

Abstract
Evidence from natural systems suggests that hybridization between animal species is more common than traditionally thought, but the overall contribution of introgression to standing genetic variation within species remains unclear for most animal systems. Here, we use targeted exon capture to sequence thousands of nuclear loci and complete mitochondrial genomes from closely related chipmunk species in the Tamias quadrivittatus group that are distributed across the Great Basin and the central and southern Rocky Mountains of North America. This recent radiation includes six overlapping, ecologically distinct species (Tamias canipes, Tamias cinereicollis, Tamias dorsalis, T. quadrivittatus, Tamias rufus, and Tamias umbrinus) that show evidence for widespread introgression across species boundaries. Such evidence has historically been derived from a handful of markers, typically focused on mitochondrial loci, to describe patterns of introgression; consequently, the extent of introgression of nuclear genes is less well characterized. We conducted a series of phylogenomic and species-tree analyses to resolve the phylogeny of six species in this group. In addition, we performed several population-genomic analyses to characterize nuclear genomes and infer coancestry among individuals. Furthermore, we used emerging quartets-based approaches to simultaneously infer the species tree (SVDquartets) and identify introgression (HyDe). We found that, in spite of rampant introgression of mitochondrial genomes between some species pairs (and sometimes involving up to three species), there appears to be little to no evidence for nuclear introgression. These findings mirror other genomic results where complete mitochondrial capture has occurred between chipmunk species in the absence of appreciable nuclear gene flow. The underlying causes of recurrent massive cytonuclear discordance remain unresolved in this group but mitochondrial DNA appears highly misleading of population histories as a whole. Collectively, it appears that chipmunk species boundaries are largely impermeable to nuclear gene flow and that hybridization, while pervasive with respect to mtDNA, has likely played a relatively minor role in the evolutionary history of this group. [Cytonuclear discordance; hyridization; introgression, phylogenomics; SVDquartets; Tamias.]
Over the last few decades, evidence has accumulated that hybridization is a widespread evolutionary process (e.g., Mallet 2005; Taylor and Larson 2019). This realization has led to the view of semipermeable species boundaries (reviewed in Harrison and Larson 2014), whereby introgression of neutral or potentially adaptive heterospecific variation is counteracted by selection against generally deleterious or hybrid incompatible alleles (Harris and Nielsen 2016; Schumer et al. 2018; Edelman et al. 2019). Hybridization can lead to an array of outcomes (reviewed by Runemark et al. 2019) including despeciation (or at least lineage fusion; e.g., Kearns et al. 2018), extinction (e.g., via genetic swamping; Todesco et al. 2016), formation of hybrid zones (see e.g., Teeter et al. [2008], for a well-studied hybrid zone in Mus), introgression of adaptation (e.g., Heliconius Genome Consortium 2012), and even hybrid speciation (e.g., Lachemilla; Morales-Briones et al. 2018). However, the overall contribution of introgression to standing genetic variation within species remains unclear for most animal systems (Good et al. 2015; examples in Bonnet et al. 2017). This gap in understanding is particularly acute for recent species radiations, where patterns of gene flow among groups of closely related species are often difficult to differentiate from patterns generated by stochastic processes (e.g., incomplete lineage sorting [ILS] Malinsky et al. 2018; He et al. 2019; Ferreira et al. 2020).
The radiation of western North American chipmunks (Sciuridae: Tamias) provide an intriguing system in which to examine the nature of species boundaries and the contribution of introgression to standing variation within and among species. The genus Tamias includes monotypic species distributed throughout Asia (Tamias sibiricus, subgenus Eutamias) and eastern North America (Tamias striatus, subgenus Tamias), punctuated by a recent radiation of at least 23 species inhabiting diverse ecosystems across western North America (subgenus Neotamias; but see Piaggio and Spicer 2001; Patterson and Norris 2016 for an alternative classification). Western chipmunk species often show complex patterns of parapatry with broadly overlapping ranges locally partitioned by ecological preference and competitive exclusion (e.g., Grinnell and Storer 1924; Brown 1971; Heller 1971). They can also often be unambiguously identified by their genital morphology, with the baculum (os penis) providing a key diagnostic character of species limits (White 1953; Sutton 1982, 1992). Chipmunk bacula evolve rapidly, presumably driven by sexual selection (e.g., Eberhard 1985; Simmons and Firman 2013; Schultz et al. 2016) and have been posited to contribute to reproductive barriers between nascent species (Patterson and Thaeler 1982; Good et al. 2003).
Despite rapid genital evolution (e.g., Patterson and Thaeler 1982) and strong ecological preferences usually defining clear species boundaries (e.g., Brown 1971), western chipmunks also show evidence of widespread introgression of mtDNA between some species. Sullivan et al. (2014) summarized work from the previous 13+ years in this system and outlined several well-studied cases of introgression. Extensive mtDNA introgression within two chipmunk subgroups has been described in a series of studies (Good and Sullivan 2001; Good et al. 2003, 2008; Hird et al. 2010; Reid et al. 2010, 2012; Sullivan et al. 2014; Good et al. 2015). Initially, asymmetric introgression from T. ruficaudus into T. amoenus was described in the northern Rocky Mountains (Good et al. 2003, 2008; Hird et al. 2010; Reid et al. 2010). Subsequent work (Reid et al. 2012; Sullivan et al. 2014; Sarver et al. 2017) documented the extent of mtDNA introgression in the central and southern Rocky Mountains, specifically among six species in the T. quadrivittatus group; mtDNA introgression appears rampant among four of the six species. The ranges of these species show extensive overlap (Fig. 1), with broad zones of parapatry occurring at transitions between montane forest communities. In particular, populations of the cliff chipmunk (T. dorsalis) appear to be particularly prone to local introgression. This relatively widespread species shares parapatric contact zones with several of the other species along transitions from pinyon-juniper woodlands to other montane forest communities and appears to be locally fixed for the mitochondrial genome of whichever congener it contacts (Sullivan et al. 2014; Supplementary Fig. S2 available on Dryad at http://dx.doi.org/10.5061/dryad.6t1g1jwws). Indeed, mtDNA haplotypes that are specific to T. dorsalis only have been found in allopatric populations that therefore do not contact any of the other species (Sullivan et al. 2014; Sarver et al. 2017; Supplementary Fig. S2 available on Dryad).
Figure 1.

Ranges of the six species of chipmunk examined here (from Sullivan et al. 2014).
In addition to the recurrent, if not widespread, hybridization in western chipmunks (Sullivan et al. 2014), mtDNA is more generally prone to extensive introgression, presumably due to interactions of strong genetic drift, purifying selection, and perhaps positive selection (Ballard and Whitlock 2004; Sloan et al. 2017). However, little work has focused on the extent of introgression across the nuclear genomes of chipmunks. Good et al. (2015) used a targeted sequence-capture approach (Bi et al. 2012, 2013) to sequence whole mtDNA genomes and  10,500 protein-coding exonic regions (
10,500 protein-coding exonic regions ( 4 megabases of target) to quantify introgression at nuclear loci in T. amoenus canicaudus, a taxon that is fixed for an ancient mitochondrial genome introgression from T. ruficaudus (Good et al. 2003, 2008). Surprisingly, they found no evidence for appreciable nuclear introgression, indicating that complete mtDNA capture likely resulted from relatively ancient and rare hybridization. One other genome-wide study (Bi et al. 2019) has detected rare nuclear introgression between paraptric populations of lodgepole (T. speciosus) and least chipmunks (T. minimus).
4 megabases of target) to quantify introgression at nuclear loci in T. amoenus canicaudus, a taxon that is fixed for an ancient mitochondrial genome introgression from T. ruficaudus (Good et al. 2003, 2008). Surprisingly, they found no evidence for appreciable nuclear introgression, indicating that complete mtDNA capture likely resulted from relatively ancient and rare hybridization. One other genome-wide study (Bi et al. 2019) has detected rare nuclear introgression between paraptric populations of lodgepole (T. speciosus) and least chipmunks (T. minimus).
Here, we use a targeted sequence-capture approach to sequence thousands of loci from 51 chipmunks across six species in the T. quadrivittatus group using the array-based targeted sequence-capture approach of Good et al. (2015; designed and characterized in Bi et al. 2012, 2013). We first use these data to evaluate the relationships among these species using a variety of species-tree techniques. We then use the same genome-scale data to investigate the population genomics of this group and assess introgression at nuclear loci.
Materials and Methods
Samples, Preparation, and Sequencing
Fifty-one T. quadrivittatus-group chipmunks (5 T. canipes, 9 T. cinereicollis, 11 T. dorsalis, 11 T. quadrivittatus, 5 T. rufus, and 10 T. umbrinus) were selected from the 231 individuals used by Sullivan et al. (2014) to characterize mtDNA introgression. For each species, we included individuals inferred to have introgressed and nonintrogressed mtDNA, the same individuals that we selected for the mitochondrial genome sequencing study of Sarver et al. (2017). We also included data from one T. striatus individual published by Bi et al. (2012) as an outgroup. We recently reported results of that experiment for the targeted assembly of whole mitochondrial genomes. Collection localities are resented in Supplementary Table S1 available on Dryad, and all specimens are deposited in the Denver Museum of Nature & Science (DMNS).
DNA was isolated from heart or liver tissue using Qiagen DNEasy DNA extraction kits. Samples were eluted into 50  L of 10 mM Tris-Cl and stored at 4
L of 10 mM Tris-Cl and stored at 4 C before use. We then performed targeted capture using custom Agilent SureSelect 1M microarrays following Bi et al. (2012, 2013). Samples were sequenced on two lanes of an Illumina HiSeq 2000 (100 bp paired-end sequencing) at the Vincent Coates Genome Sequencing Laboratory at the University of California-Berkeley. After sequencing, reads were processed using a comprehensive cleaning pipeline. PCR duplicates were removed using a custom script and were screened for quality and residual adapters using SeqyClean (http://bitbucket.org/izhbannikov/seqyclean), and overlapping reads were merged using Flash (Magoè and Salzberg 2011). Population genetic analyses used the same pipeline without Flash overlapping.
C before use. We then performed targeted capture using custom Agilent SureSelect 1M microarrays following Bi et al. (2012, 2013). Samples were sequenced on two lanes of an Illumina HiSeq 2000 (100 bp paired-end sequencing) at the Vincent Coates Genome Sequencing Laboratory at the University of California-Berkeley. After sequencing, reads were processed using a comprehensive cleaning pipeline. PCR duplicates were removed using a custom script and were screened for quality and residual adapters using SeqyClean (http://bitbucket.org/izhbannikov/seqyclean), and overlapping reads were merged using Flash (Magoè and Salzberg 2011). Population genetic analyses used the same pipeline without Flash overlapping.
A reference Tamias genome assembly has not yet been published. Therefore, reads were assembled iteratively into contigs using Assembly by Reduced Complexity (ARC, Hunter et al. 2015; http://ibest.github.io/ARC/). This approach uses a set of contigs (i.e., targets) as a starting point for assembly; here, we used the same genomic regions used to design the capture probes as reference set (see Bi et al. 2012). Cleaned reads were mapped to the sequence targets and each pool of reads was assembled de novo. The resulting assembled contigs were then used iteratively as a new target set for another round of mapping and assembly, and this process was repeated until no new reads were recruited in successive generations (see Hunter et al. 2015). ARC has the ability to recruit additional reads throughout iterations thereby increasing the length of the assembled contigs beyond the original target sequences and into flanking regions. As targets were originally designed from a multi-tissue de novo transcriptome assembly (Bi et al. 2012), which potentially includes closely related sequences from gene families or multiple transcripts from the same gene, we removed any targets that included potentially redundant exons. Individual ARC assemblies were generated for each individual. In order to capture heterozygous sites and correct assembled sequencing errors, sequencing reads were mapped to each ARC contig using BWA v0.7.10 (Li and Durbin 2010), followed by variant calling using UnifiedGenotyper in the Genome Analysis Toolkit v3.1 GATK (McKenna et al. 2010; DePristo et al. 2011). High-quality heterozygous sites, as identified through high mapping quality and depth of coverage, were translated into their corresponding IUPAC ambiguity codes and injected back into the ARC contig.
Population Genomics
For population-genomic analyses, the ARC contig set from one T. umbrinus individual (umb 600, catalogued in DMNS as ZM.11687) was arbitrarily selected to serve as a genomic reference. In order to eliminate potential artifacts associated with misassembly, the ARC assembly for this individual was pruned to include targets that produced three or fewer contigs. Genotypes were then called for each individual by comparison to this reference. Cleaned reads were aligned to the ARC assembly using Bowtie 2 v2.1.0 (Langmead and Salzberg 2012). Indels were realigned using the Genome Analysis Toolkit (GATK; McKenna et al. 2010; DePristo et al. 2011), variants were phased, and missing genotypes were imputed with BEAGLE v4.0 (Browning and Browning 2007, 2009; Browning and Yu 2009). Variants were then screened using VCFtools v0.1.12a (Danecek et al. 2011) to remove sites that violated Hardy–Weinberg Equilibrium (HWE) per species group at a  value of 0.05. Remaining variants were filtered using a minor allele frequency cutoff of 0.05.
value of 0.05. Remaining variants were filtered using a minor allele frequency cutoff of 0.05.
Individual coancestry was estimated using ADMIXTURE v1.23 (Alexander et al. 2009), with 10 rounds of crossvalidation and 10 values of K (number of genotypic clusters) after selecting a single variant from each ARC contig to account for linkage. The same single-variant data set was used for a multidimensional scaling analysis visualized in two dimensions to provide an overall assessment of population-genomic structure using PLINK v.1.07 (Purcell et al. 2007). Additionally, Weir and Cockerham’s F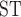 (Weir and Cockerham 1984) and average allele frequencies were estimated for all single nucleotide variants (SNVs) across all species pairs using the Genotype–Phenotype Association Toolkit
(Weir and Cockerham 1984) and average allele frequencies were estimated for all single nucleotide variants (SNVs) across all species pairs using the Genotype–Phenotype Association Toolkit  , part of vcflib (available at https://github.com/vcflib/vcflib). F
, part of vcflib (available at https://github.com/vcflib/vcflib). F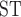 values less than zero were converted to zero. The number of heterozygotous calls, observed heterozygosity, and F
values less than zero were converted to zero. The number of heterozygotous calls, observed heterozygosity, and F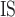 were also calculated for each species. SNV positions that were not polymorphic within a given population were removed before calculating statistics. For comparison, F
were also calculated for each species. SNV positions that were not polymorphic within a given population were removed before calculating statistics. For comparison, F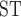 was also calculated using DnaSP (Librado and Rozas 2009) from a concatenated set of mitochondrial protein-coding genes assembled for Sarver et al. (2017).
was also calculated using DnaSP (Librado and Rozas 2009) from a concatenated set of mitochondrial protein-coding genes assembled for Sarver et al. (2017).
Phylogenetic Analyses
The final contig set produced by ARC was processed using Biostrings (Pagès et al. 2016) and native libraries in R v3.0.2 (R Core Team 2013). In particular, all results were trimmed to include only targets where ARC produced a single contig across all libraries. A multiple sequence alignment was then performed on each set of sequences using MUSCLE v3.8.31 (Edgar 2004). The resulting matrices were subsequently squared by trimming hanging ends to reduce the amount of missing data across samples.
We inferred phylogenies with this set of sequences using several approaches. First, all contigs were concatenated using Phyutility (Smith and Dunn 2008). A phylogeny was inferred using RAxML v8.0.5 (Stamatakis 2014) and a full ML search across 1000 bootstrap replicates under a GTR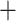 I
I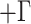 model of nucleotide sequence evolution. RAxML was used here because of the size of this data set and its computational efficiencies. De novo assemblers produce haploid references and do not incorporate heterozygous calls into resulting contigs; instead, they either use the first base encountered as the reference call or they use a majority-rule call, effectively removing heterozygous calls. Therefore, in order to assess the impact of heterozygous sites, we also constructed a data set consisting of ARC contigs without heterozygous sites (i.e., contigs resulting from assembly with no subsequent modification). This data set was subject to the same processing treatment as the data set above, and the same phylogenetic inference was performed using RAxML v8.0.5 (Stamatakis 2014). Thus, phylogenies were inferred using a one data set that is agnostic to heterozygous sites and another data set without heterozygous sites.
model of nucleotide sequence evolution. RAxML was used here because of the size of this data set and its computational efficiencies. De novo assemblers produce haploid references and do not incorporate heterozygous calls into resulting contigs; instead, they either use the first base encountered as the reference call or they use a majority-rule call, effectively removing heterozygous calls. Therefore, in order to assess the impact of heterozygous sites, we also constructed a data set consisting of ARC contigs without heterozygous sites (i.e., contigs resulting from assembly with no subsequent modification). This data set was subject to the same processing treatment as the data set above, and the same phylogenetic inference was performed using RAxML v8.0.5 (Stamatakis 2014). Thus, phylogenies were inferred using a one data set that is agnostic to heterozygous sites and another data set without heterozygous sites.
Second, we used SVDquartets (Chifman and Kubatko 2014) as implemented in PAUP* (v 4.0a build 167; Swofford 2017) using the concatenated alignment as input to infer the lineage tree of all individuals using all possible quartets and 100 bootstrap replicates. We optimized branch lengths on the SVDquartets tree using maximum likelihood under the GTR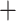 I
I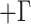 model.
model.
We then conducted a series of species-tree inferences using approaches that either infer gene trees first and then estimate the species tree or that estimate the species tree directly from the data. Inferring a species tree from gene trees is contingent on accurate gene trees (e.g., Roch and Warnow 2015). The majority of contigs were relatively short and contained fewer than 13 variable positions (Supplementary Fig. S1 available on Dryad); therefore, we binned contigs using two approaches. First, we implemented a naïve binning approach (Bayzid and Warnow 2013) in which all contigs were randomly assigned without replacement into 10 bins of equal size. Phylogenies were estimated from 25 random binnings. Model selection was performed on each contig set using DT-ModSel (Minin et al. 2003) and phylogenetic trees were estimated using Garli v2.01 (Zwickl 2006) under the selected model and a termination threshold of 0.01 for 50,000 generations. Ten independent search replicates were performed and the best tree among replicates was used for analysis. Each tree was made ultrametric using treePL (Smith and O’Meara 2012) fixing the three T. striatus samples as an outgroup with the minimum split time set to 7 Ma (Sullivan et al. 2014). Results of analyses from naïve-binning approaches were similar to, but less resolved than those from moderately informed binning (described below) and are not discussed further.
We then used a binning strategy informed by studies of syntenic groups. We approximated the chromosomal location of each ARC contig relative to the GRCm38 reference assembly of Mus musculus using BLAT (Kent 2002). Despite deep divergence between murids and sciurids (56–94 Ma; Swanson et al. 2019), chromosome painting studies (Li et al. 2004) have demonstrated moderate conservatism in karyotypes and local synteny between murids and sciurids. We required that all individuals to be assigned unambiguously to the same Mus chromosome in order for assignment to a chromosome. Contigs were then concatenated based on their chromosome assignment. Model selection, phylogenetic inference, and ultrametric transformation were performed as described above. We did not conduct statistical binning because such approaches underrepresent coalscent stochasticity. ASTRAL-III (v5.7.3; Zhang et al. 2018) was used to infer a species tree from quartets of taxa induced by the collections of gene trees generated using each binning approach. This was done for both nuclear gene trees alone and for nuclear gene trees plus the mtDNA genome tree. We used default parameters and mapped all individuals to their respective species following Sullivan et al. (2014).
Conversely, SVDquartets (Chifman and Kubatko 2014) assesses support for quartets of taxa directly from pattern frequencies in the SNP data. In doing so, this approach evaluates the support for all three possible resolutions for each quartet, which is an enormous advantage in instances where introgressive hybridization may contribute to gene-tree/species-tree discordance, as may be the case here. Specifically, by evaluating support for all three resolutions for each quartet, the approach detects deviations from site-pattern frequencies expected under the multispecies coalescent (Kubatko and Chifman 2019). Therefore, we used SVDquartets as implemented in PAUP* (v 4.0a build 167; 2017) on the same data set to estimate the species tree using all possible quartets, the multispecies coalescent, and 100 bootstrap replicates.
Assessment of Introgression
As indicated above, the invariants framework of SVDquartets and HyDe provides a well-justified approach for assessing introgression (Blischak et al. 2018; Kubatko and Chifman 2019) in a manner analogous to the ABBA-BABA test (Green et al. 2010; Durand et al. 2011). Just as for ABBA-BABA tests, the quartet resolution corresponding to the species tree is expected to have the majority of support (i.e., the invariant, or quartet score, will approach zero), whereas, in the absence of introgression, the two alterative resolutions of the quartet will show symmetrically low support (the invariants will be similarly larger than zero). Conversely, in the presence of introgression, one of the alternative resolutions should show more support than the other resolution (Kubatko and Chifman 2019). Thus, these analytical frameworks permit the detection of incongruence with the species tree exceeding that which is attributable to coalescent stochasticity (i.e., ILS).
Using the mtDNA genome tree as a guide (Sullivan et al. 2014; Sarver et al. 2017), we hypothesize multiple independent introgression events from different sources. For example, T. dorsalis has likely hybridized with T. cinereicollis at least twice (introgressed mtDNA in dor 711 and in dor 236), with T. qudrivittatus at least once (dor 217), and with T. umbrinus at least three times (dor 210, dor 582, dor 605). Furthermore, these apparent instances of introgression are geographically restricted and are likely to be independent (Sullivan et al. 2014; Sarver et al. 2017; Supplementary Figs. S2–S5 available on Dryad). Thus, we conducted separate analyses for each putative introgression event; Table 1 lists the quartet of taxa included in each of the sets of analyses. For each quartet analyzed, we calculated the support scores for each of the three possible topologies for the quartet using PAUP* (SVDQuartets evalQuartets=all showScores) to visualize the strength of support across the tree resolutions. We then input sequences into HyDe to assess the statistical support for our assessment of introgression. The Hils statistic, developed by Kubatko and Chifman (2019), quantifies the asymmetry in support between the alternative resolutions of the quartet. HyDe (Blischak et al. 2018) uses a normal distribution to infer the probability of an asymmetry as or more extreme than the test statistic being observed under coalescent stochasticity in the absence of hybridization. For each set of taxa, we contrasted quartet scores and  values from analyses of only the nuclear genome data (221,556 sites, Nuclear Data) and of the combined nuclear plus mtDNA genome sequence data (238,056 sites, All Data; 221,556 sites from the nuclear genomes plus 16,500 sites from the mtDNA genomes).
values from analyses of only the nuclear genome data (221,556 sites, Nuclear Data) and of the combined nuclear plus mtDNA genome sequence data (238,056 sites, All Data; 221,556 sites from the nuclear genomes plus 16,500 sites from the mtDNA genomes).
Table 1.
Taxa selected to assess introgression in T. dorsalis
| Supplementary | Supplementary | Supplementary | Supplementary | Supplementary | |
|---|---|---|---|---|---|
| Figure S2A | Figure S2B | Figure S2C | Figure S2D | Figure S2E | |
| available | available | available | available | available | |
| Figure 5 | on Dryad | on Dryad | on Dryad | on Dryad | on Dryad |
| dor 713 | dor 713 | dor 713 | dor 713 | dor 713 | dor 713 |
| dor 711* | dor 201* | dor 605* | dor 582* | dor 236* | dor 217* |
| cin 213 | umb 256 | umb 600 | umb 251 | cin 226 | qua 85 |
| st 11 | st 11 | st 11 | st 11 | st 11 | st 11 |
dor  T. dorsalis; cin
T. dorsalis; cin  T. cinereicollis; umb
T. cinereicollis; umb  T. umbrinus; qua
T. umbrinus; qua  T. quadrivittatus; s
T. quadrivittatus; s  , T. striatus
, T. striatus
Results
Assembly and Processing
The pruned ARC targets file included a total length of 4,003,445 bp of sequence data consisting of 7627 genes and 11,976 exons or targeted loci. ARC performed a total of 1,300,188 de novo assemblies across all targets and individuals. Additionally, 5640 assemblies were terminated due to the incorporation of a large number of reads relative to the previous ARC iteration or assembly timeout ( 20 min walltime), most likely due to the incorporation of difficult-to-resolve repetitive sequences.
20 min walltime), most likely due to the incorporation of difficult-to-resolve repetitive sequences.
For molecular phylogenetics, assembled contigs were only included in the final analysis if they were present across all individuals to reduce the effect of missing data and low confidence in placement of a sample on the gene tree. Furthermore, in order to avoid possible errors due to improper resolution of sequence order and incorrect inference of orthology (i.e., two contigs from a single gene may or may not be called in a consistent order across assemblies due to the stochastic nature of the assembly process), only genes that produced a single contig were included in downstream analyses. This resulted in 1106 loci. After alignment, this set was truncated to remove sequences that were 100% identical among all libraries and assemblies that contained an extremely large number of substitutions, which is indicative of assembly errors. This generated a working set of 1060 loci with from 0.33% to 15.2% variable sites, but the majority of loci had five or fewer variable positions (including the outgroup). The final alignment consists of 221,556 bp per individual with no missing data.
The T. umbrinus reference (umb 600) was generated by selecting targets for which ARC produced three or fewer contigs per gene. Of the 7627 capture genes, 6827 (89.5%) met this criterion; 10,088 of 11,976 (84.2%) loci were included, and 4326 (56.7%) genes were resolved as a single contig for this library. The cutoff of three or fewer contigs per gene is conservative but nevertheless recovered approximately 85% of targeted sequences.
Population Genomics
There were, on average, 4,667,279 genotyped sites per library. A total of 218,792 variable sites consisting of 214,149 SNVs and 4643 indels were identified. A small fraction of positions exhibited three or more allelic states (6277; 0.13% of all sites, 2.90% of variable sites). Two population-level filters were applied to the raw VCF; the first removed sites where fewer than 75% of individuals were genotyped, and the second removed sites with a minor allele frequency less than 1%. After filtering, 180,879 variable sites remained (176,554 SNVs and 4325 indels) for analysis. Filtering on HWE and minor allele frequency (a final filtering at 5%) resulted in a final count of 111,441 SNVs. Selection of one variant per contig resulted in a thinned data set of 7530 SNVs for population assignment analyses.
Average observed heterozygosity ranged from 0.11% to 0.14% with T. umbrinus having the lowest diversity. F estimates range from 0.0374 to 0.0755 (Table 2). Pairwise F
estimates range from 0.0374 to 0.0755 (Table 2). Pairwise F values showed clear differences among species (Table 3). Any comparison that contained T. umbrinus had a higher F
values showed clear differences among species (Table 3). Any comparison that contained T. umbrinus had a higher F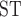 relative to other comparisons (
relative to other comparisons (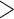 0.38). F
0.38). F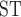 values including T. canipes, T. dorsalis, or T. rufus showed intermediate estimates. The T. quadrivittatus and T. cinereicollis estimate was the lowest (0.10). There was a stark contrast between values calculated from mitochondrial and nuclear data. In some comparisons, such as those involving T. rufus and T. canipes (which show no evidence of mitochondrial introgression; Sullivan et al. 2014; Sarver et al. 2017), F
values including T. canipes, T. dorsalis, or T. rufus showed intermediate estimates. The T. quadrivittatus and T. cinereicollis estimate was the lowest (0.10). There was a stark contrast between values calculated from mitochondrial and nuclear data. In some comparisons, such as those involving T. rufus and T. canipes (which show no evidence of mitochondrial introgression; Sullivan et al. 2014; Sarver et al. 2017), F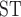 values were large and approach one in some cases. Other comparisons, especially those with T. dorsalis, have lower F
values were large and approach one in some cases. Other comparisons, especially those with T. dorsalis, have lower F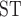 values, with some approaching 0.05.
values, with some approaching 0.05.
Table 2.
Population genetic summary statistics
| Mean | Total number of | Mean | ||
|---|---|---|---|---|
| number of | heterozygous | observed | ||
| Species | sites | genotypes | heterozygosity | F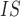
|
| T. canipes | 4,298,388 | 30,658 | 0.0014 | 0.0473 |
| T. cinereicollis | 4,793,771 | 61,382 | 0.0014 | 0.0465 |
| T. dorsalis | 4,826,024 | 73,022 | 0.0014 | 0.0683 |
| T. quadrivittatus | 4,564,067 | 67,181 | 0.0013 | 0.0479 |
| T. rufus | 4,409,452 | 29,748 | 0.0013 | 0.0374 |
| T. umbrinus | 4,781,474 | 54,540 | 0.0011 | 0.0755 |
Mean number of sites is the total length of sequence data that has a sequencing depth of at least one read averaged across all individuals in each species pool. F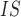 is calculated per SNP for each species pool and then averaged over all sites. Noninformative sites are removed before calculation.
is calculated per SNP for each species pool and then averaged over all sites. Noninformative sites are removed before calculation.
Table 3.
Pairwise F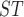 estimates
estimates
| T. canipes | T. cinereicollis | T. dorsalis | T. quadrivittatus | T. rufus | T. umbrinus | |
|---|---|---|---|---|---|---|
| T. canipes | — | 0.90039 | 0.77282 | 0.7423 | 0.94093 | 0.83481 |
| T. cinereicollis | 0.3039 | — | 0.13329 | 0.34286 | 0.86096 | 0.39919 |
| T. dorsalis | 0.3185 | 0.3005 | — | 0.0903 | 0.63409 | 0.05138 |
| T. quadrivittatus | 0.2939 | 0.1032 | 0.2899 | — | 0.59239 | 0.22374 |
| T. rufus | 0.281 | 0.2563 | 0.305 | 0.2348 | — | 0.73139 |
| T. umbrinus | 0.4012 | 0.3998 | 0.3762 | 0.3905 | 0.4017 | — |
Estimates from this study are on the lower diagonal. Sites with F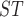 values less than zero are set equal to zero. The upper diagonal contains estimates from mitochondrial genes using the data from Sarver et al. (2017).
values less than zero are set equal to zero. The upper diagonal contains estimates from mitochondrial genes using the data from Sarver et al. (2017).
Crossvalidation with ADMIXTURE suggests the optimum number of populations is 7 (Supplementary Fig. S6 available on Dryad), though it is important to interpret other values of 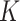 , especially when there is hierarchical structure in the data. ADMIXTURE coancestry plots (Supplementary Fig. S7 available on Dryad) revealed a progression of resolution across K values, with T. umbrinus being resolved as its own population first (
, especially when there is hierarchical structure in the data. ADMIXTURE coancestry plots (Supplementary Fig. S7 available on Dryad) revealed a progression of resolution across K values, with T. umbrinus being resolved as its own population first (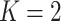 ), then T. dorsalis (
), then T. dorsalis (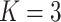 ), followed by T. rufus
), followed by T. rufus 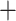 T. canipes and T. cinereicollis
T. canipes and T. cinereicollis 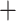 T. quadrivittatus (
T. quadrivittatus (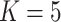 ) and a resolution of T. cinereicollis and T. quadrivittatus (
) and a resolution of T. cinereicollis and T. quadrivittatus (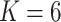 ). At
). At  populations (Fig. 4), each species was identified as a cluster and substructure was indicated within T. dorsalis. There was little indication of interspecific admixture, although a few individuals exhibited slightly less than 100% coancestry within its species.
populations (Fig. 4), each species was identified as a cluster and substructure was indicated within T. dorsalis. There was little indication of interspecific admixture, although a few individuals exhibited slightly less than 100% coancestry within its species.
Figure 4.

ADMIXTURE coancestry plot for  . Colors are indicative of species assignment, as indicated below the graph. Little shared coancestry is indicated among species and T. dorsalis exhibits geographic heterogeneity.
. Colors are indicative of species assignment, as indicated below the graph. Little shared coancestry is indicated among species and T. dorsalis exhibits geographic heterogeneity.
Multidimensional scaling revealed similar clustering of individuals into the six species (Supplementary Fig. S8 available on Dryad). T. umbrinus and T. dorsalis were separated from the other four species in multivariate space. T. rufus and T. canipes cluster cleanly but were separated by much less distance. T. cinereicollis and T. quadrivittatus showed clear clustering but nearly overlap.
Phylogenetic Inference
Analysis of the nuclear genome data produced a tree with strong support for the monophyly of each species and that recovers T. umbrinus as sister to the rest of the T. quadrivittatus group (Fig. 2, left). Strong support was recovered for the relationships among T. umbrinus, T. dorsalis, T. quadrivittatus, and T. cinereicollis. Moderate support (bipartition frequencies between 70% and 80%) was recovered for bipartitions including T. rufus and T. canipes. The species tree estimated using SVDquartets (Fig. 3) is consistent with results of the concatenated analyses. Species-tree estimation from inferred gene trees was performed using 802 loci (of the 1060 ARC contigs total) assigned to Mus chromosome locations (i.e., putative syntenic groups). The number of contigs assigned to each of the 19 mouse autosomes ranged from 26 to 66, with the exception of a single contig assigned to the X and zero assigned to the Y. Species-tree estimation using ASTRAL (with syntenic group trees as input) recovered the same relationships as the SVDquartets tree. These conclusions differ only slightly from phylogenies estimated in previously published analyses (Reid et al. 2012; Sullivan et al. 2014), and bootstrap values and posterior probabilities resolving the placement of T. canipes and T. rufus in Reid et al. (2012) were low, with their placement on the tree swapped relative to this study. Other relationships among the six 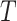 . quadrivittatus group species examined here are consistent between studies. In analyses where the 1060 ARC contigs were subject to 25 rounds of naïve binning, results were largely congruent. No binning replicate produced a species tree estimate with an RF-distance greater than two from the SVDquartets tree (Supplementary Table S2 available on Dryad).
. quadrivittatus group species examined here are consistent between studies. In analyses where the 1060 ARC contigs were subject to 25 rounds of naïve binning, results were largely congruent. No binning replicate produced a species tree estimate with an RF-distance greater than two from the SVDquartets tree (Supplementary Table S2 available on Dryad).
Figure 2.

Phylogenies of all 52 individuals included in this study; both were trees rooted using T. striatus as an outgroup. a) The nuclear genome phylogeny was estimated using the lineage option of SVDquartets. Each species is strongly supported (by bootstrap values) as monophyletic. Maximum likelihood was used to estimate branch lengths (GTR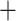 I
I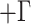 ) from the concatenated data set. b) Maximum likelihood estimate of the mtDNA genome phylogeny (as in Sarver et al. 2017) of the same individuals. Asterisks indicate 100% bootstrap support for short branches. Individuals inferred to carry introgressed mitochondrial genomes are denoted with an
) from the concatenated data set. b) Maximum likelihood estimate of the mtDNA genome phylogeny (as in Sarver et al. 2017) of the same individuals. Asterisks indicate 100% bootstrap support for short branches. Individuals inferred to carry introgressed mitochondrial genomes are denoted with an 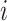 superscript.
superscript.
Figure 3.

Estimate of the species tree for the T. quadrivittatus group. Numbers above branches represent bootstrap values from SVDquartets, whereas numbers below branches represent posterior probabilities from ASTRAL-III.
Assessment of Introgression
Quartet-based analyses of introgression were consistent in showing little to no evidence of nuclear gene flow across species in analyses that included only the nuclear genome but strong evidence for introgression in analyses that included both nuclear and mtDNA genomes (Fig. 5, Supplementary Fig. S9 available on Dryad). For example, in the analyses shown in Figure 5, we selected a nonintrogressed T. dorsalis (dor 713), one T. dorsalis (dor 711) with a mitochondrial genome introgressed from T. cinereicollis, and a T. cinereicollis (cin 223) with a nearly identical mtDNA genome (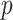 -distance
-distance 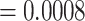 ), which we interpret as a true T. cinereicollis mtDNA genome. In the analysis of the nuclear genome, the quartet resolution congruent with the species tree received the most support, whereas the resolution congruent with the mitochondrial genome tree received the same low level of support as the third resolution (
), which we interpret as a true T. cinereicollis mtDNA genome. In the analysis of the nuclear genome, the quartet resolution congruent with the species tree received the most support, whereas the resolution congruent with the mitochondrial genome tree received the same low level of support as the third resolution (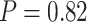 ; Fig. 5b). When the mtDNA genome was included, analysis of the same individuals demonstrated significantly more support for the resolution consistent with mtDNA introgression (
; Fig. 5b). When the mtDNA genome was included, analysis of the same individuals demonstrated significantly more support for the resolution consistent with mtDNA introgression (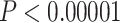 ; Fig. 5c).
; Fig. 5c).
Figure 5.

Analysis of introgression for a selected quartet of individuals. Included are a T. dorsalis with an introgressed mitochondrial genome (dor 711*), a T. dorsalis with a nonintrogressed mtGenome (dor 713), a T. cinereicollis (cin 223) that has a nearly identical mtGenome to the introgressed T. dorsalis, and outgroup T. striatus (st 11). a) Alternative resolutions of this quartet. b) In the nuclear data, there is substantial support for the resolution congruent with the species tree and symmetrically low support for the alternative resolutions, indicating no evidence of introgression in the nuclear genomes. c) In the combined nuclear 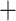 mtDNA genome data, there is strong support for the species tree and significantly more support for the resolution congruent with the mtDNA genome tree (top) than the alternative resolution (middle).
mtDNA genome data, there is strong support for the species tree and significantly more support for the resolution congruent with the mtDNA genome tree (top) than the alternative resolution (middle).
The analyses of other selected quartets that included individuals with both introgressed and nonintrogressed mtDNA showed similar results (Supplementary Fig. S9 available on Dryad). In analyses of the nuclear genome data alone, there is much support for the resolution congruent with the species tree and a symmetric lack of support for the other two resolutions. Furthermore, analysis of all quartets using HyDe indicated that there is no significant signal of introgression in the nuclear genomes across all possible quartets of these individuals. Similarly, in all analyses of both nuclear and mtDNA genomes, significant introgression is detected (Supplementary Fig. S9 available on Dryad). Therefore, we conclude that, in spite of multiple independent introgression events of mitochondrial genomes among these taxa, there is no evidence of introgression in the nuclear genome.
Discussion
In the last decade or so, increasing attention had focused on understanding the extent and genomic heterogeneity of introgression, the evolutionary dynamics of introgressing genes, and their roles for speciation and evolution (reviewed by Payseur and Rieseberg 2016; Runemark et al. 2019). Concurrently, advances in sequencing allow for genomic characterization in nonmodel systems (Bi et al. 2012; Seehausen et al. 2014; Jones and Good 2016), which has enabled insights into the evolutionary process that often cannot be obtained in model genetic systems (Hewitt 1988). Here, we investigate genomic patterns of divergence in a radiation of chipmunks that have been shown to exhibit widespread mitochondrial introgression (Good et al. 2003, 2008; Hird and Sullivan 2009; Reid et al. 2012; Sullivan et al. 2014; Sarver et al. 2017).
Given the extensive mitochondrial introgression we have demonstrated among these taxa, including the sharing of nearly identical mtDNA genomes in individual Uinta (T. umbrinus, e.g., umb 256) and cliff chipmunks (T. dorsalis, e.g., dor 201; Sarver et al. 2017), we expected to see evidence of admixture in the nuclear genomes. However, ADMIXTURE results (Fig. 4 and Supplementary Fig. S7 available on Dryad) indicated little coancestry among species. Across all species, individuals shared coancestry almost exclusively with individuals of their same species. Exceptions to this may indicate very low levels of introgression (Fig. 4), but multidimensional scaling indicates that individuals cluster only with their species as assigned by bacular morphology (Supplementary Fig. S8 available on Dryad). These results suggest that even though there has been rampant mitochondrial introgression among Tamias species, relatively little nuclear introgression has taken place. This is especially emphasized by the symmetrically low support for alternative resolutions of all quartets in the SVDquartets and HyDe analyses of the nuclear genome data (Fig. 5 and Supplementary Fig. S9 available on Dryad) and is particularly remarkable because selected quartets were chosen to include heterospecific individuals which share virtually identical mtDNA genomes, indicating relatively recent hybridization. These results are consistent with genomic data of Good et al. (2015), who found no evidence for nuclear introgression in the case of an ancient introgression (Good et al. 2008) of T. ruficaudus simulans mtDNA into T. amoenus canicaudus, likely a much older mtDNA introgression than the gene flow involved in the T. quadrivittus species group examined here (Sullivan et al. 2014).
It is possible that there is evidence of hybridization in the nuclear genome that we have not detected. Whereas we have used powerful analytical approaches, the data were generated using targeted-capture probes designed by comparison of chipmunk (T. alpinus) transcriptome data with a reference genome from Mus, a deep (likely 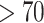 MY; Swanson et al. 2019) divergence. Thus, our data are biased for relatively conserved protein-coding genes and it is possible that our data may focus on genomic regions that are resistant to introgression. Gene-dense regions often show reduced introgression (Schumer et al. 2018; Edelman et al. 2019), and so any estimate of gene flow based on exome data is expected to be downwardly biased to some extent. However, the likelihood that large amounts of nuclear introgression exist and went undetected here is probably quite low. Our data are fairly well distributed throughout the genome, with 802 of our contigs mapping to the 19 Mus autosomes and each autosome represented from 26 to 66 contigs.
MY; Swanson et al. 2019) divergence. Thus, our data are biased for relatively conserved protein-coding genes and it is possible that our data may focus on genomic regions that are resistant to introgression. Gene-dense regions often show reduced introgression (Schumer et al. 2018; Edelman et al. 2019), and so any estimate of gene flow based on exome data is expected to be downwardly biased to some extent. However, the likelihood that large amounts of nuclear introgression exist and went undetected here is probably quite low. Our data are fairly well distributed throughout the genome, with 802 of our contigs mapping to the 19 Mus autosomes and each autosome represented from 26 to 66 contigs.
Observations of mtDNA introgression in the absence of gene flow across the nuclear genome are beginning to accumulate (Toews and Brelsford 2012; Bonnet et al. 2017), including inferences from microsatellite data (e.g., Reid et al. 2010; Pons et al. 2013; Zielinski et al. 2013) and genome-wide sequences (e.g., Good et al. 2015; McElroy et al. 2020). Several mechanisms could account for such observations (Sloan et al. 2017), including positive selection on introgressing mtDNA genomes (e.g., Melo-Ferreira et al. 2014), simple demographic effects associated with range expansion (e.g., Currat et al. 2008; Marques et al. 2017), or some combination of demographics, mate choice, and selection against hybrids (Bonnet et al. 2017).
Sarver et al. (2017) did not detect signatures of positive directional selection on the mtDNA genomes included in our current sample, and ecologically mediated positive selection on mtDNA (introgressed or otherwise) may be relatively rare in endotherms (Galtier et al. 2009). However, one intriguing selection-based hypothesis is that mtDNA introgression may often reflect selective purging of deleterious mutations (Sloan et al. 2017). For example, if an introgressed mtDNA genome harbored fewer deleterious mutations than a resident mtDNA genome, then selection could drive complete mtDNA replacement in the absence of long-term protein-coding signatures of recurrent positive directional selection. This purging model assumes that the long-term efficacy of selection is reduced for mtDNA (haploid, maternally inherited) relative to the nuclear genome (diploid, biparental inheritance; Lynch and Blanchard 1998; Neiman and Taylor 2009), an assumption that is not always supported by population genetic data (Cooper et al. 2015). In our focal species, the cliff chipmunk (T. dorsalis) appears to be the universal recipient of mtDNA from other species in the T. quadrivittatus group. If this pattern reflects selective purging of deleterious mutations, then individuals carrying the nonintrogressed T. dorsalis mtDNA genomes are predicted to be generally less fit. Putatively native mtDNA genomes from allopatric (nonntrogressed) T. dorsalis populations (Sullivan et al. 2014; Sarver et al. 2017; Supplementary Fig. S2 available on Dryad) differed from all other species by 66 fixed nucleotide differences, 56 of which were within protein-coding regions (Supplementary Table S3 available on Dryad). Only two of these differences result in nonsynonymous amino acid changes and both appear to be relatively conservative replacements. While it is possible that these subtle amino acid changes affect mitochondrial function, there is little compelling evidence for the accumulation of appreciable genetic load between the mtDNA genomes of these species.
Alternatively, it is likely that T. dorsalis (the universal recipient of mtDNA in the group; Sullivan et al. 2014) has experienced rather large-scale range expansion following the last glacial maximum (LGM). Waltari and Guralnick (2009) reconstructed the LGM distribution of T. dorsalis by layering their current ecological niche model onto LGM climatic reconstructions. Suitable habitat was likely only available for T. dorsalis in the southern portion of its current range, where it may have been isolated from other chipmunk species. Under some nonequilibrium conditions thought to be common under range expansions, introgression is generally predicted to be strongly asymmetric from resident species into invading species, and this effect should be strongest for mtDNA and other genomic elements subject to increased genetic drift (Currat et al. 2008). Thus, it is entirely plausible that simple demographic effects associated with post-Pleistocene range expansion could explain the observed pattern of extensive and recurrent mtDNA introgression. However, we note that a recent simulation study (Bonnet et al. 2017) found that demography alone (e.g., sex-biased dispersal, spatial expansion) is not expected to often produce rampant cytonuclear discordance, and genetic drift does so under a relatively narrow range of conditions (e.g., when hybridization is very rare).
Our results indicate that hybridization has made a negligible contribution to overall genomic variation within and among species of Tamias endemic to the Central and Southern Rocky Mountains. Although these species represent only about one-quarter of the diversity of the western chipmunks (subgenus Neotamias, Thorington and Hoffmann 2005), this radiation also represents perhaps the strongest evidence for recurrent introgressive hybridization of mtDNA in Tamias (Sullivan et al. 2014). Furthermore, our findings echo results from T. amoenus canicaudus; a group of yellow-pine chipmunks that is fixed for an ancient mtDNA genome introgression from T. ruficaudus (Good et al. 2008) but shows no evidence for nuclear gene flow (Good et al. 2015). Collectively, it appears that chipmunk species boundaries are largely impermeable to nuclear gene flow and that hybridization has played a relatively minor role in the evolutionary history of this group.
As the field of phylogenetics has shifted towards a predominantly genomic view, researchers have shifted towards treating mtDNA as a single manifestation of the coalescent process. However, inferences derived from mtDNA continue to have a disproportionately large influence on some perceptions of population histories, phylogenetic relationships, and speciation (e.g., Hill 2016). In some systems, mtDNA introgression has proven to be an effective indicator of appreciable nuclear gene flow among closely related species (e.g., Ferreira et al. 2020), including hybridization events that have seeded local adaptation (Jones et al. 2018, 2020). In contrast, the lack of evidence for mtDNA introgression between modern humans and Neandertals (Currat and Excoffier 2004; Briggs et al. 2009) contributed to initial skepticism of the now widely accepted history of gene flow between these populations (Green et al. 2010; Kuhwilm et al. 2016; Sankararaman et al. 2016). In chipmunks, mitochondrial DNA emerges as a molecular canary in the mineshaft of genome evolution—especially sensitive to the vagaries of genetic drift and natural selection (Ballard and Whitlock 2004; Galtier et al. 2009) and, it seems, highly prone to rare events that seem to have little, if any, influence population structure characteristic of their nuclear genomes.
The current study represents the first genomic-scale study in Central and Southern Rocky Mountains chipmunks and is one of just a few in chipmunks (see Bi et al. 2012, 2013; Good et al. 2015). Here, however, we only consider species that span 24% of the diversity of the genus and did not include two other species frequently assigned to the T. quadrivittatus species group: T. bulleri (endemic to a small area in the Sierra Madre Occidental of Mexico) and T. palmeri (endemic to the Spring Mountains of Nevada). Although our central goal was a phylogenomic assessment of introgression in this species group, our results also provide insights into phylogenetic relationships among its members. Our analysis supports aspects of previously estimated multilocus relationships using reproductive protein genes (Reid et al. 2012; Sullivan et al. 2014). This includes T. umbrinus and T. dorsalis as successive sister species to a larger group that includes T. canipes, T. cinereicollis, T. rufus, and T. quadrivittatus. Inclusion of the missing two additional species typically assigned to the group, T. bulleri and T. palmeri, might alter relationships within the T. quadrivittatus species group. Interestingly, in spite of no apparent recent mtDNA introgression involving T. canipes and T. rufus (they are the only taxa that are monophyletic with respect to the mtDNA genome tree; Fig. 2 right; Sullivan et al. 2014; Sarver et al. 2017), the phylogenetic placement of both differ between mtDNA and nuclear data. Interestingly, in the HyDe analyses of all taxa and including both nuclear and mtDNA genome data, even individuals of T. rufus are implicated as hybrids (data not shown). Thus, like T. amoenus canicaudus (Good et al. 2008, 2015), T. rufus may have fixed on an ancient introgression of mtDNA. Future work will focus on analyzing genus-wide genomic data to arrive at more general conclusions about the architecture of nature of divergence in this system. Specifically, the assembly and phylogenomic approaches implemented here can be used across species to provide resolution across the genus and build on approaches using few loci (e.g., Reid et al. 2012). A resolved phylogeny, in concert with population-genomic estimates of divergence, gene flow, and population structure, will result in a comprehensive characterization of this natural system.
In spite of many years of research on divergence of chipmunks in western North America (e.g., Howell 1929; Sutton and Nadler 1969; Levenson et al. 1985; Piaggio and Spicer 2001; Sullivan et al. 2014), several questions remain to be addressed. It may be that some mechanism, such as selection against hybrids, prevents nuclear introgression in the face of such recent mtDNA introgression. In T. sibiricus, male reproductive success is correlated with range size, and males of intermediate size may be inferior competitors for large territories (Marmet et al. 2012). However, it is unclear whether this holds for western North American chipmunks.
Similarly, patterns of hybrid fertility (e.g., Haldane’s Rule, biased backcrossing, etc.) are nearly completely unknown in this system. Chipmunks engage in scramble competition and are reciprocally polygamous (Schulte-Hostedde et al. 2004); thus, it is possible that sexual selection plays an important role in species divergence in this system. Breeding studies will be required to address these issues, although these will be challenging given that chipmunks are monestrous.
Conclusion
Here, we use targeted sequence capture to sequence nuclear loci from a radiation of six species of chipmunks in the T. quadrivittatus group. Using phylogenomic approaches, we were able to produce a phylogeny using a variety of techniques that resolves the species tree of this group. Furthermore, we document a lack of nuclear introgression in the face of substantial mitochondrial introgression in this group, consistent with the results of Good et al. (2015) for another instance of mtDNA capture. Future work will characterize this system further using additional analyses and increased, genus-wide sampling
Acknowledgements
The authors would like to thank the following individuals for assistance in the field over several years: K. Bell, W. Bell, I. Demboski, M. Fraker, D. Good, P. Good, J. Harper, A. Hornsby, S. Poler, and A. Runck. Ke Bi, Sara Keeble, and Dan Vanderpool assisted with lab work and/or development of targeted captures protocols. This research was conducted in compliance with University of Idaho Animal Care and Use Committee, under protocol UIACUC-2005-40.
Contributor Information
Brice A J Sarver, Department of Biological Sciences, University of Idaho, Moscow, ID 83844, USA; Institute for Bioinformatics and Evolutionary Studies (IBEST), University of Idaho, Moscow ID 83844, USA.
Nathanael D Herrera, Division of Biological Sciences, University of Montana, Missoula, MT 59812, USA.
David Sneddon, Department of Biological Sciences, University of Idaho, Moscow, ID 83844, USA.
Samuel S Hunter, Institute for Bioinformatics and Evolutionary Studies (IBEST), University of Idaho, Moscow ID 83844, USA; UC-Davis Genome Center, Davis, CA 95616, USA.
Matthew L Settles, UC-Davis Genome Center, Davis, CA 95616, USA.
Zev Kronenberg, Pacific Biosciences, Menlo Park, CA 94025, USA.
John R Demboski, Department of Zoology, Denver Museum of Nature & Sciences, Denver, CO 80205, USA.
Jeffrey M Good, Division of Biological Sciences, University of Montana, Missoula, MT 59812, USA; Wildlife Biology Program, University of Montana, Missoula, Montana 59812, USA.
Jack Sullivan, Department of Biological Sciences, University of Idaho, Moscow, ID 83844, USA; Institute for Bioinformatics and Evolutionary Studies (IBEST), University of Idaho, Moscow ID 83844, USA.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.6t1g1jwws.
Funding
Funding was provided by a seed grant from the University of Idaho Research Foundation, the NSF EPSCoR program (NSF cooperative agreement number EPS-9720634), the Institute for Bioinformatics and Evolutionary Studies (IBEST) at the University of Idaho (by NIH NCRR 1P20RR016454-01; NIH NCRR 1P20RR016448-01; NSF EPS-809935), NSF DEB-0717426 (J.S.), NSF DEB-0716200 (J.R.D.), NSF Cooperative Agreement No. DBI-0939454 (J.S.), and the Denver Museum of Nature & Science. J.S. and B.A.J.S. received funding through BEACON, an NSF-funded Center the Study of Evolution in Action (DBI-0939454). J.M.G. and N.D.H. received funding through UNVEIL, a research and training network funded by NSF EPSCoR (OIA-1736249), and through an NSF grant (DBI-1561748). The development of exome capture assays in chipmunks were developed in part through a grant from the Gordon and Betty Moore Foundation (GBMF2983). A grant from the National Institute of General Medical Sciences (R01GM098536 to J.M.G.) supported the development of experimental protocols in mice that were utilized in the current study. Additional instrumentation and laboratory support were provided by the University of Montana Genomics Core, supported by a grant from the M. J. Murdock Charitable Trust. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Data Deposition
Raw sequence data for this project has been deposited at the NCBI Sequence Read Archive under BioProject accession PRJNA347372.
References
- Alexander D.H., Novembre J., Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard J.W.O., Whitlock M.C. 2004. The incomplete natural history of mitochondria. Mol. Ecol. 13:729-744. [DOI] [PubMed] [Google Scholar]
- Bayzid M.S., Warnow T. 2013. Naive binning improves phylogenomic analyses. Bioinformatics 29:2277–2284. [DOI] [PubMed] [Google Scholar]
- Bi K., Linderoth T., Vanderpool D, Good J.M., Nielsen R., Moritz C. 2013. Unlocking the vault: next-generation museum population genomics. Mol. Ecol. 22:6018–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K., Vanderpool D., Singhal S., Linderoth T., Moritz C., Good J.M. 2012. Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics 13:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blischak P., Chifman J, Wolfe A.D., Kubatko L.S. 2018. HyDe: a Python package for genome-scale hybridization detection. Syst. Biol. 67: 821-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet T., Leblois R., Roussett F., Crochet P.-A. 2017. A reassessment of explanations for discotdant introgressions of mitochondrial and nuclear genomes. Evolution 71:2140-2158. [DOI] [PubMed] [Google Scholar]
- Briggs A.W., Good J.M., Green R.E., Krause J., Maricic T., Stenzel U., Lalueza-Fox C., Rudan P., Brajkovic D., Kucan Z., Gusic I., Schmitz R., Doronichev V.B., Golovanova L.V., de la Rasilla M., Fortea J., Rosas A., Paabo S. 2009. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325:318-321. [DOI] [PubMed] [Google Scholar]
- Brown J.H. 1971. Mechanisms of competitive exclusion between two species of chipmunks. Ecology 52:305-311. [Google Scholar]
- Browning B.L., Browning S.R. 2009. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 84:210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning S.R., Browning B.L. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81:1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning B.L., Yu Z. 2009. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. Am. J. Hum. Genet. 85:847–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifman J., Kubatko L.S. 2014. Quartet inference from SNP data under the coalescent model. Bioinformatics 30:3317-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper B.S., Burrus C.R., Ji C., Hahn M.W., Montooth K.L. 2015. Similar efficacies of selection shape mitochondrial and nuclear genes in both Drosophila melanogaster and Homo sapiens. G3: Genes, Genomes, Genetics 5:2165-2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currat M., Excoffier L. 2004. Modern humans did not admix with Neanderthals during their range expansion into Europe. PLoS Biol. 2:2264–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currrat M., Ruedi M., Petit R.J., Excoffier L. 2008. The hidden side of invasions: massive introgression by local genes. Evolution 62:1908-1920. [DOI] [PubMed] [Google Scholar]
- Danecek P., Auton A., Abecasis G., Albers C., Banks E., DePristo M., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., McVean G., Durbin R. 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M., Banks E., Poplin R., Garimella K.V, Maguire J.R., Hartl C., Philippakis A., del Angel G., Rivas M., Hanna M., McKenna A., Fennell T.J., Kernytsky A.M., Sivachenko A.Y., Cibulskis K., Gabriel S.B., Altshuler D., Daly M.J. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand E.Y., Patterson N., Reich D., Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28:2239-2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhard W.G. 1985. Sexual selection and animal genetalia. Cambridge (MA): Harvard University Press. [Google Scholar]
- Edelman N.B., Frandsen P.B., Miyagi M., Clavijo B., Davey J., Dickow R.B., García-Accinelli G., Van Belleghem S.M., Patterson N., Neafsey D.E., Challis R., Kumar S., Moreira G.P.R., Salazar C., Chouteau M., Counterman B.A., Papa R., Blaxter M., Reed R.D., Dasmahapatra K.K., Kronforst M., Joron M., Jiggins C.D., McMillan W.O., Di Palma F., Blumberg A.J., Wakeley J., Jaffe D., Mallet J. 2019. Genomic architecture and introgression shape a butterfly radiation. Science 366:594-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira M.S., Jones M.R., Callahan C.M., Farelo L., Tolesa Z., Suchentrunk F., Boursot P., Mills L.S., Alves P.C., Good J.M., Melo-Ferreira J. 2020. The legacy of recurrent introgression during the radiation of hares. BioRxiv 2020.06.19.160283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N., Nabholz B., Glemin S., Hurst G.D.D. 2009. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol. Ecol. 18:4541-4550. [DOI] [PubMed] [Google Scholar]
- Good J.M., Sullivan J. 2001. Phylogeography of red-tailed chipmunks (Tamias ruficaudus), a northern Rocky Mountains endemic. Mol. Ecol. 10:2683-2696. [DOI] [PubMed] [Google Scholar]
- Good J.M., Demboski J.R., Nagorsen D.W., Sullivan J. 2003. Phylogeography and introgressive hybridization: chipmunks (genus Tamias) in the northern Rocky Mountains. Evolution 57:1900–1916. [DOI] [PubMed] [Google Scholar]
- Good J.M., Hird S., Reid N., Demboski J.R., Steppan S.J., Martin-Nims T.R., Sullivan J. 2008. Ancient hybridization and mitochondrial capture between two species of chipmunks. Mol. Ecol. 17:1313–1327. [DOI] [PubMed] [Google Scholar]
- Good J.M., Vanderpool D., Keeble S., Bi K. 2015. Negligible nuclear introgression despite complete mitochondrial capture between two species of chipmunks. Evolution 69:1961-1972. [DOI] [PubMed] [Google Scholar]
- Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H., Hansen N.F., Durand E.Y., Malaspinas A.S., Jensen J.D., Marques-Bonet T., Alkan C., Prüfer K., Meyer M., Burbano H.A., Good J.M., Schultz R., Aximu-Petri A., Butthof A., Höber B., Höffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E.S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Ž., Gušic I., Doronichev V.B., Golovanova L.V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R.W., Johnson P.L.F., Eichler E.E., Falush D., Birney E., Mullikin J.C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Pääbo S. 2010. A draft sequence of the Neandertal genome. Science 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell J., Storer T.I. 1924. Animal life in the Yosemite. Berkeley: University of California Press. [Google Scholar]
- Harris K., Nielsen R. 2016. The genetic cost of Neanderthal introgression. Genetics 203:881-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison R.G., Larson E.L. 2014. Hybridization, introgression, and the nature of species boundaries. J. Heredity 105:795-809. [DOI] [PubMed] [Google Scholar]
- He F., Pasam R., Shi F., Kant S., Keeble-Gagnere G., Kay P., Forrest K., Fritz A., Hucl P., Wiebe K., Knox R., Cuthbert R., Pozniak C., Akhunova A., Morrell P.L., Davies J.P., Webb S.R., Spangenberg G., Hayes B., Daetwyler H., Tibbits J., Hayden M., Akhunov E. 2019. Exome sequencing highlights the role of wild-relative introgression in shaping the adaptive landscape of the wheat genome. Nat. Gen. 51:896-904. [DOI] [PubMed] [Google Scholar]
- Heliconius Genome Consortium. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 478:94-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller H.C. 1971. Altitudinal zonation of chipmunks (Eutamias): interspecific aggression. Ecology 52:312-319. [Google Scholar]
- Hewitt G.M. 1988. Hybrid zones—natural laboratories for evolutionary studies. Trends Ecol. Evol. 3:158-167. [DOI] [PubMed] [Google Scholar]
- Hill G.E. 2016. Mitonuclear coevolution as the genesis of speciation and the mitochondrial DNA barcode gap. Ecol. Evol. 6:5831-5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird S., Sullivan J. 2009. Assessment of gene flow across a hybrid zone in red-tailed chipmunks (Tamias ruficaudus). Mol. Ecol. 18:3097–109. [DOI] [PubMed] [Google Scholar]
- Hird S., Reid N., Demboski J.R., Sullivan. J. 2010. Introgression at differentially aged hybrid zones in red-tailed chipmunks. Genetica 138:869–83. [DOI] [PubMed] [Google Scholar]
- Howell A.H. 1929. Revision of the American chipmunks (genera Tamias and Eutamias). N. Am. Fauna 52:1-256. [Google Scholar]
- Hunter S.S., Lyon R.T., Sarver B.A., Hardwick K., Forney L.J., Settles M.L. 2015. Assemble by reduced complexity (ARC): a hybrid approach for targeted assembly of homologous sequences. bioRxiv, doi.org/ 10.1101/014662. [DOI] [Google Scholar]
- Jones M.R., Good J.M. 2016. Targeted capture in evolutionary and ecological genomics. Mol. Ecol. 85:185-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M.R., Mills L.S., Alves P.C., Callahan C.M., Alves J.M., Lafferty D.J.R., Jiggins F.M., Jensen J.D., Melo-Ferreira J., Good J.M. 2018. Adaptive introgression underlies polymorphic seasonal camouflage in snowshoe hares. Science 360:1355-1358. [DOI] [PubMed] [Google Scholar]
- Jones M.R., Mills L.S., Jensen J.D., Good J.M. 2020. The origin and spread of locally adaptive seasonal camouflage in snowshoe hares. Am Nat. 196:316–332. [DOI] [PubMed] [Google Scholar]
- Kearns A.M., Restani M., Szabo I., Schroder-Nielsen A., Kim J.A., Richardson H.M., Marzluff J.M., Fleischer R.C., Johnsen A., Omland K.E. 2018. Genomic evidence of speciation reversal in ravens. Nat. Commun., 9:906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. 2002. BLAT—the BLAST-Like Alignment Tool. Genome Res. 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubatko L.S., Chifman J. 2019. An invariants-based method for efficient identification of hybrid species from large-scale genomic data. BMC Evol. Biol. 19:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlwilm M., Gronau I., Hubisz M.J., de Filippo C., Prado-Martinez J., Kircher M., Fu Q., Burbano H.A., Lalueza-Fox C., de la Rasilla M., Rosas A., Rudan P., Brajkovic D., Kucan Ž., Gušic I., Marques-Bonet T., Andrés A.M., Viola B., Pääbo S., Meyer M., Siepel A., Castellano S. 2016. Ancient gene flow from early modern humans into Eastern Neanderthals. Nature 530:429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson H., Hoffmann R.S., Nadler C.F., Deutsch L., Freeman S.D. 1985. Systematics of the Holarctic chipmunks (Tamias). J. Mammal., 66:219-242. [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics, 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., O’Brien P.C.M., Biltueva L., Fu B., Wang J., Nie W., Ferguson-Smith M., Graphodatsky A.S., Yang F. 2004. Evolution of genome organizations of squirrels (Sciuridae) revealed by cross-species chromosome painting. Chromosome Res. 12:317–35. [DOI] [PubMed] [Google Scholar]
- Librado P.J.R., Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25. 25:1451–1452. [DOI] [PubMed] [Google Scholar]
- Lynch M., Blanchard J.L. 1998. Deleterious mutation accumulation in organelle genomes. Genetica 103:29–39. [PubMed] [Google Scholar]
- Magoè T., Salzberg S.L. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinsky M., Svardal H., Tyers A.M., Miska E.A., Genner M.J., Turner G.F., Durbin R. 2018. Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nat. Ecol. Evol. 2:1940–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J. 2005. Hybridization as an invasion of the genome. Trends Ecol. Evol. 20:229–37. [DOI] [PubMed] [Google Scholar]
- Marmet J., Pisanu B., Chapuis J.-L., Jacob G., Baudry E. 2012. Factors affecting male and female reproductive success in a chipmunk (Tamias sibiricus) with a scramble competition mating system. Behav. Ecol. Sociobiol. 66:1449–1457. [Google Scholar]
- Marques J.P., Farelo L., Vilela J., Vanderpool D., Alves P.C., Good J.M., Boursot P., Melo-Ferreira J. 2017. Range expansion underlies historical introgressive hybridization in the Iberian hare. Sci. Rep. 7:40788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy K., Black A., Dolman G., Horton P., Pedler L., Campbell C.D., Drew A., Joseph L. 2020. Robbery in progress: Historical museum collections bring to light a mitochondrial capture within a bird species widespread across southern Australia, the Copperback Quail-thrush Cinclosoma clarum. Ecol Evol. 10:6785–6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Ferreira J., Vilela J., Fonsecs M.M., da Fonseca R.R., Boursot P., Alves P.C. 2014. The elusive nature of adaptive mitochondrial DNA evolution of and arctic lineage prone to frequent introgression. Genome Biol. Evol. 6:886-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin V., Abdo Z., Joyce P., Sullivan J. 2003. Performance-based selection of likelihood models for phylogeny estimation. Syst. Biol. 52:674–683. [DOI] [PubMed] [Google Scholar]
- Morales-Briones D.F., Liston A., Tank D.C. 2018. Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the Neotropical genus Lachemilla (Rosaceae). 2018. New Phytol. 218:1668-1684. [DOI] [PubMed] [Google Scholar]
- Neiman M., Taylor D.R. 2009. The causes of mutation accumulation in mitochondrial genomes. Proc. Royal Soc. B 276:1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès H., Aboyoun, P. Gentleman R., DebRoy S. 2016. Biostrings: string objects representing biological sequences, and matching algorithms. R package version 2.40.2. [Google Scholar]
- Patterson B.D., Norris R.W. 2016. Towards a uniform nomenclature for ground squirrels: the status of the Holarctic chipmunks. Mammalia 80:241-251. [Google Scholar]
- Patterson B.D., Thaeler C.S. 1982. The mammalian baculum: hypotheses on the nature of bacular variability. J. Mammal. 63:1-15. [Google Scholar]
- Payseur B.A., Rieseberg L.H. 2016. A genomic perspective on hybridization and speciation. Mol. Ecol. 25:2337-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Piaggio A.J., Spicer G.S. 2001. Molecular phylogeny of the chipmunk genus Tamias inferred from the mitochondrial cytochrome
 and cytochrome oxidase II gene sequences. Mol. Phylogenet. Evol. 20:335-350. [DOI] [PubMed] [Google Scholar]
and cytochrome oxidase II gene sequences. Mol. Phylogenet. Evol. 20:335-350. [DOI] [PubMed] [Google Scholar] - Pons J.M., Sonsthagen S., Dove C., Crochet P.A. 2013. Extensive mitochondrial introgression in North American Great Black-backed Gulls (Larus marinus) from the American Herring Gull (Larus smithsonianus) with little nuclear DNA impact. Heredity 112:226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M. J., Sham P.C. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2013. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Reid N., Demboski J.R., Sullivan J. 2012. Phylogeny estimation of the radiation of western North American chipmunks (Tamias) in the face of introgression using reproductive protein genes. Syst. Biol. 61:44–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid N., Hird S., Schulte-Hostedde A., Sullivan J. 2010. Examination of nuclear loci across a zone of mitochondrial introgression between Tamias ruficaudus and T. amoenus. J. Mammal. 91:1389–1400. [Google Scholar]
- Roch S., Warnow T. 2015. On the robustness to gene tree estimation error (or lack thereof) of coalescent-based species tree methods. Syst. Biol. 64:663-676. [DOI] [PubMed] [Google Scholar]
- Runemark A., Vallejo-Marin M., Meier J.I. 2019. Eukaryote hybrid genomes. PLoS Genet. 15: e1008404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S., Patterson N., Reich D. 2016. The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr. Biol. 26;1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarver B.A.J., Demboski J.R., Good J.M., Forshee N., Hunter S.L., Sullivan J. 2017. Comparative mitochondrial phylogenomic assessment of introgression among several species of chipmunks (Tamias). Genome Biol. Evol. 9:1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Hostedde A.I., Millar J.S., Gibbs H.L. 2004. Sexual selection and matting patterns in a mammal with female-biased sexual dimorphism. Behav. Ecol. 15:351-356. [Google Scholar]
- Schultz N.G., Lough-Stevens M., Abreu E., Orr T., Dean M.D. 2016. The baculum was gained and lost multiple times during mammalian evolution. Integ. Comp. Biol. 56: 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M., Xu C., Powell D.L., Durvasula A., Skov L., Holland S., Blazier J.C., Sankararaman S., Andolfato P., Rosenthal G.G., Prezeworski M. 2018. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360:656-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehausen O., Butlin R.K., Keller I., Wagner C.E., Boughman J.W., Hohenlohe P.A., Peichel C.L., Saetre G-P., Bank C., Brännström A., Brelsford A., Clarkson C.S., Eroukhmanoff F., Feder J.L., Fischer M.C., Foote A.D., Franchini P., Jiggins C.D., Jones F.C., Lindholm A.K., Lucek K., Maan M.E., Marques D., Martin S.H., Matthews B., Meier J.I., Möst M., Nachman M.W., Nonaka E., Rennison D.J., Schwarzer J., Watson E.T., Westram A. M., Widmer A. 2014. Genomics and the origin of species. Nat. Rev. Genet. 15:176–92. [DOI] [PubMed] [Google Scholar]
- Simmons L.W., Firman R.C. 2013. Experimental evidence for the evolution of the mammalian baculum by sexual selection. Evolution 68:276–83. [DOI] [PubMed] [Google Scholar]
- Sloan B.B., Havird J.C., Sharbrough J. 2017. The on-again, off-again relationship between mitochondrial genomes and species boundaries. Mol. Ecol. 26:2212-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.A, Dunn C.W. 2008. Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24:715–716. [DOI] [PubMed] [Google Scholar]
- Smith S.A., O’Meara B.C. 2012. treePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 28:2689–90. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley P., Ramm S.A., Sherborne A.L., Thom M.D.F., Paterson S., Hurst J.L. 2013. Baculum morphology predicts reproductive success of male house mice under sexual selection. BMC Biol. 11:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan J., Demboski J.R., Bell K.C., Hird S., Sarver B.A.J., Reid N., Good J.M. 2014. Divergence-with-gene-flow within the recent chipmunk radiation (Tamias). Heredity 113:185-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton D.A. 1982. The female genital bone of chipmunks, genus Eutamias. Southwestern Nat. 27:393-402. [Google Scholar]
- Sutton D.A. 1992. Tamias amoenus. Mammal. Species 390:1-8. [Google Scholar]
- Sutton D.A., Nadler C.F. 1969. Chromosomes of the North American chipmunk genus Eutamias. J. Mammal. 50:524-535. [Google Scholar]
- Swanson M.T., Oliveros C.H., Esselstyn J.A. 2019. A phylogenomic rodent tree reveals the repeated evolution of masseter architectures. Proc. R. Soc. B, 286: 2019.0672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D.L. 2017. PAUP* version 4.0 a167. Sunderland, MA: Sinauer. [Google Scholar]
- Taylor S.A., Larson E.L. 2019. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nature Ecol. Evol. 3:170-177. [DOI] [PubMed] [Google Scholar]
- Teeter K.C., Payseur B.A., Harris L.W., Bakewell M.A., Thibodeau L.M., O’Brien J.E., Krenz J.G., Sans-Fuentes M.A., Nachman M.W., Tucker P.K. 2008. Genome-wide patterns of gene flow across a house mouse hybrid zone. Genome Res. 18:67-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorington R.W., Jr., Hoffmann R.S. 2005. Family Sciuridae. pp. 754-818, in Mammal Species of the World. A Taxonomic and Geographic Reference (3rd ed) (Wilson D. E. and Reeder D. M., eds.). Johns Hopkins University Press, 2 vols., 2142 pp. [Google Scholar]
- Todesco M., Pascual M.A., Owens G L., Ostevik K.L., Moyers B.T., Hubner S., Heredia S.M., Hahn M.A., Caseys C., Bock D.G., Rieseberg L.J. 2016. Hydridization and extinction. Evol. Appl. 9:892-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toews D.P., Brelsford L.J. 2012. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21:3907–3930. [DOI] [PubMed] [Google Scholar]
- Waltari E., Guralnick R.P. 2009. Ecological niche modelling of montane mammals in the Great Basin, North America: examining past and present connectivity of species across basins and ranges. J. Biogeogr. 36:148-161. [Google Scholar]
- Weir B., Cockerham C. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. [DOI] [PubMed] [Google Scholar]
- White J.A. 1953. The baculum in the chipmunks of western North America. Univ. Kansas Publ. Museum of Natural History 5:611-631. [Google Scholar]
- Wu C.-I. 2001. The genic view of the process of speciation. J. Evol. Biol. 14:851–865. [Google Scholar]
- Yahner R.H. 1978. The adaptive nature of the social system and behavior in the eastern chipmunk, Tamias striatus. Behav. Ecol. Sociobiol. 3:397–427. [Google Scholar]
- Zhang C., Rabiee M., Sayyari E., Mirarab S. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinf. 19(suppl 6):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski P., Nadachowska-Brzyska K., Wielstra B., Szkotak R., Cogǎlniceanu S.D., Babik W. 2013. No evidence for nuclear introgression despite complete mtDNA replacement in the Carpathian newt (Lissotriton montandoni). Mol. Ecol. 22:1884–1903. [DOI] [PubMed] [Google Scholar]
- Zwickl D.J. 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Austin, Taxas: The University of Texas at Austin. [Google Scholar]