

Invasive mucinous adenocarcinoma (IMA) is defined by the WHO classification as a primary lung adenocarcinoma with tumor cells showing goblet cell- or columnar cell-morphology (Fig. 1) with abundant intracytoplasmic mucin1. Due to its distinctive clinical features, i.e., peripheral location and a high frequency of multifocal, multilobular, and bilateral occurrence it has been defined as a distinctive entity with dismal outcome for many years and formerly been referred to as mucinous bronchioloalveolar carcinoma. Previous studies provided evidence for molecular features distinct from non-mucinous adenocarcinomas, with frequent KRAS mutations resembling RAS alterations in gastrointestinal tumors and oncogenic fusions in KRAS wild-type IMAs, as well as distinct clinical characteristics such as predominant recurrences in the lungs and a more aggressive phenotype for NRG1-rearranged tumors2,3.
Fig. 1. Histology of invasive mucinous adenocarcinoma (IMA) of the lung.
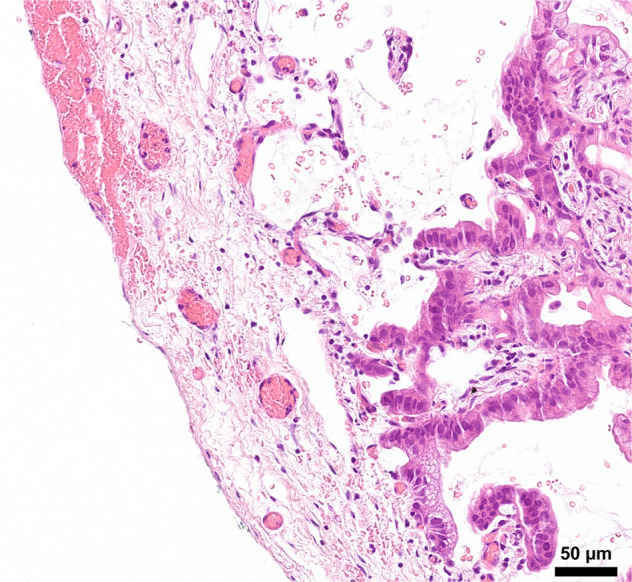
This case shows both columnar and goblet cell differentiation, presented as a multifocal bilateral tumor and revealed a truncal KRASG12D oncogenic driver mutation. The pleural surface is shown in the left side indicating the peripheral tumor location.
Two manuscripts provide deep genomic insights and focus on the clonal relationship of multifocal IMAs using whole-exome sequencing4 and DNA-based next-generation sequencing of a large targeted gene panel5. Kim and colleagues from South Korea analyzed East Asian patients and report that, despite its multilocular presentation, IMAs share early and clonal initiating driver events, such as KRAS, NKX2-1, TP53, or ARID1A mutations. Overall tumor mutational burden was low, but still intratumoral heterogeneity was detected. Interestingly the predominant mutational signatures were signature 1 (endogenous mutational process), signature 2 and 13 (APOBEC activity), and signature 6 (DNA mismatch repair) but not related with smoking signature. Consistently, all patients were either never-smokers or had ceased smoking in their past history. This is somewhat surprising as IMAs are believed to be associated with demographic smoking habits, not different from non-mucinous lung adenocarcinomas1. The most frequent clonal oncogenic drivers in this study were activating KRAS mutations G12D, G12S, G12V, and Q61H. This is quite different from non-mucinous adenocarcinomas in East Asian patients where the most frequent drivers are activating mutations in EGFR exons 19 or 216. Interestingly, the authors showed that multiple IMAs arising in the background of usual interstitial pneumonia (UIP) revealed distinct genomic features and hence represented multiple independent primary tumors. Thus, clonal multilocular and multiple primary IMAs in UIP seem to represent different origins of an otherwise morphologically similar tumor entity, mostly driven by clonal KRAS mutations. Further studies are needed to determine the risk of multiple IMAs in UIP.
Yang and colleagues from New York sequenced IMAS using the MSK-Assay “Integrated Mutation Profiling of Actionable Cancer Targets” for profiling more than 400 genes by DNA-based NGS and 62 genes for RNA-based NGS. Overall genomic analyses provided sufficient variants for calculating clonality versus non-clonality with significant confidence. All but one of 24 tumors revealed clonal KRAS G12D or G12V mutations and very few other driver alterations as a F11R-NRG1 fusion, a non-canonical BRAFK483E mutation and an ERBB2 exon20 insertion. Only three of their tumors revealed a KRASG12C mutation. The NRG1 fusion is quite interesting as NRG1 fusion events were originally identified from an IMA7. Thus, all oncogenic driver alterations appear to converge in a pathway of EGFR/HER2 receptor activation or downstream alterations in the KRAS/BRAF signaling cascade. Smoking habits were not reported in detail, however, the overall smoking burden in this cohort was 13 pack years, indicating that smoking may occur in patients with IMAs but may not be the main driver of tumorigenesis. This data is also supported by previous studies reporting different genomic profiles between mucinous and non-mucinous adenocarcinomas and low association with smoking2,3. Interestingly, none of the patients revealed radiological signs of interstitial lung disease (ILD), and hence the two cohorts from Korea and the US may reflect different epidemiological backgrounds.
The most striking feature of both studies remains the strong difference in types of KRAS mutations between IMAs and non-mucinous adenocarcinomas. While in non-mucinous carcinomas, KRASG12C is the most frequent driver with an incidence of almost 45%8, more than 90% of IMAs reveal KRAS G12D,V or other rare activating mutations. The mystery of entity-related KRAS mutations has been noted previously, for example as striking differences in KRAS mutation types between pancreatic, colonic, and non-mucinous lung adenocarcinomas. It is very likely that the establishment and outgrowth of adenocarcinomas may be dependent on different KRAS mutations under the control of specific homing factors. Obviously, G12C is not favored by the local milieu in the peripheral lung and further experimental mouse models studying the local tissue-specific requirements are needed to address this question. In any case, the successful clinical development of KRASG12C-specific inhibitors9,10 will not change the dismal outcome of most patients with IMAs. Also, early detection is unlikely to prevent disease progression as early airborne spread frequently prevents local surgery. Thus, current development of panKRAS inhibitors11 remains the cornerstone for possible future treatment options.
In summary, both studies by Kim (this issue) and Yang underline the importance of deep molecular profiling of all lung cancers currently summarized under the broad umbrella of “lung adenocarcinoma”. This umbrella comprises a broad spectrum of quite different tumor entities and molecular profiling will not only lead to more precise and effective therapies in advanced stages but also better diagnose the precise tumor entity with its specific risk profile for progression and need for adjuvant therapies in locally confined resected tumors12. Thus, pathologists need to integrate morphological features and genomic profiles for precision diagnostics in lung cancers both in advanced and in localized stages.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Competing interests
The author declares no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO Classification of Tumors. Thoracic Tumors Vol. 5, 5th edn (IARC, 2021).
- 2.Shim HS, et al. Unique genetic and survival characteristics of invasive mucinous adenocarcinoma of the lung. J. Thorac. Oncol. 2015;10:1156–1162. doi: 10.1097/JTO.0000000000000579. [DOI] [PubMed] [Google Scholar]
- 3.Chang JC, et al. Comprehensive molecular and clinicopathologic analysis of 200 pulmonary invasive mucinous adenocarcinomas identifies distinct characteristics of molecular subtypes. Clin. Cancer Res. 2021;27:4066–4076. doi: 10.1158/1078-0432.CCR-21-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim, M. et al. Genomic characteristics of invasive mucinous adenocarcinoma of the lung with multiple pulmonary sites of involvement. Mod. Pathol. 10.1038/s41379-021-00872-0 (2021). [DOI] [PMC free article] [PubMed]
- 5.Yang SR, et al. Invasive mucinous adenocarcinomas with spatially separate lung lesions: analysis of clonal relationship by comparative molecular profiling. J. Thorac. Oncol. 2021;16:1188–1199. doi: 10.1016/j.jtho.2021.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet. 2020;52:177–186. doi: 10.1038/s41588-019-0569-6. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Cuesta L, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Disco. 2014;4:415–422. doi: 10.1158/2159-8290.CD-13-0633. [DOI] [PubMed] [Google Scholar]
- 8.Scheffler M, et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J. Thorac. Oncol. 2019;14:606–616. doi: 10.1016/j.jtho.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skoulidis F, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 2021;384:2371–2381. doi: 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kessler D, Gerlach D, Kraut N, McConnell DB. Targeting son of sevenless 1: the pacemaker of KRAS. Curr. Opin. Chem. Biol. 2021;62:109–118. doi: 10.1016/j.cbpa.2021.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Saleh, M. M. et al. Comprehensive Analysis of TP53 and KEAP1 Mutations and their Impact on Survival in Localized and Advanced Stage Non-Small Cell Lung Cancer. J. Thorac. Oncol. 10.1016/j.jtho.2021.08.764 (2021). Online ahead of print. [DOI] [PubMed]