

Abstract
Associations between the human gut microbiome and expression of host illness have been noted in a variety of conditions ranging from gastrointestinal dysfunctions to neurological deficits. Machine learning (ML) methods have generated promising results for disease prediction from gut metagenomic information for diseases including liver cirrhosis and irritable bowel disease, but have lacked efficacy when predicting other illnesses. Here, we review current ML methods designed for disease classification from microbiome data. We highlight the computational challenges these methods have effectively overcome and discuss the biological components that have been overlooked to offer perspectives on future work in this area.
Keywords: bioinformatics, host–microbe interactions, machine learning, microbiome
Introduction
The collection of microscopic organisms residing in the intestinal tract is commonly referred to as the gut microbiome [1]. This community of microorganisms is associated with the well-being of the host [2–7], yet the specific roles and contributions of the individual microbes towards disease are often unknown [8, 9]. The advent of high-throughput sequencing along with pioneering work on population-level analysis of the human gut microbiome from the Human Microbiome Project (HMP) [10, 11], Belgian Flemish Gut Flora Project [12] and METAgenomics of the Human Intestinal Tract (MetaHIT) Project [13] have all broadened our understanding of host–microbiome interactions and raise the possibility that the gut microbiome could be an avenue for new forms of medical interventions.
Microbiome data is especially enticing in human health research as it has the potential to explain medical mysteries that current clinical information has not been able to resolve. Host DNA for example can be used to calculate disease risk, but the static nature of this material means it cannot be used to measure the current health state of an individual [14]. Microbiome information is still unique to each individual [15, 16], yet has proven to change with many common illnesses and infections, providing a real-time snapshot into the health state of the host [17, 18]. Microbiome information is also intriguing from a data science perspective since it may not be just the presence of specific microbes that influences disease expression, but rather microbe abundances, the phylogenetic relationship between microbes, or the communication between microbes and their environment [19, 20].
A natural first question is if the data and descriptive statistics from the gut microbial community for any particular disease contain predictive power as to the disease state of the host. If so, a stool sample could be applied directly as a non-invasive clinical diagnostic tool provided the accuracy is robust and reproducible. But more broadly, as with any data science discipline, exploring and parsing the relationship between variables and outcome is the route to additional discovery. In this review, we present a survey of machine learning (ML) models designed specifically for this purpose: classification of host disease status based on features derived from DNA sequencing of the gut microbiome.
A standard study design for investigating associations between microbes and host health is a case-control study, illustrated in Figure 1.
Figure 1. Standard workflow for determining microbiome-disease associations through a case-control study or ML model.
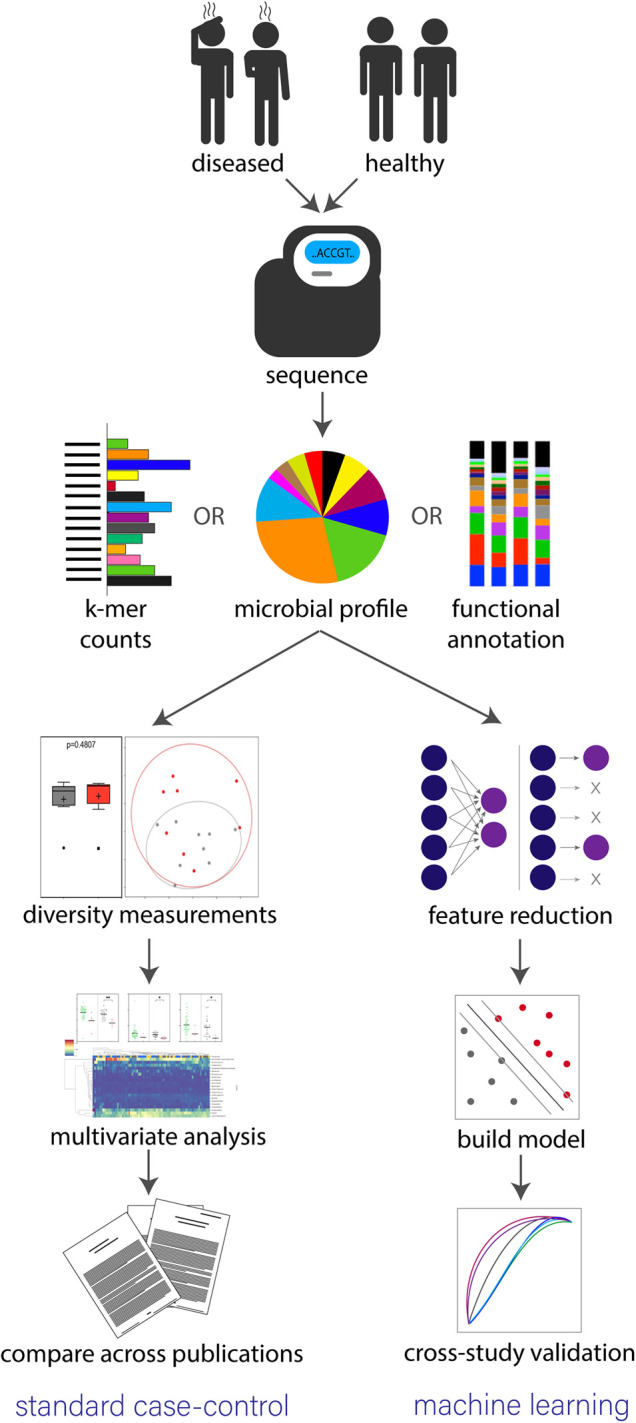
Both approaches begin by separating study participants into diseased and healthy cohorts, collecting samples, then performing high-throughput sequencing. Sequencing is completed through either a WGS or 16S approach then reads are converted to either k-mer counts [21], microbial profiles or functional annotations. In a standard case-control study (left path) alpha diversity, beta diversity and multivariate analysis are used to establish statistically significant differences between the two cohorts. A manual literature review is then performed to determine if findings are consistent across various studies. However, in a standard ML approach, features are extracted from sequence information and a model is constructed to detect trends separating the two groups. Cross-study validation is then performed by calculating accuracy in classification results from other test data sets.
First, subjects are separated into two equally sized cohorts based on disease state (sick and healthy) with the subjects chosen to make the two groups as closely matched as possible in terms of other potentially confounding variables. The gut microbiome for each participant is then established by sequencing a fecal sample through either a whole genome sequencing (WGS) or 16S rRNA approach. In WGS, all genetic material is sequenced providing a complete understanding of all cells in the sample with limited bias [22, 23]. A more cost-effective alternative is to perform targeted amplicon sequencing for the 16S gene, which restricts results so that only the microbes’ abundances relative to each other can be assumed [24, 25]. The resulting data are processed using various computational pipelines [21, 26–30] to distill the large volume of unstructured sequences into more manageable descriptive data that can be mined for signal.
A common first analysis is a basic comparison to test for statistically significant differences in the average community of the two groups. This is analogous to an ANOVA test in single-variable statistics, though in practice it is done using summary metrics like alpha diversity, beta diversity or multiple-hypothesis tests for differences in a larger set of values (i.e. microbe abundance) with an appropriate software [31, 32]. This is not a predictive modeling exercise per se, but establishing that there is a non-zero average difference in the communities is a primitive version of classification. Variations of these statistical models have been valuable for finding trends in the study-specific associations between microbes and a particular disease [33–36], but have been unsuccessful in terms of disease diagnosis or prevention because individual studies often report inconsistent findings [19, 37–41], raising reproducibility questions. Many researchers are voicing concerns with these statistical tests due to their ability to introduce bias [42, 43].
ML models are enticing for microbiome-phenotype classification tasks because, theoretically, disease profiles and biomarkers can be identified with only limited prior knowledge of the underlying system [44]. In addition, ML methods have shown success in other avenues of microbiome analysis, such as taxonomic classification [26, 45]. The potential scope of features is virtually limitless, as we will discuss later, but the approaches presented in this review rely on a few usual suspects: species-level relative abundance, a set of strain-specific markers from MetaPhlAn2 [27], k-mer profiles, and operating taxonomic units (OTUs). To produce relative abundance or biomarker detection results, a reference database is used to assign sequences as the closest match. K-mer profile generation skips the sequence classification step however by simply counting the frequency of all substrings of length k. Methods utilizing this technique have shown great success in reducing computational complexity while still producing accurate results in a range of microbiome analysis tasks [30, 46, 47]. In OTU clustering, similar sequences are grouped together then assigned either a consensus sequence or a reference-based taxonomic classification. A more recent form of clustering is one involving amplicon sequence variant (ASV), which includes an error reduction step to establish exact sequences and filters out reads based on a confidence threshold [48]. Any of these features can be fed into an off-the-shelf ML algorithm for supervised learning (i.e. random forest [49], support vector machines [50], neural networks [51]). Model output is then a simple yes/no disease classification with the potential to extract the most influential features through either built-in methods or post-processing pipelines [52], which can provide clinical value once interpreted [53, 54]. Accuracy can be validated on some kind of holdout sample, although in practice a more robust method is to apply the model to a totally separate study under different conditions, which truly tests the generalizability of the model. This cross-study validation step is challenging, however, and is not always completed.
In this article, we review several ML approaches for disease prediction from metagenomic data and their strategies for overcoming computational and biological challenges. We have chosen seven methods for inclusion, for two key reasons: (1) they are all recent additions to the literature that made specific design decisions putting them beyond simple off-the-shelf models, (2) six of the seven have been evaluated on the same set of benchmark case-control studies (Table 1), which allows us to assess these methods based on a common benchmark data set. Following this, we will discuss the major challenges that remain, as well as prospective developments and advances.
Table 1. Summary statistics for discussed data sets. Here, ‘x’ denotes use of data set in method publication.
| Disease | Cases | Controls | MetAML | PopPhy-CNN | Met2Img | MetaPheno | DeepMicro | MVIB |
|---|---|---|---|---|---|---|---|---|
| Liver cirrhosis | 118 | 114 | x | x | x | – | x | x |
| IBD | 25 | 85 | x | – | x | – | x | x |
| Obesity | 164 | 89 | x | x | x | x | x | x |
| Type 2 diabetes | 170 | 174 | x | x* | x | x | x | x |
x*: reported results for disease include additional samples (53 case, 43 controls).
Current approaches
The HMP and MetaHIT projects spurred the development of several 16S and WGS microbiome foundational tools including: taxonomic classification, phylogenetic placement, functional annotation and clustering [27, 30, 55–63]. These tools and data sets have been used in both single trial and encompassing meta-analyses studies for detecting microbiome trends, or lack there of, in relation to disease [37, 38, 40]. The methods covered in this section (Table 2), along with several others [54, 71–78], take this idea one step further and aim to establish generalizable models for discovering consistent microbiome-phenotype trends across individual studies.
Table 2. A selection of machine learning methods for disease classification from metagenomic sequences. Best AUC here denotes the highest AUC value reported in publication for specified data set.
| Software | Model input | Model description | Best AUC | Novelty | |||
|---|---|---|---|---|---|---|---|
| Cirr. | IBD | T2D | Obes. | ||||
| MetAML 2016 [64] | sp. rel. ab. or strain markers | Parameter sweep for 4 classifiers (SVM, RF, Lasso, ENet) with 3 feature selection methods (RF n most important, Lasso, ENet) | 0.96 SVM | 0.91 SVM | 0.76 SVM | 0.66 SVM | Foundational cross-validation test data and framework; first parameter sweep of metagenome disease prediction from off-the-shelf ML models |
| PopPhy-CNN 2020 [65] | OTU rel. ab. | PhyloT tree construction; populated with input OTU rel. ab.; transformed to 2D matrix; CNN with ELU | 0.95 | N/A | 0.69 | 0.67 | CNN with spatial quantitative relationship in input taxonomy data; novel alg for selecting most important features from first convolutional layer |
| Met2Img 2018 [66] | sp. or genus rel. ab. | Rel. ab. binned, colored, and visualized with Fill-up or t-SNE; 24x24 px (or smaller) images input into CNN with ReLU | 0.91 Fillup SPB | 0.87 Fillup SPB | 0.68 tSNE QTF | 0.69 tSNE SPB | Colored pixel visualization for microbiome profile; explores 3 binning methods (PR, QTF, SPB) with color and gray colormaps |
| MicroPheno 2018 [67] | 16S raw seqs | Find subsample size for stable k-mer profile; find best k; input k-mers to DNN (MLP w/ ReLU), RF, or multi-class linear SVM | N/A | N/A | N/A | N/A | 16S sequences; k-mer distribution from shallow sub-samples outperformed OTU features; first 16S deep learning metagenome-phenotype exploration |
| MetaPheno 2019 [68] | sp. rel. ab. or raw seqs | Jelly-fish k-mer counts; identify sig. k-mers with cohort p-values; apply hyper-parameter grid search models | N/A | N/A | 0.76 gcF, k-mer | 0.65 gcF, rel. ab. | Review of current methods; compares features: k-mers and rel. ab. with classifiers: SVM, RF, XGBoost, gcForest, AE-pretained DNN (AutoNN) |
| DeepMicro 2020 [69] | sp. rel. ab. or strain markers | Low-dimensional profile representation from autoencoder; input into MLP with ReLU or hyper-parameter grid SVM or RF | 0.94 SVM CAE | 0.96 SVM SAE | 0.76 MLP CAE | 0.67 RF DAE | 4 autoencoders (shallow, deep, variational, convolutional) to reduce microbiome dimension; combines with MLP, SVM, and RF param. sweep |
| MVIB 2021 [70] | sp. rel. ab. and strain markers | MLP for each modality (rel. ab., strain marker, metabolomics); Information Bottleneck theory to learn joint stochastic encoding | 0.93 D | 0.94 J;T | 0.76 J;T | 0.67 D | Combine multiple heterogeneous data modalities; explore default and joint pre-processing (D,J); optional triple margin loss extension (T) |
MetAML
The metagenomic prediction analysis based on machine learning (MetAML) [64] software laid the groundwork for detecting microbiome-phenotype associations by generating the first validated toolbox for disease prediction from shotgun metagenomes. MetAML established a computational ML framework for metagenome-based prediction tasks with implemented classifiers: support vector machine (SVM), random forest (RF), Lasso [79] and Elastic Net (ENet) [80]. MetAML also established the quantitative assessment to evaluate accuracy of each model and its ability to translate to the general population through cross-validation (prediction strength on metagenomic data) and cross-study (generalization of model on different studies) analyses. Results are measured with accuracy metrics: overall accuracy (OA), precision, recall, F1 and area under the curve (AUC). The MetAML framework was evaluated on metagenomic case-control datasets from five different diseases: inflammatory bowel disease (IBD), obesity, type-2 diabetes (T2D), liver cirrhosis and colorectal cancer. Features for tested models were generated from either species-level profiles or strain-specific presence markers based on results from MetaPhlAn2 [27] and further feature selection was also conducted with Lasso, ENet and RF embedded feature selection to give emphasis to features with greater discrepancy between cohorts.
MetAML reported AUC scores over 0.88 for liver cirrhosis, colorectal cancer and IBD prediction, which highlighted the potential for disease detection from gut metagenomic data. Additionally, this exploratory analysis showed improved results when healthy cohorts were included in training models, features were extracted from a lower taxonomic rank (strain-specific markers), and methods of feature reduction were implemented. Despite these promising results, T2D and obesity datasets reported lower AUC scores (<0.80), encouraging researchers to explore alternative approaches to improve upon MetAML and yield better disease prediction results for these datasets.
PopPhy-CNN
PopPhy-CNN [65] aimed to improve classification accuracy of the liver cirrhosis, type 2 diabetes and obesity datasets from the MetAML package with a convolutional neural network (CNN) [81, 82] learning framework, where each layer uses the exponential linear unit (ELU) activation function. This method uses genus- and species-level relative abundances as well as a phylogenetic tree to empower the neural network to explore both quantitative characteristics from metagenomic data and spatial relationships from the taxonomic tree. The novelty of this approach lies in the use of a taxonomic relativity between microbes and a custom-built feature extraction algorithm, yet results did not show significant improvement over MetAML across tested datasets.
Met2Img
Met2Img [66] also built a CNN, but this time with a rectified linear unit (ReLU) activation function. This approach incorporated a creative feature extraction step where each sample is transformed into an image containing colored pixels representing the various microbes and their relative quantities. Images are generated by one of two different methods: phylogenetic sorting using Fill-up or a t-distributed stochastic neighbor embedding (t-SNE) visualization method. The resulting images are then used as features for the neural network. Met2Img reports improved accuracy over the MetAML RF model for three of the diseases (liver cirrhosis, IBD and obesity), but little to no change for the remaining diseases (colorectal cancer and T2D).
MicroPheno
MicroPheno [67] simplified the sequencing step of the pipeline by utilizing k-mers from short-read 16S rRNA data, rather than shotgun metagenomic sequences, in a deep learning model. MicroPheno extracts k-mers from shallow sub-samples and includes hidden layer dropout from its multi-layer-perceptrons (MLP) [83] neural network architecture, allowing for a computationally inexpensive pipeline. When applied to a sample set consisting of samples from different human body sites, an F1 score of over 0.90 was reported. However, this number dropped to 0.75 when applied to a Crohn’s disease dataset. While this oversimplified pipeline may prevent the model from accurate classification for complex samples, it did raise the idea of k-mer based feature extraction.
MetaPheno
LaPierre et al. [68] compared and contrasted existing metagenomic methods that used MetAML datasets in their publication results in an evaluation called MetaPheno. The authors hypothesize that classification accuracy falls short on T2D and obesity datasets due to overfitting, and specifically explore ways to improve upon these results. They implemented k-mer-based feature extraction as shown in MicroPheno, but this time k-mers were derived from shotgun metagenomic data rather than subsampled from 16S. The MetaPheno pipeline is completed by counting k-mer frequencies with Jelly-fish [21], extracting significant k-mers through a statistical model, then applying a machine learning model (SVM, RF, XGBoost [84], gcForest [85] or an autoencoder-pretrained deep neural network (DNN) [83, 86, 87]). Results showed that no single model outperformed others in all metrics and that the explored methods of feature reduction were unsuccessful in drastically improving accuracy over MetAML findings. The authors ultimately were not successful with T2D and obesity; they speculate that prediction using only metagenomic reads may not be possible and perhaps there is an upper limit on the accuracy that can be achieved this input data. They recommend future work in methods utilizing host genomic or additional multi-omic data sources alongside metagenomic data, then building a deep learning model such as a similarity network fusion [88].
DeepMicro
Following the publication of MetaPheno, DeepMicro [69] was released as another deep learning method evaluated on the datasets from MetAML, specifically focused on feature extraction. DeepMicro experiments with converting high-dimensional microbiome data to low-dimensional representations through an autoencoder (AE) [87]. Datasets from all 5 MetAML diseases were tested with both species-level relative abundance and strain-level marker output from MetaPhlAn2, with four different autoencoders (shallow (SAE), deep (DAE), variational (VAE) and convolutional (CAE)), and with three different classification algorithms (SVM, RF and MLP). In the results, the best performing autoencoder is highly dependent on the problem complexity and intrinsic properties of the input data. Additionally, incorporating healthy controls into the model worsened performance, a contrast with findings from MetAML. Still, the best DeepMicro approach outperformed the best MetAML approach in all but one of the tested diseases (colorectal cancer), highlighting the importance of effective feature extraction techniques for metagenomic data. However, and yet again, the AUC score for obesity is still only 0.674, leaving plenty of room for future success in this application.
MVIB
ML approaches to accurately classify obesity and T2D is still an open area of research that continues to improve as new ML techniques are developed. One up-and-coming method is multimodal variation information bottleneck (MVIB) [70], which takes advantage of both the species-level abundance and strain-level markers from MetaPhlAn2 output by computing a joint stochastic encoding from both profiles. MVIB is a multimodal generalization of the Deep Variational Information Bottleneck [89], which allows a model to learn a joint encoding from heterogeneous input data modalities with a deep neural network. MVIB reports an improvement over DeepMicro with VAE in each of the test datasets, emphasizing the value in obtaining multiple sources of information for each sample in a single model. This is especially valuable to this line of research as it sets a foundation for incorporating additional input parameters and clinical data which could potentially improve the classification accuracy of future models. In addition, the authors experiment with adding a joint collection pre-processing step, where input abundances and markers were made homogeneous across all diseases, as well as transfer learning from non-targeted disease data sets.
Open challenges
Catering ML models to detect disease patterns from microbiome data presents a range of challenges (Table 3). The first is the standard ‘big-p, little-n’ issue, where the number of variables in the input data dwarf the number of samples available. Importantly, this particular challenge is the defining challenge of phenotype classification from microbiome features. Sequencing costs combined with logistical challenges of sample collection and patient recruitment limit the number of available samples that will be available for a given study. Increasing the size of training data has long been a reliable way to improve model performance in most disciplines, but that is commonly not an option for microbiome studies. Nonetheless, this situation often results in overfitting, yielding a suboptimal model and in turn preventing the model from accurate classification on test data.
Table 3. Challenges presented by microbiome data as input for ML models and the approaches taken by discussed methods to tackle these challenges.
| Large feature space; small sample size |
| MetAML feature selection with Lasso, ENet, or RF n most important |
| PopPhy-CNN feature selection with novel alg; network regularization |
| DeepMicro autoencoder for low-dimensionality representation; early stopping |
| MVIB stochastic probabilistic encoders |
| MicroPheno shallow subset of 16S k-mers; early stopping; dropout hidden layers |
| MetaPheno select k-mer counts from 1000 most significant k-mers |
| Met2Img convert profile to binned image; early stopping |
| Presence of novel species |
| MicroPheno & MetaPheno raw sequence input data (k-mers) |
| Temporal fluctuations in microbe abundances |
| MetAML include multiple samples from a single test subject |
| MVIB combine abundance and marker profiles for each sample |
To overcome this issue, each of the methods discussed above includes a feature reduction step. Both MetAML and PopPhy-CNN select only features that are considered the most important for the model. MetaPheno also uses this ideology by embedding an algorithm to select the 1000 features with the smallest p-value between case and control groups. DeepMicro explores four different autoencoders, which are used in the model to learn low-dimensional representations from complete microbiome profiles. MVIB incorporates a stochastic encoder for each input data modality. MetaPheno and MicroPheno both use k-mer counts and reduce complexity in their neural networks with hidden layer dropout tactics. Met2Img takes an entirely unique approach and converts microbial quantities into binned images, which essentially transforms each sample into a single feature.
A second challenge encountered is the presence of novel species. This challenge highlights the limitations of reference-based feature computation, where the most similar database entry is used for labeling and therefore reads can only be classified as an existing database entry. This results in novel microbes either misclassified as an incorrect organism or grouped together in an ‘unclassified’ category. Both MicroPheno and MetaPheno overcome this challenge by using k-mer count profiles, avoiding the classification step entirely. An alternative approach would be use of a reference-free and therefore database-agnostic method [90].
A third challenge presented is the fluctuating nature of microbial communities. Although the gut microbiome in healthy adults is shown to be relatively stable over time [91], more substantial fluctuations have been observed in subjects exhibiting illness [92–94]. This dynamic environment raises concern since the most informative time to collect samples is not yet established and, beyond this, the true separation between disease states may lie in the changes that occur over time [95]. MetAML takes this knowledge into account by incorporating samples from various stages throughout a participant’s illness in the liver cirrhosis and T2D test data sets. MVIB aims to gain a more thorough representation of each changing environments by including multiple profiles (species-level abundance and strain-level markers) for each input samples. However, none of the methods presented exploit longitudinal patient samples from both a healthy and diseased state and therefore cannot truly explore temporal changes in an individual that arise with disease onset. Although this would introduce a layer of complexity to the approach illustrated in Figure 1, it would provide a comprehensive tactic to account for the gut microbe community changes that have been observed in disease.
Future research directions
Despite repeatedly observed associations of the gut microbiota in T2D and obesity studies, the presented methods have encountered difficulty providing accurate phenotype classification for these diseases [34, 35, 96–102]. This raises the question: do the current feature sets, exclusively derived from metagenomic data, inherently limit predictive power for some phenotypes? It may be that microbes are only one of several contributing factors in the condition, or it may be that the microbes do not contribute at all but rather respond in a consistent way to the environmental change brought about by the disease. A major source of future advancement in phenotype-prediction would be the result of discovering new data sources or feature types that have complementary predictive power, then utilizing the appropriate model structures for leveraging additional information. Here, we consider two biological factors that interact with the microbiome to have a combined effect on host health (host genetics and microbe-derived metabolites) and explore their potential to act as complementary predictors in microbiome-disease relations.
Gut microbiome and host genetics
Because gut microbes impact host health due to interactions with host cells [19], incorporation of host DNA may improve future models. In addition, associations between gut microbes and host genes have been detected in a variety of studies [103–108]. One case where inclusion of host genomic information strengthened findings was in a study where Ryan et al. [109] examined the colonic microbiota in relation to IBD through host transcriptomics, epigenomics and genetics data. Authors concluded that while the microbiota appeared to be linked to the disease, there was no evidence of a distinct microbial diagnostic signature, likely due to heterogeneous host–microbe interactions. They then included epithelial DNA methylation into the algorithm, which yielded better classification results.
Gut microbiome and metabolites
Metabolites are the intermediate or end product of interactions between microbes and host cells [110]. Short-chain fatty acids (SCFA) and bile acids are metabolites that can have beneficial or damaging impacts on the host tissue [111] and have shown associations with a range of illnesses [102, 112–116]. This promotes the idea of incorporating metabolite information in disease state analyses. Jeffery et al. found that only the fecal metabolome, rather than the microbiome alone, could distinguish between IBD patients that expressed the bile acid malabsorption phenotype. In a separate study, Sanna et al. [117, 118] leveraged SCFA levels in addition to gut microbial data to discover an association with risk of T2D, which is likely due to the fact that microbe-derived SCFA acts an additional energy source and therefore can increase likelihood for obesity. In both these studies, inclusion of microbe-derived metabolites was essential for observing associations between the microbiome and disease states. Promising results produced from leveraging both these data types have led to the development of further studies and software tools for effective combination of metabolite and microbiome information as input data [70, 119–122].
Microbiome-based models for obesity classification
Current microbiome-based models for obesity classification will likely be improved by incorporating variables containing additional signal [123, 124]. Increased blood glucose levels heavily influences obesity [125], but is difficult to control since foods elicit vastly different responses across individuals [126–128]. However, Zeevi et al. showed great success in developing an ML algorithm for predicting postprandial blood glucose levels by integrating blood parameters, dietary habits, anthropometrics, physical activity and gut microbiome data. This model was constructed from a 800-person cohort, then tested on a separate 100-person cohort where it accurately predicted personalized glucose response after consumption of different foods. In addition, the model was adjusted and applied in a blinded randomized controlled dietary intervention, where participants effectively altered their gut microbiota to successfully lower postprandial responses based on personalized diet recommendations from the algorithm. This advancement in blood glucose response prediction suggests the possibility of a similarly accurate model for obesity classification.
Another avenue for future research in phenotype prediction models is the integration of the causal relationship between microbiota and host disease, given one exists. This relationship is likely to differ between illnesses and is currently difficult to ascertain in many cases [41, 129]. Plausibly addressing causality is challenging since the fine control over experimental conditions required can only be conducted in animals. Given microbial communities vary substantially across hosts, conclusions in animal models often are not transferable to humans [130], even with increased validation from replication of findings across different animal hosts. As the causal relationship of gut microbes becomes further established for each illness, leveraging this information appears promising for future disease classification ML models.
Conclusion
Recent advances in ML methods have opened the door to deciphering the intricate role of gut microbes in host health and disease. Methods have proven successful in classification of multiple illnesses using solely metagenomic information due to their inherent ability to handle multi-dimensional data and identify trends with little upfront knowledge. Published methods have explored a broad range of standard model platforms including random forest, deep neural networks and support vector machines as well as unconventional approaches to overcome challenges presented by microbial communities. While notable accuracy of irritable bowel disease and liver cirrhosis classification has been reported, less success has been observed for obesity [64, 68, 70]. After exhausting model types and parameter settings through trial-and-error, the current limiting factor appears to be due to unknown causal roles for microbes and lack of further influential features. Additional clinical data, including but not limited to human genetics, metabolomes and lifestyle factors, combined with microbial information and the appropriate feature reduction technique shows promise for improved disease prediction accuracy in future ML algorithms for these complex gut microbiota relationships.
Summary
Several ML methods have been developed for host disease detection from microbiome sequences with various classifiers and feature reduction approaches.
Methods have proven successful in predicting liver cirrhosis and IBD from gut metagenomic samples, but have not shown such accuracy with obesity prediction.
Future perspectives for improvement in disease detection algorithms include incorporating additional biological factors as input into ML models, such as host genomics, microbiome-derived metabolite levels or blood glucose response predictions.
Acknowledgements
We would like to thank members of the Savidge lab, Tor Savidge, Qinglong Wu, Charlie Seto, and the Villapol lab, Sonia Villapol and Sirena Soriano, for insightful discussion specific to disease detection from the gut microbiome.
Abbreviations
- IBD
inflammatory bowel disease
- ML
machine learning
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Funding
This work was funded in part by the C3.ai Digital Transformation Institute COVID-19 award. K.D.C. and T.J.T. were supported in part by a NIH grant from NIAID P01-AI152999. M.G.N. was funded by a fellowship from the National Library of Medicine Training Program in Biomedical Informatics and Data Science (T15LM007093, PI: Kavraki).
Author Contribution
K.D.C. performed the literature review. K.D.C., M.G.N. and T.J.T. contributed to writing the manuscript. All authors read and approved the manuscript.
References
- 1.Ursell, L.K., Metcalf, J.L., Parfrey, L.W. and Knight, R. (2012) Defining the human microbiome. Nutr. Rev. 70, S38–S44 10.1111/nure.2012.70.issue-s1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clemente, J.C., Ursell, L.K., Parfrey, L.W. and Knight, R. (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 10.1016/j.cell.2012.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brody, H. (2020) The gut microbiome. Nature 577, S5–S5 10.1038/d41586-020-00194-2 [DOI] [PubMed] [Google Scholar]
- 4.Shreiner, A.B., Kao, J.Y. and Young, V.B. (2015) The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 31, 69–75 10.1097/MOG.0000000000000139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sekirov, I., Russell, S.L., Antunes, L.C.M. and Finlay, B.B. (2010) Gut microbiota in health and disease. Physiol. Rev. 90, 859–904 10.1152/physrev.00045.2009 [DOI] [PubMed] [Google Scholar]
- 6.Yan, Y., Nguyen, L.H., Franzosa, E.A. and Huttenhower, C. (2020) Strain-level epidemiology of microbial communities and the human microbiome. Genome Med. 12, 71 10.1186/s13073-020-00765-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeevi, D., Korem, T., Godneva, A., Bar, N., Kurilshikov, A., Lotan-Pompan, M.. et al. (2019) Structural variation in the gut microbiome associates with host health. Nature 568, 43–48 10.1038/s41586-019-1065-y [DOI] [PubMed] [Google Scholar]
- 8.Nagpal, R., Kumar, M., Yadav, A., Hemalatha, R., Yadav, H., Marotta, F.. et al. (2016) Gut microbiota in health and disease: an overview focused on metabolic inflammation. Benef. Microbes 7, 181–194 10.3920/bm2015.0062 [DOI] [PubMed] [Google Scholar]
- 9.Oniszczuk, A., Oniszczuk, T., Gancarz, M. and Szymańska, J. (2021) Role of gut microbiota, probiotics and prebiotics in the cardiovascular diseases. Molecules 26, 1172 10.3390/molecules26041172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Human Microbiome Project Consortium. (2012) A framework for human microbiome research. Nature 486, 215–221 10.1038/nature11209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lloyd-Price, J., Mahurkar, A., Rahnavard, G., Crabtree, J., Orvis, J., Hall, A.B.. et al. (2017) Strains, functions and dynamics in the expanded human microbiome project. Nature 550, 61–66 10.1038/nature23889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falony, G., Joossens, M., Vieira-Silva, S., Wang, J., Darzi, Y., Faust, K.. et al. (2016) Population-level analysis of gut microbiome variation. Science 352, 560–564 10.1126/science.aad3503 [DOI] [PubMed] [Google Scholar]
- 13.Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K.S., Manichanh, C.. et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler, D.A., Srinivasan, M., Egholm, M., Shen, Y., Chen, L., McGuire, A.. et al. (2008) The complete genome of an individual by massively parallel DNA sequencing. Nature 452, 872–876 10.1038/nature06884 [DOI] [PubMed] [Google Scholar]
- 15.Zhu, A., Sunagawa, S., Mende, D.R. and Bork, P. (2015) Inter-individual differences in the gene content of human gut bacterial species. Genome Biol. 16, 82 10.1186/s13059-015-0646-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodrich, J.K., Waters, J.L., Poole, A.C., Sutter, J.L., Koren, O., Blekhman, R.. et al. (2014) Human genetics shape the gut microbiome. Cell 159, 789–799 10.1016/j.cell.2014.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durack, J. and Lynch, S.V. (2019) The gut microbiome: relationships with disease and opportunities for therapy. J. Exp. Med. 216, 20–40 10.1084/jem.20180448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris, V.C., Haak, B.W., Boele van Hensbroek, M. and Wiersinga, W.J. (2017) The intestinal microbiome in infectious diseases: the clinical relevance of a rapidly emerging field. Open Forum Infect. Dis. 4, ofx144 10.1093/ofid/ofx144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cani, P.D. (2018) Human gut microbiome: hopes, threats and promises. Gut 67, 1716–1725 10.1136/gutjnl-2018-316723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohajeri, M.H., Brummer, R.J.M, Rastall, R.A., Weersma, R.K., Harmsen, H.J.M, Faas, M.. et al. (2018) The role of the microbiome for human health: from basic science to clinical applications. Eur. J. Nutr. 57, 1–14 10.1007/s00394-018-1703-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marçais, G. and Kingsford, C. (2011) A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 10.1093/bioinformatics/btr011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van El, C.G., Cornel, M.C., Borry, P., Hastings, R.J., Fellmann, F., Hodgson, S.V.. et al. (2013) Whole-genome sequencing in health care. Eur. J. Hum. Genet. 21, 580–584 10.1038/ejhg.2013.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Browne, P.D., Nielsen, T.K., Kot, W., Aggerholm, A., Gilbert, M.T.P., Puetz, L.. et al. (2020) GC bias affects genomic and metagenomic reconstructions, underrepresenting GC-poor organisms. GigaScience 9, giaa008 10.1093/gigascience/giaa008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woese, C.R., Kandler, O. and Wheelis, M.L. (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl Acad. Sci. U.S.A. 87, 4576–4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jo, J.H., Kennedy, E.A. and Kong, H.H. (2016) Bacterial 16S ribosomal RNA gene sequencing in cutaneous research. J. Invest. Dermatol. 136, e23–e27 10.1016/j.jid.2016.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolyen, E., Rideout, J.R., Dillon, M.R., Bokulich, N.A., Abnet, C.C., Al-Ghalith, G.A.. et al. (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Truong, D.T., Franzosa, E.A., Tickle, T.L., Scholz, M., Weingart, G., Pasolli, E.. et al. (2015) MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903 10.1038/nmeth.3589 [DOI] [PubMed] [Google Scholar]
- 28.Curry, K.D., Wang, Q., Nute, M.G., Tyshaieva, A., Reeves, E., Soriano, S.. et al. (2021) Emu: specieslevel microbial community profiling for full-length nanopore 16S reads, bioRxiv p.2021.05.02.442339
- 29.Albin, D., Nasko, D., Elworth, R.A.L., Lu, J., Balaji, A., Diaz, C.et al. (2019) SeqScreen: a biocuration platform for robust taxonomic and biological process characterization of nucleic acid sequences of interest. In 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), pp. 1729–1736, IEEE 10.1109/BIBM47256.2019.8982987 [DOI]
- 30.Wood, D.E. and Salzberg, S.L. (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 10.1186/gb-2014-15-3-r46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mallick, H., Rahnavard, A., McIver, L.J., Ma, S., Zhang, Y., Nguyen, L.H.. et al. (2021) multivariable association discovery in population-scale meta-omics studies. bioRxiv p. 2021.01.20.427420
- 32.Paulson, J.N., Stine, O.C., Bravo, H.C. and Pop, M. (2013) Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202 10.1038/nmeth.2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin, N., Yang, F., Li, A., Prifti, E., Chen, Y., Shao, L.. et al. (2014) Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64 10.1038/nature13568 [DOI] [PubMed] [Google Scholar]
- 34.Turnbaugh, P.J., Ley, R.E., Mahowald, M.A., Magrini, V., Mardis, E.R. and Gordon, J.I. (2006) An obesityassociated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- 35.Thaiss, C.A., Itav, S., Rothschild, D., Meijer, M.T., Levy, M., Moresi, C.. et al. (2016) Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature 540, 544–551 10.1038/nature20796 [DOI] [PubMed] [Google Scholar]
- 36.Treangen, T.J., Wagner, J., Burns, M.P. and Villapol, S. (2018) Traumatic brain injury in mice induces acute bacterial dysbiosis within the fecal microbiome. Front. Immunol. 9, 2757 10.3389/fimmu.2018.02757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duvallet, C., Gibbons, S.M., Gurry, T., Irizarry, R.A. and Alm, E.J. (2017) Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 8, 1784 10.1038/s41467-017-01973-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sze, M.A. and Schloss, P.D. (2016) Looking for a signal in the noise: revisiting obesity and the microbiome. mBio 7, e01018-16 10.1128/mBio.01018-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walters, W.A., Xu, Z. and Knight, R. (2014) Meta-analyses of human gut microbes associated with obesity and IBD. FEBS. Lett. 588, 4223–4233 10.1016/j.febslet.2014.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finucane, M.M., Sharpton, T.J., Laurent, T.J. and Pollard, K.S. (2014) A taxonomic signature of obesity in the microbiome? Getting to the guts of the matter. PLoS ONE 9, e84689 10.1371/journal.pone.0084689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wade, K.H. and Hall, L.J. (2020) Improving causality in microbiome research: can human genetic epidemiology help? Wellcome Open Res. 4, 199 10.12688/wellcomeopenres [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willis, A.D. (2019) Rarefaction, alpha diversity, and statistics. Front. Microbiol. 10, 2407 10.3389/fmicb.2019.02407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nearing, J.T., Douglas, G.M., Hayes, M., MacDonald, J., Desai, D., Allward, N.. et al. (2021) Microbiome differential abundance methods produce disturbingly different results across 38 datasets. bioRxiv p. 2021.05.10.443486
- 44.Moreno-Indias, I., Lahti, L., Nedyalkova, M., Elbere, I., Roshchupkin, G., Adilovic, M.. et al. (2021) Statistical and machine learning techniques in human microbiome studies: contemporary challenges and solutions. Front. Microbiol. 12, 635781 10.3389/fmicb.2021.635781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dilthey, A.T., Jain, C., Koren, S. and Phillippy, A.M. (2019) Strain-level metagenomic assignment and compositional estimation for long reads with metamaps. Nat. Commun. 10, 3066 10.1038/s41467-019-10934-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gardner, S.N. and Hall, B.G. (2013) When whole-genome alignments just won’t work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS ONE 8, e81760 10.1371/journal.pone.0081760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ondov, B.D., Treangen, T.J., Melsted, P., Mallonee, A.B., Bergman, N.H., Koren, S.. et al. (2016) Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 10.1186/s13059-016-0997-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Callahan, B.J., McMurdie, P.J. and Holmes, S.P. (2017) Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643 10.1038/ismej.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho, T.K. (1998) The random subspace method for constructing decision forests. IEEE Trans. Pattern Anal. Mach. Intell. 20, 832–844 10.1109/34.709601 [DOI] [Google Scholar]
- 50.Cortes, C. and Vapnik, V. (1995) Support-vector networks. Mach. Learn. 20, 273–297 10.1023/A:1022627411411 [DOI] [Google Scholar]
- 51.Hopfield, J.J. (1982) Neural networks and physical systems with emergent collective computational abilities. Proc. Natl Acad. Sci. U.S.A. 79, 2554–2558 10.1073/pnas.79.8.2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lundberg, S.M., Erion, G., Chen, H., DeGrave, A., Prutkin, J.M., Nair, B.. et al. (2020) From local explanations to global understanding with explainable AI for trees. Nat. Mach. Intell. 2, 56–67 10.1038/s42256-019-0138-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong, C.W., Yost, S.E., Lee, J.S., Gillece, J.D., Folkerts, M., Reining, L.. et al. (2021) Analysis of gut microbiome using explainable machine learning predicts risk of diarrhea associated with tyrosine kinase inhibitor neratinib: a pilot study. Front. Oncol. 11, 604584 10.3389/fonc.2021.604584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gou, W., Ling, C.w., He, Y., Jiang, Z., Fu, Y., Xu, F.. et al. (2021) Interpretable machine learning framework reveals robust gut microbiome features associated with type 2 diabetes. Diabetes Care 44, 358–366 10.2337/dc20-1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang, Q., Garrity, G.M., Tiedje, J.M. and Cole, J.R. (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Westcott, S.L. and Schloss, P.D. (2015) De Novo clustering methods outperform reference-based methods for assigning 16S rRNA gene sequences to operational taxonomic units. PeerJ 3, e1487 10.7717/peerj.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brady, A. and Salzberg, S.L. (2009) Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat. Methods 6, 673–676 10.1038/nmeth.1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Treangen, T.J., Koren, S., Sommer, D.D., Liu, B., Astrovskaya, I., Ondov, B.. et al. (2013) MetAMOS: a modular and open source metagenomic assembly and analysis pipeline. Genome Biol. 14, R2 10.1186/gb-2013-14-1-r2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ounit, R., Wanamaker, S., Close, T.J. and Lonardi, S. (2015) CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics 16, 236 10.1186/s12864-015-1419-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edgar, R.C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 61.Ghodsi, M., Liu, B. and Pop, M. (2011) DNACLUST: accurate and efficient clustering of phylogenetic marker genes. BMC Bioinformatics 12, 271 10.1186/1471-2105-12-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsen, F.A., Kodner, R.B. and Armbrust, E.V.. 2010 Pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11, 538 10.1186/1471-2105-11-538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen, N.P., Mirarab, S., Liu, B., Pop, M. and Warnow, T. (2014) TIPP: taxonomic identification and phylogenetic profiling. Bioinformatics (Oxford, England) 30, 3548–3555 10.1093/bioinformatics/btu721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pasolli, E., Truong, D.T., Malik, F., Waldron, L. and Segata, N. (2016) Machine learning meta-analysis of large metagenomic datasets: tools and biological insights. PLoS Comput. Biol. 12, e1004977 10.1371/journal.pcbi.1004977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reiman, D., Metwally, A.A., Sun, J. and Dai, Y. (2020) PopPhy-CNN: a phylogenetic tree embedded architecture for convolutional neural networks to predict host phenotype from metagenomic data. IEEE J. Biomed. Health Inform. 24, 2993–3001 10.1109/JBHI.6221020 [DOI] [PubMed] [Google Scholar]
- 66.Nguyen, T.H., Prifti, E., Sokolovska, N. and Zucker, J. (2019) Disease prediction using synthetic image representations of metagenomic data and convolutional neural networks. In Proceedings of The 13th IEEE-RIVF International Conference on Computing and Communication Technologies, pp. 231–236, IEEE 10.1109/RIVF.2019.8713670 [DOI] [Google Scholar]
- 67.Asgari, E., Garakani, K., McHardy, A.C. and Mofrad, M.R.K. (2018) MicroPheno: predicting environments and host phenotypes from 16S rRNA gene sequencing using a k-mer based representation of shallow sub-samples. Bioinformatics 34, i32–i42 10.1093/bioinformatics/bty296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.LaPierre, N., Ju, C.J.T., Zhou, G. and Wang, W. (2019) MetaPheno: a critical evaluation of deep learning and machine learning in metagenome-based disease prediction. Methods 166, 74–82 10.1016/j.ymeth.2019.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh, M. and Zhang, L. (2020) DeepMicro: deep representation learning for disease prediction based on microbiome data. Sci. Rep. 10, 6026 10.1038/s41598-020-63159-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grazioli, F., Siarheyeu, R., Pileggi, G. and Meiser, A. (2021) Microbiome-based disease prediction with multimodal variational information bottlenecks. bioRxiv p. 2021.06.08.447505v3 [DOI] [PMC free article] [PubMed]
- 71.Wirbel, J., Zych, K., Essex, M., Karcher, N., Kartal, E., Salazar, G.. et al. (2021) Microbiome meta-analysis and cross-disease comparison enabled by the SIAMCAT machine learning toolbox. Genome Biol. 22, 93 10.1186/s13059-021-02306-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rahman, M.A. and Rangwala, H. (2018) RegMIL: phenotype classification from metagenomic data. In Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics BCB ’18, pp. 145–154, Association for Computing Machinery, New York, NY
- 73.Lo, C. and Marculescu, R. (2019) MetaNN: accurate classification of host phenotypes from metagenomic data using neural networks. BMC Bioinformatics 20, 314 10.1186/s12859-019-2833-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Queyrel, M., Prifti, E., Templier, A. and Zucker, J.D. (2021) Towards end-to-end disease prediction from raw metagenomic data. bioRxiv p. 2020.10.29.360297
- 75.Martino, C., Shenhav, L., Marotz, C.A., Armstrong, G., McDonald, D., Vázquez-Baeza, Y.. et al. (2021) ContextAware dimensionality reduction deconvolutes gut microbial community dynamics. Nat. Biotechnol. 39, 165–168 10.1038/s41587-020-0660-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang, F. and Zou, Q. (2020) mAML: an automated machine learning pipeline with a microbiome repository for human disease classification. Database 2020, baaa050 10.1093/database/baaa050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu, H., Cai, L., Li, D., Wang, X., Zhao, S., Zou, F.. et al. (2018) Metagenomics biomarkers selected for prediction of three different diseases in Chinese population. Biomed. Res. Int. 2018, 2936257 10.1155/2018/2936257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ditzler, G., Polikar, R. and Rosen, G. (2015) Multi-layer and recursive neural networks for metagenomic classification. IEEE Trans. Nanobioscience. 14, 608–616 10.1109/TNB.2015.2461219 [DOI] [PubMed] [Google Scholar]
- 79.Tibshirani, R. (1996) Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Methodol.) 58, 267–288 10.1111/j.2517-6161.1996.tb02080.x [DOI] [Google Scholar]
- 80.Zou, H. and Hastie, T. (2005) Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 67, 301–320 10.1111/rssb.2005.67.issue-2 [DOI] [Google Scholar]
- 81.LeCun, Y., Boser, B., Denker, J., Henderson, D., Howard, R., Hubbard, W.. et al. (1990) Handwritten digit recognition with a back-propagation network. In Advances in Neural Information Processing Systems, vol. 2. Morgan-Kaufmann, Denver, CO [Google Scholar]
- 82.Krizhevsky, A., Sutskever, I. and Hinton, G.E. (2012) ImageNet classification with deep convolutional neural networks. In Advances in Neural Information Processing Systems, vol.25. Curran Associates, Inc 10.1145/3065386 [DOI] [Google Scholar]
- 83.Svozil, D., Kvasnicka, V. and Pospichal, J. (1997) Introduction to multi-layer feed-forward neural networks. Chemometr. Intell. Lab. Syst. 39, 43–62 10.1016/S0169-7439(97)00061-0 [DOI] [Google Scholar]
- 84.Chen, T. and Guestrin, C. (2016) XGBoost: a scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 785–794, ACM, New York, NY, USA 10.1145/2939672.2939785 [DOI] [Google Scholar]
- 85.Zhou, Z.H. and Feng, J. (2019) Deep forest. Natl. Sci. Rev. 6, 74–86 10.1093/nsr/nwy108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vincent, P., Larochelle, H., Lajoie, I., Bengio, Y., Manzagol, P.A.. Stacked denoising autoencoders: Learning useful representations in a deep network with a local denoising criterion. J. Machine Learning Res. 11, 38 [Google Scholar]
- 87.Hinton, G.E. and Salakhutdinov, R.R. (2006) Reducing the dimensionality of data with neural networks. Science 313, 504–507 10.1126/science.1127647 [DOI] [PubMed] [Google Scholar]
- 88.Wang, B., Mezlini, A.M., Demir, F., Fiume, M., Tu, Z., Brudno, M.. et al. (2014) Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 11, 333–337 10.1038/nmeth.2810 [DOI] [PubMed] [Google Scholar]
- 89.Alemi, A.A., Fischer, I., Dillon, J.V. and Murphy, K. (2019) Deep variational information bottleneck, arXiv:1612.00410 [cs, math]
- 90.Balaji, A., Sapoval, N., Elworth, R.L., Segarra, S. and Treangen, T.J.. 2020) KOMB: taxonomy-oblivious characterization of metagenome dynamics via k-core decomposition, bioRxiv
- 91.Fassarella, M., Blaak, E.E., Penders, J., Nauta, A., Smidt, H. and Zoetendal, E.G. (2021) Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut 70, 595–605 10.1136/gutjnl-2020-321747 [DOI] [PubMed] [Google Scholar]
- 92.Halfvarson, J., Brislawn, C.J., Lamendella, R., Vázquez-Baeza, Y., Walters, W.A., Bramer, L.M.. et al. (2017) Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2, 1–7 10.1038/nmicrobiol.2017.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tap, J., Ruppé, E. and Derrien, M. (2021) The human gut microbiota in all its states: from disturbance to resilience. In Reference Module in Food Science, Elsevier; 10.1016/B978-0-12-819265-8.00039-5 [DOI] [Google Scholar]
- 94.Lozupone, C.A., Stombaugh, J.I., Gordon, J.I., Jansson, J.K. and Knight, R. (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 10.1038/nature11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vázquez-Baeza, Y., Gonzalez, A., Xu, Z.Z., Washburne, A., Herfarth, H.H., Sartor, R.B.. et al. (2018) Guiding longitudinal sampling in IBD cohorts. Gut 67, 1743–1745 10.1136/gutjnl-2017-315352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tilg, H. and Kaser, A. (2011) Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Invest. 121, 2126–2132 10.1172/JCI58109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ley, R.E. (2010) Obesity and the human microbiome. Curr. Opin. Gastroenterol. 26, 5–11 10.1097/MOG.0b013e328333d751 [DOI] [PubMed] [Google Scholar]
- 98.Turnbaugh, P.J. and Gordon, J.I. (2009) The core gut microbiome, energy balance and obesity. J. Physiol. 587, 4153–4158 10.1113/jphysiol.2009.174136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sharma, S. and Tripathi, P. (2019) Gut microbiome and type 2 diabetes: where we are and where to go? J. Nutr. Biochem. 63, 101–108 10.1016/j.jnutbio.2018.10.003 [DOI] [PubMed] [Google Scholar]
- 100.Sanmiguel, C., Gupta, A. and Mayer, E.A. (2015) Gut microbiome and obesity: a plausible explanation for obesity. Curr. Obes. Rep. 4, 250–261 10.1007/s13679-015-0152-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F.. et al. (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60 10.1038/nature11450 [DOI] [PubMed] [Google Scholar]
- 102.Baothman, O.A., Zamzami, M.A., Taher, I., Abubaker, J. and Abu-Farha, M. (2016) The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis. 15, 108 10.1186/s12944-016-0278-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hughes, D.A., Bacigalupe, R., Wang, J., Rühlemann, M.C., Tito, R.Y., Falony, G.. et al. (2020) Genome-wide associations of human gut microbiome variation and implications for causal inference analyses. Nat. Microbiol. 5, 1079–1087 10.1038/s41564-020-0743-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bonder, M.J., Kurilshikov, A., Tigchelaar, E.F., Mujagic, Z., Imhann, F., Vila, A.V.. et al. (2016) The effect of host genetics on the gut microbiome. Nat. Genet. 48, 1407–1412 10.1038/ng.3663 [DOI] [PubMed] [Google Scholar]
- 105.Wang, J., Kurilshikov, A., Radjabzadeh, D., Turpin, W., Croitoru, K., Bonder, M.J.. et al. (2018) Meta-analysis of human genome-microbiome association studies: the mibiogen consortium initiative. Microbiome 6, 101 10.1186/s40168-018-0479-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blekhman, R., Goodrich, J.K., Huang, K., Sun, Q., Bukowski, R., Bell, J.T.. et al. (2015) Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 16, 191 10.1186/s13059-015-0759-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Turpin, W., Espin-Garcia, O., Xu, W., Silverberg, M.S., Kevans, D., Smith, M.I.. et al. (2016) Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 48, 1413–1417 10.1038/ng.3693 [DOI] [PubMed] [Google Scholar]
- 108.Goodrich, J.K., Davenport, E.R., Beaumont, M., Jackson, M.A., Knight, R., Ober, C.. et al. (2016) Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19, 731–743 10.1016/j.chom.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ryan, F.J., Ahern, A.M., Fitzgerald, R.S., Laserna-Mendieta, E.J., Power, E.M., Clooney, A.G.. et al. (2020) Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease. Nat. Commun. 11, 1512 10.1038/s41467-020-15342-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Venes, D.E. (1940) Taber’s Cyclopedic Medical Dictionary, 23 ed., F.A. Davis, Philadelphia [Google Scholar]
- 111.Iadanza, E., Fabbri, R., Bašić-ČiČak, D., Amedei, A. and Telalovic, J.H. (2020) Gut microbiota and artificial intelligence approaches: a scoping review. Health Technol. 10, 1343–1358 10.1007/s12553-020-00486-7 [DOI] [Google Scholar]
- 112.Bauermeister, A., Mannochio-Russo, H., Costa-Lotufo, L.V., Jarmusch, A.K. and Dorrestein, P.C. (2021) Mass spectrometry-based metabolomics in microbiome investigations. Nat. Rev. Microbiol. 1–18 10.1038/s41579-021-00621-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sanna, S., van Zuydam, N.R., Mahajan, A., Kurilshikov, A., Vich Vila, A., Võsa, U.. et al. (2019) Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 51, 600–605 10.1038/s41588-019-0350-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shore, S.A. and Cho, Y. (2016) Obesity and asthma: microbiome-metabolome interactions. Am. J. Respir. Cell Mol. Biol. 54, 609–617 10.1165/rcmb.2016-0052PS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jeffery, I.B., Das, A., O’Herlihy, E., Coughlan, S., Cisek, K., Moore, M.. et al. (2020) Differences in fecal microbiomes and metabolomes of people with vs without irritable bowel syndrome and bile acid malabsorption. Gastroenterology 158, 1016–1028.e8 10.1053/j.gastro.2019.11.301 [DOI] [PubMed] [Google Scholar]
- 116.Bauer, E. and Thiele, I. (2018) From metagenomic data to personalized in silico microbiotas: predicting dietary supplements for Crohn’s disease. npj Syst. Biol. Appl. 4, 1–9 10.1038/s41540-018-0063-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.De la Cuesta-Zuluaga, J., Mueller, N.T., Álvarez-Quintero, R., Velásquez-Mejía, E.P., Sierra, J.A., Corrales-Agudelo, V.. et al. (2019) Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometabolic disease risk factors. Nutrients 11, 51 10.3390/nu11010051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murugesan, S., Nirmalkar, K., Hoyo-Vadillo, C., García-Espitia, M., Ramírez-Sánchez, D. and GarcíaMena, J. (2018) Gut microbiome production of short-chain fatty acids and obesity in children. Eur. J. Clin. Microbiol. Infect. Dis. 37, 621–625 10.1007/s10096-017-3143-0 [DOI] [PubMed] [Google Scholar]
- 119.Shoaie, S., Ghaffari, P., Kovatcheva-Datchary, P., Mardinoglu, A., Sen, P., Pujos-Guillot, E.. et al. (2015) Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 22, 320–331 10.1016/j.cmet.2015.07.001 [DOI] [PubMed] [Google Scholar]
- 120.Shaffer, M., Thurimella, K., Quinn, K., Doenges, K., Zhang, X., Bokatzian, S.. et al. (2019) AMON: annotation of metabolite origins via networks to integrate microbiome and metabolome data. BMC Bioinformatics 20, 614 10.1186/s12859-019-3176-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reiman, D., Layden, B.T. and Dai, Y. (2021) MiMeNet: exploring microbiome-metabolome relationships using neural networks. PLoS Comput. Biol. 17, e1009021 10.1371/journal.pcbi.1009021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yin, X., Altman, T., Rutherford, E., West, K.A., Wu, Y., Choi, J.. et al. (2020) A comparative evaluation of tools to predict metabolite profiles from microbiome sequencing data. Front. Microbiol. 11, 595910 10.3389/fmicb.2020.595910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xavier, R.J. (2021) Translating the human microbiome: a path to improving health. Genome Med. 13, 78 10.1186/s13073-021-00896-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eetemadi, A., Rai, N., Pereira, B.M.P., Kim, M., Schmitz, H. and Tagkopoulos, I. (2020) The computational diet: a review of computational methods across diet, microbiome, and health. Front. Microbiol. 11, 393 10.3389/fmicb.2020.00393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sherwin, R.S., Fisher, M., Hendler, R. and Felig, P. (1976) Hyperglucagonemia and blood glucose regulation in normal, obese and diabetic subjects. N. Engl. J. Med. 294, 455–461 10.1056/NEJM197602262940901 [DOI] [PubMed] [Google Scholar]
- 126.Zeevi, D., Korem, T., Zmora, N., Israeli, D., Rothschild, D., Weinberger, A.. et al. (2015) Personalized nutrition by prediction of glycemic responses. Cell 163, 1079–1094 10.1016/j.cell.2015.11.001 [DOI] [PubMed] [Google Scholar]
- 127.Asnicar, F., Berry, S.E., Valdes, A.M., Nguyen, L.H., Piccinno, G., Drew, D.A.. et al. (2021) Microbiome connections with host metabolism and habitual diet from 1098 deeply phenotyped individuals. Nat. Med. 27, 321–332 10.1038/s41591-020-01183-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Korem, T., Zeevi, D., Zmora, N., Weissbrod, O., Bar, N., Lotan-Pompan, M.. et al. (2017) Bread affects clinical parameters and induces gut microbiome-associated personal glycemic responses. Cell Metab. 25, 1243–1253.e5 10.1016/j.cmet.2017.05.002 [DOI] [PubMed] [Google Scholar]
- 129.Marcos-Zambrano, L.J., Karaduzovic-Hadziabdic, K., Loncar Turukalo, T., Przymus, P., Trajkovik, V., Aasmets, O.. et al. (2021) Applications of machine learning in human microbiome studies: a review on feature selection, biomarker identification, disease prediction and treatment. Front. Microbiol. 12, 634511 10.3389/fmicb.2021.634511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Walter, J., Armet, A.M., Finlay, B.B. and Shanahan, F. (2020) Establishing or exaggerating causality for the gut microbiome: lessons from human microbiota-associated rodents. Cell 180, 221–232 10.1016/j.cell.2019.12.025 [DOI] [PubMed] [Google Scholar]