

ABSTRACT
For clinical application by dendritic cell (DC)-based cancer immunotherapy, a proper adjuvant system to elicit a strong anticancer immune response is needed. Here, we investigated the potential of chorismate mutase (TBCM, Rv1885c), a putative Mycobacterium tuberculosis (TB) virulence factor, as an immunoadjuvant in DC-based tumor immunotherapy. First, we found that TBCM functionally activated DCs by upregulating costimulatory molecules, increasing the secretion of proinflammatory cytokines, enhancing migration and inducing the Th1-type immune response in a dose-dependent manner via TLR4-mediated signaling. In addition, subcutaneous injection of TBCM-activated DCs loaded with cell lysates led to reduced tumor mass, enhanced mouse survival and lowered tumor incidence in lung carcinoma (LLC) cell-bearing mice. This is mainly mediated by functional cytotoxic T lymphocyte-mediated oncolytic activity and inhibition of cancer proliferation- and metastasis-related genes. Moreover, TBCM-induced DCs can also generate memory CD4 T cells and exert long-term tumor prevention effects. In conclusion, our findings suggest that TBCM (Rv1885c), a novel TLR4 agonist, could be used as an immunoadjuvant for DC-based cancer immunotherapy.
KEYWORDS: Mycobacterium tuberculosis, chorismate mutase (TBCM), TLR4 agonist, dendritic cells, cancer immunotherapy, adjuvant
Introduction
Dendritic cells (DCs) are professional antigen-presenting cells (APCs) capable of presenting tumor antigens to naïve, nonprimed T cells to activate the tumor antigen-specific immune response.1,2 DCs comprising functional heterogeneous subsets could have distinct phenotypes, immunogenic or tolerogenic, depending on their inflammatory cytokines within the local milieu and simulation strength via pathogen-associated molecular patterns (PAMPs).3–5 Immunogenic DCs can inhibit tumor progression via both proliferation and activation of functional tumor-specific T cells, such as type I helper T (Th1) cells and cytotoxic T lymphocytes (CTLs).6,7 In contrast, tolerogenic DCs can lead to even cancer progression.8,9 DC-based vaccines have been extensively investigated as a feasible approach for cancer immunotherapy to enhance tumor antigen-specific immune responses.10–12 However, most clinical outcomes of DC-based vaccines have been disappointing.13 One major challenge of its clinical application is the limitation of adjuvants currently used in clinical trials to elicit DC activation. Therefore, the development of more effective adjuvants to induce Th1-type responses is urgently needed for cancer immunotherapeutic strategies.14–16
Pattern recognition receptors (PRRs) can lead to both maturation and activation of APCs via recognition by pathogen-associated molecular patterns (PAMPs) from external pathogens or damage-associated molecular patterns (DAMPs) from damaged tissues.17 Therefore, major efforts have been made to find a novel proper PAMP as an immunoadjuvant capable of sufficiently activating DCs for cancer immunotherapy. Of these PRRs, a total of four types of pattern recognition receptors (PRRs) have been identified, including C-type lectin receptors (CLRs), RIG I-like receptors (RLRs), NOD-like receptors (NLRs), and Toll-like receptors (TLRs).18,19 Of these, TLR4 is a member of TLRs first found in humans that can recognize lipopolysaccharide (LPS), a component present in gram-negative bacteria,20 and significant efforts have also been focused on the potential of its ligands as immunoadjuvants for DC-based vaccines.
As cell wall components and proteins of Mycobacterium spp. can lead to Th1 skewing immune responses,21–23 antitumor immunotherapeutic effects based on Mycobacterium spp. have been widely studied.24 Notably, Mycobacterium bovis Bacillus Calmette-Guérin (BCG) therapy for nonmuscle invasive bladder cancer (NMIBC) is one of the few examples of successful immunotherapy in clinical use.25 Furthermore, heat-killed (HK) Mycobacterium vaccae,26 Mycobacterium indicus pranii (MIP)27 and Mycobacterium paragordonae (Mpg)28 have also been demonstrated to exert anticancer effects in both in vitro and in vivo models. In addition to whole cell-based approaches, two different components of the BCG vaccine, heparin-binding hemagglutinin protein (HBHA)29 and heat shock protein X (HspX),30 have been reported to activate DCs mainly via TLR4 pathways, and DC-based vaccines using them as adjuvants have been proven to induce effective anticancer effects.
Chorismate mutase (CM) plays a central role in the shikimate pathway for the biosynthesis of aromatic amino acids in bacteria, fungi and higher plants, including phenylalanine, tyrosine, and tryptophan, by catalyzing the conversion of chorismate to prephenate.31 There are two putative genes for CM: Rv1885c and Rv0948c. Of these, the secretory form, TBCM (encoded by Rv1885c), is assumed to play a key role in the pathogenesis of tuberculosis,32 and it has low sequence homology among known CM. Therefore, it has gained increased attention as an interesting target for the discovery of antitubercular agents. However, very little is known about the pathogenic role of TBCM in TB pathogenesis. During research regarding its pathogenic role in TB infections, we unexpectedly found that TBCM can induce DC maturation and activation in BMDCs in a TLR4-dependent manner (Figures 1 and 8). Therefore, we hypothesized that TBCM could be effectively used as an immunoadjuvant for DC-based cancer immunotherapy.
Figure 1.
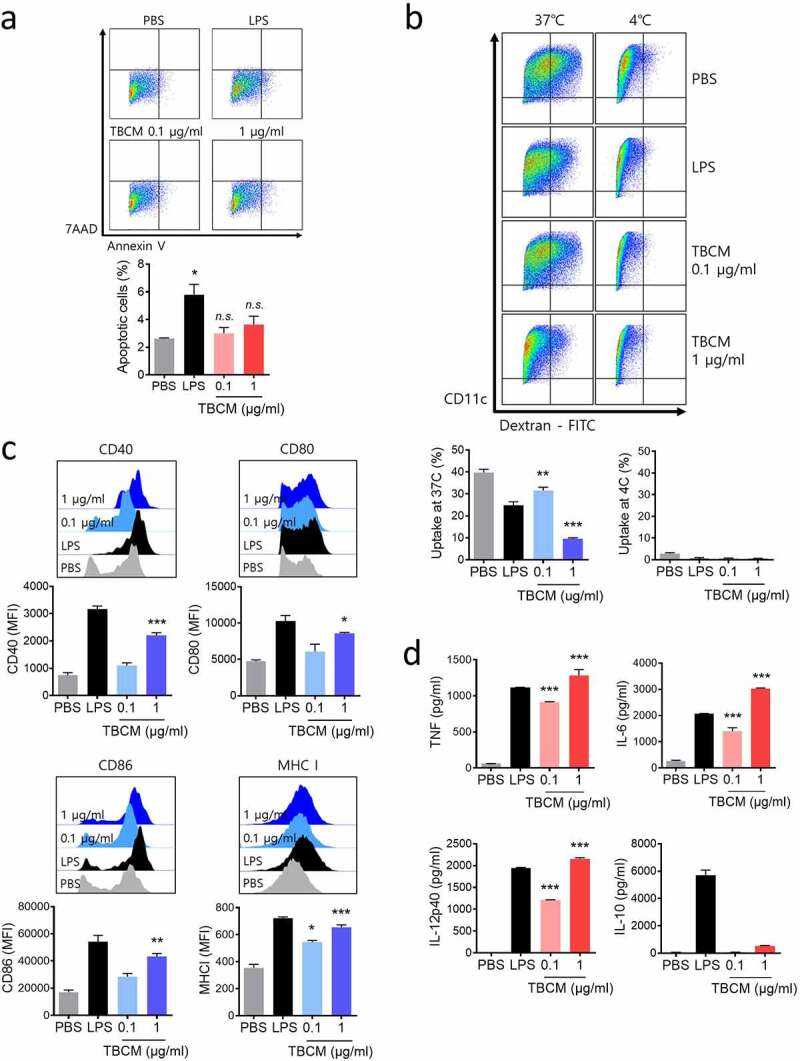
TBCM induces phenotypic and functional maturation in DCs. (a) Viability of BMDCs treated with TBCM proteins. BMDCs were incubated with TBCM (0.1 or 1 μg/ml) or LPS (0.1 μg/ml) for 24 h, and cells were then assessed by apoptosis assay. (b) DCs were activated with TBCM or LPS for 24 h (see A). The activated DCs were incubated with dextran-FITC at 37°C or 4°C for 30 min and analyzed for dextran uptake by flow cytometry. (c) BMDCs were stimulated for 24 h with TBCM or LPS (see A) and analyzed for the expression of surface markers by flow cytometry. (d) Cytokines in the culture supernatant were measured by ELISA. Significant differences (*p 0.05, **p 0.01, and ***p 0.001) among the different groups are shown in the related figures, and the data are presented as the means s.e.m. of three independent experiments.
Figure 2.
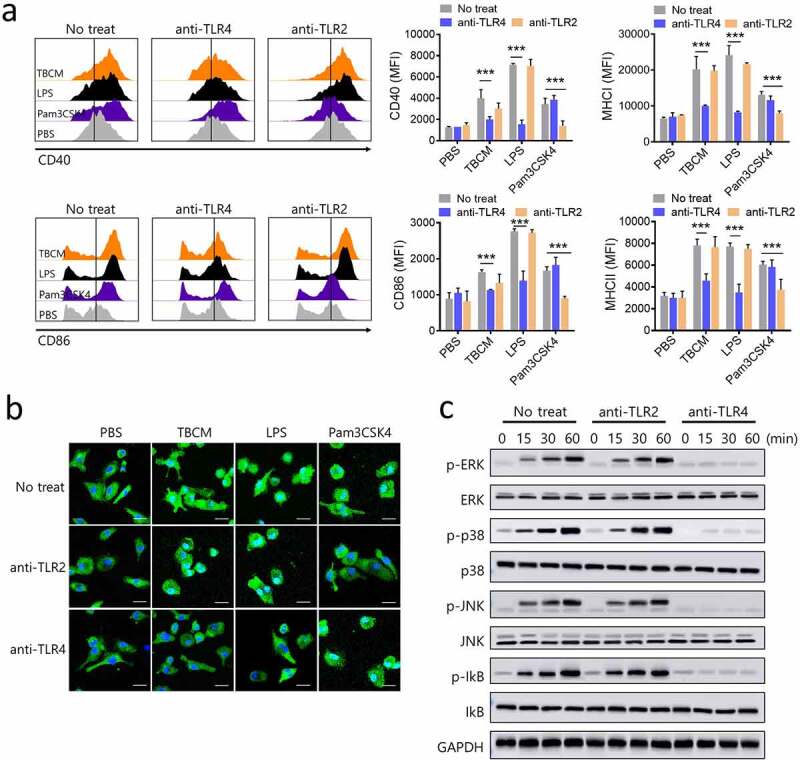
TBCM induces DC activation via activation of MAPKs and NF-kB in DCs in a TLR4–dependent manner. BMDCs were incubated with anti-TLR2 IgG or anti-TLR4 IgG prior to incubation with TBCM (1 μg/ml), LPS (0.1 μg/ml) or Pam3CSK4 (0.1 μg/ml). (a) After 24 h incubation with TBCM, LPS or Pam3CSK4, the surface expression of CD40, CD86, MHC class I and MHC class II was assessed by flow cytometry. (b) The effects of TBCM on the cellular localization of the p65 subunit of NF-kB in DCs were assessed. After 12 h of stimulation with TBCM, LPS or Pam3CSK4, the intracellular localization of NF-kB p65 was determined by immunofluorescence. (c) DCs were harvested at the indicated time points, and then the DC lysates were subjected to SDS-polyacrylamide gel and an immunoblot analysis was conducted. Significant differences (*p 0.05, **p 0.01, and ***p 0.001) among the different groups are shown in the related figures, and the data are presented as the means s.e.m. of three independent experiments.
Figure 3.
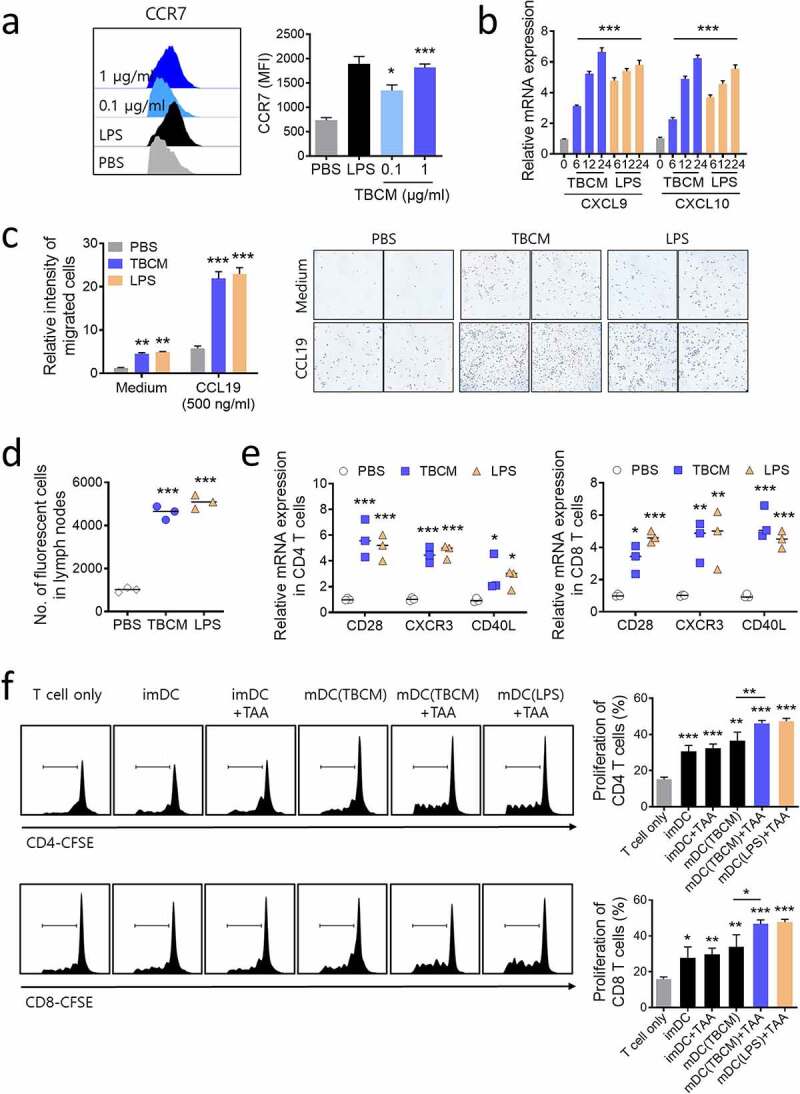
TBCM enhances the migration of DCs in vitro and in vivo, and TBCM-induced DCs stimulate T cell proliferation. BMDCs were incubated with TBCM (0.1 or 1 μg/ml) or LPS (0.1 μg/ml). (a) After 24 h incubation, the surface expression of CCR7 was determined by flow cytometry. (b) DCs were harvested at the indicated time points. mRNA extracted from DCs was assessed by RT–qPCR. (c) DCs were subjected to an in vitro Transwell chemotaxis assay in which movement toward media alone or media containing CCL19 (500 ng/ml) was measured. (d) After 24 h incubation, CFSE-labeled DCs were injected into the mouse footpad, and CFSE-positive cells were detected in the inguinal lymph nodes by flow cytometry 72 h after the injection. (e) One week after the footpad injection of TBCM-treated DCs, mRNA of CD4+ and CD8+ T cells isolated from inguinal lymph nodes was assessed by RT–qPCR. (f) BMDCs were incubated with LLC lysates (tumor-associated antigens; TAAs) at a ratio of 3:1 tumor cell equivalents in the presence or absence of TBCM (1 μg/ml) or lipopolysaccharide (0.1 μg/ml) for 24 h. CD4+ and CD8+ T cells isolated from mouse spleen were stained with CFSE and cocultured with TAA-, TBCM- or LPS-treated DCs (T cell:DC = 10:1) for 96 h. Thereafter, T cell proliferation was assessed by flow cytometry. Significant differences (*p 0.05, **p 0.01, and ***p 0.001) among the different groups are shown in the related figures, and the data are presented as the means s.e.m. of three or four independent experiments.
Figure 4.
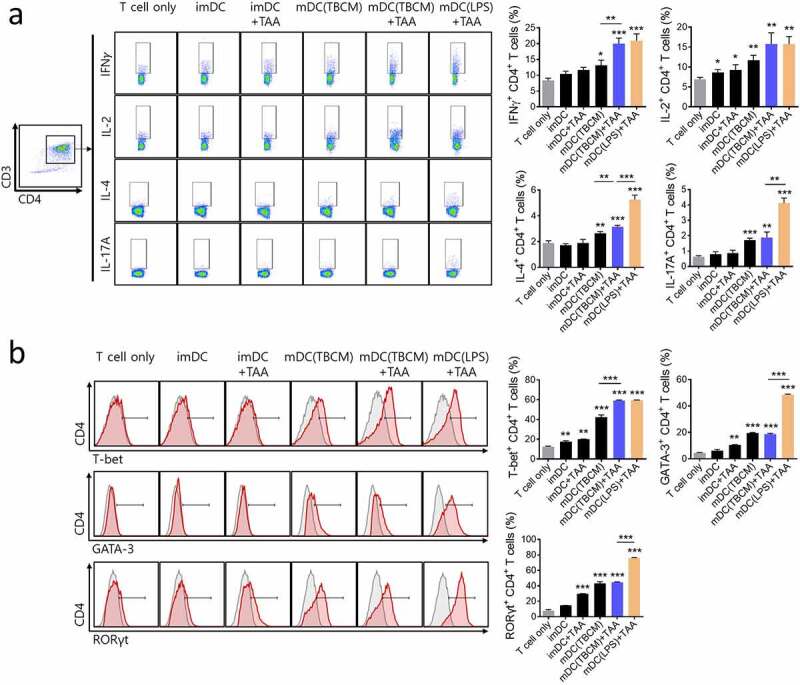
TBCM-induced DCs stimulate enhanced polarization of Th1 cells. BMDCs were incubated with TBCM (0.1 or 1 μg/ml) or LPS (0.1 μg/ml) in the presence or absence of LLC lysates (tumor-associated antigens; TAAs) for 24 h. DCs were then cocultured with CD4+ T cells (DC:T cell = 1:10) isolated from mouse spleen for 72 h. (a) Intracellular cytokine production in CD4+ T cells was assessed by flow cytometry. (b) On Day 3 of coculturing, the expression of Th1-specific regulator, T-bet, Th2-development transcription factor, GATA-3, and Th17-development regulator RORγt in CD4+ T cells was assessed by flow cytometry. Significant differences (*p 0.05, **p 0.01, and ***p 0.001) among the different groups are shown in the related figures, and the data are presented as the means s.e.m. of four independent experiments.
Figure 5.
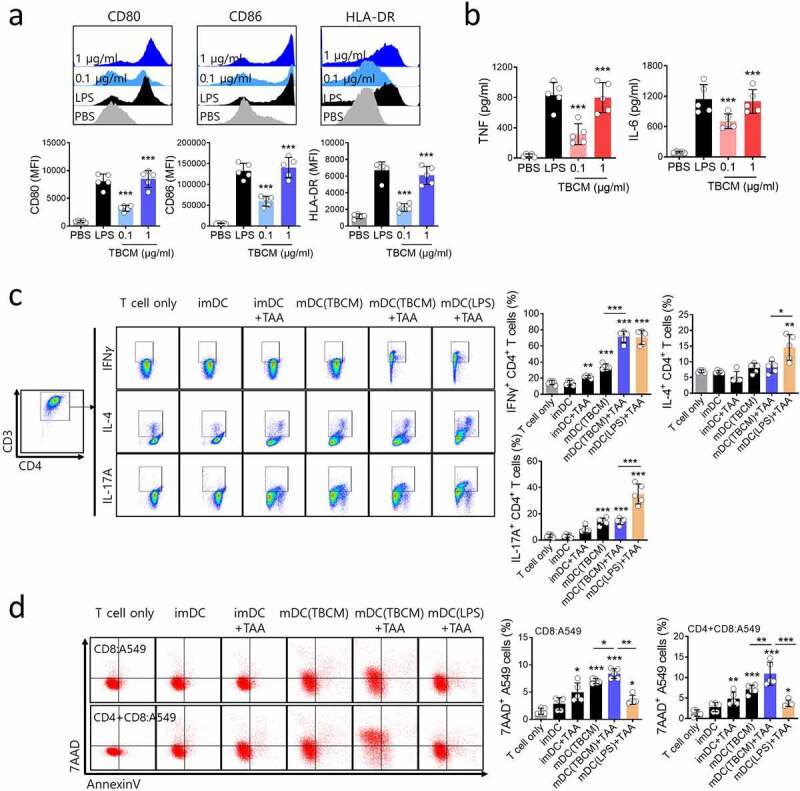
TBCM-induced human DCs enhances antigen-specific T cell cytotoxic response against human cancer cell. (a) Human DCs were activated for 24 h with TBCM (1 μg/ml) or LPS (0.1 μg/ml) and analyzed for the surface expression marker by flow cytometry. (b) Cytokine levels in the culture supernatant were measured by ELISA. (c) A549 cell lysates (tumor-associated antigens, TAAs)-, TBCM- or LPS-activated human DCs were cocultured with CD4+ T cells (DC:T cell = 1:10) isolated from PBMCs for 72 h. Then, intracellular cytokine production in CD4+ T cells were assessed by flow cytometry. (d) TAAs-, TBCM- or LPS-activated human DCs were cocultured with CD8+ T cells (DC:T cell = 1:10) in the presence or absence of CD4+ T cells for five days. Thereafter, the cells were cocultured with CFSE-labeled A549 cells (E:T = 5:1) for 48 h, and antigen-specific cytotoxicity of T cells were measured by cell apoptosis assay. Significant differences (*p 0.05, **p 0.01, ***p 0.001) among the different groups are shown in the related figures, and the data are presented as the means s.e.m. of five independent experiments.
Figure 6.
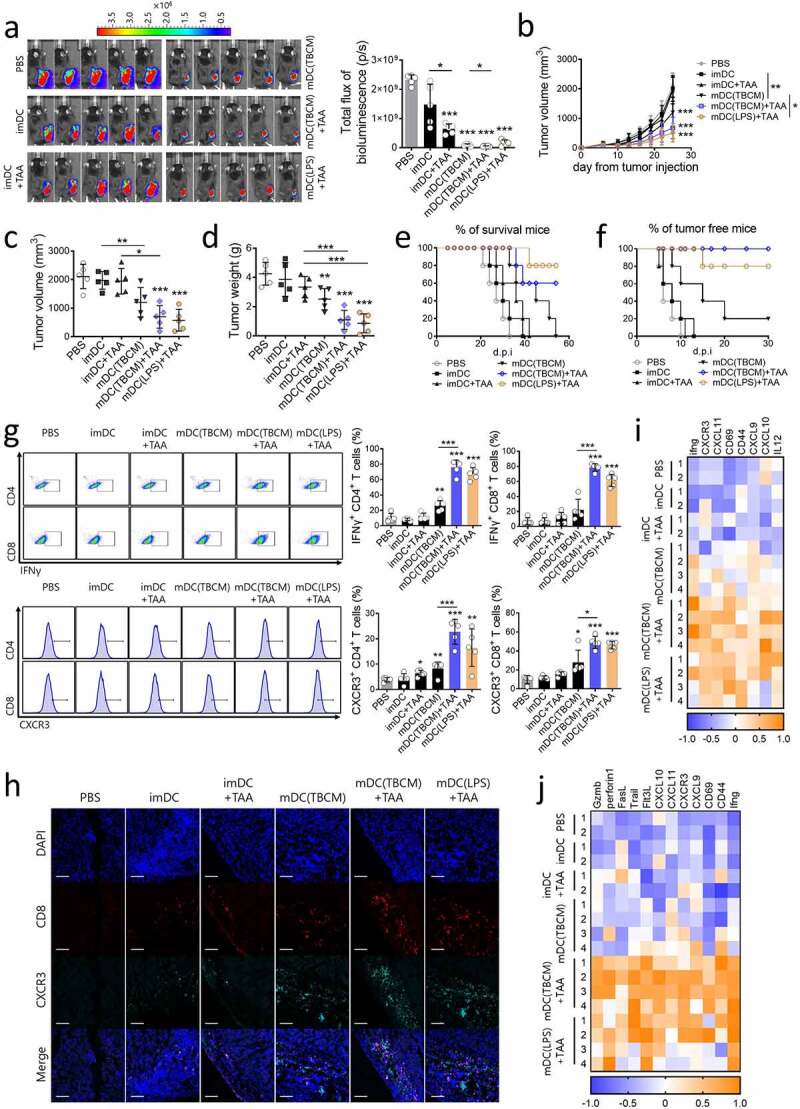
TBCM-induced DCs exert therapeutic effects in lung cancer via induction functional CD4 and CD8 T cells in vivo. (a) C57BL/6 mice were subcutaneously injected with LLC cancer cells and vaccinated with DCs via footpad injection twice at intervals of one week. Representative pictures show luciferase-positive LLC cancer cells on Day 25 after cancer cell injection. (b) Growth curve shows tumor volume as the mean SD at the indicated time points. (c,d) Tumor volume and weight measured on Day 25 after the cancer cell injection. (e) C57BL/6 mice were treated with LLC and DCs (see a) and observed for survival. (f) C57BL/6 mice were subcutaneously injected with LLC cancer cells seven days after the last DC injection. Tumor incidence is shown. (g) C57BL/6 mice were treated with LLC and DCs (see a). On Day 25 after cancer cell injection, tumor-infiltrating DCs and lymphocytes were assessed by flow cytometry. (h) CXCR3+ CD8+ T cells in paraffin-embedded tumor tissue were detected by immunofluorescence staining. (i,j) The transcription level of mRNA in tumor-draining lymph nodes and tumor-infiltrating lymphocytes was quantified by RT–qPCR. Significant differences (*p 0.05, **p 0.01, and ***p 0.001) among the different groups are shown in the related figures, and the data are presented as the means s.e.m. of five independent experiments.
Figure 7.
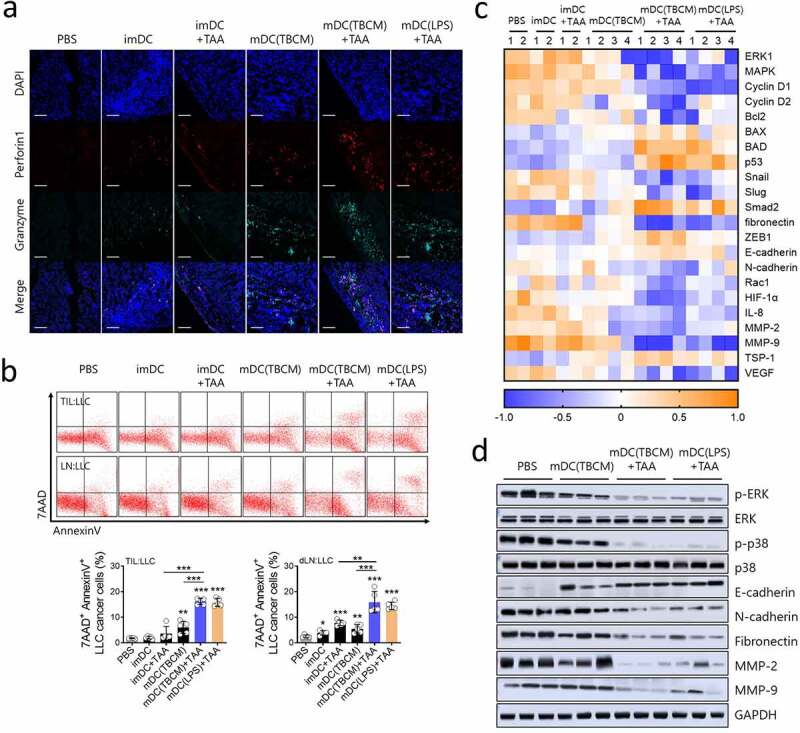
TBCM-induced DCs suppress tumor progression through recruitment of functional CTLs and inhibit expression of metastasis related genes in the tumor microenvironment. C57BL/6 mice were subcutaneously injected with LLC cancer cells and vaccinated with DCs via footpad injection twice at intervals of one week. (a) Cytolytic protein perforin-1 and granzyme in tumor tissue were observed by immunofluorescence staining. (b) On Day 25 after the cancer cell injection, tumor-infiltrating lymphocytes and tumor-draining lymph nodes were cocultured with CFSE-labeled LLC cancer cells (E:T = 5:1), and cytotoxicity against LLC cells was assessed by flow cytometry. (c) The transcription level of mRNA in primary tumor cells was quantified by RT–qPCR. (d) Protein expression of ERK, epithelial-mesenchymal transition (EMT)-related proteins, and matrix metalloproteinases MMP-2 and MMP-9 in primary tumor cells was assessed by immunoblotting assay. Significant differences (*p 0.05, **p 0.01, and ***p 0.001) among the different groups are shown in the related figures, and the data are presented as the means s.e.m. of five independent experiments.
Figure 8.
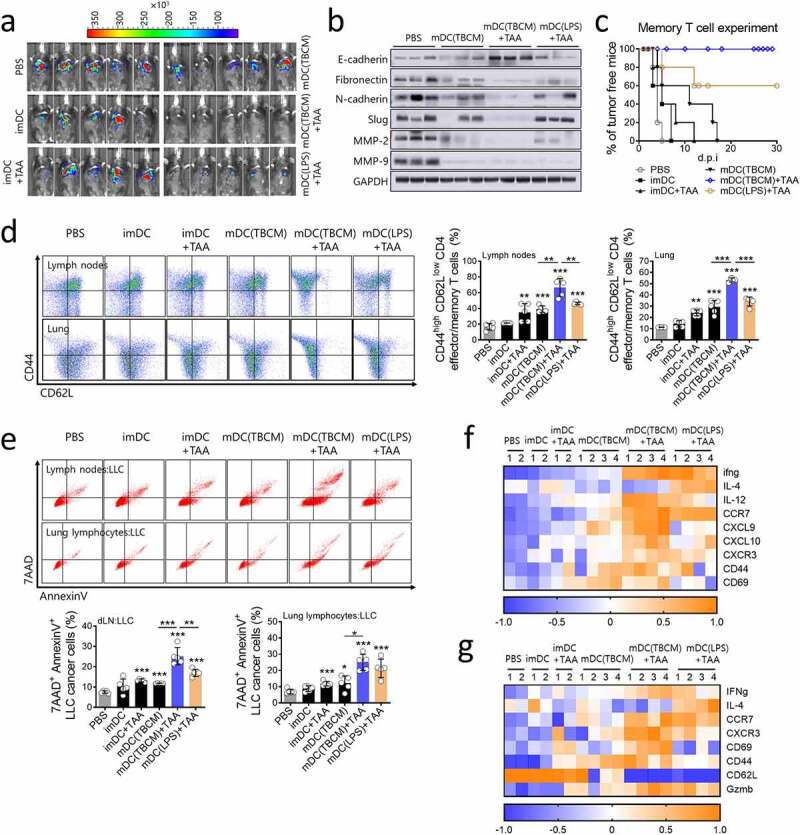
TBCM-induced DCs generate memory T cells and exert sustained tumor prevention effects. (a) C57BL/6 mice were vaccinated with BMDCs twice at intervals of one week. Seven weeks after the last DC injection, LLC cancer cells were injected intravenously. Seven days after the cancer cell injection, mice were observed for tumor size measurement using an IVIS imaging system 100 and sacrificed for tissue analysis. Representative pictures show luciferase-positive LLC cancer cells one week after cancer cell injection. (b) Protein expression of EMT-related proteins and matrix metalloproteinases MMP-2 and MMP-9 in lung tissue was assessed by immunoblotting assay. (c) After mice were vaccinated with DCs twice at intervals of one week, mice were subcutaneously injected with LLC cancer cells and observed for tumor incidence. (d) One week after cancer cell injection (see a), the population of CD44high CD62Llow CD4+ T cells in the lymph nodes and lung was measured by flow cytometry. (e) One week after cancer cell injection (see a), lung lymphocytes and lymph nodes were cocultured with CFSE-labeled LLC cancer cells (E:T = 5:1), and cytotoxicity against LLC cells was assessed by flow cytometry. (f,g) The transcription level of mRNA in lymph nodes and lung lymphocytes was quantified by RT–qPCR. Significant differences (*p 0.05, **p 0.01, and ***p 0.001) among the different groups are shown in the related figures, and the data are presented as the means s.e.m. of five independent experiments.
In this study, we investigated the effects of TBCM on the maturation and activation of mouse and human dendritic cells in vitro. Furthermore, we also investigated the role of TBCM as an immunoadjuvant for DC-based cancer immunotherapy. Finally, we investigated whether TBCM can induce cancer prevention via enhanced generation of memory CD4 T cells in an in vivo model.
Materials and methods
Mice
Six- to seven-week-old specific pathogen-free (SPF) female C57BL/6 mice were purchased from Orient Bio and maintained under SPF conditions in accordance with the animal care guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University (Approval No. SNU-200210-2) and Institutional Biosafety Committee of Seoul National University (Approval No. SNUIBC-R200302-1).
In vivo experiments
Tumor cell lysates (tumor-associated antigens; TAAs) were prepared by five cycles of liquid nitrogen and 37°C water bath freeze-thawing using LLC cancer cells (1 × 107 cells/ml in PBS). Cellular debris was removed by centrifugation, and the lysate solution was passed through a 0.2 μm filter and stored at – 80°C. BMDCs were incubated with LLC lysates at a ratio of 3:1 tumor cell equivalents in the presence or absence of TBCM (1 μg/ml) or lipopolysaccharide (0.1 μg/ml; Escherichia coli serotype 0111:B4, Sigma, USA) for 24 h at 37°C in 5% CO2.
For the tumor treatment and survival experiments, C57BL/6 mice were subcutaneously injected with LLC, B16F10 or EO771 cancer cells (1 × 106 cells/mouse, respectively) and vaccinated with BMDCs (1 × 106 cells/50 μl PBS) via footpad injection twice at intervals of one week. The tumor size was measured once every 2 or 3 days using calipers and calculated following the formula: tumor volume (mm3) = (longest diameter shortest diameter2)/2. The mice were observed until the tumor diameter was over 3 mm. In the tumor prevention experiment, mice were subcutaneously injected with LLC cancer cells (1 × 106 cells/mouse) seven days after the last DC injection and observed for tumor incidence. For the memory T cell experiment, mice were vaccinated with BMDCs twice at intervals of one week. Seven weeks after the last DC injection, LLC cancer cells (1 × 106 cells/100 μl PBS) were injected intravenously. Seven days after the cancer cell injection, mice were observed for tumor size measurement using an IVIS imaging system 100 (Xenogen, USA).
Results
TBCM induces phenotypic and functional maturation in DCs.
Prior to studying the immunological functions in DCs, we assessed whether TBCM affects the viability of mouse bone marrow-derived dendritic cells (BMDCs), same as previous study.33 DC viability was not affected after incubation with TBCM at a concentration up to 1 μg/ml (Figure 1a). DC maturation is essential for DC functions in antigen-presenting cells (APCs), demonstrated by downregulated endocytosis and increased cytokine levels. TBCM-treated BMDCs exhibited a significantly decreased population of dextran+ CD11c+ cells compared with untreated BMDCs (Figure 1b). In addition, the surface expression level of co-stimulatory molecules CD40, CD80, CD86 and MHC class I was upregulated in DCs following TBCM stimulation in a dose-dependent manner (Figure 1c). Co-expression of MHCII and CD80, CD83 or CD86 were also observed in TBCM-treated DCs (Supplementary Figure S1A). Furthermore, TBCM induced BMDCs to secrete TNF-α, IL-6, IL-10 and IL-12p40 in a dose-dependent manner (Figure 1d). Meanwhile, we assessed LPS contamination of TBCM protein and found that the effects of TBCM in DCs were not as a result of endotoxin contamination from E. coli (Supplementary Figure S2). Collectively, these results demonstrate that TBCM promotes DC activation through phenotypic and functional maturation.
TBCM induces DC activation via activation of MAPKs and NF-kB in DCs in a TLR4-dependent manner.
Toll-like receptors (TLRs) play crucial roles in the innate immune system by recognizing pathogen-associated molecular pattern derived from various microbes. The role of TLRs in APCs is particularly important.34 Thus, we examined the TLR-dependency for DC activation and maturation. To identify the TLRs on DCs that interact with TBCM, DCs were incubated with anti-TLR2 IgG or anti-TLR4 IgG and then treated with TBCM. The expression of surface co-stimulatory molecules and secretion of pro-inflammatory cytokine were significantly induced in untreated DCs and DCs blocked with TLR2 after TBCM treatment. In contrast, these effects were significantly decreased in DCs blocked with TLR4, indicating that TBCM functions as a TLR4 agonist in DCs (Figure 2a and Supplementary Figure S3B). As DC maturation mediated by mycobacterial antigens involves mitogen-activated protein kinases (MAPKs) and nuclear factor (NF)-kB signaling pathways,35,36 we examined whether TBCM activates MAPKs and NF-kB in DCs. TBCM induced nuclear translocation of NF-kB p65 from the cytosol in untreated DCs, and DCs blocked with TLR2 but not in DCs blocked with TLR4 (Figure 2b). Additionally, TBCM-induced phosphorylation of p38 MAPK, including p38, ERK and JNK, was significantly decreased after TLR4 blockade (Figure 2c and Supplementary Figure S3A). In addition to these results, TBCM induces the activation of human monocytic THP-1 cells via interaction with TLR4 (Supplementary Figure S4). These results indicated that TBCM induces DC maturation in a TLR4-dependent manner, leading to increased expression of cell surface molecules and proinflammatory cytokines.
TBCM enhances the migration of DCs in vitro and in vivo, and TBCM-induced DCs stimulate T cell proliferation.
As CCR7 is an important factor for the migration of DCs to lymphoid organs,37,38 the increased levels of CCR7 surface expression and mRNA expression of CXCL9 and CXCL10 in TBCM-treated DCs were examined (Figure 3a,b). We then performed an in vitro Transwell migration assay to analyze the DC migratory capacity in response to chemokine ligand 19 (CCL19), which is the ligand for CCR7. Increased migratory capacity of DCs was observed in the TBCM-treated DCs, which is consistent with the upregulation of CCR7 expression (Figure 3c). Furthermore, we found that the number of CFSE+ DCs in the inguinal lymph nodes was significantly increased in the TBCM-treated group compared with the untreated group via an in vivo migration test (Figure 3d). Additionally, increased mRNA levels of CXCR3, CD40L and CD28 in CD4+ and CD8+ T cells were induced by TBCM-treated DC injection, indicating that TBCM-treated DCs producing CXCL9 and CXCL10 may interact with CD4+ and CD8+ T cells in the lymph nodes (Figure 3e).
Mature DCs interact with T cells and cross-present antigens to T cells.39 Thus, we examined whether TBCM-mediated DC stimulates T cell proliferation. CFSE-labeled CD4+ and CD8+ T cells were cocultured with BMDCs that were incubated with LLC lysates in the presence of TBCM. These T cells displayed increased proliferation compared with T cells cocultured with BMDCs that were incubated with LLC lysates or with TBCM (Figure 3f). Compared with CD4+ and CD8+ T cells cocultured with DCs that were treated with LLC lysates, increased levels of IL-2 and IFN-γ were observed as a consequence of the priming of T cells by DCs that were incubated with LLC lysates in the presence of TBCM (Supplementary Figure S5A). These observations demonstrated that TBCM elicits optimal DC activation, resulting in the proliferation of CD4+ and CD8+ T cells.
TBCM-induced DCs stimulate enhanced polarization of Th1 cells.
As TBCM increased the expression of DC maturation markers involved in the antigen presentation to T cells, we assessed the ability of TBCM-treated DC to stimulate T cell cytokine production and polarization. TBCM-treated DCs increased the secretion of Th1-type cytokines IFN-γ and IL-2 in CD4 T cells; however, the Th2- and Th17-type cytokines IL-4 and IL-17A did not (Figure 4a and Supplementary Figure S5B). Additionally, TBCM-treated DCs slightly increased the cytokine levels of IFN-γ and IL-2 in CD8+ T cells (Supplementary Figure S5C). When CD4+ T cells were cocultured with BMDCs stimulated with TBCM, the expression of Th1-specific regulator T-bet was elevated, similar to that observed in BMDCs treated with LPS. However, upon TBCM stimulation, the expression of GATA-binding protein 3 (GATA-3), that is critical for Th2 cell development, and RAR-related orphan receptor gamma t (RORγt), the Th17 development regulator, was not increased in DCs compared with those upon LPS treatment (Figure 4b). These results suggested that DCs activated by TBCM differentiate CD4+ T cells toward Th1 immunity.
TBCM-induced human DCs enhances antigen-specific T cell cytotoxic response against human cancer cell.
Next, we assessed the effects of TBCM on human DCs. In the same way as TBCM induces mouse BMDC maturation, TBCM induces phenotypic and functional maturation in human DCs in a dose-dependent manner (Figure 5a,b). Also, TBCM-activated DCs loaded with A549 lysates (TAAs) stimulate enhanced polarization of Th1 CD4+ T cells, but not Th2 or Th17 cells (Figure 5c). Furthermore, TBCM-activated DCs loaded with TAAs stimulate generation of CD8+ T cells capable of killing A549 cancer cells (Figure 5d). Therefore, these results indicated that TBCM-activated DCs have enhanced ability to prime and activate antigen-specific T cell cytotoxic response against human cancer cells.
TBCM-induced DCs exert therapeutic effects in lung cancer via induction of functional CD4 and CD8 T cells in vivo.
We next assessed the anticancer effects of mature DCs activated by TBCM as a DC vaccine. Mice were subcutaneously injected with LLC cancer cells and vaccinated with TBCM-treated DCs via footpad injection twice at intervals of one week. TBCM- or LPS-treated mature DCs loaded with tumor-associated antigens (TAAs) significantly inhibited tumor progression and long-term survival compared with all other groups (Figure 6a–e). Additionally, inflammatory cytokine production in serum and the cytokine secretion response of the spleen against LLC cells were significantly enhanced in the groups treated with TBCM- or LPS-activated DCs loaded with TAAs (Supplementary Figure S6). Furthermore, all of the mice vaccinated with TBCM-activated DCs loaded with TAAs remained tumor-free for 30 days, indicating that TBCM-activated DC vaccines could prevent tumorigenesis (Figure 6f). Then, we investigated whether the antitumor effect in the treatment model was correlated with the T cell responses induced by TBCM-activated DCs loaded with the TAA vaccine. Infiltration of IFNγ-releasing CD4+ and CD8+ T cells and IL-12-releasing DCs in the tumor microenvironment was noticeably induced after administration of TBCM- or LPS-activated DCs loaded with TAAs compared with all other groups (Figure 6g and Supplementary Figure S6). Furthermore, recent evidence suggests that the dominant chemokines for recruitment of effector CD8+ T cells are those that engage the chemokine receptor CXCR3.40 It has been reported that the CXCR3 ligands CXCL9 and CXCL10 are not expressed in tumors lacking a CD8+ T cell infiltrate.41 TBCM-activated DCs loaded with the TAA vaccine increased the infiltration of CXCR3+ CD8+ T cells into tumor tissue, which might kill tumor cells (Figure 6h). These trends were also observed in B16F10 and EO771 tumor-bearing mouse model (Supplementary Figure S7,8). Additionally, as the transcription of mRNA in both dLNs and TILs tended to be similar, most effector T cells observed in tumors may originate from lymph nodes where TBCM-activated DCs injected through footpad injection had reached (Figure 6i,j). Therefore, these results demonstrated that administration of TBCM-activated DCs loaded with TAAs effectively induced the infiltration of effector T cells capable of killing tumor cells into tumor tissue.
TBCM-induced DCs suppress tumor progression through recruitment of functional CTLs and inhibit the expression of metastasis-related genes in the tumor microenvironment.
As TBCM-activated DCs loaded with TAAs induced cell-mediated immune responses in the tumor microenvironment and lymph nodes, phenotypic differences in tumors were observed. The pore-forming molecule perforin-1 and the proapoptotic protease granzyme B were induced by the administration of TBCM-activated DCs loaded with the TAA vaccine, indicating that the cytolytic immune response of CTLs induced by TBCM-activated DCs loaded with TAAs can lead to granule exocytosis of cancer cells (Figure 7a). Additionally, 7AAD+ Annexin V+ LLC cancer cells were significantly increased by the TILs and tumor-draining lymph nodes induced by TBCM-activated DCs loaded with the TAA vaccine (Figure 7b). The transcription levels related to cell proliferation, apoptosis and epithelial mesenchymal transition (EMT) were significantly decreased in response to TBCM-activated DCs loaded with TAAs (Figure 7c). Additionally, TBCM-activated DCs loaded with the TAA vaccine suppressed the protein expression of the ERK/p38 signaling pathway, EMT factors N-cadherin and Fibronectin, and matrix metalloproteinases MMP-2 and MMP-9 in primary tumor cells (Figure 7d). Together, these results indicated that TBCM-induced DCs suppress tumor progression through recruitment of functional CTLs and inhibit the expression of proliferation- or metastasis-related genes in the tumor microenvironment.
TBCM-induced DCs generate memory T cells and exert sustained prevention effects.
To investigate whether TBCM-activated DCs loaded with the TAA vaccine generate memory T cells, cancer cells were intravenously injected seven weeks after DC vaccination. Seven days after the cancer cell injection, tumor prevention effects were the most pronounced in TBCM-activated DCs loaded with the TAA vaccine (Figure 8a). The protein expression levels of EMT factors and MMPs in lung tissue were also suppressed in TBCM-activated DCs loaded with TAAs compared with all other groups (Figure 8b). Of note, TBCM-activated DCs loaded with the TAA vaccine had a greater sustained tumor prevention effects than LPS-activated DCs loaded with the TAA vaccine (Figure 8c). Importantly, an increased population of effector/memory CD4+ T cells in the lung and lymph nodes and induced lymphocytes capable of killing cancer cells were observed in response to TBCM-activated DCs loaded with TAAs (Figure 8d,e and Supplementary Figure S9). Additionally, TBCM-activated DCs loaded with TAAs induced immune cell activation after cancer cell injection (Figure 8f,g). These results indicated that TBCM-induced DCs exert sustained tumor prevention effects via the generation of memory T cells.
Discussion
In the present study, we sought to examine whether TBCM, a putative secreted pathogenic factor of tuberculosis, can act as an immunoadjuvant effectively used for DC-based cancer immunotherapy. First, we found that TBCM treatment led to increased expression of the costimulatory molecules CD40, CD80 and CD86, as well as MHC class I (Figure 1c), enhanced the production of proinflammatory cytokines such as TNF-a, IL-6, and IL-12 (Figure 1d) and reduced endocytic capacity at a concentration of 1 μg/ml in DCs (Figure 1a). Of note, production of IL-10, an immunosuppressive cytokine, was rarely induced by TBCM treatment compared with LPS treatment (Figure 1d), suggesting the merit of the former in differentiation into immunogenic DCs. Our Ab blocking test also proved that the maturation of DCs by TBCM is dependent on the MAPK and NF-kB pathways mediated by TLR4 but not TLR2 (Figure 2). These results strongly demonstrate that TBCM can act as a novel TLR4 agonist capable of DC maturation and activation.
To further prove the adjuvant effect of TBCM, we tested the DC migration capacity of TBCM. Our data indicated that TBCM treatment led to increased levels of CCR7 expression and mRNA expression of CXCL9 and CXCL10 in DCs (Figure 3b), enhanced DC migratory capacity against CCL19 in an in vitro Transwell migration assay (Figure 3c) and increased CFSE+ DCs in the draining lymph nodes in an in vivo test (Figure 3d), suggesting that TBCM can enhance the migration capacity of DCs in vitro and in vivo. Moreover, our ex vivo coculture tests showed that TBCM-treated DCs pulsed with TAAs also led to strong proliferation of CD4+ and CD8+ T cells (Figure 3f) and increased levels of IL-2 and IFN-γ (Supplementary Figure S5A), suggesting a positive role of TBCM in the optimal induction of cancer-specific T cell responses.
In addition, we also found that TBCM-treated DCs induced enhanced production of the Th1-type cytokines IFN-γ and IL-2 but not the Th2- and Th17-type cytokines IL-4 and IL-17A (Figure 4a) and enhanced production of Th1-specific regulator, T-bet, but not Th2 and Th17 specific regulator, GATA-3 and RORγt in CD4 T cells (Figure 4b), suggesting that the adjuvant activity of TBCM can induce a Th1-type immune response. Of note, LPS-treated DCs induced Th2- or Th17-type cytokine and transcription factor (GATA-3 and RORγt) as well as Th1-type cytokine and T bet expression (Figure 4b). The underlying mechanism regarding the disparity between TBCM and LPS in inducing the immune response should be investigated in future studies.
Our findings also showed that subcutaneous injection of TBCM-activated DCs loaded with TAA led to reduced tumor mass, enhanced mouse survival and lowered tumor incidence in lung carcinoma (LLC) cell-bearing mice (Figure 6a-f). We also found that the anticancer effect of TBCM was mainly due to the functional CXCR3+ CD8+ T cell-mediated CTL response infiltrating the tumor microenvironment (Figure 6g,h) and inhibiting cancer proliferation- and metastasis-related genes such as ERK, epithelial-mesenchymal transition (EMT)-related proteins, and matrix metalloproteinases MMP-2 and MMP-9 (Figure 7d)
It has been reported that memory CD4 T cells can play a pivotal role in anticancer immune responses.42–44 Our cancer prevention mouse model using intravenously injected LLC cells showed that TBCM-activated DCs loaded with TAAs had a greater preventive effect than LPS-activated DCs loaded with the TAA vaccine (Figure 8), which can potentially be by enhanced generation of CD44high CD62Llow CD4+ T cells in lung and lymph nodes (Figure 8d), further supporting the feasibility of TBCM as an immunoadjuvant in cancer prevention as well as cancer treatment.
Several Mycobacterium tuberculosis-derived proteins showed immunostimulatory properties as TLR4-mediated adjuvants or therapeutic effects in tumor-bearing mice.30,33,45 Among them, TBCM (Rv1885c) has several merits. For example, Rv1917c strongly induces secretion of IL-10 and Th2 immunity which favors Mtb infection.46 In contrast, TBCM activates polarization of Th1 cells, but not Th2 and Th17 cells. Moreover, among Mtb-derived proteins, TBCM showed long-term tumor prevention effects for the first time (Figure 8). To understand the function of memory T cells, additional animal experiments are needed in the future study.
In summary, our data proved that TBCM (Rv1885c) can drive DC maturation and activation in a TLR4-dependent manner via enhanced expression of costimulatory molecules, enhanced production of proinflammatory cytokines, enhanced DC migration capacity and enhanced Th1 skewing activity. TBCM can also exert strong anticancer or cancer preventive effects mainly via functional CD8 T cell-mediated CTL responses, generation of memory CD4 T cells and inhibition of the expression of proliferation- or metastasis-related genes in tumor microenvironments. In conclusion, for the first time, we proved that TBCM (Rv1885c) is a novel potent TLR4 agonist with a potential immunoadjuvant for DC-based cancer immunotherapy.
Author contributions
Bum-Joon Kim and Hyein Jeong conceived of the presented idea and developed the theory. Hyein Jeong and So-Young Lee carried out murine BMDC assays and animal experiments. Hyejun Seo performed protein expression. Dong Hyun Kim supported animal experiments and Duhyung Lee supported human PBMC assays. Bum-Joon Kim supervised overall experiments and wrote the manuscript. Hyein Jeong wrote the manuscript and discussed the results. All authors contributed to the article and approved the submitted version.
Supplementary Material
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Shurin MR. Dendritic cells presenting tumor antigen. Cancer Immunol Immunother. 1996;43:158–14. doi: 10.1007/s002620050317. [DOI] [PubMed] [Google Scholar]
- 2.Heath WR, Belz GT, Behrens GMN, Smith CM, Forehan SP, Parish IA, Davey GM, Wilson NS, Carbone FR, Villadangos JA, et al. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunological Rev. 2004;199(1):9–26. doi: 10.1111/j.0105-2896.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 3.Butcher MJ, Galkina EV.. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front Physiol. 2012;3:44. doi: 10.3389/fphys.2012.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J, Dai X, Hsu C, Ming C, He Y, Zhang J, Wei L, Zhou P, Wang C-Y, Yang J, et al. Discrimination of heterogeneity of bone marrow-derived dendritic cells. Med Rep. 2017;16(5):6787–6793. doi: 10.3892/mmr.2017.7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Zhao LB, Chang S, Ming C-S, Yang J, Gong N-Q. Dynamic changes of phenotypes and secretory functions during the differentiation of pre-DCs to mature DCs. J Huazhong Univ Sci Technol Med Sci. 2017;37:191–196. doi: 10.1007/s11596-017-1714-z. [DOI] [PubMed] [Google Scholar]
- 6.Shi GN, Zhang CN, Xu R, Niu J-F, Song H-J, Zhang X-Y, Wang -W-W, Wang Y-M, Li C, Wei X-Q, et al. Enhanced antitumor immunity by targeting dendritic cells with tumor cell lysate-loaded chitosan nanoparticles vaccine. Biomaterials. 2017;113:191–202. doi: 10.1016/j.biomaterials.2016.10.047. [DOI] [PubMed] [Google Scholar]
- 7.Schnurr M, Galambos P, Scholz C, Then F, Dauer M, Endres S, Eigler A. Tumor cell lysate-pulsed dendritic cells induce a T-cell response against pancreatic carcinoma cells: an in vitro model for the assessment of tumor vaccines. Cancer Res. 2001;61:6445–6450. [PubMed] [Google Scholar]
- 8.Shurin MR, Shurin GV, Lokshin A, Yurkovetsky ZR, Gutkin DW, Chatta G, Zhong H, Han B, Ferris RL. Intratumoral cytokines/chemokines/growth factors and tumor infiltrating dendritic cells: friends or enemies? Cancer Metastasis Rev. 2006;25(3):333–356. doi: 10.1007/s10555-006-9010-6. [DOI] [PubMed] [Google Scholar]
- 9.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 10.Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T cells in patients with malignant glioma. Cancer Res. 2004;64(14). doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 11.Prins RM, Craft N, Bruhn KW, Khan-Farooqi H, Koya RC, Stripecke R, Miller JF, Liau LM. The TLR-7 agonist, Imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176(1):157–164. doi: 10.4049/jimmunol.176.1.157. [DOI] [PubMed] [Google Scholar]
- 12.Jung NC, Lee JH, Chung KH, Kwak YS, Lim D-S. Dendritic cell-based immunotherapy for solid tumors. Translational Oncol. 2018;11(3):686–690. doi: 10.1016/j.tranon.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use dendritic cells for cancer therapy. Lancet Oncol. 2014;15:e257–67. doi: 10.1016/S1470-2045(13)70585-0. [DOI] [PubMed] [Google Scholar]
- 14.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 15.Hartung E, Becker M, Bachem A, Reeg N, Jäkel A, Hutloff A, Weber H, Weise C, Giesecke C, Henn V. Induction of potent CD8 T cell cytotoxicity by specific targeting of antigen to cross-presenting dendritic cells in vivo via murine or human XCR1. J Immunol. 2015;194:1069–1079. doi: 10.4049/jimmunol.1401903. [DOI] [PubMed] [Google Scholar]
- 16.Mizumoto Y, Hemmi H, Katsuda M, Miyazawa M, Kitahata Y, Miyamoto A, Nakamori M, Ojima T, Matsuda K, Nakamura M, et al. Anticancer effects of chemokine-directed antigen delivery to a cross-presenting dendritic cell subset with immune checkpoint blockade. Br J Cancer. 2020;122:1185–1193. doi: 10.1038/s41416-020-0757-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCall KD, Muccioli M, Benencia F. Toll-like receptors signaling in the tumor microenvironment. Tumor Microenvironment Advances in Exp Med Biol. 2020;1223:81–97. [DOI] [PubMed] [Google Scholar]
- 18.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 20.Ulevitch R, Tobias PS. Recognition of gram-negative bacteria and endotoxin by the innate immune system. Current Opinion Immunol. 1999;11(1):19–22. doi: 10.1016/S0952-7915(99)80004-1. [DOI] [PubMed] [Google Scholar]
- 21.Xu Y, Yang E, Huang Q, Ni W, Kong C, Liu G, Li G, Su H, Wang H. PPE57 induces activation of macrophages and drives Th1 type immune responses through TLR2. J Mol Med. 2015;93:645–662. doi: 10.1007/s00109-014-1243-1. [DOI] [PubMed] [Google Scholar]
- 22.Tima HG, Huygen K, Romano M. Innate signaling by mycobacterial cell wall components and relevance for development of adjuvants for subunit vaccines. Expert Rev Vaccines. 2016;15(11):1409–1420. doi: 10.1080/14760584.2016.1187067. [DOI] [PubMed] [Google Scholar]
- 23.Quesniaux V, Fremond C, Jacobs M, Parida S, Nicolle D, Yeremeev V, Bihl F, Erard F, Botha T, Drennan M, et al. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes and Infection. 2004;6(10):946–959. doi: 10.1016/j.micinf.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 24.Noguera-Ortega E, Guallar-Garrido S, Julian E. Mycobacteria-based vaccines as immunotherapy for non-urological cancer. cancers. 2020;12(7):1802. doi: 10.3390/cancers12071802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pettenati C, Ingersoll MA. Mechanisms of BCG immunotherapy and its outlook for bladder cancer. Nat Rev Urol. 2018;15:615–625. doi: 10.1038/s41585-018-0055-4. [DOI] [PubMed] [Google Scholar]
- 26.Fowler DW, Copier J, Wilson N, Dalgleish AG, Bodman-Smith MD. Mycobacteria activate gammadelta T cell antitumor responses via cytokines from type 1 myeloid dendritic cells: a mechanism of action for cancer immunotherapy. Cancer Immunol Immunother. 2012;61:535–547. doi: 10.1007/s00262-011-1121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar P, Das G, Bhaskar S. Mycobacterium indicus pranii therapy induce tumor regression in Myd88- and TLR2-dependent manner. BMC Res Notes. 2019;12:648. doi: 10.1186/s13104-019-4679-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SY, Yang SB, Choi YM, Oh S-J, Kim B-J, Kook Y-H, Kim B-J. Heat-killed Mycobacterium paragordonae therapy exerts an anti-cancer immune response via enhanced immune cell-mediated oncolytic activity in xenograft mice model. Cancer Lett. 2020;472:142–150. doi: 10.1016/j.canlet.2019.12.028. [DOI] [PubMed] [Google Scholar]
- 29.Jung ID, Jeong SK, Lee CM, Noh KT, Heo DR, Shin YK, Yun CH, Koh WJ, Akira S, Whang J, et al. Enhanced efficacy of therapeutic cancer vaccines produced by co-treatment with Mycobacterium tuberculosis Heparin-binding hemagglutinin, a novel TLR4 agonist. Cancer Res. 2011;71:2858-2870. doi: 10.1158/0008-5472.CAN-10-3487. [DOI] [PubMed] [Google Scholar]
- 30.Jung ID, Shin SJ, Lee MG, Kang TH, Han HD, Lee SJ, Kim WS, Kim HM, Park WS, Kim HW, et al. Enhancement of tumor-specific T cell-mediated immunity in dendritic cell-based vaccines by mycobacterium tuberculosis heat shock protein X. J Immunol. 2014;193:1233–1245. doi: 10.4049/jimmunol.1400656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SK, Reddy SK, Nelson BC, Vasquez GB, Davis A, Howard AJ, Patterson S, Gilliland GL, Ladner JE, Reddy PT, et al. Biochemical and structural characterization of the secreted chorismate mutase (RV1885c) from Mycobacterium tuberculosis H37Rv: an *AroQ enzyme not regulated by the aromatic amino acids. J Bacteriol. 2020;188(24):8638-8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JY, Kim BJ, Koo HK, Kim J, Kim J-M, Kook Y-H, Kim B-J. Diagnostic potential of IgG and IgA responses to Mycobacterium tuberculosis antigens for discrimination among active tuberculosis, latent tuberculosis infection, and non-infected individuals. microorganisms. 2020;8(7):979. doi: 10.3390/microorganisms8070979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi HH, Kwon KW, Han SJ, Kang SM, Choi E, Kim A, Cho SN, Shin SJ, et al. PPE39 of the Mycobacterium tuberculosis strain Beijing/K induces Th1-cell polarization through dendritic cell maturation. J Cell Sci. 2019;132(17). doi: 10.1242/jcs.228700. [DOI] [PubMed] [Google Scholar]
- 34.Harding CV, Boom WH. Regulation of antigen presentationby Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol. 2010;8:296–307. doi: 10.1038/nrmicro2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bansal K, Sinha AY, Ghorpade DS, Togarsimalemath SK, Patil SA, Kaveri SV, Balaji KN, Bayry J, et al. Src homology 3-interacting domain of Rv1917c of Mycobacterium tuberculosis induces selective maturation of human dendritic cells by regulating PI3K-MAPK-NF-kB signaling and drives Th2 immune responses. J Biol Chem. 2010;47:36511–36522. doi: 10.1074/jbc.M110.158055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byun EH, Kim WS, Kim JS, Jung ID, Park YM, Kim HJ, Cho SN, Shin SJ, et al. Mycobacterium tuberculosis Rv0577, a novel TLR2 agonist, induces maturation of dendritic cells and drives Th1 immune response. FASEB J. 2012;26(6):2695–2711. doi: 10.1096/fj.11-199588. [DOI] [PubMed] [Google Scholar]
- 37.Hirao M, Onai N, Hiroishi K, Watkins SC, Matsushima K, Robbins PD, Lotze MT, Tahara H. CC chemokine receptor-7 on dendritic cells is induced after interaction with apoptotic tumor cells: critical role in migration from the tumor site to draining lymph nodes. Cancer Res. 2000;60:2209–2217. [PubMed] [Google Scholar]
- 38.Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99(1):23–33. doi: 10.1016/S0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 39.Breedveld A, Kormelink TG, Egmond M, de Jong EC. Granulocytes as modulators of dendritic cell function. J Leukoc Biol. 2017;102(4):1003–1016. doi: 10.1189/jlb.4MR0217-048RR. [DOI] [PubMed] [Google Scholar]
- 40.Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. Non-redundant requirement for CXCR3 signaling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6:7458. doi: 10.1038/ncomms8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastasis associated with CD8+ T-cell recruitment. Cancer Res. 2009;69(7):3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Broderick L, Yokota SJ, Reineke J, Mathiowitz E, Stewart CC, Barcos M, Kelleher RJ, Bankert RB. Human CD4+ effector memory T cells persisting in the microenvironment of lung cancer xenografts are activated by local delivery of IL-12 to proliferate, produce IFN-γ, and eradicate tumor cells. J Immunol. 2005;174(2):898–906. doi: 10.4049/jimmunol.174.2.898. [DOI] [PubMed] [Google Scholar]
- 43.Ning ZK, Hu CG, Huang C, Liu J, Zhou T-C, Zong Z. Molecular subtypes and CD4+ memory T cell-based signature associated with clinical outcomes in gastric cancer. Front Oncol. 2020;10:626912. doi: 10.3389/fonc.2020.626912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caserta S, Borger JG, Zamoyska R. Central and effector memory CD4 and CD8 T cell responses to tumor-associated antigens. Critical Rev Immunol. 2012;32:97–126. doi: 10.1615/CritRevImmunol.v32.i2.10. [DOI] [PubMed] [Google Scholar]
- 45.Jung BG, Wang X, Yi N, Ma J, Turner J, Samten B. Early secreted antigenic target of 6-kDa of Mycobacteirum tuberculosis stimulates IL-6 production by macrophages through activation of STAT3. Sci Rep. 2017;7:40984. doi: 10.1038/srep40984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bansal K, Sinha AY, Ghorpade DS, Togarsimalemath SK, Patil SA, Kaveri SV, Balaji KN, Bayry J. Src homology 3-interacting domain of Rv1917c of Mycobacterium tuberculosis induces selective maturation of human dendritic cells by regulating PI3K-MAPK-NFkappaB signaling and drives Th2 immune responses. J Biol Chem. 2010;285:36511–36522. doi: 10.1074/jbc.M110.158055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.