

Abstract
Osteoporosis can be caused by a multitude of factors and is defined by a decrease in bone density and mass caused by the destruction of bone microstructure, resulting in increased bone brittleness. Thus, it is a systemic bone disease in which patients are prone to fracture. The role of ferroptosis in the pathogenesis of osteoporosis has become a topic of growing interest. In this review, we discuss the cell morphology, basic mechanisms of ferroptosis, the relationship between ferroptosis and osteoclasts and osteoblasts, as well as the relationship between ferroptosis and diabetic osteoporosis, steroid-induced osteoporosis, and postmenopausal osteoporosis. Emerging biomedical research has provided new insights into the roles of ferroptosis and osteoporosis, such as in cellular function, signaling pathways, drug inhibition, and gene silencing. The pathophysiology and mechanism of ferroptosis and osteoporosis need to be further studied and elucidated to broaden our understanding of iron metabolism and immune regulation. Studies using animal models of osteoporosis in vivo and cell models in vitro will help clarify the relationship between ferroptosis and osteoporosis and provide research ideas for the elucidation of new mechanisms and development of new technologies and new drugs for the treatment of osteoporosis in the future.
1. Introduction
Cell death includes apoptosis, pyroptosis, necrosis, autophagy, ferroptosis, and other death mechanisms. Before the concept of ferroptosis was revealed, one study showed that the regulation of iron metabolism and the maintenance of iron homeostasis have indispensable biological roles in the human pathophysiological process. In 2003, Dolma et al. [1] found that the small molecule erastin can induce RAS mutations in tumor cells, leading to cell death in a manner different from traditional apoptosis. In 2008, using high-throughput small-molecule screening technology, it was found that Ras selective lethal small molecules could kill human foreskin fibroblasts (BJeLR) in a nonapoptotic manner [2]; however, neither apoptosis inhibitors nor necrostatin inhibitors (Necrostatin-1) [3] could reverse cell death induced by erastin and RSLs. In contrast, the antioxidant vitamin E and the iron-chelating agent deferoxamine mesylate (DFO) can inhibit cell death [4], indicating that ferroptosis is an iron-dependent cell death process. In 2012, Dixon et al. named this process erastin-induced cell death with distinct morphological, genetic, and biochemical characteristics [5].
Iron, an essential trace element for humans and a necessary substance for life, plays a vital role in many biochemical processes, including oxygen transport, enzymatic reactions, and immune reactions. With accumulating research, various physiological and pathological processes, such as tumor, Parkinson's disease, atherosclerosis, viral infection, osteoporosis, immune response, and ischemia-reperfusion injury, have been found to be related to ferroptosis [6–8], and ferroptosis is expected to become a new research direction for disease treatment.
Interestingly, a growing number of studies have reported a relationship between ferroptosis and osteoporosis. Here, we summarize the basic pathological features of ferroptosis and the relationship between ferroptosis and osteoclasts and osteoblasts. We also summarize the relationship between ferroptosis and osteoporosis and show how ferroptosis regulates diabetic osteoporosis (DOP), glucocorticoid-induced osteoporosis (GIOP), and postmenopausal osteoporosis (PMOP).
2. Morphological Characteristics of Ferroptosis
Ferroptosis was delineated as a type of regulated cell death (RCD) by the Nomenclature Committee on Cell Death in 2018. It is initiated by oxidative perturbations in the intracellular microenvironment, which are constitutively controlled by glutathione peroxidase 4 (GPX4) and can be inhibited by iron-chelating agents and lipophilic antioxidants [9]. Ferroptosis is distinguished from apoptosis [10], necrosis [11], autophagy [12], and pyroptosis [13] by a varying set of morphological characteristics, inducing factors, and regulatory pathways.
Cancer cells undergoing ferroptosis are generally round, small, and scattered [1]. In human prepuce fibroblast BJeLR cells treated with erastin, several effects were observed: mitochondria atrophied and decreased in number, membrane density increased, the normal structure of mitochondrial cristae was destroyed, nuclear size was normal but lacked chromatin aggregation, and cell membranes were blistered without rupturing [5, 14–16]. These morphological features can help to distinguish ferroptosis from other modes of cell death, such as apoptosis, necrosis, pyroptosis, and autophagy.
3. Mechanisms and Regulation of Ferroptosis
3.1. Iron Metabolism and Ferroptosis
Iron is involved in the synthesis of various important proteases and is an essential element in the life activities of the body [17, 18]. Iron overload caused by abnormal iron metabolism is one of the main characteristics of ferroptosis. Binding of ferric acid to transferrin is the main mechanism for circulation of iron in the bloodstream. Circulating iron enters cells by binding to transferrin receptor 1 (TFR1) on the cell membrane, where the six-transmembrane epithelial antigen of prostate 3 (STEAP3) reduces ferric iron to ferrous iron. Finally, divalent metal transporter 1 (DMT1) releases divalent iron into the labile iron pool (LIP) in the cytoplasm. LIP enables the active uptake of free iron in the cytoplasm, as well as recovery of iron in ferritin and mitochondria, and large quantities of LIP are present in lysosomes [19]. Lysosomes are therefore considered the main organelles responsible for cellular ferroptosis [20]. Excess bivalent iron is transported extracellularly by ferroportin 1 (FPN1) and stored in ferritin heavy chain 1 and ferritin light chain 1 (FTL1) [21, 22].
Under physiological conditions, ferritin provides a strong buffer that regulates physiological responses to iron deficiency and excess, maintaining homeostasis [23]. Under pathological conditions, iron overload can induce ferroptosis by producing ROS through Fenton and Haber–Weiss reactions [24, 25]. Studies have shown that DFO, an iron chelator, can inhibit ferroptosis caused by intracellular iron overload [26, 27]. In addition, a high iron diet can lead to serious heart damage, and the ferroptosis inhibitor ferrostatin-1 mitigates the damage caused by ferroptosis [28]. Under physiological conditions, mitochondrial ferritin (FtMt) regulates the free iron content in the mitochondria and maintains normal mitochondrial iron metabolism. FtMt overexpression can reverse erastin-induced ferroptosis both in vivo and in vitro [29, 30]. In neurological diseases, mitochondrial transferrin mitoferrin 1/2 (Mfrn1/2) on the inner mitochondrial membrane is found to be destroyed, resulting in abnormal iron metabolism in the mitochondria [31].
Iron metabolism has been implicated in the occurrence and development of ferroptosis. However, the role of systemic iron regulation in the cellular role of iron deposits and ferritinophagy remains elusive. Whether systemic iron levels fully determine the effect of ferroptosis on the disease remains to be clarified.
3.2. Ferroptosis Mediated by P53
P53 is an important tumor suppressor gene with important roles in cell cycle inhibition, apoptosis, tumorigenesis, and aging [32–34]. mRNA and protein expression levels of SLC7A11 were significantly decreased after the upregulation of p53 gene expression, which confirmed that SLC7A11 is a new target of the p53 gene [35, 36]. p53-silenced H1299 cells were treated with ROS, and there was no change in cell activity. In contrast, cells with activated p53 that were treated with ROS had a 90% death rate. After adding the ferroptosis inhibitor, ferrostatin-1, the cell death rate decreased by approximately 40%, indicating that p53 can induce ferroptosis [37]. Recent studies have shown that P53 can inhibit the uptake of cystine by system xc− by downregulating the expression of the SLC7A11 subunit, resulting in a decrease in cystine-dependent glutathione peroxidase activity and cell antioxidant capacity and an increase in lipid ROS, leading to cellular ferroptosis [38].
3.3. Voltage-Dependent Anion Channels with Ferroptosis
Mitochondria, the main regulators of oxidative phosphorylation, play an important role in oxidative stress and are major producers of ROS [39, 40]. Iron can reach the mitochondrial matrix by passing through the outer mitochondrial membrane and inner mitochondrial membrane, subsequently regulating the physiological functions of important organelles in the mitochondria [41]. Mitochondria also play an important role in the regulation of ferroptosis [42]. Under erastin induction, the voltage-dependent anion channel protein 2/3 (VDAC2/3) on the outer mitochondrial membrane is opened, leading to iron accumulation in the mitochondria. However, the detailed mechanism underlying the role of erastin and VDAC2/3 is still being explored [43].
3.4. Ferroptosis Induced by Inhibition of GPX4 and Cysteine-Glutamate Transporter Receptors (System xc−)
GPX4, an important specific marker of ferroptosis [44], can reduce lipid peroxides to lipid alcohols and hydrogen peroxide to water during ferroptosis [45]. Both GPX4 knockout and the use of the small molecule inhibitor RSL3 antagonism against GPX4 could effectively induce ferroptosis [46–49]. L-glutathione (GSH) is composed of glycine, glutamate, and cysteine and is an important antioxidant in the oxidative stress response, widely present in cells in the form of reduced GSH and oxidized glutathione (GSSG) [50]. The degradation of lipid peroxides by GPX4 requires GSH for the provision of electrons to complete the process [20]. GSH synthesis requires intracellular uptake of cysteine, which is mediated by the sodium-dependent system xc– (also named the cystine/glutamate antiporter), a disulfide-linked heterodimer composed of a heavy chain (4F2hc, gene name SLC3A2), and a light chain (xCT, gene name SLC7A11). The sodium-dependent system xc– transports extracellular cystine into the cell and further converts it to cysteine, which is then used in GSH biosynthesis [5, 51–55]. Studies have shown that selective inhibition of system xc− leads to a decrease in intracellular GSH, which aggravates ROS accumulation and eventually leads to ferroptosis [56, 57]. Although a regulatory mechanism between GXP4 and ferroptosis is known to exist, the roles of GXP4 in different RCD and the mechanism of information transduction pathways remain unclear.
3.5. Ferroptosis Mediated by Lipid Peroxidation
Lipid peroxidation is another key factor in ferroptosis. Recent studies [58] have shown that lipid peroxides can destroy the stability of the lipid bilayer, causing disintegration of the cell membrane. Lipidomics analyses have indicated that both AA and adrenic acid containing phosphatidyl ethanolamine are lipid products of ferroptosis, which can undergo spontaneous peroxidation in the presence of hydroxyl radicals (produced by the Fenton reaction between redox-active iron divalent and hydrogen peroxide) [46, 59, 60]. Polyunsaturated fatty acids (PUFAs) are prone to lipid peroxidation, owing to the presence of highly active hydrogen atoms in methylene bridges. Hydroxyl radicals can directly interact with PUFAs in membrane phospholipids through chain reactions to form lipid peroxides, which attack the cytomembrane and trigger morphological changes in ferroptosis [61, 62]. Derivatives resulting from the decomposition of lipid peroxides, including 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), can react with nucleic acids and proteins, leading to further cell destruction [63, 64]. These derivatives can also be used as important molecular markers for the detection of ferroptosis and lipid peroxidation. In addition, divalent iron can be used as a cofactor of lipoxygenase (LOX) to catalyze lipid peroxidation of PUFAs [65]. Recent studies have shown [62] that both lysophosphatidylcholine acyltransferase 3 (LPCAT3) and ACSL4 are involved in lipid peroxidation of membrane PUFAs, and they serve as important molecular markers of ferroptosis.
In the upper panel, we can see that ferroptosis involves multiple signaling pathways and their regulators (Figure 1). Understanding these signaling molecules and their transduction pathways is of great significance in the pathophysiology of ferroptosis.
Figure 1.
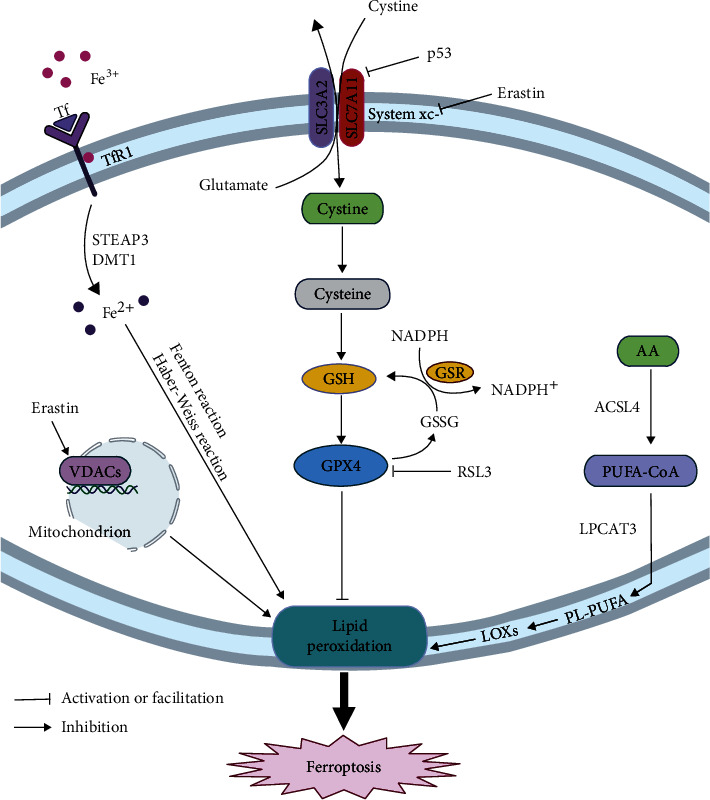
The mechanism of ferroptosis. Circulating iron enters cells by binding to TFR1 on the cell membrane, where the STEAP3 reduces ferric iron to ferrous iron. DMT1 releases the divalent iron into a labile iron pool in the cytoplasm, and iron overload can induce ferroptosis by producing ROS through the Fenton and Haber–Weiss reactions. The sodium-dependent system xc– transports extracellular cystine into the cell and further converts it to cysteine. The selective inhibitor of system xc− leads to a decrease in intracellular GSH, which aggravates ROS accumulation and eventually leads to ferroptosis; P53 can inhibit the uptake of cystine by system xc− via downregulating the expression of the SLC7A11 subunit, resulting in a decrease in cystine-dependent glutathione peroxidase activity and cell antioxidant capacity and an increase in lipid ROS, leading to ferroptosis of cells; RSL3 can induce ferroptosis by antagonizing GPX4; Hydroxyl radicals can directly interact with PUFAs in membrane phospholipids through chain reactions to form lipid peroxides, inducing ferroptosis.
3.6. Ferroptosis with Osteoclasts and Osteoblasts
Over the last two decades, the relationship between iron and osteoporosis has attracted increasing attention. Studies have reported that disorders of iron metabolism, including iron deficiency and iron overload, can lead to osteoporosis [66–70]. The homeostasis and integrity of bone tissue are maintained by maintaining a balance between osteoclastic and osteogenic activities, and the remodeling process of bone tissue is a continuous cycle. Osteoclasts mainly play the role of bone resorption, whereas osteoblasts mainly play the role of bone reconstruction, such as the formation, mineralization, and secretion of osteocytes. They mutually restrict and balance the metabolism of bone tissues [71, 72]. Song et al. found that FA complementation group D2 (FANCD2) suppresses erastin-induced ferroptosis in bone mesenchymal stem cells (BMSCs), and FANCD2 reduces iron accumulation and lipid peroxidation in ferroptosis [73]. This is due to the multidirectional differentiation potential of BMSCs. These results suggest that ferroptosis may also occur during the targeted differentiation of BMSCs under certain circumstances.
3.7. Ferroptosis May Occur in Osteoclasts
Osteoclasts are large multinucleated cells formed by the fusion of mononuclear macrophage lineage or BMSCs by the inductive form of receptor activator of nuclear factor-kappa B ligand (RANKL) and perform the function of bone resorption. Iron ions can promote osteoclast differentiation and bone resorption by producing ROS [74]. The iron chelator DFO inhibits osteoclast formation in vitro [75]. Liu et al. found the iron-starvation response and ferritinophagy under normoxia in the process of osteoclast differentiation confirmed the involvement of ferroptosis; following RANKL stimulation, MDA and prostaglandin endoperoxide synthase 2 (PTGS2) gene expression in bone marrow-derived macrophages (BMDMs) were increased, GSH and iron levels in the culture medium supernatant decreased, and iron accumulation in mitochondria was observed [76].
3.8. Ferroptosis May Occur in Osteoblasts
Osteoblasts play an important role in bone regeneration and play a leading role in the synthesis, secretion, and mineralization of the bone matrix [77]. Previous studies have shown that the inhibitory effect of iron on the osteogenic differentiation of MSCs is proposed, and iron overload in mice is associated with increased ferritin and decreased RUNX family transcription factor 2 (RUNX2) levels in compact bone osteoprogenitor cells [69]. A high dose of dexamethasone (10 μM dexamethasone) may activate osteoblasts to induce ferroptosis by downregulating GPX4 and system xc− [78]. Subsequent studies found that GPX4 was significantly reduced, ROS levels were increased in MC3T3 cells induced by high glucose, and mitochondria were generally smaller and less tubular, with a darker-stained membrane with distinctly disrupted inner membrane folding. In addition, the ability of MC3T3 to differentiate into osteoblasts and the formation of mineralized nodules was decreased in a high glucose environment, and similar phenomena were observed in osteoblasts in mice [79, 80].
Based on the results of the above studies, we hypothesized that ferroptosis of osteoclasts would reduce the occurrence of bone resorption, while ferroptosis of osteoblasts would lead to reduced bone formation.
4. Potential Relationship between Ferroptosis and Osteoporosis
Osteoporosis is a metabolic bone disease, involving an imbalance between the bone resorptive functions of osteoclasts and bone forming functions of osteoblasts. This imbalance leads to loss of bone mass and strength, resulting in the increased risk of fragility fractures and a progressive decrease in healing ability following fractures [81–83]. Global rates of hip fractures, vertebral fractures, and wrist fractures caused by osteoporosis have increased, emerging as a major global public health problem [84] with approximately 8.9 million people worldwide experiencing osteoporotic fractures each year [85]. It is estimated that by 2050, hip fractures in elderly men will increase by 310%, and hip fractures in elderly women will increase by 240% [86]. Preventing and effectively treating the occurrence and development of osteoporosis are therefore an urgent priority in global health.
4.1. Ferroptosis and DOP
Approximately 1 in 11 adults worldwide have diabetes, and 90% have type 2 diabetes [87]. Diabetes mellitus is often associated with osteoporosis [88] that this is often associated to multiple factors. It has been suggested that the factors contributing to reduced bone formation include oxidative stress caused by high blood sugar and accumulation of advanced glycation end products (AGEs) in collagen [88, 89]. Other studies suggest that low concentrations of insulin and insulin-like growth factor 1 (IGF-1) may affect osteogenic activity and lead to osteoporosis [90]. Antidiabetic drugs, such as thiazolidinedione, have been shown to negatively affect bone metabolism and fracture risk [91, 92]. However, the detailed pathological mechanism is not fully understood and is still being explored. In addition, patients with diabetes often have complications such as vision loss and neuropathy, which can increase the risk of falls and fractures. An in-depth study of the pathological mechanism of DOP would help to improve the prediction of DOP risk, as well as enable timely and reasonable prevention and treatment of brittle fractures caused by osteoporosis.
Iron metabolism is often disturbed in patients with diabetes [93–95]. Iron is also a strong oxidant that can promote the production of many reactive oxygen free radicals. Indicators of iron metabolism (transferrin, ferritin, hepcidin, transferrin receptor, etc.) can directly or indirectly affect the occurrence and development of type 2 diabetes [94]. Ferroptosis results in the production of abundant ROS through the Fenton reaction, which causes the accumulation of lipid peroxides and cell damage [96]. Wang et al. [80] detected the expression of FtMt and the occurrence of ferroptosis in the bone tissue of a (T2DOP) rat model of type 2 diabetes. They found that overexpression of FtMt reduced oxidative stress induced by excess iron under high glucose conditions to inhibit the occurrence of ferroptosis in osteoblasts, while the silencing of FtMt induced mitochondrial autophagy of T2DOP through the ROS/PINK1/Parkin signaling pathway. This suggests that FtMt may be a potential target for the treatment of T2DOP. Meanwhile, it was found that ferroptosis of osteoblasts increased following mitochondrial activation by carbonyl cyanide-M-chlorophenyl-hydrazine (CCCP, a mitochondrial agonist). Additionally, several other effects were observed: decreased expression of GPX4, osteocalcin (OCN), alkaline phosphatase (ALP), and osteoprotegerin (OPG), decreased mineralized nodules, increased ROS levels, and increased lipid peroxide. In contrast, treatment of the CCCP group with ferroptosis inhibitors was able to rescue ferroptosis. Ma et al. [79] found that high glucose induced ferroptosis in the bone tissue of a T2DOP rat model by increasing the consumption of ROS/lipid peroxidation/GSH. More importantly, melatonin (N-acetyl-5-methoxytryptamine) significantly reduced the level of ferroptosis by activating the Nrf2/HO-1 signaling pathway in vivo and in vitro and improved the osteogenic ability of MC3T3-E1 cells. Based on these studies, we speculated that the occurrence of T2DOP was correlated with iron homeostasis imbalance and ferroptosis in osteoblasts. Nonetheless, the detailed mechanisms require further investigation.
4.2. Ferroptosis and GIOP
Ferroptosis is a recently discovered form of cell death characterized by lipid peroxidation caused by the downregulation of GPX4 and system xc−. It is involved in GIOP. Yang et al. [97] found that high-dose and long-term use of steroid hormones can alter antioxidant capacity, reduce the activity and function of osteoblasts, and lead to osteoporosis and osteonecrosis. Endothelial cell-secreted exosomes (EC-Exos) are important mediators of cell-to-cell communication and are involved in many physiological and pathological processes. By inhibiting ferritin-phagocytosis-dependent ferroptosis, EC-Exos reversed the inhibitory effect of glucocorticoid-induced osteoblasts on osteogenesis. Lu et al. [78] established a GIOP model with a high dose of dexamethasone and found that high-dose dexamethasone (10 μM) can induce ferroptosis of osteoblasts, possibly by downregulating GPX4 and system xc−. KEGG-based gene set enrichment analysis was performed to demonstrate the activation of the ferroptosis pathway. Extracellular vesicles extracted from bone marrow-derived endothelial progenitor cells inhibited activation of the ferroptosis pathway by restoring GPX4 and system xc−. The changes in the expression of ferroptosis markers, such as SLC3A2, SLC7A11, and GPX4, were further confirmed using RNA-seq. EPC-EVS reversed dexamethasone-induced changes in cysteine and oxidative damage markers, such as MDA, GSH, and glutathione disulfide (GSSG), and improved skeletal parameters in mice. EPC-EVS reversed dexamethasone-induced changes in cysteine and oxidative damage markers, such as MDA, GSH, and GSSG, and improved skeletal parameters in mice. They suggested that EPC-EVS prevents glucocorticoid-induced osteoporosis in mice by inhibiting the ferroptotic pathway of osteoblasts. However, further research is needed to elucidate the ferroptosis mechanisms associated with GIOP.
4.3. Ferroptosis and PMOP
PMOP is caused by estrogen deficiency in postmenopausal women. Estrogen deficiency is associated with insufficient differentiation of osteoblasts and increased activity of osteoclasts, ultimately leading to decreased bone mass and increased bone fragility [98]. A significant association between low serum iron levels and PMOP has been reported [66, 99]. Abraham et al. [100] found that dietary iron (or related factors) may have a protective effect against bone loss in the postmenopausal spine. Ni et al. [76] found a correlation between ferroptosis and RANKL-induced osteoclast differentiation and iron-starvation response, and ferritinophagy promoted the ferroptosis of osteoclasts induced by RANKL. In vivo, the HIF-1α-specific inhibitor 2-methoxyestradiol (2ME2) was found to prevent bone loss in OVX mice. The authors, therefore, proposed that the induction of ferroptosis of osteoclasts by targeting HIF-1α and ferritin could be an alternative treatment for osteoporosis.
Together, these findings show that patients with osteoporosis often experience iron metabolism disorders, oxidative stress, and lipid peroxidation, leading to ferroptosis (Figure 2). This suggests that the regulation of ferroptosis of osteoclasts or osteoblasts may provide a potential therapeutic strategy for osteoporosis (Table 1).
Figure 2.
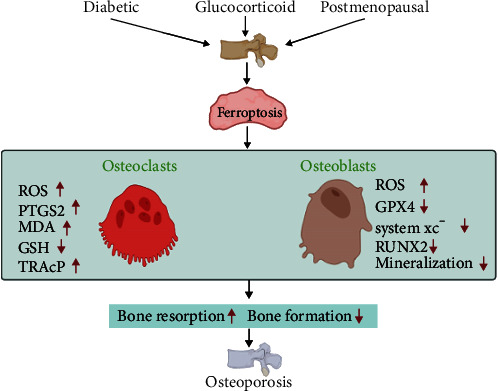
Diabetic, glucocorticoid, and postmenopausal induce ferroptosis in osteoclasts and osteoblasts. The subsequent loss of bone mass contributes to osteoporosis. RUNX2: Runt-related transcription factor 2; TRAcP: tartrate resistance acid phosphatase.
Table 1.
Interventions and reagents targeting ferroptosis for osteoporosis.
| Intervention methods or reagents | Mechanism | Effects on cells | Reference |
|---|---|---|---|
| 2ME2 (2-methoxyestradiol) | Targeting HIF-1α and ferritin | Inducing the ferroptosis of osteoclasts | [76] |
| EPC-EVs | Restoring GPX4 and system xc− levels | Inhibiting ferroptotic pathway of osteoblasts | [78] |
| Melatonin | Activating the Nrf2/ho-1 signaling | Reducing ferroptosis in MC3T3-E1 | [79] |
| Silencing FtMt | Inducing mitophagy via ROS/PINK1/Parkin pathway | Inhibiting ferroptosis of osteoblasts | [80] |
| CCCP (mitophagy agonist) | Activating mitochondria | Promoting ferroptosis of osteoblasts | [80] |
| EC-exos | Inhibiting ferritin-phagocytosis-dependent ferroptosis | Reversing the inhibitory effect of glucocorticoid on osteoblasts | [97] |
5. Conclusion
Owing to consistent research efforts, osteoporosis has developed from a highly disabling disease to a disease that can be managed. Existing drugs and biologics marketed to treat osteoporosis have some clinical efficacy, but are associated with side effects including cancer, osteonecrosis of the jaw, and adverse effects on liver and kidney function. Improved antiosteoporosis treatment is therefore a significant priority for medical researchers and clinicians.
In this review, we have summarized the cell morphology, cell characteristics, and pathogenesis of ferroptosis, as well as the relationship between ferroptosis and osteoporosis. Ferroptosis is an iron-dependent nonapoptotic form of RCD, which is closely related to the pathophysiological processes of various human diseases. It is accompanied by disorders of iron metabolism and energy metabolism, upregulation of inflammation and oxidative stress, and functional impairment of important organelles in cells.
Elucidating the molecular mechanisms of ferroptosis and osteoporosis can provide substantial insights into the field of bone metabolism and immunity, enabling the discovery of new therapeutics with fewer side effect for the prevention and treatment of osteoporosis. At present, these encouraging research results have generated great interest in further exploring the mechanisms underlying iron-dependent cell death and osteoporosis. The interaction and crosstalk among osteoclasts, osteoblasts, and osteocytes should be considered in the treatment of osteoporosis. Research on ferroptosis is still at a relatively early stage, and the specific mechanism, nodal molecules, and related signaling pathways of ferroptosis remain unclear. Further study is required to explore these newly discovered mechanisms, their associated signaling pathways and molecular targets, to develop effective treatment methods. The use of osteoporosis models in castrated mice, diabetic mice, and aging mice will help to determine the relationship between osteoporosis and ferroptosis and guide research for further understanding and effectively treating osteoporosis.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (82073539 and 81773432), the Foundation of The First Affiliated Hospital of Chengdu Medical College (CYFY-GQ35), the Project of Sichuan Provincial Department of Science and Technology (22MZGC0226), and the Foundation of Chengdu Medical College (CYZYB21-12).
Contributor Information
Xiaoping Yu, Email: cyggwsyxp@sina.com.
Ji Tu, Email: ji.tu@student.unsw.edu.au.
Zhengdong Zhang, Email: doctorzzd@vip.qq.com.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors' Contributions
PL, XY, JT, and ZZ generated the ideas, reviewed the publications, and wrote the manuscript. WW, ZL, and YL participated in the discussions.
References
- 1.Dolma S., Lessnick S. L., Hahn W. C., Stockwell B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell . 2003;3(3):285–296. doi: 10.1016/S1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 2.Yang W. S., Stockwell B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chemistry & Biology . 2008;15(3):234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werthmoller N., Frey B., Wunderlich R., Fietkau R., Gaipl U. S. Modulation of radiochemoimmunotherapy-induced B16 melanoma cell death by the pan-caspase inhibitor zVAD-fmk induces anti-tumor immunity in a HMGB1-, nucleotide- and T-cell-dependent manner. Cell Death & Disease . 2015;6(5, article e1761) doi: 10.1038/cddis.2015.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y., Zeng X., Lu D., Yin M., Shan M., Gao Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Human Reproduction . 2021;36(4):951–964. doi: 10.1093/humrep/deaa363. [DOI] [PubMed] [Google Scholar]
- 5.Dixon S. J., Lemberg K. M., Lamprecht M. R., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell . 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mao C., Liu X., Zhang Y., et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature . 2021;593(7860):586–590. doi: 10.1038/s41586-021-03539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen S., Aki T., Unuma K., Uemura K. Chemically induced models of Parkinson's disease: history and perspectives for the involvement of ferroptosis. Frontiers in Cellular Neuroscience . 2020;14, article 581191 doi: 10.3389/fncel.2020.581191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Y., Zhang Z., Wang C., Lu D. Ferroptotic cell death: new regulatory mechanisms for metabolic diseases. Endocrine, Metabolic & Immune Disorders Drug Targets . 2021;21(5):785–800. doi: 10.2174/1871530320666200731175328. [DOI] [PubMed] [Google Scholar]
- 9.Galluzzi L., Vitale I., Aaronson S. A., et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death and Differentiation . 2018;25(3):486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirzayans R., Murray D. Do TUNEL and other apoptosis assays detect cell death in preclinical studies? International Journal of Molecular Sciences . 2020;21(23):p. 9090. doi: 10.3390/ijms21239090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J., Ye J., Kong W., Zhang S., Zheng Y. Programmed cell death pathways in hearing loss: a review of apoptosis, autophagy and programmed necrosis. Cell Proliferation . 2020;53(11, article e12915) doi: 10.1111/cpr.12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galluzzi L., Baehrecke E. H., Ballabio A., et al. Molecular definitions of autophagy and related processes. The EMBO Journal . 2017;36(13):1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu P., Zhang Z., Li Y. Relevance of the pyroptosis-related inflammasome pathway in the pathogenesis of diabetic kidney disease. Frontiers in Immunology . 2021;12, article 603416 doi: 10.3389/fimmu.2021.603416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu H., Guo P., Xie X., Wang Y., Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. Journal of Cellular and Molecular Medicine . 2017;21(4):648–657. doi: 10.1111/jcmm.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Y., Song X., Sun X., et al. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochemical and Biophysical Research Communications . 2016;473(4):775–780. doi: 10.1016/j.bbrc.2016.03.052. [DOI] [PubMed] [Google Scholar]
- 16.Han C., Liu Y., Dai R., Ismail N., Su W., Li B. Ferroptosis and its potential role in human diseases. Frontiers in Pharmacology . 2020;11:p. 239. doi: 10.3389/fphar.2020.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura T., Naguro I., Ichijo H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochimica et Biophysica Acta - General Subjects . 2019;1863(9):1398–1409. doi: 10.1016/j.bbagen.2019.06.010. [DOI] [PubMed] [Google Scholar]
- 18.van Swelm R. P. L., Wetzels J. F. M., Swinkels D. W. The multifaceted role of iron in renal health and disease. Nature Reviews Nephrology . 2020;16(2):77–98. doi: 10.1038/s41581-019-0197-5. [DOI] [PubMed] [Google Scholar]
- 19.Torii S., Shintoku R., Kubota C., et al. An essential role for functional lysosomes in ferroptosis of cancer cells. The Biochemical Journal . 2016;473(6):769–777. doi: 10.1042/BJ20150658. [DOI] [PubMed] [Google Scholar]
- 20.Stockwell B. R., Friedmann Angeli J. P., Bayir H., et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell . 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drakesmith H., Nemeth E., Ganz T. Ironing out ferroportin. Cell Metabolism . 2015;22(5):777–787. doi: 10.1016/j.cmet.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou R. P., Chen Y., Wei X., et al. Novel insights into ferroptosis: implications for age-related diseases. Theranostics . 2020;10(26):11976–11997. doi: 10.7150/thno.50663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abbaspour N., Hurrell R., Kelishadi R. Review on iron and its importance for human health. Journal of Research in Medical Sciences: The Official Journal of Isfahan University of Medical Sciences . 2014;19(2):164–174. [PMC free article] [PubMed] [Google Scholar]
- 24.Yu H., Yang C., Jian L., et al. Sulfasalazine‑induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncology Reports . 2019;42(2):826–838. doi: 10.3892/or.2019.7189. [DOI] [PubMed] [Google Scholar]
- 25.Kajarabille N., Latunde-Dada G. O. Programmed cell-death by ferroptosis: antioxidants as mitigators. International Journal of Molecular Sciences . 2019;20(19):p. 4968. doi: 10.3390/ijms20194968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H., An P., Xie E., et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology . 2017;66(2):449–465. doi: 10.1002/hep.29117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng H. F., Yue L. X., Wang N. N., et al. Mitochondrial iron overload-mediated inhibition of Nrf2-HO-1/GPX4 assisted ALI-induced nephrotoxicity. Frontiers in Pharmacology . 2020;11, article 624529 doi: 10.3389/fphar.2020.624529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang X., Cai Z., Wang H., et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circulation Research . 2020;127(4):486–501. doi: 10.1161/CIRCRESAHA.120.316509. [DOI] [PubMed] [Google Scholar]
- 29.Gao M., Yi J., Zhu J., et al. Role of mitochondria in ferroptosis. Molecular Cell . 2019;73(2):354–363.e3. doi: 10.1016/j.molcel.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X., Yu C., Kang R., Tang D. Iron metabolism in ferroptosis. Frontiers in Cell and Development Biology . 2020;8, article 590226 doi: 10.3389/fcell.2020.590226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward D. M., Cloonan S. M. Mitochondrial iron in human health and disease. Annual Review of Physiology . 2019;81(1):453–482. doi: 10.1146/annurev-physiol-020518-114742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ong A. L. C., Ramasamy T. S. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Research Reviews . 2018;43:64–80. doi: 10.1016/j.arr.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 33.McCubrey J. A., Demidenko Z. N. Recent discoveries in the cycling, growing and aging of the p53 field. Aging . 2012;4(12):887–893. doi: 10.18632/aging.100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leveille E., Johnson N. A. Genetic events inhibiting apoptosis in diffuse large B cell lymphoma. Cancers . 2021;13(9):p. 2167. doi: 10.3390/cancers13092167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang L., Kon N., Li T., et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature . 2015;520(7545):57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu B., Kon N., Chen D., et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nature Cell Biology . 2019;21(5):579–591. doi: 10.1038/s41556-019-0305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang L., Hickman J. H., Wang S. J., Gu W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle . 2015;14(18):2881–2885. doi: 10.1080/15384101.2015.1068479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lei G., Zhang Y., Hong T., et al. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene . 2021;40(20):3533–3547. doi: 10.1038/s41388-021-01790-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen-Goodspeed M., Lukan A. N., Dessauer C. W. Modeling of Gαs and Gαi regulation of human type V and VI adenylyl cyclase∗. The Journal of Biological Chemistry . 2005;280(3):1808–1816. doi: 10.1074/jbc.M409172200. [DOI] [PubMed] [Google Scholar]
- 40.Klionsky D. J., Abdel-Aziz A. K., Abdelfatah S., et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) Autophagy . 2021;17:1–382. doi: 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paul B. T., Manz D. H., Torti F. M., Torti S. V. Mitochondria and iron: current questions. Expert Review of Hematology . 2017;10(1):65–79. doi: 10.1080/17474086.2016.1268047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takashi Y., Tomita K., Kuwahara Y., et al. Mitochondrial dysfunction promotes aquaporin expression that controls hydrogen peroxide permeability and ferroptosis. Free Radical Biology & Medicine . 2020;161:60–70. doi: 10.1016/j.freeradbiomed.2020.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y., Luo M., Zhang K., et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nature Communications . 2020;11(1):p. 433. doi: 10.1038/s41467-020-14324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang W. S., SriRamaratnam R., Welsch M. E., et al. Regulation of ferroptotic cancer cell death by GPX4. Cell . 2014;156(1-2):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brandes R. P., Weissmann N., Schroder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radical Biology & Medicine . 2014;76:208–226. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 46.Friedmann Angeli J. P., Schneider M., Proneth B., et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature Cell Biology . 2014;16(12):1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sui X., Zhang R., Liu S., et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Frontiers in Pharmacology . 2018;9:p. 1371. doi: 10.3389/fphar.2018.01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye J., Jiang X., Dong Z., Hu S., Xiao M. Low-concentration PTX and RSL3 inhibits tumor cell growth synergistically by inducing ferroptosis in mutant p53 hypopharyngeal squamous carcinoma. Cancer Management and Research . 2019;Volume 11:9783–9792. doi: 10.2147/CMAR.S217944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vuckovic A. M., Bosello Travain V., Bordin L., et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14‐3‐3ε. FEBS Letters . 2020;594(4):611–624. doi: 10.1002/1873-3468.13631. [DOI] [PubMed] [Google Scholar]
- 50.Forman H. J., Zhang H., Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Molecular Aspects of Medicine . 2009;30(1-2):1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lewerenz J., Hewett S. J., Huang Y., et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxidants & Redox Signaling . 2013;18(5):522–555. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koppula P., Zhuang L., Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein & Cell . 2021;12(8):599–620. doi: 10.1007/s13238-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim J. K. M., Delaidelli A., Minaker S. W., et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proceedings of the National Academy of Sciences of the United States of America . 2019;116(19):9433–9442. doi: 10.1073/pnas.1821323116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poltorack C. D., Dixon S. J. Understanding the role of cysteine in ferroptosis: progress & paradoxes. The FEBS Journal . 2021;10 doi: 10.1111/febs.15842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tu H., Tang L. J., Luo X. J., Ai K. L., Peng J. Insights into the novel function of system xc- in regulated cell death. European Review for Medical and Pharmacological Sciences . 2021;25(3):1650–1662. doi: 10.26355/eurrev_202102_24876. [DOI] [PubMed] [Google Scholar]
- 56.Proneth B., Conrad M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death and Differentiation . 2019;26(1):14–24. doi: 10.1038/s41418-018-0173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou S. Y., Cui G. Z., Yan X. L., et al. Mechanism of ferroptosis and its relationships with other types of programmed cell death: insights for potential interventions after intracerebral hemorrhage. Frontiers in Neuroscience . 2020;14, article 589042 doi: 10.3389/fnins.2020.589042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gaschler M. M., Stockwell B. R. Lipid peroxidation in cell death. Biochemical and Biophysical Research Communications . 2017;482(3):419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haeggstrom J. Z., Funk C. D. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chemical Reviews . 2011;111(10):5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 60.Shah R., Shchepinov M. S., Pratt D. A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Central Science . 2018;4(3):387–396. doi: 10.1021/acscentsci.7b00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan H. F., Zou T., Tuo Q. Z., et al. Ferroptosis: mechanisms and links with diseases. Signal Transduction and Targeted Therapy . 2021;6(1):p. 49. doi: 10.1038/s41392-020-00428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doll S., Proneth B., Tyurina Y. Y., et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature Chemical Biology . 2017;13(1):91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng H., Stockwell B. R. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biology . 2018;16(5, article e2006203) doi: 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ayala A., Munoz M. F., Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Medicine and Cellular Longevity . 2014;2014 doi: 10.1155/2014/360438.360438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang W. S., Kim K. J., Gaschler M. M., Patel M., Shchepinov M. S., Stockwell B. R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proceedings of the National Academy of Sciences of the United States of America . 2016;113(34):E4966–E4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.D'Amelio P., Cristofaro M. A., Tamone C., et al. Role of iron metabolism and oxidative damage in postmenopausal bone loss. Bone . 2008;43(6):1010–1015. doi: 10.1016/j.bone.2008.08.107. [DOI] [PubMed] [Google Scholar]
- 67.Okabe H., Suzuki T., Uehara E., Ueda M., Nagai T., Ozawa K. The bone marrow hematopoietic microenvironment is impaired in iron-overloaded mice. European Journal of Haematology . 2014;93(2):118–128. doi: 10.1111/ejh.12309. [DOI] [PubMed] [Google Scholar]
- 68.Che J., Yang J., Zhao B., et al. The effect of abnormal iron metabolism on osteoporosis. Biological Trace Element Research . 2020;195(2):353–365. doi: 10.1007/s12011-019-01867-4. [DOI] [PubMed] [Google Scholar]
- 69.Balogh E., Tolnai E., Nagy B., Jr., et al. Iron overload inhibits osteogenic commitment and differentiation of mesenchymal stem cells via the induction of ferritin. Biochimica et Biophysica Acta . 1862;2016:1640–1649. doi: 10.1016/j.bbadis.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 70.Cheng Q., Zhang X., Jiang J., et al. Postmenopausal iron overload exacerbated bone loss by promoting the degradation of type I collagen. BioMed Research International . 2017;2017 doi: 10.1155/2017/1345193.1345193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao H., Ito Y., Chappel J., Andrews N. W., Teitelbaum S. L., Ross F. P. Synaptotagmin VII regulates bone remodeling by modulating osteoclast and osteoblast secretion. Developmental Cell . 2008;14(6):914–925. doi: 10.1016/j.devcel.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kolodziejska B., Stepien N., Kolmas J. The influence of strontium on bone tissue metabolism and its application in osteoporosis treatment. International Journal of Molecular Sciences . 2021;22(12):p. 6564. doi: 10.3390/ijms22126564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Song X., Xie Y., Kang R., et al. FANCD2 protects against bone marrow injury from ferroptosis. Biochemical and Biophysical Research Communications . 2016;480(3):443–449. doi: 10.1016/j.bbrc.2016.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jia P., Xu Y. J., Zhang Z. L., et al. Ferric ion could facilitate osteoclast differentiation and bone resorption through the production of reactive oxygen species. Journal of Orthopaedic Research . 2012;30(11):1843–1852. doi: 10.1002/jor.22133. [DOI] [PubMed] [Google Scholar]
- 75.Ishii K. A., Fumoto T., Iwai K., et al. Coordination of PGC-1β and iron uptake in mitochondrial biogenesis and osteoclast activation. Nature Medicine . 2009;15(3):259–266. doi: 10.1038/nm.1910. [DOI] [PubMed] [Google Scholar]
- 76.Ni S., Yuan Y., Qian Z., et al. Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radical Biology & Medicine . 2021;169:271–282. doi: 10.1016/j.freeradbiomed.2021.04.027. [DOI] [PubMed] [Google Scholar]
- 77.Florencio-Silva R., Sasso G. R., Sasso-Cerri E., Simoes M. J., Cerri P. S. Biology of bone tissue: structure, function, and factors that influence bone cells. BioMed Research International . 2015;2015 doi: 10.1155/2015/421746.421746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu J., Yang J., Zheng Y., Chen X., Fang S. Extracellular vesicles from endothelial progenitor cells prevent steroid- induced osteoporosis by suppressing the ferroptotic pathway in mouse osteoblasts based on bioinformatics evidence. Scientific Reports . 2019;9(1):p. 16130. doi: 10.1038/s41598-019-52513-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma H., Wang X., Zhang W., et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxidative Medicine and Cellular Longevity . 2020;2020 doi: 10.1155/2020/9067610.9067610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang X., Ma H., Sun J., et al. Mitochondrial ferritin deficiency promotes osteoblastic ferroptosis via mitophagy in type 2 diabetic osteoporosis. Biological Trace Element Research . 2022;200(1):298–307. doi: 10.1007/s12011-021-02627-z. [DOI] [PubMed] [Google Scholar]
- 81.Compston J. E., McClung M. R., Leslie W. D. Osteoporosis. Lancet . 2019;393(10169):364–376. doi: 10.1016/S0140-6736(18)32112-3. [DOI] [PubMed] [Google Scholar]
- 82.Yamamoto Y., Chiba T., Dohmae S., Higashi K., Nakazawa A. Osteoporosis medication after fracture in older adults: an administrative data analysis. Osteoporosis International . 2021;32(6):1245–1246. doi: 10.1007/s00198-021-05973-9. [DOI] [PubMed] [Google Scholar]
- 83.de Wit M., Cooper C., Tugwell P., et al. Practical guidance for engaging patients in health research, treatment guidelines and regulatory processes: results of an expert group meeting organized by the World Health Organization (WHO) and the European Society for Clinical and Economic Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (ESCEO) Aging Clinical and Experimental Research . 2019;31(7):905–915. doi: 10.1007/s40520-019-01193-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sozen T., Ozisik L., Calik Basaran N. An overview and management of osteoporosis. European Journal of Rheumatology . 2017;4(1):46–56. doi: 10.5152/eurjrheum.2016.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hernlund E., Svedbom A., Ivergard M., et al. Osteoporosis in the European Union: medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA) Archives of Osteoporosis . 2013;8(1-2):p. 136. doi: 10.1007/s11657-013-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bass M. A., Sharma A., Nahar V. K., et al. Bone mineral density among men and women aged 35 to 50 years. The Journal of the American Osteopathic Association . 2019;119(6):357–363. doi: 10.7556/jaoa.2019.064. [DOI] [PubMed] [Google Scholar]
- 87.Zheng Y., Ley S. H., Hu F. B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nature Reviews Endocrinology . 2018;14(2):88–98. doi: 10.1038/nrendo.2017.151. [DOI] [PubMed] [Google Scholar]
- 88.Anagnostis P., Paschou S. A., Gkekas N. N., et al. Efficacy of anti-osteoporotic medications in patients with type 1 and 2 diabetes mellitus: a systematic review. Endocrine . 2018;60(3):373–383. doi: 10.1007/s12020-018-1548-x. [DOI] [PubMed] [Google Scholar]
- 89.Farr J. N., Drake M. T., Amin S., Melton L. J., 3rd, McCready L. K., Khosla S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. Journal of Bone and Mineral Research . 2014;29(4):787–795. doi: 10.1002/jbmr.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Epstein S., LeRoith D. Diabetes and fragility fractures -- a burgeoning epidemic? Bone . 2008;43(1):3–6. doi: 10.1016/j.bone.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 91.Napoli N., On behalf of the IOF Bone, Diabetes Working Group, et al. Mechanisms of diabetes mellitus-induced bone fragility. Nature Reviews Endocrinology . 2017;13(4):208–219. doi: 10.1038/nrendo.2016.153. [DOI] [PubMed] [Google Scholar]
- 92.Eller-Vainicher C., Cairoli E., Grassi G., et al. Pathophysiology and management of type 2 diabetes mellitus bone fragility. Journal Diabetes Research . 2020;2020, article 7608964 doi: 10.1155/2020/7608964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rajpathak S. N., Crandall J. P., Wylie-Rosett J., Kabat G. C., Rohan T. E., Hu F. B. The role of iron in type 2 diabetes in humans. Biochimica et Biophysica Acta . 2009;1790(7):671–681. doi: 10.1016/j.bbagen.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 94.Liu J., Li Q., Yang Y., Ma L. Iron metabolism and type 2 diabetes mellitus: a meta-analysis and systematic review. Journal of Diabetes Investigation . 2020;11(4):946–955. doi: 10.1111/jdi.13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Simcox J. A., McClain D. A. Iron and diabetes risk. Cell Metabolism . 2013;17(3):329–341. doi: 10.1016/j.cmet.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Forcina G. C., Dixon S. J. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics . 2019;19(18, article e1800311) doi: 10.1002/pmic.201800311. [DOI] [PubMed] [Google Scholar]
- 97.Yang R. Z., Xu W. N., Zheng H. L., et al. Exosomes derived from vascular endothelial cells antagonize glucocorticoid- induced osteoporosis by inhibiting ferritinophagy with resultant limited ferroptosis of osteoblasts. Journal of Cellular Physiology . 2021;236(9):6691–6705. doi: 10.1002/jcp.30331. [DOI] [PubMed] [Google Scholar]
- 98.Eastell R. Treatment of postmenopausal osteoporosis. The New England Journal of Medicine . 1998;338(11):736–746. doi: 10.1056/NEJM199803123381107. [DOI] [PubMed] [Google Scholar]
- 99.Okyay E., Ertugrul C., Acar B., Sisman A. R., Onvural B., Ozaksoy D. Comparative evaluation of serum levels of main minerals and postmenopausal osteoporosis. Maturitas . 2013;76(4):320–325. doi: 10.1016/j.maturitas.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 100.Abraham R., Walton J., Russell L., et al. Dietary determinants of post-menopausal bone loss at the lumbar spine: a possible beneficial effect of iron. Osteoporosis International . 2006;17(8):1165–1173. doi: 10.1007/s00198-005-0033-6. [DOI] [PubMed] [Google Scholar]