak stage and homozygous minor allele carriers (Fig. 1 c, AA–AG p = 0.024, AA–GG p = 3.3 × 10−5, AG–GG p = 7.2 × 10−5). Analysis of data from each independent genotyping array separately confirmed that the signal was apparent in both subsets of the data (Infinium OmniExpress-24, n = 440, β = 0.27, SE = 0.05, p = 1.11 × 10−6, λ = 1.03, Global screening array n = 207, β = 0.20, SE = 0.08, p = 1.42 × 10−2, λ = 1.01, Supplementary Table 2 and Fig. 5a–d, online resource). The individual summary statistics derived from the separate chip analysis were then combined to run a meta-analysis, and the resulting p value was similar to the combined analyses, as well as in agreement in the direction of effect tested allele (p = 5.61 × 10−8).
Fig. 1.
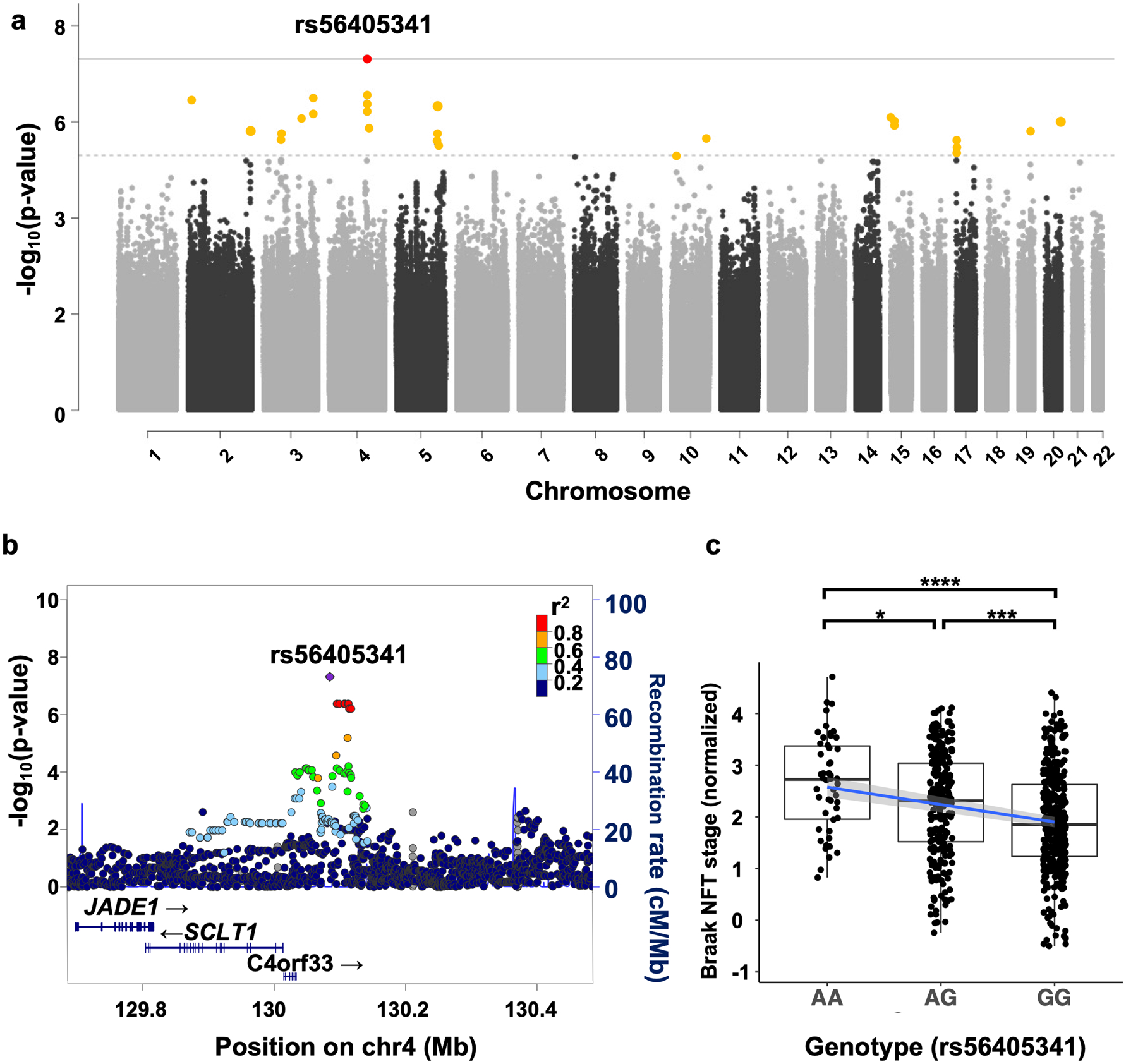
Genome-wide association study (GWAS) in primary age-related tauopathy. a Quantitative trait GWAS was performed using normalized Braak neurofibrillary tangle stage with age, sex, principal components (PCs), and genotyping SNP array as covariates (n = 647). The threshold for genome-wide significance (p < 5 × 10−8) is indicated by the solid grey line; the suggestive line (p < 5 × 10−6) is indicated by the dotted line. b LocusZoom plot shows a strong signal with multiple SNPs in linkage disequilibrium on chromosome 4q28.2. The x axis is the base pair position, and the y axis is the – log10 of the p value for the association with Braak stage. The blue line represents the recombination rate. c Association between single-nucleotide polymorphism (SNP), rs56405341 and Braak tangle stage (adjusted for age and sex). Pairwise comparisons using Wilcoxon rank sum test, AA–AG p = 0.024, AA–GG p = 3.3 × 10−5, AG–GG p = 7.2 × 10−5
Table 2.
Associations with Braak stage in PART (p < 5.0E − 06, n = 647)
| Chr | Base pair | SNP | Genes | A1 | Beta | SE | L95 | U95 | t statistic | p |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 11,935,322 | rs78580932 | GREB1, NTSR2, LPIN1, MIR4262 | C | − 0.54 | 0.10 | −0.76 | −0.34 | −5.15 | 3.55E-07 |
| 2 | 235,768,810 | rs74600760 | ARL4C, SH3BP4 | T | − 0.53 | 0.11 | −0.74 | −0.32 | −4.86 | 1.48E-06 |
| 3 | 61,518,166 | rs111610564 | FHIT, PTPRG | C | − 0.49 | 0.10 | −0.70 | −0.30 | −4.82 | 1.81E-06 |
| 3 | 140,216,029 | rs349509 | CLSTN2, CLSTN2-AS1, TRIM42 | A | − 0.77 | 0.16 | −1.08 | −0.47 | −4.97 | 8.64E-07 |
| 3 | 179,019,705 | rs13323081 | PIK3CA, KCNMB3, ZNF639, MFN1 | G | 0.24 | 0.05 | 0.15 | 0.33 | 5.17 | 3.16E-07 |
| 4 | 130,085,480 | rs56405341 | C4orf33, SCLT1, JADE1 | A | 0.25 | 0.05 | 0.16 | 0.35 | 5.52 | 4.82E-08 |
| 4 | 137,329,297 | rs77506227 | – | A | − 0.79 | 0.16 | −1.12 | −0.48 | −4.88 | 1.34E-06 |
| 5 | 150,473,104 | rs6579838 | GPX3, TNIP1, ANXA6 | C | 0.23 | 0.05 | 0.14 | 0.32 | 5.10 | 4.57E-07 |
| 10 | 124,487,608 | rs150945906 | DMBT1, C10orf120, DMBT1P1 | A | − 0.64 | 0.14 | −0.91 | −0.38 | −4.77 | 2.31E-06 |
| 15 | 24,515,682 | rs147462127 | PWRN2, PWRN1 | T | − 0.84 | 0.17 | −1.18 | −0.51 | −4.98 | 8.37E-07 |
| 15 | 36,865,219 | rs185845364 | C15orf41, CSNK1A1P1 | G | − 0.79 | 0.16 | −1.11 | −0.48 | −4.94 | 9.79E-07 |
| 17 | 1,879,293 | rs12946465 | RPA1, RTN4RL1, DPH1, OVCA2 | C | − 0.21 | 0.05 | −0.31 | −0.13 | −4.75 | 2.51E-06 |
| 19 | 45,719,790 | rs10415392 | TRAPPC6A, BLOC1S3, EXO3L2, MARK4 | T | 0.34 | 0.07 | 0.21 | 0.48 | 4.85 | 1.59E-06 |
| 20 | 56,754,110 | rs78923929 | C20orf85, PPP4R1L, RAB22A | A | − 0.65 | 0.13 | −0.92 | −0.40 | −4.95 | 9.76E-07 |
Genome-wide significant difference in bold, chr = chromosome, SNP = single nucleotide polymorphism, SE = standard error
Fig. 5.
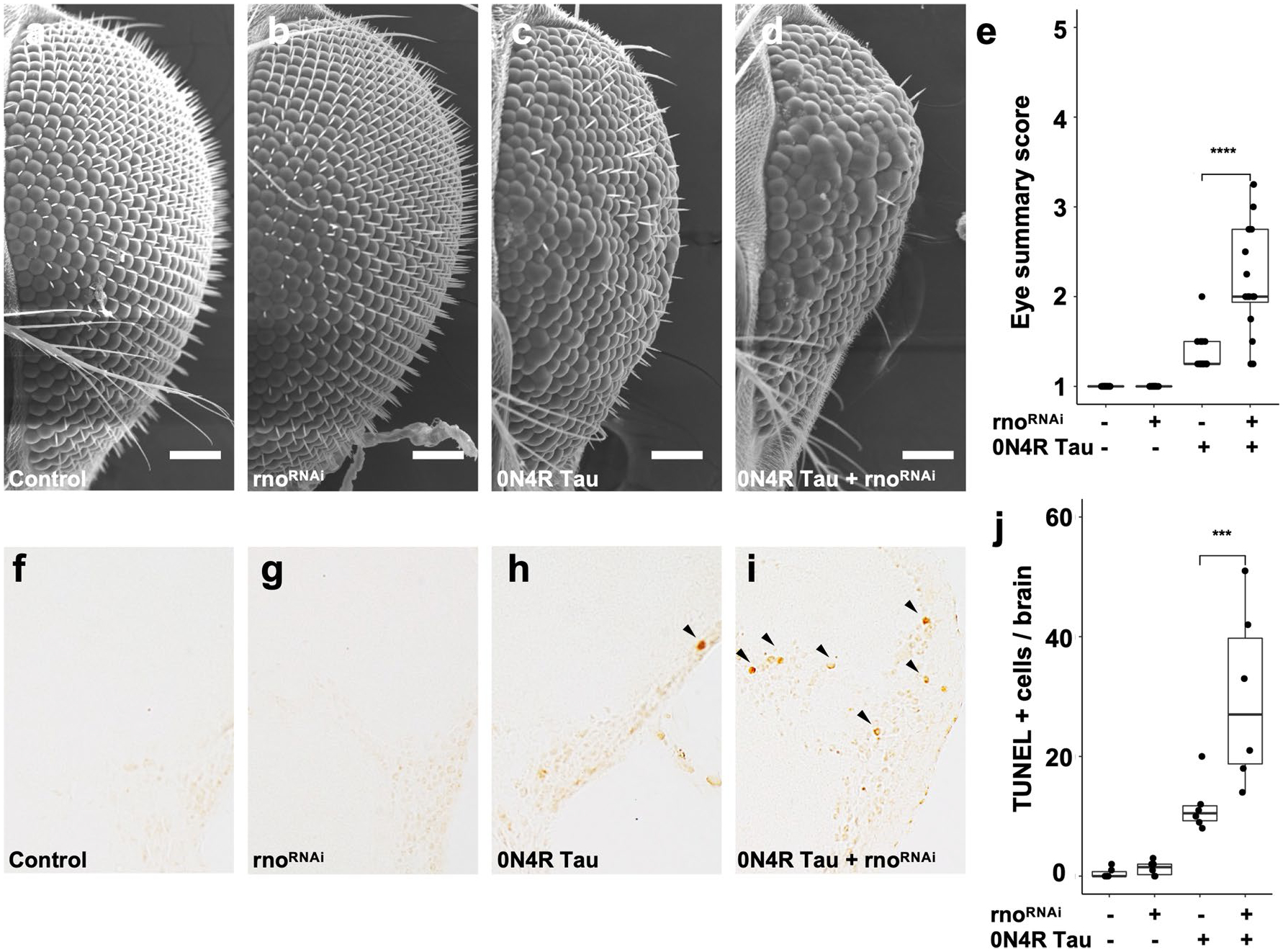
RNAi-mediated knockdown of the JADE1 ortholog rhinoceros (rno) enhances tau-induced toxicity in Drosophila. a–e Scanning electron micrograph images showing rnoRNAi significantly enhances the tau-induced rough eye phenotype (size, roughness, shape, and conical shape, p = 8.7 × 10−5, n = 16 per genotype). Scale bar 100 μm. f–j RnoRNAi significantly increases the number of terminal deoxynucleotidyl transferase dUTP nick end-labels (TUNEL) in tau transgenic Drosophila compared to tau expressed alone (p = 0.008, n = 6 per genotype). TUNEL was performed at day 10 of adulthood and quantified in the entire fly brain. Representative images are shown from the cortex. An equal number of male and female flies were used for each experiment
Replication in an independent cohort proved challenging given the lack of data sets containing similarly neuropathologically ascertained individuals with PART. To our knowledge, the only available autopsy based study performed in a cohort of AD subjects using Braak as a quantitative trait identified two other SNPs (rs4975209 R2 = 0.0072, D′ = 0.0967 and rs10009321 R2 = 0.0091, D′ = 0.1061) in a region distant (≈ 150,000 bp) from 4q28.2, and consequently these SNPS are not in linkage disequilibrium with our lead SNP rs56405341 [6]. Another case/control AD GWAS used a cerebral spinal fluid (CSF) Aβ42/40 ratio threshold to dichotomize their study into 2 groups (normal and abnormal Aβ42/40 ratios) and found another unique SNP in the region (rs13129839) which was genome-wide suggestive (p = 9.0 × 10−6), had a positive (protective) odds ratio (OR = 0.043), and was in high R2 and D′ with our lead as well as supporting SNPs (> 0.8 and 0.1, respectively) [37]. It should be noted the Aβ42/40 ratio in the CSF is highly relevant in the context of PART given Aβ42/40 ratios can be used as a proxy for lower levels of brain amyloid levels which is observed in PART. Taken together, these two independent genetic studies show signals near the 4q28.2 loci, with the latter using the same quantitative trait described here (Braak stage), and the former accounting for amyloidosis but lacking a measurement of tau burden.
We then examined candidate SNPs previously found to be associated with AD and progressive supranuclear palsy (PSP) in prior GWAS studies to explore convergent and divergent genetic risk (Table 3, Supplementary table 5, online resource). Of the 52 candidates investigated, we found five also suggestively associated with Braak stage in PART. rs12590654, which is in the SLC24A4 locus, had the highest significance level (p = 0.001). rs1582763, rs2081545, rs7935829, within the MS4A6A locus (p = 0.01, 0.01, 0.02, respectively). The remaining AD SNP, rs7657553, was within HS3ST1 (p = 0.02) locus. We found two variants that overlapped with PSP risk: rs242557 (p = 0.02) in the MAPT locus, and rs7571971 (p = 0.03) in the EIF2AK3 locus. In summary, seven of the 52 probed risk AD and PSP SNPs showed significant associations (p < 0.05) in PART; however, they do not survive multiple test corrections.
Table 3.
Significant overlapping PART genetic associations with AD and PSP
| Associated disease | Chr | Base pair | SNP | Gene | A1 | Beta | SE | L95 | U95 | t statistic | p |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AD | 14 | 92,938,855 | rs12590654 | SLC24A4 | A | − 0.14 | 0.044 | −0.23 | −0.05 | −3.19 | 0.0015 |
| AD | 11 | 60,021,948 | rs1582763 | MS4A6A | A | − 0.11 | 0.044 | −0.19 | −0.02 | −2.47 | 0.0138 |
| AD | 11 | 59,958,380 | rs2081545 | MS4A6A | A | − 0.10 | 0.044 | −0.19 | −0.02 | −2.36 | 0.0185 |
| AD | 4 | 11,723,235 | rs7657553 | HS3ST1 | A | 0.11 | 0.048 | 0.01 | 0.20 | 2.24 | 0.0255 |
| PSP | 17 | 44,019,712 | rs242557 | MAPT | A | 0.10 | 0.047 | 0.01 | 0.20 | 2.24 | 0.0256 |
| AD | 11 | 59,942,815 | rs7935829 | MS4A6A | G | − 0.10 | 0.043 | −0.18 | −0.01 | −2.21 | 0.0274 |
| PSP | 2 | 88,895,351 | rs7571971 | EIF2AK3 | T | − 0.10 | 0.047 | −0.19 | −0.01 | −2.09 | 0.0373 |
AD = Alzheimer disease, PSP = progressive supranuclear palsy, Chr = chromosome, SNP = single nucleotide polymorphism
Next, we refocused on the nominal association at the 4q28.2 locus. While the strength of this association is relatively weak, it passed the most conservative Bonferroni threshold for genome-wide significance; therefore, we investigated the 4q28.2 locus as previous studies have shown that many nominal associations are replicated in subsequent studies [74]. Examination of RNA expression quantitative trait loci (eQTL) using the Religious Orders Study (ROS) and Memory and Aging Project (MAP) data yielded a significant lead SNP (rs56405341) eQTL (p = 0.038) for JADE1 (Fig. 2a) [25, 88]. As our GWAS quantitative trait was specific to tau pathology and our SNP eQTL was only modestly significant, we then examined mRNA expression levels of genes in the locus using a novel single-cell soma RNA sequencing data set which measured transcriptomic changes specifically in tangle-bearing neurons and non-tangle-bearing neurons (Fig. 2b–d). Using this data set, we first looked at the overall change in JADE1 expression and found that tangle-bearing excitatory neurons significantly differentially upregulated JADE1 compared to non-tangle-bearing excitatory neurons (p = 1.04 × 10−61). Conversely C4orf33 and SCLT1, the other genes in the locus, had low levels of expression regardless of cell type and tangle status. Given that the cell-type composition is slightly different in each type of neuronal group, we then looked at individual cell subclusters and observed two subpopulations of excitatory neurons in which JADE1 was significantly differentially expressed (adjusted p = 4.55 × 10−15, 7.82 × 10−8). We did not observe differences in JADE1 expression in the remaining neuronal populations due to low cell numbers which decreased our statistical power (Supplemental Fig. 2a). We also examined the relative average expression and percentage of cells expressing JADE1 and observed increases in both metrics in tangle-containing neuronal populations compared to non-tangle-containing neuronal populations (Fig. 2e–g). SCLT1 and C4orf33 had negligible expression levels in a substantially smaller percentage of neurons (Supplemental Fig. 5b, c). Taken together, these data suggest increased JADE1 mRNA expression is a characteristic of tangle-bearing neurons.
Fig. 2.
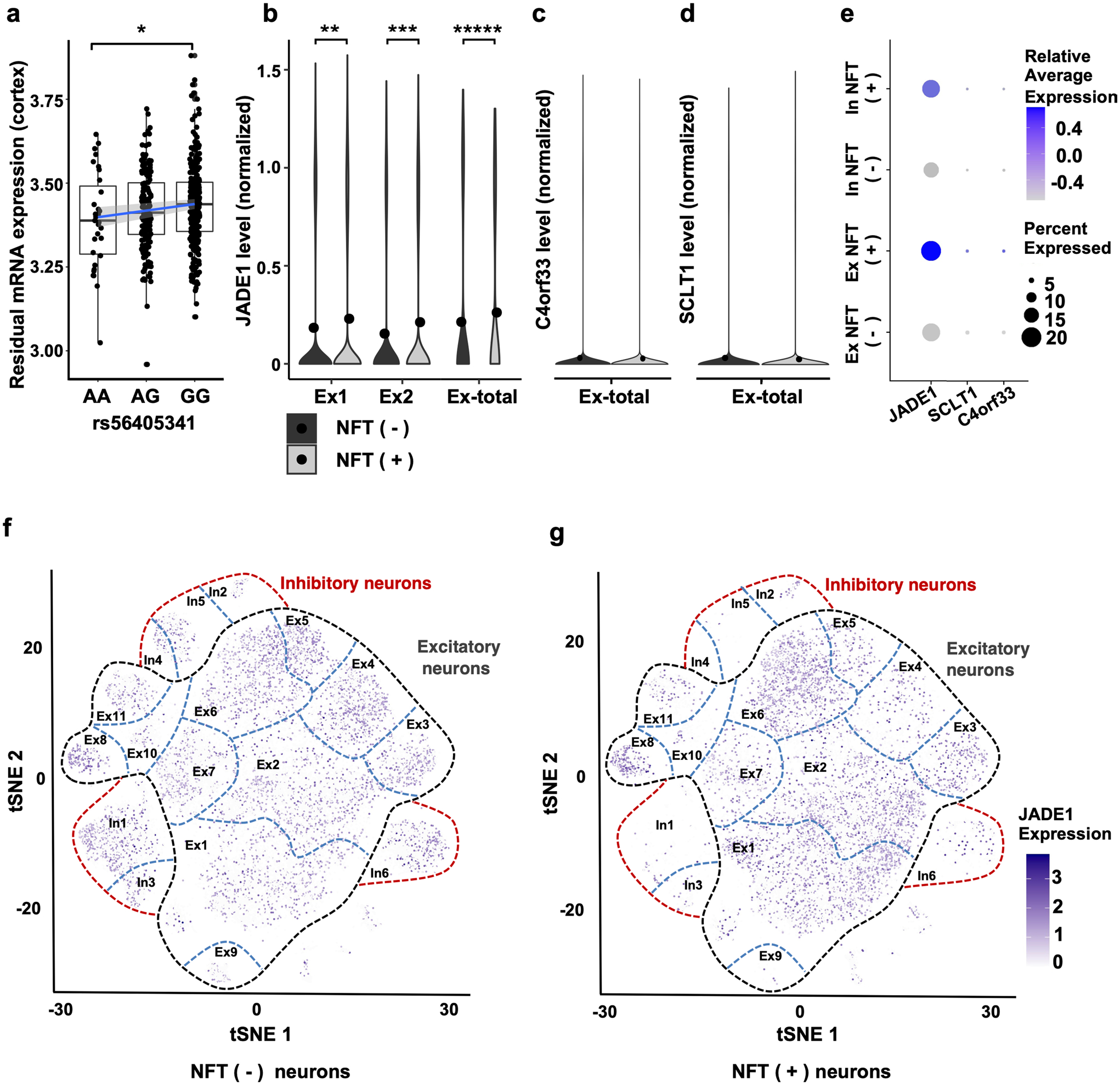
Bulk and single-cell sequencing reveals JADE1 mRNA is modulated by rs56405341 and upregulated in tangle-bearing neurons. a Bulk RNA sequencing data yielded a significant lead SNP (rs56405341) eQTL (p = 0.038) in the dorsolateral prefrontal cortex of 452 postmortem human brain samples. Expression of JADE1 decreases with in subjects carrying the minor allele. Single cell sequencing data of populations of neurons with and without neurofibrillary tangles isolated from human post-mortem brain samples separated using fluorescence-activated cell sorting and transcriptomic profiles from single-cell RNA-sequencing were subsequently analyzed. b In 2 unique excitatory neuronal populations (Ex1 and Ex2) JADE1 mRNA was significantly differentially expressed in the tangle bearing neurons (adjusted p = 7.82 × 10−8, 4.55 × 10−15); in comparing the overall population of tangle-bearing excitatory neurons (Ex-total) to non-tangle bearing neurons the difference is highly significant (adjusted p = 1.04 × 10−61). c, d Other two genes in the locus, C4orf33 and SCLT1, were overall nominally expressed in both excitatory neuronal groups, as well as subclusters (Supplementary Fig. 5 b, c, online resource). e Dot plot showing average relative expression and percent expression of the candidate genes in the locus. Both JADE1 relative average expression and percentage of cells expressed was higher than C4orf33 and SCLT1. f, g t-Distributed stochastic neighbor embedding (tSNE) plots showing the different populations neurons, tangle bearing status, and relative expression of JADE1 in neuronal subpopulations
Given our evidence of a genetic signal in the 4q28.2 locus, and single cell transcriptomic data indicating modulation of JADE1 expression in neurons with NFT pathology, we conducted an immunohistochemical study using specific antisera to JADE1 in our collection of post-mortem tauopathy brain tissue (Fig. 3). We assessed tauopathies that are known to involve preferentially tau isoforms with 3 micro-tubule-binding domain repeats (3R), 4 microtubule-binding domain repeats (4R) or a mixture of the two [50]. We found strong and specific JADE1 immunopositivity in structures morphologically indicative of mature aggregate containing intracellular NFT in not only PART, but also the other mixed 3R/4R tauopathies (i.e., AD and chronic traumatic encephalopathy, Fig. 3a–f). In the 4R tauopathies (PSP, corticobasal degeneration, and argyrophilic grain disease), NFT were also immunopositive for JADE1 (Fig. 3g–l). Notably, gliofibrillary pathology in these diseases (i.e., aging-related tau astrogliopathy, tufted astrocytes, and astrocytic plaques) were also immunopositive. Surprisingly, no signal was detected in Pick bodies in Pick disease (PiD), a predominately 3R tauopathy (Fig. 3m, n). Double-labeling experiments showed that early pre-NFT that consist predominately of prefibrillary soluble oligomers were negative for JADE1 suggesting that this factor begins to coalesce into NFT at the transition to the aggregate stage (Fig. 3o). To ensure the antibody was specifically targeting the JADE1 protein rather than binding non-specifically to NFT, we added blocking peptides with the JADE1 antibody and did not observe any staining (Fig. 3p). These findings indicate that JADE1 protein expression is localized specifically in mature NFTs. Furthermore, translocation of JADE1 to NFTs does not occur in PiD, the only 3R tauopathy examined, suggesting selective tau isoform interactions.
Fig. 3.
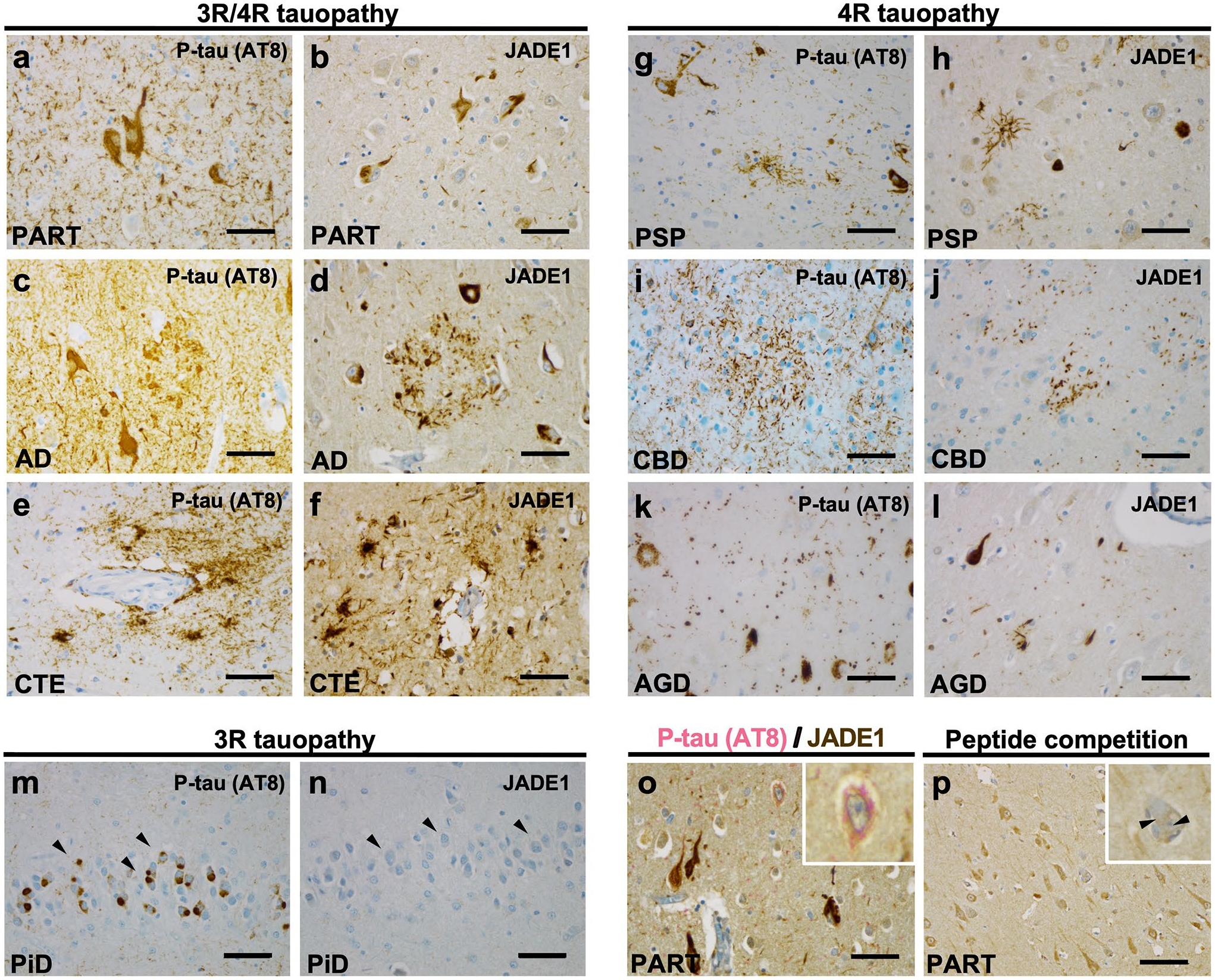
Selective immunolabeling of tau aggregates containing tau with four microtubule-binding domain repeats (4R), but not three (3R), in post-mortem human tauopathy brains with antisera targeting JADE1 protein. Immunohistochemical staining with phospho-tau (p-tau) specific antisera (AT8) and JADE1 specific antisera demonstrates neurofibrillary tangle (NFT) formation marked by the presence of JADE1 in specific populations of neurons and glia. a, b Primary age-related tauopathy (PART, n = 3) NFTs contain JADE1 positive staining in the soma and neurites in the entorhinal cortex. c, d Alzheimer disease (AD, n = 3) individual with Aβ and AT8-positive neuritic plaques and NFTs (subiculum) also display JADE1 immunopositivity in dystrophic neurites and NFTs. e, f Chronic traumatic encephalopathy (CTE, n = 3) contains positive p-tau staining around a blood vessel in the depth of a neocortical sulcus that is immunopositive for JADE1. g, h AT8 positive tufted astrocytes, oligodendroglial coiled bodies, and NFTs are positive in the subthalamic nucleus in an individual with progressive supranuclear palsy (PSP, n = 3) which are also in immunopositive for JADE1. i, j Astrocytic plaques in corticobasal degeneration (CBD, n = 3) and extensive thread-like pathology positive for p-tau and JADE1 in the neocortex. k, l In the cornu ammonis 1 (CA1) sector in an individual with argyrophilic grain disease (AGD, n = 3), abundant grains that are immunopositive for p-tau and JADE1 are evident. m, n Pick disease (PiD), a 3R tauopathy, with Pick bodies in the dentate gyrus that are immunopositive for p-tau but negative for JADE1. o Double staining of a PART entorhinal cortex showing the absence of JADE1 (brown) staining in early pre-tangles, but the presence of p-tau (pink, see inset). p Negative control using a peptide competition despite the presence of a tangle (inset). Scale bar, 100 μm
We then biochemically examined JADE1 protein expression by immunoblot using lysates derived from the entorhinal cortex and hippocampus proper (cornu ammonis 1–4 and dentate gyrus) of PART and AD individuals. JADE1 exists as 2 isoforms, JADE1 long (JADE1L) and JADE1 short (JADE1S), both of which contain proline, glutamic acid, serine, threonine (PEST) domains and 2 PHD fingers. However, the long form is 333 amino acids longer and contains an additional PEST domain as well as a nuclear localization signal (Fig. 4a). Immunoblot analysis revealed expression of JADE1S in both brain regions and diseases, but no bands were observed at the expected molecular weight for JADE1L, indicating the signal observed in IHC may be specific to the short isoform of JADE1 (Fig. 4b).
Fig. 4.
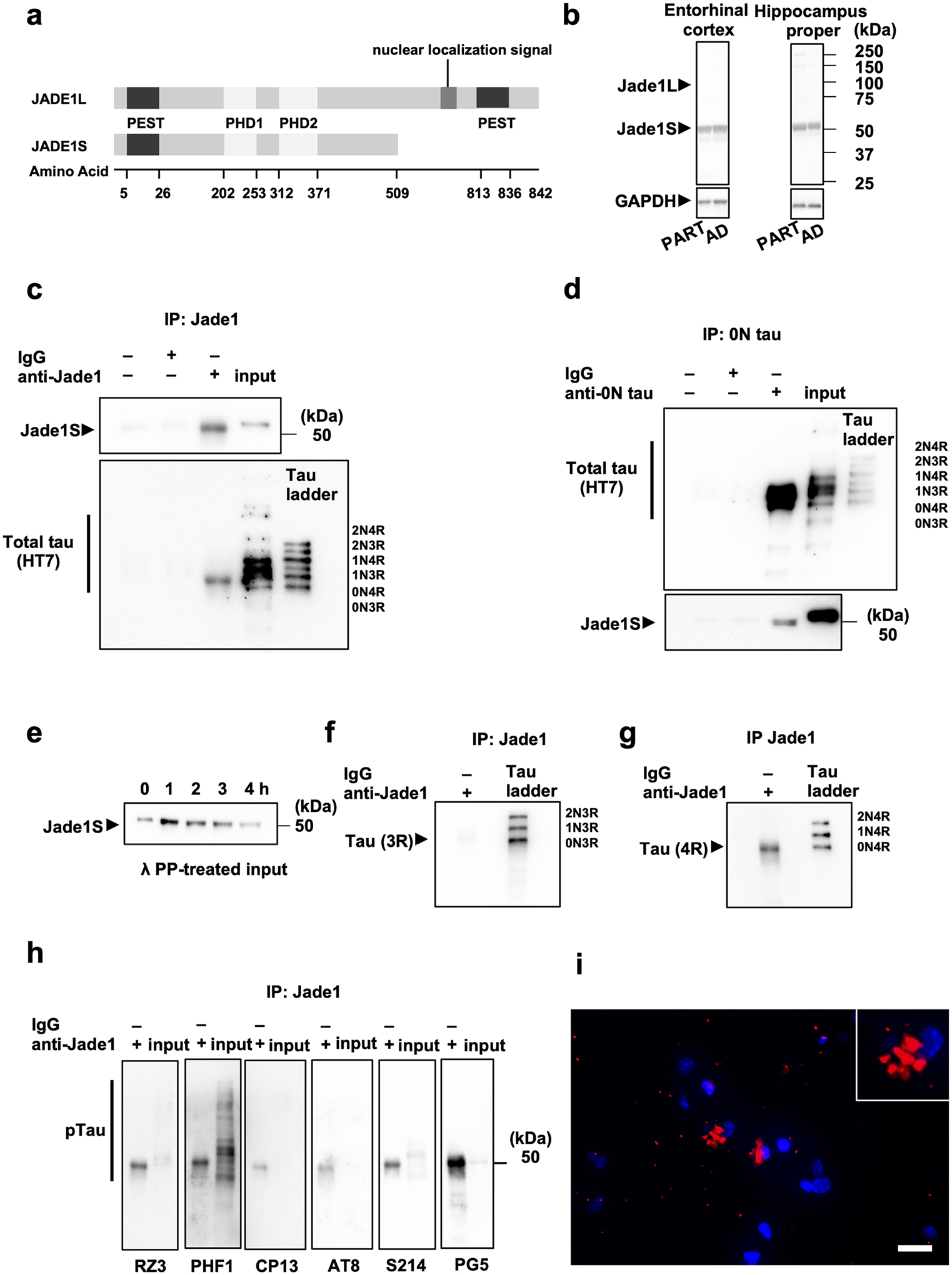
JADE1S protein interacts with tau containing four microtubule-binding domain repeats (4R) but not 3R in post-mortem human brain tissue (a) Schematic of the two JADE1 isoforms, JADE1S and JADE1L. b Representative immunoblot using antisera targeting JADE1 in entorhinal cortex and cornu ammonis in individuals with primary age-related tauopathy (PART) and Alzheimer disease (AD) shows JADE1S but not JADE1L at the expected molecular weight. GAPDH was used as a loading standard. c Immunoprecipitation using JADE1 antisera co-immunoprecipitates tau the with a molecular weight near the 0N4R isoform (40 kDa). d Reverse immunoprecipitation using 0N tau antisera co-immunoprecipitates the JADE1S isoform. e Pulled down form of JADE1S molecular weight shifts downward after treatment with lambda protein phosphatase. f, g Co-immunoprecipitated tau with JADE1 stained with C-terminal isoform specific anti-tau antisera are the 0N4R isoform and not the 3R isoform. h Co-immunoprecipitated tau was positively stained with a panel of phospho-tau specific antibodies with the most signal coming from pThr231 (RZ3), pSer396/pSer404 (PHF1), pSer214 (S214), and pSer409 (PG5). i Proximity–ligation assay showing positive fluorescence signal (red) around the nucleus of neurons (blue) indicating the close association between JADE1 and 0N tau detected using the corresponding two primary antibodies in the soma (inset). Scale bar, 20 μm
Cytoplasmic colocalization of JADE1 with NFTs immunohistochemically raises the possibility that they form a functional complex in tauopathy brains. To examine this, co-immunoprecipitation experiments were conducted using postmortem brain lysate from individuals with PART. We first immunoprecipitated JADE1 and then performed immunoblots using antisera to tau and observed a ~ 45 kDa isoform that corresponds to 0N4R (Fig. 4c). We then reverse co-immunoprecipitated using 0N tau antisera and observed a ~ 50 kDa single band when probing with our JADE1 antisera (Fig. 4d). We treated our pellets with protein phosphatase and observed a slight band shift (Fig. 4e), indicating the JADE1 bound to tau is phosphorylated. Immunoblotting isoform specific anti-tau antisera (3R and 4R tau) confirmed that the coimmunoprecipitated tau was predominately 4R, thus 0N4R (Fig. 4f, g). To further validate our findings, we repeated the same coimmunoprecipitation experiments using a non-commercially available JADE1S-specific antibody raised against a different epitope and observed the same results [106]. We then ran immunoblots on the immunoprecipitated JADE1 that we probed using a panel of phospho-tau site-specific antibodies. The strongest signals relative to the input were with pThr231 (RZ3) and pSer409 (PG5), of which both have been reported to mark pre- and intercellular tangles, but not mature tangles [5] (Fig. 4h). Finally, to confirm the interaction of JADE1 and tau, proximity–ligation assays (PLA) were performed in fixed hippocampus from PART individuals. JADE1 and tau antibodies were labeled with oligonucleotides. The red fluorescent signal indicates the close proximity (maximum 30–40 nm) of these two proteins (Fig. 4i). Controls did not show signal (Supplementary Fig. 7e, f, online resource). Therefore, JADE1S likely forms a complex with 0N4R tau in tangle bearing neurons.
Finally, given the association between JADE1 and 0N4R tau, we asked whether JADE1 plays a functional role in tau pathology. We used a Drosophila model that overexpresses human mutant 0N4R tau [100] as well as RNAi-mediated reduction of rhinoceros (rno), the highest matched JADE1 Drosophila ortholog. We blindly evaluated the fly eye phenotype using a semi-quantitative assessment of size, roughness, overall shape, and conical shape and observed a significant increase in severity between tau transgenic Drosophila and tau transgenic Drosophila with rno knockdown (Fig. 5a–e, p = 8.7 × 10−5). We did not observe significant differences between rnoRNAi and controls in the absence of transgenic tau. To assess toxicity, we quantified TUNEL-positive cells throughout the Drosophila brain. We find that rno knockdown significantly enhances neurotoxicity in tau transgenic Drosophila but not controls based on TUNEL staining at day 10 of adulthood (p = 0.008, Fig. 5f–i). These data provide in vivo evidence that JADE1/rno loss plays a mechanistic role in promoting neurotoxicity in tauopathy and suggest that proper functioning of JADE1/rno is protective.
Discussion
Genome-wide association studies (GWAS) have enabled advances in our understanding of sporadic tauopathies [17, 35, 42, 49, 81, 95, 102]. Yet, in the context of the growing numbers of genes associated with Alzheimer disease (AD) [44], direct links with tau proteinopathy have been challenging to pinpoint as association signals show minimal overlap with factors classically implicated in tauopathy (e.g., proteostasis, tau protein kinases, prions, etc.) [34, 50, 59]. This is not surprising given the ubiquity and heterogeneity of tau and other pathological changes in the aging human brain that are variably associated with cognitive impairment [63]. While dementia is the phenotype focused on in most genetic studies, it is non-specific. Here, we performed an autopsy-based neuropathology-based GWAS, which minimizes classification errors and other issues, assembling the largest cohort of post-mortem brain tissues from aged individuals devoid of Aβ neuritic plaque pathology with a goal of identifying factors independently associated with primary age-related tauopathy (PART). In doing so, we sought to obtain genetic evidence that might clarify the controversial relationship between PART and AD, which are neuropathologically similar. We failed to find an association with APOE ε4, the strongest common risk allele for sporadic late-onset AD, but did observe modest signals in other candidate risk loci. We also found a novel, albeit nominal, genome-wide significant association at the chromosome 4q28.2 locus. Our data indicate that among genes in this locus, only the gene for apoptosis and differentiation in epithelia 1 (JADE1), a member of a small family of multifunctional adaptor proteins implicated in renal and other cancers [19, 104–106], is upregulated in tangle-bearing neurons at both the mRNA and protein levels. This accumulation of JADE1 protein in NFT is not specific to PART but occurs in AD and all tauopathies with accumulation of 4R isoforms, but not in Pick disease, a 3R tauopathy. We also show that JADE1 binds 0N4R tau, an isoform proposed to be a critical driver of tau pathology [86, 98]. Finally, experiments in Drosophila show that reducing expression of the JADE1 homolog rhinoceros (rno) exacerbates tau-induced neurotoxicity in vivo. Together, these findings strongly argue that JADE1 is a factor broadly capable of protecting neurons from neurofibrillary cell death that links PART to the tauopathic component of AD.
We confirm that the genetics of PART has a partial overlap with sporadic late-onset AD and replicated the consistent finding showing the lack of a signal in the APOE locus despite its strong association with AD [4, 44, 52, 54, 58]. It has been shown previously that individuals with PART have a higher APOE ϵ2 allele frequency which distinguishes PART from AD both neuropathologically and genetically [41, 45, 79]. We have reported that the frequency of the APOE ε4 allele is lower in PART [83] and other studies have found similar results in independent cohorts [7, 61]. It should be noted that these values can fail to reach significance when comparing groups with varying degrees of neuritic plaque pathology [9, 61]. Recent studies in mice and humans have indicated that the APOE ε4 allele may exacerbate tau pathology independently of Aβ plaques [87], while other human studies failed to show an interaction [1, 30]. These results reinforce prior evidence that PART occurs independently of APOE ε4 irrespective of Aβ plaque pathology. Alternatively, given the higher APOE ϵ2 and lower APOE ϵ4 allele frequency observed in PART, this enrichment could suggest a functional epistatic interaction with JADE1 and APOE which also provides context to the lack of signal in the observed in the 4q28.2 locus in other large AD GWAS studies.
17q21.31 MAPT locus is the strongest genetic risk factor for PSP [35], which we and others had previously reported is associated with PART [43, 83]. The MAPT H1 haplotype has also been associated with AD [66, 82, 93]. However, this region has a complex haplotype structure and may be more important in specific AD subgroups given the modest signal and variable findings in these association studies [11, 92]. Intriguingly, in one AD GWAS using clinically ascertained individuals, removal of APOE ε4 carriers enhanced signals in the 17q21.31 locus [46]. In the present study, there was only a modest association of MAPT with PART. This result may stem from differences in cohort selection in case–control studies focusing on pathological extremes, while our study design included a range of pathological severity, specifically including mild to moderately affected individuals. Together, these data highlight that further investigation of the role of 17q21.31 MAPT locus in PART is warranted.
Beyond APOE and MAPT, we found four additional association signals in PART that overlap with either AD or PSP. Eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3) encodes an endoplasmic reticulum (ER) membrane protein critical for the unfolded protein response (UPR) [91, 94]. Activation of the UPR has been observed and positively correlated with tau pathology, but not with Aβ plaque burden, in the hippocampus of aged cognitively normal individuals [94]. Solute carrier family 24 member 4 (SLC24A4), a gene in the locus most strongly associated with PART and AD, is a member of the potassium-dependent sodium/calcium exchanger protein family and is involved in neural development; however, little is known about its possible function in AD [55, 103]. We identified an association of PART with the membrane spanning 4-domains A6A (MS4A6A) locus, which contains the binding regions for the transcription factor PU.1 which is selectively expressed in brain microglia and macrophages [29]. The last overlapping genetic locus contains heparan sulfate-glucosamine 3-sulfotransferase 1 (HS3ST1), which has been suggested to modulate heparan sulfate proteoglycans as receptors for the spreading of tau [36, 54]. Taken together, these data are compatible with the hypothesis that the candidate genes we identified in our GWAS are possible modulators of tau pathology.
We identified a novel locus on chromosome 4q28.2 associated with PART pathology. This locus was previously associated with Braak NFT stage in an AD autopsy GWAS, as well as a second GWAS focusing on dichotomized CSF Aβ positivity [6, 37]. This prompted us to focus on this locus for further validation and functional studies. Because we only identified a nominally significant expression quantitative trait locus signal to JADE1, and given that our trait was tangle-specific, we reasoned that modulation of mRNA expression of genes in the locus might also be cell-type specific. This hypothesis was also motivated by the increase in genetic to transcriptomic associations found in cell specific populations in other contexts [47, 56, 107]. Our results indicate that of the genes in the 4q28.2 locus, only JADE1 mRNA was significantly and differentially expressed in tangle-bearing neurons. Our immunohistochemical studies revealed JADE1 protein accumulation in both neuronal and glial tau-immunopositive cells, validating these findings. Thus, JADE1 is most likely responsible for the GWAS signal at this locus; however, further genetic and expressions studies on perturbated cell populations are required in the future to validate these findings.
Our immunohistochemical studies indicate that JADE1 is potentially involved broadly in 4R and mixed 3R/4R tauopathies. We observed immunopositivity not only in PART tangles, but also in tangles of tauopathies with aggregates containing 4R tau and in mixed tauopathies with aggregates containing both 3R and 4R tau. The absence of staining in Pick disease, the only tauopathy with 3R tau aggregates examined, was surprising. Our biochemical studies suggest that JADE1 protein specifically interacts with 0N4R tau that is phosphorylated on epitopes known to be hyperphosphorylated in NFT. Our proximity ligation assay confirms the direct interaction between JADE1 protein and tau. Studies using cryo-EM and mass spectrometry have shown the ultra-structure of tau aggregates at unprecedented resolution, and it has been reported that 0N4R has a unique single β-sheet conformation for the fibril core [27, 32, 84]. Intriguingly, recent mass spectrometry profiling studies of human post-mortem brain tissues have suggested that changes in 0N4R tau isoform specifically is an early event in tauopathy [98]. Double-labeling experiments indicate that JADE1 increases shortly after the pre-tangle stage, accumulating alongside insoluble tau aggregates during the transition to the intracellular tangle stage possibly reflecting a reactive/protective compensatory role [14].
Our findings provide clues as to how JADE1 may be functioning in tauopathy. JADE1S protein has been previously implicated as a renal tumor suppressor involved in apoptosis, an inhibitor of Wnt signaling by ubiquitylating β-catenin, and an inhibitor of pro-survival kinase AKT and interactor with cell cycle regulators [12, 13, 19, 75, 90, 105]. Of the two JADE1 isoforms, the short form lacks a nuclear localization signal, consistent with the cytoplasmic localization of the protein we observed by immunohistochemistry. This prompted us to hypothesize that JADE1S functions in the cytoplasm to modulate tauopathy. Our in vivo studies in which we reduced rno (the closest JADE1 ortholog) levels in Drosophila overexpressing mutant human 0N4R tau significantly enhanced the tau-induced rough eye phenotype as well as TUNEL-positive cells, a marker of apoptotic DNA fragmentation. While JADE1 has been shown to promote apoptosis in some contexts, RNAi knockdown experiments suggest that proper functioning of JADE1 may be neuroprotective. Other studies have demonstrated that loss of rno function attenuates apoptosis in Drosophila [96]. Because previous studies have found JADE1 to be stabilized by the von Hippel–Lindau tumor suppressor which is a component of an E3 ubiquitin–protein ligase activity and JADE1 is itself a single-subunit ubiquitin ligase for beta-catenin, JADE1 may be working with 0N4R tau through a similar mechanism to promote ubiquitin-mediated clearance of tau [57, 89, 106].
Limitations of our study include small sample size for GWAS standards; however, it should be noted that it is still the largest study of its kind and limited by the availability of tissue that meet our diagnostic criteria for PART, recognizing that PART is currently only reliably diagnosed postmortem. To amass sufficient donors, our study relies on neuropathological assessments performed at multiple centers that could introduce batch effects (although none were detected). In addition, we were only able to identify a modestly significant eQTL signal between our lead SNP and JADE1 mRNA expression. However, our analysis of perturbed single soma RNA sequencing stratified by the presence or absence of NFTs allude to the fact that our lead SNP could be modulating JADE1 expression specifically in tangle containing neurons. Our attempt to replicate our findings only identified candidate SNPs (p < 5 × 10−8, > 5 × 10−6) which were not in strong linkage disequilibrium with our lead SNP. We hypothesize this is likely a product of these studies containing subjects with amyloid pathology, and for these reasons future replication is required to validate our findings. Finally, while Braak staging is highly reproducible across neuropathologists and institutions, with one report showing that across brains and raters the kappa score was greater than 0.90 [67], it is a semiquantitative (ordinal) variable. Braak NFT staging is also modeled after a subset of AD cases with patterns of NFT formation that may differ somewhat from the pattern seen in PART. Furture studies using a more quantitative approach to measuring tau burden that we have shown more closely aligns with functional clinical measures in PART may reveal additional candidates [39].
In conclusion, drug development efforts are increasingly targeting tau due to the lack of clinical efficacy of Aβ modulating therapeutic approaches [22]. Here, by focusing on individuals with PART who lack Aβ neuritic plaques, we enriched our cohort for signals related to tau proteinopathy. Our analysis provides additional evidence that PART overlaps with but has considerable differences from AD. This interdisciplinary approach led to the identification of JADE1, which interacts with 0N4R tau, and is protective in vivo. Additional studies in experimental models are necessary to further validate these findings. Further understanding the genetics of PART will provide opportunities for rationally designed therapeutics for tauopathies.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the neuropathology core of the Massachusetts Alzheimer Disease Research Center, the Biosample Management Repository at Genentech/Roche, the brain repository at UCI, Knight Alzheimer Disease Research Center Neuropathology Core at Washington University School of Medicine, the BioRepository and Integrated Neuropathology Laboratory and Precision Neuropathology Core at the University of Washington, the neurodegenerative disease brain bank at the University of California San Francisco, the Neuropathology Brain Bank & Research Core at Mount Sinai, the Newcastle Brain Tissue Resource, and the following people: Ryan Cassidy Bohannan, Chad Caraway, Allison Beller, Kim Howard, Suresh Selvaraj, Ward Ortmann, Ping Shang, Jeff Harris, and Chan Foong. The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org).
Funding
IMSSM: R01 AG054008, R01 NS095252, R01 AG060961, and R01 NS086736 Rainwater Charitable Foundation, Genentech/Roche, Alexander Saint-Amand Fellowship (JFC), F32 AG056098 and P30 AG066514 (KF), P50 AG005138 and P30 AG066514 (VH, JFC, MS, SG, AG, PRH), 75N95019C00049 (VH), K99 AG070109 (SJA), U01 AG058635 (AG, EM, AER) BU / MSSM / MAYO: R01 AG062348 (AM JFC DD), UMC: P50AG008702 (JPV AFT), BU: U54 NS115266 (AM), R01 CA079830 (HTC), UPENN: P30 AG010124, P01 AG017586 and U19 AG062418 (JQT), P30 AG072979 and P01 AG066597 (EBL), R01 AG066152 (CTM), PITT: R01 AG066152 P30 AG066468 (JK), Banner: U24 NS072026 and P30 AG019610 The Arizona Department of Health Services, and the Michael J. Fox Foundation for Parkinson’s Research (TB), Johns Hopkins: P50 AG05146 (JCT), U Iowa: K23 NS109284, The Roy J. Carver Foundation, the Carver College of Medicine, and the Williams-Cannon Foundation (MMH), Northwestern: P30 AG013854 (MEF), Emory: P30 NS055077 and P50 AG025688 (MG), OHSU: P30 AG08017 (RW), UTSW: Win-spear Family Center for Research on the Neuropathology of Alzheimer Disease (CWIII), Toronto: Rossy Foundation and by the Safra Foundation (GGK), Stanford: R01AG059848 (IC), MADRC: P50 AG05134, P30 AG062421 (BTH), RUSH: ADC grant AG10161 and MAP grant (JS), UCI: R01AG021055 and P50AG016573 (CHK, MMC), P01AG000538 (WP), UCSD: P30 AG062429 01 and P50 AG005131 (RR), UK: P30 AG028383 (PN), U Washington: P50 AG005136, P30 AG066509, U01 AG006781, U19 066567 and the Nancy and Buster Alvord Endowment (CDK), Washington U / Knight ADRC: P30 AG066444, P01 AG003991, 01 AG026276 (RJP), Newcastle: UK Medical Research Council (G0400074), by Brains for Dementia research, a joint venture between Alzheimer’s Society and Alzheimer’s Research UK and by the NIHR Newcastle Biomedical Research Centre awarded to the Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University (JA), Other: J.M.R. Barker Foundation, The McCune Foundation.
Footnotes
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00401-021-02379-z.
Conflict of interest JFC, DWD, MF, PRH, GGK, EBL, PTN, JQT, are editorial board members and JA is editor in chief of Acta Neuropathologica, but were not involved in the editorial handling of this article. JH, TB, are employees of Genentech (a subsidiary of Roche) and hold stocks/stock options in FH-LR Ltd. AG is on the Scientific advisory board for Genentech and consultant for AbbVie. All other authors declare no relevant conflicts.
References
- 1.Abner EL, Neltner JH, Jicha GA, Patel E, Anderson SL, Wilcock DM et al. (2018) Diffuse amyloid-beta plaques, neurofibrillary tangles, and the impact of APOE in elderly persons’ brains lacking neuritic amyloid plaques. J Alzh Dis 64:1307–1324. 10.3233/Jad-180514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ (2008) The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J Neurosci Methods 172:250–254. 10.1016/j.jneumeth.2008.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT (2010) Data quality control in genetic case-control association studies. Nat Protoc 5:1564–1573. 10.1038/nprot.2010.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrews SJ, Fulton-Howard B, Goate A (2020) Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol 19:326–335. 10.1016/S1474-4422(19)30435-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 103:26–35. 10.1007/s004010100423 [DOI] [PubMed] [Google Scholar]
- 6.Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ et al. (2014) Genome-Wide Association Meta-analysis of Neuropathologic Features of Alzheimer’s disease and related dementias. Plos Genet. 10.1371/journal.pgen.1004606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell WR, An Y, Kageyama Y, English C, Rudow GL, Pletnikova O et al. (2019) Neuropathologic, genetic, and longitudinal cognitive profiles in primary age-related tauopathy (PART) and Alzheimer’s disease. Alzheimers Dement 15:8–16. 10.1016/j.jalz.2018.07.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC et al. (2006) Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66:1837–1844. 10.1212/01.wnl.0000219668.47116.e6 [DOI] [PubMed] [Google Scholar]
- 9.Besser LM, Crary JF, Mock C, Kukull WA (2017) Comparison of symptomatic and asymptomatic persons with primary age-related tauopathy. Neurology 89:1707–1715. 10.1212/WNL.0000000000004521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Besser LM, Mock C, Teylan MA, Hassenstab J, Kukull WA, Crary JF (2019) Differences in cognitive impairment in primary age-related tauopathy versus alzheimer disease. J Neuropath Exp Neur 78:219–228. 10.1093/jnen/nly132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boettger LM, Handsaker RE, Zody MC, McCarroll SA (2012) Structural haplotypes and recent evolution of the human 17q21.31 region. Nat Genet 44:881–882. 10.1038/ng.2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boone DK, Weisz HA, Bi M, Falduto MT, Torres KEO, Willey HE et al. (2017) Evidence linking microRNA suppression of essential prosurvival genes with hippocampal cell death after traumatic brain injury. Sci Rep 7:6645. 10.1038/s41598-017-06341-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borgal L, Habbig S, Hatzold J, Liebau MC, Dafinger C, Sacarea I et al. (2012) The ciliary protein nephrocystin-4 translocates the canonical Wnt regulator Jade-1 to the nucleus to negatively regulate beta-catenin signaling. J Biol Chem 287:25370–25380. 10.1074/jbc.M112.385658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braak E, Braak H, Mandelkow EM (1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 87:554–567. 10.1007/BF00293315 [DOI] [PubMed] [Google Scholar]
- 15.Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16:271–278. 10.1016/0197-4580(95)00021-6 [DOI] [PubMed] [Google Scholar]
- 16.Braak H, Del Tredici K (2014) Are cases with tau pathology occurring in the absence of A beta deposits part of the AD-related pathological process? Acta Neuropathol 128:767–772. 10.1007/s00401-014-1356-1 [DOI] [PubMed] [Google Scholar]
- 17.Broce I, Karch CM, Wen N, Fan CC, Wang YP, Tan CH et al. (2018) Immune-related genetic enrichment in frontotemporal dementia: an analysis of genome-wide association studies. Plos Med. 10.1371/journal.pmed.1002487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen JA, Chen Z, Won H, Huang AY, Lowe JK, Wojta K et al. (2018) Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol Neurodegener 13:41. 10.1186/s13024-018-0270-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI et al. (2008) Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol 10:1208–1216. 10.1038/ncb1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke L, Fairley S, Zheng-Bradley XQ, Streeter I, Perry E, Lowy E et al. (2017) The international genome sample resource (IGSR): a worldwide collection of genome variation incorporating the 1000 genomes project data. Nucleic Acids Res 45:D854–D859. 10.1093/nar/gkw829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Combs B, Kanaan NM (2017) Exposure of the amino terminus of tau is a pathological event in multiple tauopathies. Am J Pathol 187:1222–1229. 10.1016/j.ajpath.2017.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Congdon EE, Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14:399–415. 10.1038/s41582-018-0013-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I et al. (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A et al. (2016) Next-generation genotype imputation service and methods. Nat Genet 48:1284–1287. 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D et al. (2018) A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci Data 5:180142. 10.1038/sdata.2018.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derisbourg M, Leghay C, Chiappetta G, Fernandez-Gomez FJ, Laurent C, Demeyer D et al. (2015) Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci Rep-Uk. 10.1038/srep09659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dregni AJ, Mandala VS, Wu H, Elkins MR, Wang HK, Hung I et al. (2019) In vitro 0N4R tau fibrils contain a monomorphic beta-sheet core enclosed by dynamically heterogeneous fuzzy coat segments. Proc Natl Acad Sci U S A 116:16357–16366. 10.1073/pnas.1906839116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duyckaerts C, Braak H, Brion JP, Buee L, Del Tredici K, Goedert M et al. (2015) PART is part of Alzheimer disease. Acta Neuropathol 129:749–756. 10.1007/s00401-015-1390-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Efthymiou AG, Goate AM (2017) Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12:43. 10.1186/s13024-017-0184-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA (2016) Association of APOE with tau-tangle pathology with and without beta-amyloid. Neurobiol Aging 37:19–25. 10.1016/j.neurobiolaging.2015.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK et al. (2015) MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16:278. 10.1186/s13059-015-0844-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ et al. (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547:185–190. 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Folstein MF, Robins LN, Helzer JE (1983) The mini-mental state examination. Arch Gen Psychiatry 40:812. [DOI] [PubMed] [Google Scholar]
- 34.Frost B, Ollesch J, Wille H, Diamond MI (2009) Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem 284:3546–3551. 10.1074/jbc.M805627200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoglinger GU, Melhem NM, Dickson DW, Sleiman PMA, Wang LS, Klei L et al. (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43:699–U125. 10.1038/ng.859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmes BB, Devos SL, Kfoury N, Li M, Jacks R, Yanamandra K et al. (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. P Natl Acad Sci USA 110:E3138–E3147. 10.1073/pnas.1301440110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong S, Prokopenko D, Dobricic V, Kilpert F, Bos I, Vos SJB et al. (2020) Genome-wide association study of Alzheimer’s disease CSF biomarkers in the EMIF-AD multimodal biomarker discovery dataset. Trans Psychiatry 10:403. 10.1038/s41398-020-01074-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H et al. (1998) Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- 39.Iida MA, Farrell K, Walker JM, Richardson TE, Marx G, Bryce CH et al. (2021) Predictors of cognitive impairment in primary age-related tauopathy: an autopsy study. bioRxiv. 10.1101/2021.06.08.447553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikeda K, Akiyama H, Arai T, Oda T, Kato M, Iseki E et al. (1999) Clinical aspects of “senile dementia of the tangle type” - A subset of dementia in the senium separable from late-onset Alzheimer’s disease. Dement Geriatr Cogn 10:6–11. 10.1159/000017091 [DOI] [PubMed] [Google Scholar]
- 41.Ikeda K, Akiyama H, Arai T, Sahara N, Mori H, Usami M et al. (1997) A subset of senile dementia with high incidence of the apolipoprotein E epsilon2 allele. Ann Neurol 41:693–695. 10.1002/ana.410410522 [DOI] [PubMed] [Google Scholar]
- 42.Jabbari E, Koga S, Valentino RR, Reynolds RH, Ferrari R, Tan MMX et al. (2021) Genetic determinants of survival in progressive supranuclear palsy: a genome-wide association study. Lancet Neurol 20:107–116. 10.1016/S1474-4422(20)30394-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janocko NJ, Brodersen KA, Soto-Ortolaza AI, Ross OA, Lies-inger AM, Duara R et al. (2012) Neuropathologically defined subtypes of Alzheimer’s disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol 124:681–692. 10.1007/s00401-012-1044-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S et al. (2019) Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51:404–413. 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jellinger KA, Attems J (2007) Neurofibrillary tangle-predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol 113:107–117. 10.1007/s00401-006-0156-7 [DOI] [PubMed] [Google Scholar]
- 46.Jun G, Ibrahim-Verbaas CA, Vronskaya M, Lambert JC, Chung J, Naj AC et al. (2016) A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 21:108–117. 10.1038/mp.2015.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK et al. (2017) A unique microglia type associated with restricting development of alzheimer’s disease. Cell 169(1276–1290):e1217. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 48.Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ et al. (2003) Neuropathology of cognitively normal elderly. J Neuropath Exp Neur 62:1087–1095. 10.1093/jnen/62.11.1087 [DOI] [PubMed] [Google Scholar]
- 49.Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A et al. (2015) Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 10.1038/ncomms8247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kovacs GG (2015) Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 41:3–23. 10.1111/nan.12208 [DOI] [PubMed] [Google Scholar]
- 51.Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H et al. (2016) Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131:87–102. 10.1007/s00401-015-1509-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC et al. (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51:414–430. 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lafirdeen ASM, Cognat E, Sabia S, Hourregue C, Lilamand M, Dugravot A et al. (2019) Biomarker profiles of Alzheimer’s disease and dynamic of the association between cerebrospinal fluid levels of beta-amyloid peptide and tau. PLoS ONE 14:e0217026. 10.1371/journal.pone.0217026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45:1452–1458. 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larsson M, Duffy DL, Zhu G, Liu JZ, Macgregor S, McRae AF et al. (2011) GWAS findings for human iris patterns: associations with variants in genes that influence normal neuronal pattern development. Am J Hum Genet 89:334–343. 10.1016/j.ajhg.2011.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH et al. (2021) Trem2 restrains the enhancement of tau accumulation and neurodegeneration by beta-amyloid pathology. Neuron 109(1283–1301):e1286. 10.1016/j.neuron.2021.02.010 [DOI] [PubMed] [Google Scholar]
- 57.Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W (1999) The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Gene Dev 13:1822–1833. 10.1101/gad.13.14.1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD et al. (2018) GWAS on family history of Alzheimer’s disease. Transl Psychiatry 8:99. 10.1038/s41398-018-0150-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C et al. (2013) Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res Rev 12:289–309. 10.1016/j.arr.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 60.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A et al. (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48:1279–1283. 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McMillan CT, Lee EB, Jefferson-George K, Naj A, Van Deerlin VM, Trojanowski JQ et al. (2018) Alzheimer’s genetic risk is reduced in primary age-related tauopathy: a potential model of resistance? Ann Clin Transl Neur 5:927–934. 10.1002/acn3.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brown-lee LM et al. (1991) The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486 [DOI] [PubMed] [Google Scholar]
- 63.Mitchell TW, Mufson EJ, Schneider JA, Cochran EJ, Nissanov J, Han LY et al. (2002) Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer’s disease. Ann Neurol 51:182–189. 10.1002/ana.10086 [DOI] [PubMed] [Google Scholar]
- 64.Morris JC (1993) The clinical dementia rating (CDR): current version and scoring rules. Neurology 43:2412–2414 [DOI] [PubMed] [Google Scholar]
- 65.Mungas D, Tractenberg R, Schneider JA, Crane PK, Bennett DA (2014) A 2-process model for neuropathology of Alzheimer’s disease. Neurobiol Aging 35:301–308. 10.1016/j.neurobiolaging.2013.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Myers AJ, Kaleem M, Marlowe L, Pittman AM, Lees AJ, Fung HC et al. (2005) The H1c haplotype at the MAPT locus is associated with Alzheimer’s disease. Hum Mol Genet 14:2399–2404. 10.1093/hmg/ddi241 [DOI] [PubMed] [Google Scholar]
- 67.Nagy Z, VatterBittner B, Braak H, Braak E, Yilmazer DM, Schultz C et al. (1997) Staging of Alzheimer-type pathology: an interrater-intrarater study. Dement Geriatr Cogn 8:248–251. 10.1159/000106639 [DOI] [PubMed] [Google Scholar]
- 68.Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D et al. (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18:1091–1102. 10.1016/S1474-4422(19)30320-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Santacruz K et al. (2009) Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol 68:774–784. 10.1097/NEN.0b013e3181aacbe9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ et al. (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381. 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS et al. (2007) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 66:1136–1146. 10.1097/nen.0b013e31815c5efb [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Novak M, Jakes R, Edwards PC, Milstein C, Wischik CM (1991) Difference between the tau-protein of alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies-423 and antibodies-7.51. P Natl Acad Sci USA 88:5837–5841. 10.1073/pnas.88.13.5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Otero-Garcia M, Xue Y-Q, Shakouri T, Deng Y, Morabito S, Allison T et al. (2020) Single-soma transcriptomics of tangle-bearing neurons in Alzheimer’s disease reveals the signatures of tau-associated synaptic dysfunction. bioRxiv. 10.1101/2020.05.11.088591 [DOI] [Google Scholar]
- 74.Panagiotou OA, Ioannidis JPA, Project G-WS (2012) What should the genome-wide significance threshold be? Empirical replication of borderline genetic associations. Int J Epidemiol 41:273–286. 10.1093/ije/dyr178 [DOI] [PubMed] [Google Scholar]
- 75.Panchenko MV (2016) Structure, function and regulation of jade family PHD finger 1 (JADE1). Gene 589:1–11. 10.1016/j.gene.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909. 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 77.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP et al. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26:2336–2337. 10.1093/bioinformatics/btq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007) PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ et al. (2020) Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 10.1038/s41467-019-14279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Robinson JL, Geser F, Corrada MM, Berlau DJ, Arnold SE, Lee VM et al. (2011) Neocortical and hippocampal amyloid-beta and tau measures associate with dementia in the oldest-old. Brain 134:3708–3715. 10.1093/brain/awr308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanchez-Contreras MY, Kouri N, Cook CN, Serie DJ, Heckman MG, Finch NA et al. (2018) Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol Neurodegener. 10.1186/s13024-018-0267-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sanchez-Juan P, Moreno S, de Rojaso I, Hernandez I, Valero S, Alegret M et al. (2019) The MAPT H1 haplotype is a risk factor for Alzheimer’s disease in APOE epsilon 4 non-carriers. Front Aging Neurosci. 10.3389/fnagi.2019.00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Santa-Maria I, Haggiagi A, Liu XM, Wasserscheid J, Nelson PT, Dewar K et al. (2012) The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol 124:693–704. 10.1007/s00401-012-1017-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scheres SH, Zhang W, Falcon B, Goedert M (2020) Cryo-EM structures of tau filaments. Curr Opin Struct Biol 64:17–25. 10.1016/j.sbi.2020.05.011 [DOI] [PubMed] [Google Scholar]
- 85.Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA (2009) The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis 18:691–701. 10.3233/Jad-2009-1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sealey MA, Vourkou E, Cowan CM, Bossing T, Quraishe S, Gram-menoudi S et al. (2017) Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol Dis 105:74–83. 10.1016/j.nbd.2017.05.003 [DOI] [PubMed] [Google Scholar]
- 87.Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao LZ, Luo WJ et al. (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549:523–524. 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sieberts SK, Perumal TM, Carrasquillo MM, Allen M, Reddy JS, Hoffman GE et al. (2020) Large eQTL meta-analysis reveals differing patterns between cerebral cortical and cerebellar brain regions. Scient Data. 10.1038/s41597-020-00642-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Silva MC, Ferguson FM, Cai Q, Donovan KA, Nandi G, Patnaik D et al. (2019) Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife. 10.7554/eLife.45457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Siriwardana NS, Meyer R, Havasi A, Dominguez I, Panchenko MV (2014) Cell cycle-dependent chromatin shuttling of HBO1-JADE1 histone acetyl transferase (HAT) complex. Cell Cycle 13:1885–1901. 10.4161/cc.28759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smith HL, Mallucci GR (2016) The unfolded protein response: mechanisms and therapy of neurodegeneration. Brain 139:2113–2121. 10.1093/brain/aww101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L et al. (2012) Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat Genet 44:872–873. 10.1038/ng.2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Strickland SL, Reddy JS, Allen M, N’songo A, Burgess JD, Corda MM et al. (2020) MAPT haplotype-stratified GWAS reveals differential association for AD risk variants. Alzheimers Dement 16:983–1002. 10.1002/alz.12099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, Lee VMY et al. (2013) The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Com. 10.1186/2051-5960-1-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K (2016) The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 18:421–430. 10.1038/gim.2015.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Voas MG, Rebay I (2003) The novel plant homeodomain protein rhinoceros antagonizes Ras signaling in the Drosophila eye. Genetics 165:1993–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Walker JM, Richardson TE, Farrell K, Iida MA, Foong C, Shang P et al. (2021) Early selective vulnerability of the CA2 hippocampal subfield in primary age-related tauopathy. J Neuropath Exp Neur 80:102–111. 10.1093/jnen/nlaa153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wesseling H, Mair W, Kumar M, Schlaffner CN, Tang S, Beerepoot P et al. (2020) Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 183(1699–1713):e1613. 10.1016/j.cell.2020.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191. 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M et al. (2001) Tauopathy in drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714. 10.1126/science.1062382 [DOI] [PubMed] [Google Scholar]
- 101.Yamada M (2003) Senile dementia of the neurofibrillary tangle type (tangle-only dementia): neuropathological criteria and clinical guidelines for diagnosis. Neuropathology 23:311–317. 10.1046/j.1440-1789.2003.00522.x [DOI] [PubMed] [Google Scholar]
- 102.Yokoyama JS, Karch CM, Fan CC, Bonham LW, Kouri N, Ross OA et al. (2017) Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol 133:825–837. 10.1007/s00401-017-1693-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu L, Chibnik LB, Srivastava GP, Pochet N, Yang J, Xu J et al. (2015) Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol 72:15–24. 10.1001/jamaneurol.2014.3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zeng LL, Bai M, Mittal AK, El-Jouni W, Zhou J, Cohen DM et al. (2013) Candidate tumor suppressor and pVHL partner Jade-1 binds and inhibits AKT in renal cell Carcinoma. Cancer Res 73:5371–5380. 10.1158/0008-5472.Can-12-4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou MI, Foy RL, Chitalia VC, Zhao J, Panchenko MV, Wang HM et al. (2005) Jade-1, a candidate renal tumor suppressor that promotes apoptosis. P Natl Acad Sci USA 102:11035–11040. 10.1073/pnas.0500757102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou MI, Wang H, Ross JJ, Kuzmin I, Xu C, Cohen HT (2002) The von Hippel-Lindau tumor suppressor stabilizes novel plant homeodomain protein Jade-1. J Biol Chem 277:39887–39898. 10.1074/jbc.M205040200 [DOI] [PubMed] [Google Scholar]
- 107.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR et al. (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26:131–142. 10.1038/s41591-019-0695-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.