

Abstract
Pancreatic adenocarcinoma is a lethal disease that is projected to become the second most common cause of cancer deaths by 2030. The role of adjuvant therapy after surgical resection has been established by several clinical trials to prolong survival and improve outcomes. Multiagent chemotherapy seems to be the most promising approach to counteract early recurrence and improve survival; however, in the era of precision medicine, patient selection and individualized therapy seems to hold the key to desirable superior outcomes. Several cancer susceptibility genes have been proven to be associated with an increased risk of pancreatic cancer, both familial and sporadic cases. The role of genomic profiling for germline variants has been extensive and of limited clinical value, considering their low prevalence in pancreatic ductal adenocarcinoma (PDAC). However, an accumulating body of evidence from several studies in the past decade have successfully shown a recognizable value of germline variants in risk assessment and patient stratification. Recently, anti‐PD‐1 therapy (pembrolizumab) has been FDA‐approved for use in solid malignancies with a Mismatch repair deficiency or high Microsatellite instability. Several trials have evaluated the role of poly (ADP‐ribose) polymerase (PARP) inhibitors in patients harboring germline BRCA1/2 mutations. Finally, germline variants in DNA damage response genes and particularly deleterious ones have the potential to guide therapy after surgical resection and serve as biomarkers to predict survival. The dire need to address challenges for applying precision medicine in real‐life clinical settings for PDAC patients lies in further characterizing the genetic and molecular processes through translational research.
Keywords: DDR genes, germline variants, pancreatic cancer, precision medicine
The role of genomic profiling for germline variants has been extensive and of limited clinical value, considering their low prevalence in pancreatic ductal adenocarcinoma. However, an accumulating body of evidence from several studies in the past decade that have successfully shown a recognizable value of germline variants in risk assessment and patient stratification. Germline variants in DNA damage response genes and particularly deleterious ones have the potential to guide therapy after surgical resection and serve as biomarkers to predict survival.
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a lethal disease largely attributed to late presentation, early recurrence, and resistance to treatment. Pancreatic cancer remains the fourth leading cause of cancer‐related deaths in both males and females, with an increased estimated number of new cases and deaths compared to 2020. 1 According to the most recent cancer statistics in the United States, the estimated number of new pancreatic cancer cases in 2021 is 60,430 cases with 48,220 estimated deaths. 1 At this rate, it is projected that PDAC will become the second most common cause of all cancer‐related deaths by 2030. 2
Despite the advancement in diagnostic modalities and surgical techniques, only 20% of patients diagnosed with pancreatic adenocarcinoma have resectable disease at the time of diagnosis. 3 Further understanding the aggressive biology and the delayed presentation of patients with PDAC is crucial to identify methods for early detection. In that regard, Yu et al 4 evaluated 13,131 patients from the National Cancer Institute's Surveillance, Epidemiology and End Results database in an attempt to evaluate the rate of progression of PDAC. The results showed that the rapid progression of tumor size from low to advanced stage occurs once lesions become clinically detectable. It also showed that progression from T1 stage to T4 occurred within only 14 mo.
Consequently, the role of a comprehensive treatment plan became apparently essential to achieve curative therapy with a lower chance of early recurrence and systemic spread. This is where multimodal chemotherapy in the neoadjuvant and/or adjuvant setting comes into play, offering patients an increased chance of curative resection and improved survival outcomes. 5 Unfortunately, tumor heterogeneity and resistance to therapy have deemed the available treatments very limited at achieving desirable outcomes. 6 Particularly, the genetic and epigenetic modifications within the tumor parenchyma and its microenvironment create different phenotypic variants and molecular subtypes. 6 The resulting tumor heterogeneity can influence the response to therapy and clonal evolution of cancer cells.
The fact that the majority of patients with PDAC recur after surgery provides evidence that certain populations of cancer cells persist locally after resection or systemically where they would then develop into clinically detectable lesions. As such, systemic chemotherapy agents, administered before or after surgery, also play a role in clonal selection and expansion of resistant cancer cell populations. 7 Although the mechanism for clonal selection and resistance is poorly understood, further understanding of the biology and genetic basis of the evolution of PDAC would allow us to develop and administer targeted therapies in a personalized fashion. In this review we present a timeline of the progression in the evidence supporting the role of adjuvant therapy in the treatment of PDAC, as well as a review of germline mutations in cancer susceptibility genes and their potential role in early detection and targeted therapy for pancreatic cancer in the precision medicine era.
2. ADJUVANT CHEMOTHERAPY IN PANCREATIC ADENOCARCINOMA
The role of adjuvant chemotherapy in pancreatic cancer has witnessed an accumulation of evidence from several studies since the early 1990s (Table 1). The European Study Group of Pancreatic Cancer (ESPAC‐1) was the first multicenter trial to bring forth the benefit of 5‐fluorouracil (5‐FU)‐based chemotherapy as an adjuvant therapy after curative surgical resection. The results showed that the median overall survival (OS) was 19.7 mo in the chemotherapy group compared to 14 mo in the group without adjuvant chemotherapy (P = .005). 8 In 2004, the ESPAC group published long‐term follow‐up results with a 5‐y OS of 21.1% in the adjuvant therapy group compared to 8.4% in the surgery alone group (P = .009), confirming the survival benefit of adjuvant chemotherapy after surgical resection in pancreatic adenocarcinoma. 9
TABLE 1.
Survival outcome from randomized controlled trials with adjuvant therapy for PDAC after surgical resection
| Trial | Country | Treatment arms | Median OS (mo) | HR | P value | 5‐y OS (%) |
|---|---|---|---|---|---|---|
| ESPAC‐1 | Europe | 5 FU + Folinic acid | 19.7 | 0.66 | .005 | 21.1 |
| Surgery alone | 14 | 8 | ||||
| CONKO‐001 | Germany and Austria | Gemcitabine | 22.8 | 0.76 | .01 | 20.7 |
| Surgery alone | 20.2 | 10.4 | ||||
| ESPAC‐3 | Europe | 5 FU + Folinic acid | 23.1 | 0.94 | .39 | 15.9 |
| Gemcitabine | 23.6 | 17.5 | ||||
| JASPAC‐01 | Japan | S‐1 prodrug | 46.5 | 0.57 | <.001 | 44.1 |
| Gemcitabine | 25.5 | 24.4 | ||||
| PRODIGE 24 | France and Canada | mFOLFIRINOX | 54.5 | 0.64 | .003 | 63.4 a |
| Gemcitabine | 35 | 48.6 a | ||||
| APACT | North America, Europe, Australia | nab‐Paclitaxel + Gemcitabine | 40.5 | 0.82 | .0045 | — |
| Gemcitabine | 36.2 | — |
Abbreviations: HR, hazard ratio; OS, overall survival.
3‐y OS (%).
The long‐term results of the CONKO‐001 (Charite Onkologie 001) randomized trial published in 2013 reported the impact of adjuvant therapy on survival when compared to surgery alone. Adjuvant therapy following curative surgical resection increased the median disease‐free survival (DFS) from 6.7 mo with surgery alone to 13.4 mo (P < .001). 10 The median OS was 22.8 mo in the group receiving adjuvant gemcitabine therapy compared to 20.2 mo in the group undergoing surgical resection without adjuvant chemotherapy. 10 Furthermore, the 5‐y OS increased from 10.4% in the surgery alone group to 20.7% in the adjuvant therapy group as well as 10‐y overall survival at 12.2% and 7.7%, respectively. 11 Overall, the strength of the CONKO‐001 trial lies in the multicenter trial design, which accrued patients from 88 different hospitals, including community hospitals, in Germany and Austria, allowing generalization of the already established role of adjuvant therapy in PDAC.
Although ESPAC‐3 reported no significant difference in OS between a combination of 5‐FU and folinic acid compared to gemcitabine, the Japanese JASPAC‐01 trial randomized 385 patients into an S‐1 group (fluorouracil‐based prodrug) and a gemcitabine group. 12 The results showed that 5‐y OS in the S‐1 group was 44.1% compared to 24.4% in the gemcitabine group; however, the efficacy of S‐1 prodrug on Western and non‐Japanese populations limits the generalizability of these findings. 13
In 2018, the PRODIGE trial was a landmark trial that reported findings revolutionizing the role of combination chemotherapy as an adjuvant treatment of PDAC based on the success of fluorouracil, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) in the metastatic setting. Although the modified FOLOFIRNOX group was associated with a higher incidence of therapy‐related toxicity, the median OS was 54.4 mo compared to 35 mo in the gemcitabine group (P = .003). 14 More recently, the APACT phase III study aimed at evaluating the efficacy and safety of a combination of nab‐paclitaxel (ABRAXANE) with gemcitabine vs gemcitabine alone in surgically resected pancreatic cancer showing a survival benefit of nab‐paclitaxel combined with gemcitabine with an OS of 40.5 mo vs 36.2 mo in gemcitabine alone (P = .045). 15 , 16 Long‐term results with additional OS follow‐up will offer a better assessment of the role of ABRAXANE with gemcitabine combination therapy in adjuvant therapy.
3. DNA DAMAGE AND DNA REPAIR
On a large scale, DNA damage can occur due to exogenous agents such as exposure to ionizing radiation or due to endogenous cellular processes (replication error, hydrolysis, nitrogen base oxidation, etc). If the resultant damage involves a single DNA strand, a single‐strand break (SSB) can occur. In cases where SSBs are due to a single base modification, base‐excision repair (BER) excises and replaces the inappropriate nucleotide with the correct one (Figure 1). If a DNA strand break involves a larger DNA segment, causing distortion in the DNA helix, nucleotide‐excision repair (NER) removes and resynthesizes a sequence of several nucleotides. Mismatch repair is another repair mechanism that is mostly responsible for detecting and repairing erroneous base pairings that occur during replication. Mismatch repair (MMR) proteins such as MLH1, MSH2, MSH6, PMS2, and Mut complex are involved in recognition, cleavage, and incorporation of the correct nucleotide. 17
FIGURE 1.
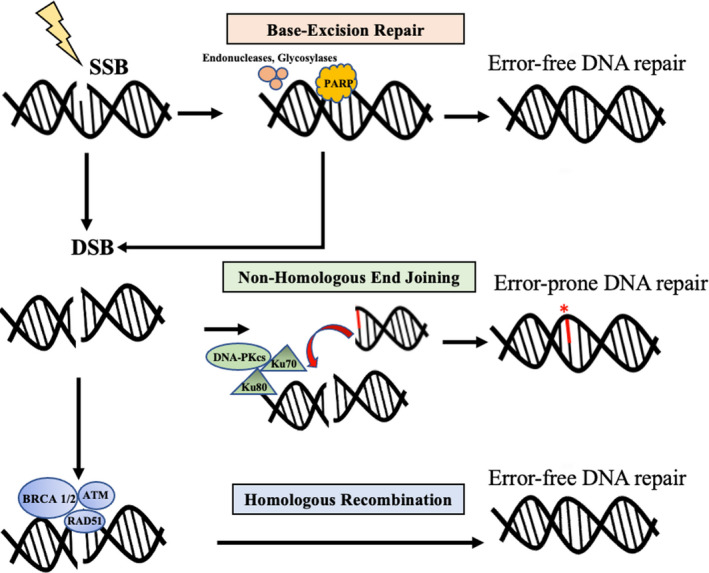
DNA damage and mechanisms involved in DNA damage repair of single‐strand (SSB) and double‐strand breaks (DSBs)
When an unrepaired SSB is encountered by a replication fork, an SSB can transform into a double‐strand break (DSB). In this case, DSBs can be repaired by either Homologous Recombination (HR) or Non‐homologous End Joining (NHEJ) (Figure 2). HR between homologous DNA segments is the “preferred” mechanism, since it is less likely to cause errors in DSB repair, whereas NHEJ is more error‐prone. The key proteins involved in recognition and repair of double‐stranded DNA breaks via the homologous recombination repair (HRR) pathway are BRCA1, BRCA2, PALB2, and ATM; those involved in NHEJ include DNA protein kinases (DNA‐PKcs), Ku70/80 heterodimer, DNA polymerases, and DNA ligases. 18
FIGURE 2.
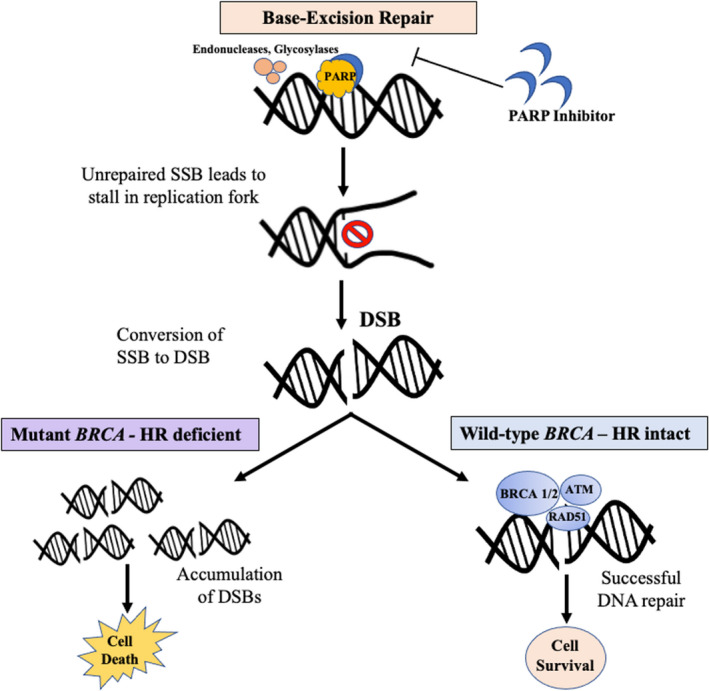
Mechanism of action of poly (ADP‐ribose) polymerase (PARP) inhibitors in homologous recombination (HR) deficient (mutant BRCA) and HR intact (wildtype BRCA) patients
Repair of DSBs by HR occurs during the S/G2 phase of the cell cycle; during this phase, BRCA proteins are also highly expressed. 19 Several studies have also shown that other proteins from known cancer susceptibility genes such as ATM, RAD51, CHK2, and others are also regulated in tandem with BRCA proteins and associated with a similar role where different pathways work together to repair DNA damage. Mutations in the HRR pathway genes will cause abnormal HRR functions, leading to genome instability. BRCA proteins play an important role in HR during DNA repair; thus, cells that are deficient in BRCA have impaired HR. This would lead the cell to seek other methods such as NHEJ or single‐strand annealing, which are more prone to error leading to structural variation and chromosomal instability. 20 During NHEJ, the single‐strand overhangs of the broken ends are removed before end‐joining, which leads to small insertions or deletions. If the repair fails and a DSB occurs at one end of the DNA segment, NHEJ would fuse that segment with any other nonadjacent double‐strand broken end that is available, which can ultimately lead to large deletions or translocations and structural variation.
Poly (ADP‐ribose) polymerase (PARP) is an enzyme that is involved in repair of SSB through base excision repair. Normal cells rely on both SSB repair by PARP and DSB repair by HR (the prominent role of BRCA 1 and 2), which would maintain genomic integrity and survival. 17 Cancer cells with mutant BRCA rely on PARP to avoid stalls in the replication fork and converting an SSB to a DSB. A PARP inhibitor would inhibit the role of PARP in SSB repair, deliberately leading to a stall in the replication fork and creation of a DSB. In the case of normal cells with an intact BRCA protein, DSB can be repaired through HR; however, in BRCA‐deficient cancer cells, DSBs cannot be repaired, leading to their accumulation and cell death (in this case, tumor cell death). In the past few years, several clinical trials have evaluated the role of different PARP inhibitors (olaparib, velaparib, and rucaparib) on disease response rate and survival in advanced PDAC, showing promising results. For instance, O'Reilly et al demonstrated an improved median OS of 23.3 mo for PDAC cases with germline BRCA1/2 treated with a combination of velaparib, cisplatin, and gemcitabine compared to a median OS of 11 mo among those harboring a wildtype BRCA1/2. The results of the most prominent clinical trials on PARP inhibitors are summarized in Table 2. 21 , 22 , 23 , 24 , 25 , 26
TABLE 2.
Clinical trials evaluating the role of PARP inhibitors in advanced PDAC with germline BRCA1/2 mutations
| Clinical trial | Mutation (N) | PDAC stage | PARPi | Treatment | Disease response rate | Survival (mo) |
|---|---|---|---|---|---|---|
| Kaufman et al (2015) | gBRCA1/2 (23) | Stage III/IV | Olaparib | Olaparib after prior Gem therapy |
Tumor response: 5/23 (21.7%) Stable disease ≥8 wk: 8/23 (35%) |
mPFS: 4.6 mo mOS: 9.8 mo |
| Lowery et al (2017) | gBRCA1/2 (16) | Stage III/IV | Velaparib | Velaparib after prior platinum therapy (14/16) | No confirmed response |
mPFS: 1.7 mo mOS: 3.1 mo |
| O'Reilly et al (2018) |
gBRCA1/2 (9) wtBRCA1/2 (7) |
Stage III/IV | Velaparib | Velaparib + Cis + Gem | gBRCA1/2:7/9 (77.8%) |
gBRCA1/2:
wtBRCA1/2:
|
| Shroff et al (2018) |
gBRCA1/2 (16) sBRCA1/2 (3) |
Locally advanced/ metastatic disease | Rucaparib | Rucaparib after previous chemotherapy | PR or stable disease ≥12 wk: 6/19 (31.6%) | NM |
| Golan et al (2019) | gBRCA1/2 (154) | Stage IV | Olaparib |
1. Olaparib after platinum‐based therapy (92) 2. Placebo after platinum‐based therapy (62) |
NM |
1. Olaparib:
2. Placebo:
|
| O'Reilly et al (2021) | gBRCA/PALB2 (50) | Stage III/IV | Velaparib |
1. Cis + Gem w Velaparib 2. Cis + Gem w/o Velaparib |
1. With Velaparib: 74.1% 2. Without Velaparib: 65.2% |
1. Cis + Gem w Velaparib:
2. Cis + Gem w/o Velaparib:
|
Abbreviations: gBRCA1/2, germline BRCA1/2; mOS, median overall survival; mPFS, median progression‐free survival; NM, not mentioned; PR, partial response; sBRCA1/2, somatic BRCA1/2.
4. PRECISION MEDICINE AND CANCER SUSCEPTIBILITY GENES
Genetic mutations and aberrant target pathways have played an important goal in the direction of precision treatment of pancreatic cancer. Immunotherapy drugs targeting immune checkpoints in the tumor microenvironment have long been researched in solid tumors. The most popular immune system checkpoint is the programmed cell death protein 1/programmed death‐ligand 1 (PD‐1/PD‐L1) where the PD‐1 receptor on T‐cells binds to the PD‐L1 on cancer cells, leading to suppression of T‐cell activation. 27 Targeting PD‐1 allows the inhibition of this interaction, which would prevent T‐cell apoptosis and promote tumor growth inhibition instead.
Pembrolizumab (anti‐PD‐1 antibody) was recently FDA approved for patients with unresectable or metastatic solid tumors with mismatch repair deficiency (dMMR) or MSI‐H. 28 With 1%–2% of patients with PDAC having dMMR or MSI‐H, immunotherapy holds enough promise to improve outcomes, especially when combined with other cytotoxic chemotherapy regimens where anti‐PD‐1 therapy has been shown to enhance the efficacy of other therapies. 29 Clinical trials investigating other antibody‐based targeted immunotherapies such as anti‐CTLA‐4, anti‐EGFR as well as others targeting specific cytokines are currently underway. 30
Several susceptibility genes have been well established in familial pancreatic cancer, including BRCA2 (most frequent; 5%–10% of familial pancreatic cancer cases), ATM (second most frequent; 2%–3% of familial cases), PALB2, CDKN2A, PRSS1, STK11, MLH1, and MSH2 genes. 31 , 32 , 33 , 34 , 35 These pathogenic germline mutations have also been reported in up to 3%–5% of sporadic pancreatic cancer cases (Table 3). 36 , 37 , 38 , 39 , 40 , 41
TABLE 3.
Recent studies reporting frequency of most common germline mutations in sporadic pancreatic adenocarcinoma
| N |
BRCA2 N (%) |
BRCA1 N (%) |
ATM N (%) |
PALB2 N (%) |
CDKN2A N (%) |
TP53 N (%) |
MLH1 N (%) |
MSH2 N (%) |
MSH6 N (%) |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Grant et al (2015) | 290 | 2 (0.69) | 1 (0.34) | 3 (1.03) | — | — | 1 (0.34) | 1 (0.34) | 2 (0.68) | 1 (0.34) |
| Shindo et al (2017) | 854 | 12 (1.41) | 3 (0.35) | 10 (1.17) | 2 (0.23) | 1 (0.11) | 1 (0.11) | 2 (0.23) | — | — |
| Hu et al (2018) | 2999 | 57 (1.9) | 18 (0.6) | 60 (2) | 12 (0.4) | 9 (0.30) | 6 (0.2) | 3 (0.1) | 1 (0.03) | 6 (0.2) |
| Brand et al (2018) | 298 | 4 (1.34) | 4 (1.34) | 10 (3.36) | 1 (0.33) | 1 (0.33) | 1 (0.33) | — | — | 1 (0.33) |
| Yurgelun et al (2019) | 289 | 4 (1.38) | 3 (1.04) | 4 (1.38) | 1 (0.34) | 2 (0.69) | 1 (0.34) | — | 1 (0.34) | 2 (0.69) |
| Rapposelli et al (2021) | 60 | 3 (5) | 1 (1.67) | 2 (3.33) | 1 (1.66) | — | — | — | — | — |
| Mutation frequency (%) | 4790 | 82 (11.7) | 30 (5.3) | 89 (12.2) | 17 (2.9) | 13 (1.4) | 10 (1.3) | 6 0.68) | 4 (1.1) | 10 (1.5) |
A recent study from Johns Hopkins Hospital evaluated 854 patients with sporadic pancreatic adenocarcinoma to determine the prevalence of germline mutations and pancreatic cancer susceptibility genes. 36 After DNA extraction, next‐generation sequencing (NGS) was performed with a 32‐gene panel including 10 known pancreatic cancer susceptibility genes (BRCA2, ATM, PALB2, BRCA1, CDKN2A, MLH1, MSH2, PRSS1, STK11, and TP53), a list of seven known cancer susceptibility genes (MSH6, PMS2, CDH1, RAD51C, RAD51D, BUB1B, and FANCJ), and candidate pancreatic cancer susceptibility genes, some of which are FANCA, FANCC, FANCG, FANCL, ARID1A, RECQL4, and others.
Of the 854 patients with pancreatic adenocarcinoma, 33 (3.9%) had a deleterious germline mutation (12 with germline BRCA2 mutation, 10 with ATM, two with PALB2, two with MLH1, one with CDKN2A, and one with TP53). In addition, 2/254 (0.2%) had a mutation in one of the candidate pancreatic cancer susceptibility genes (one of each BUB1B and BUB3) as well as 3/854 (0.3%) harbored a deleterious mutation in other known cancer susceptibility genes (CDH1, RAD51D, and RAD51B). When compared to a cohort of 339 patients with diagnoses other than pancreatic adenocarcinoma, 5/339 (1.5%) had a deleterious germline mutation compared to the 33% in the pancreatic adenocarcinoma cohort (P = .02). 36 The value of this study lies in demonstrating the relatively high yield of deleterious germline mutations in patients diagnosed with PDAC lacking any family history of pancreatic cancer.
Identifying these germline mutations in individuals diagnosed with pancreatic cancer would offer their relatives the chance for screening and early detection, as well as prevention strategies. Furthermore, patients harboring a BRCA mutation can receive targeted therapy with poly (ADP‐ribose) polymerase inhibitors or platinum‐based therapies. The National Comprehensive Cancer Network 42 guidelines for genetic testing candidates for gene testing include individuals with a close relative with pancreatic cancer, those who are of Ashkenazi Jewish descent with pancreatic cancer, and individuals with a close blood relative with ovarian cancer or young‐onset breast cancer. 42 Extrapolating these indications to the findings of the previously mentioned study would greatly underestimate the patients who could potentially have therapeutically targetable mutations.
5. PRECISION MEDICINE AND DNA DAMAGE REPAIR GENES
A recently published work by Pishvaian et al performed a retrospective analysis of the survival status of pancreatic cancer patients in the Know Your Tumor (KYT) initiative. Among the 677 patients who were finally included in the analysis, 189 patients had molecular changes with therapeutic guiding significance, and 46 of them received matched treatment. 43 The matched treatments in the study included administering several targeting therapies, some of which are immune checkpoint inhibitor therapy to patients with dMMR, HER2 antibody therapy to patients with abnormal amplification or activation of HER2, and PARP inhibitor to patients with homologous recombination DNA damage response and repair pathway genetic changes and others. The results showed that the OS of patients who received molecularly matched therapy was significantly longer than that of patients who did not receive matched therapy (2.58 y vs 1.51 y, HR = 0.42, P = .0004), and also significantly longer than patients without molecular changes with therapeutic guidance (2.58 y vs 1.32 y, HR = 0.34, P < .0001). 43
Golan et al recently published results from the landmark Pancreas Cancer Olaparib Ongoing (POLO) trial with great promise to change practice in pancreatic cancer. The POLO trial was based on evidence from previous studies showing that PARP inhibitors can offer better outcomes in patients with germline BRCA mutation compared to those with the wildtype BRCA gene. The trial aimed at evaluating the efficacy of olaparib maintenance therapy in patients harboring a germline BRCA1 and BRCA2 mutation and a diagnosis of metastatic PDAC. The results showed that maintenance PARP inhibitor therapy (olaparib) significantly increased progression‐free survival in patients with metastatic PDAC compared to placebo (7.4 mo vs 3.8 mo, P = .004); however, it did not show any benefit in prolonging OS. 25 The results from this trial remain inspiring and promising to show that variation in DNA damage repair (DDR) genes may influence the response to certain chemotherapy regimens that is a gateway to the era of precision therapy in pancreatic cancer therapy.
The question that remains to be answered is if a large cohort of DDR genes would influence the response to chemotherapy in patients with sporadic pancreatic cancer and potentially influence decision‐making in the clinical setting. Our recently published article aimed at evaluating the potential impact of 22 DDR genes on survival outcomes in patients with sporadic PDAC as well as their possible role in sensitizing these patients to chemotherapy regimens, specifically platinum‐based chemotherapy. A cohort of 854 patients diagnosed with sporadic PDAC between 2000 and 2015 at the Johns Hopkins hospital were selected. All patients underwent NGS to identify germline variants with a 32‐gene panel including 22 DDR genes. The final cohort included in the analysis consisted of 210 patients with one or more germline DDR mutation (19 deleterious variants, 103 VUS variants) and 375 patients with wildtype pancreatic cancer susceptibility genes. 44
Among the 122 patients with germline DDR gene variants, 14/19 (73.6%) deleterious patients and 77/103 (74.7%) VUS (variant of unknown significance) carriers underwent adjuvant chemotherapy. In the wildtype cohort (N = 375), 274 (73.1%) underwent adjuvant therapy. On multivariable analysis, the presence of germline variants (both deleterious and VUS carriers) was an independent predictor of worse DFS when compared to wildtype (HR = 0.462, P = .002 and HR = 0.784, P = .045, respectively). However, only deleterious variants and not VUS variants were independently predictive of a worse OS (HR = 0.517, P = .014 and HR = 0.875, P = .275, respectively). 44 Hypothesizing that patients with these DDR variants would benefit more from receiving adjuvant therapy, patients receiving neoadjuvant therapy were excluded and the two cohorts were further stratified into those undergoing adjuvant therapy and those without adjuvant therapy; DFS and OS were compared in patients harboring DDR variants and wildtype status. On the one hand, among patients undergoing adjuvant therapy, deleterious variant carriers (n = 13) had improved DFS and OS and VUS variant carriers (n = 65) had significantly improved DFS when compared to wildtype. On the other hand, no significant improvement in DFS or OS was noted in patients who did not undergo adjuvant therapy.
6. DISCUSSION
The advances in cancer genomics research as well as the deep understanding of carcinogenic pathways in the tumor microenvironment and the immune system have paved the way for the precision medicine era. The clinical applications of this individualized medicine approach have unveiled a great potential for early screening, risk prediction, and targeted therapies. However, the heterogeneity of pancreatic adenocarcinoma mandates that before precision drugs can be applied to clinical practice, it is important to gain an in‐depth understanding of the pathways governing tumor initiation, progression, and invasion. Only then can we identify targets that are actionable and offer patients the chance of receiving individualized therapies that “match” their tumor profile. Consequently, larger‐scale prospective clinical studies would provide support for precision medicine and formulate drug application guidelines for precision treatment.
The molecular landscape of PDAC has received respectable investigational efforts directed towards understanding this lethal disease both at the genomic and transcriptomic level. The somatic mutations of the four “mountain” or driver genes, namely, KRAS, TP53, SMAD4, and CDKN2A, are now recognized as the fundamental genetic alterations governing the carcinogenesis of PDAC from initiation to progression and dissemination. The role of germline mutations in cancer susceptibility genes has been strongly associated with familial predisposition; however, evidence supporting their significance in sporadic cases of pancreatic adenocarcinoma has recently begun to emerge.
Based on the different studies presented in this review article, pathogenic germline mutations have also been reported in up to 3%–5% of sporadic pancreatic cancer cases. First, identification of germline DDR gene variants in patients diagnosed with sporadic pancreatic cancer can be of clinical importance by offering their relatives genetic testing for determination of high‐risk individuals and detection of cancerous lesions early. 45 Second, germline genetic data can be a useful tool in risk assessment of early recurrence and potentially the role of adjuvant therapy after surgical resection. Third, the potential of identifying actionable gene mutations would justify the importance of routine genetic testing for all patients diagnosed with PDAC.
Germline mutations in BRCA1/2 genes have been proven to be of great value in patient stratification for individualized therapy. Patients diagnosed with ovarian cancer bearing germline BRCA gene mutations were shown to have a superior response to platinum‐based chemotherapy compared to other chemotherapeutic drugs as well as a response to PARP inhibition. 46 , 47 The role of PARP inhibitors in pancreatic cancer patients with germline mutations has been investigated by multiple clinical trials, including olaparib (ClinicalTrials.gov Identifier: NCT01078662, NCT02184195) and veliparib (NCT01585805, NCT02890355). 48 , 49 , 50 The results of the phase III POLO trial showed great promise for the clinical benefit of olaparib maintenance therapy for patients with germline BRCA mutations and metastatic pancreatic cancer. However, the proportion of patients with sporadic PDAC harboring a germline BRCA mutation who would benefit from receiving PARP inhibitors as a targeted therapy is low. As such, early results from trials investigating novel generations of PARP inhibitors have demonstrated efficacy of these therapies extending beyond germline BRCA variants to include germline PALB2 as well. 50 A recent study by Blair et al showed that out of 658 patients identified over a 15‐y period with resected PDAC and no identifiable family history, only 22 (3%) patients had a germline BRCA1 or BRCA 2 mutation. 51 On a bright note, these patients were identified to have a worse OS compared to matched patients with BRCA1/BRCA2 wildtype genes (20.2 vs 27.8 mo, respectively, P = .034) with a significant improvement in survival among those who received platinum‐based adjuvant therapy compared to alternative chemotherapy (31.0 vs 17.8 vs 9.3 mo, respectively, P < .001). 51
The clinical application of precision medicine is not without challenges. Although Pishvaian et al identified about a quarter of patients with actionable genetic alterations, only 4% of the more than 1000 patients included in the KYT initiative actually received a molecularly matched therapy. 43 Thus, continued efforts are genetic testing and research to identify novel markers for targeted therapies and risk stratification is essential. Our recently published work evaluating 22 DDR genes showed that sporadic PDAC patients with deleterious DDR gene mutations would significantly benefit from receiving systemic adjuvant therapy after surgical resection. Multi‐institutional prospective studies and clinical trials are warranted to further evaluate the responsiveness of germline DDR variants to different chemotherapeutic regimens in the both the adjuvant as well as neoadjuvant settings. Further studies aimed at understanding the role of mutations in pancreatic cancer susceptibility genes other than BRCA such as ATM and PALB2 are necessary to identify their potential in risk assessment and targeting. 41 Although large gene panels are spanned with uncertain variants and variants of unknown significance, further research and analysis of germline genetic data will render that ambiguity temporary and offer potentially useful tools in risk stratification and patient selection. Thus, exploring VUS variants and their clinical significance could offer new opportunities to further characterize the role of germline mutations in precision medicine. Consequently, once the value of these different genetic determinants is identified, larger genetic datasets will be organized in more focused and easily applied gene panels for clinical use. Finally, an important limitation to be considered is the low frequency of germline mutations detected in PDAC in spite of the growing body of evidence in the literature reporting the feasibility of detecting these deleterious mutations. In fact, a challenge of molecular profiling in general extends to detecting germline variants as the actionability of these biomarkers as well as their cost‐effectiveness is still unknown and not guaranteed.
7. CONCLUSION
The field of genomic profiling and pancreatic cancer genetics is on the rise and has shown promising results so far in identifying susceptibility genes that can greatly contribute to our endeavors in precision therapy. The accumulating evidence presented in this review emphasizes the necessity for further research dedicated to the discovery of novel biomarkers in cancer susceptibility genes and their germline variants. Genomic profiling to detect germline variants, and DDR genes in particular, is of great importance as a prognostic biomarker and potential target for individualized therapy in sporadic PDAC. Although most pathogenic variants discovered so far are rare, their established role so far has been undeniably valuable. The application of precision medicine in real‐time clinical settings has its own challenges; however, there should be no doubt in the scientific community that the future of pancreatic cancer treatment is in our hands to fully characterize the genetic, epigenetic, and molecular determinants of its aggressive nature to better serve a population in need of targeted therapy.
DISCLOSURE
Conflict of interest: The authors declare no conflicts of interest for this article.
Funding: None.
Author Contributions: Draft of the manuscript, SS, AB and JY; data collection, SS; approval of the final manuscript, all authors; supervision of the study, JY.
Shoucair S Baker AR Yu J. Germline Variants in DNA Damage Repair Genes: An Emerging Role in the Era of Precision Medicine in Pancreatic Adenocarcinoma. Ann Gastroenterol Surg.2022;6:7–16. 10.1002/ags3.12514
REFERENCES
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21. [DOI] [PubMed] [Google Scholar]
- 3. Strobel O, Hank T, Hinz U, Bergmann F, Schneider L, Springfeld C, et al. Pancreatic cancer surgery: the New R‐status counts. Ann Surg. 2017;265(3):565–73. [DOI] [PubMed] [Google Scholar]
- 4. Yu J, Blackford AL, Dal Molin M, Wolfgang CL, Goggins M. Time to progression of pancreatic ductal adenocarcinoma from low‐to‐high tumour stages. Gut. 2015;64(11):1783–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Strobel O, Neoptolemos J, Jager D, Buchler MW. Optimizing the outcomes of pancreatic cancer surgery. Nat Rev Clin Oncol. 2019;16(1):11–26. [DOI] [PubMed] [Google Scholar]
- 6. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12(7):487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Makohon‐Moore A, Iacobuzio‐Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16(9):553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neoptolemos JP, Dunn JA, Stocken DD, Almond J, Link K, Beger H, et al. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet. 2001;358(9293):1576–85. [DOI] [PubMed] [Google Scholar]
- 9. Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350(12):1200–10. [DOI] [PubMed] [Google Scholar]
- 10. Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative‐intent resection of pancreatic cancerA randomized controlled trial. JAMA. 2007;297(3):267–77. [DOI] [PubMed] [Google Scholar]
- 11. Oettle H, Neuhaus P, Hochhaus A, Hartmann JT, Gellert K, Ridwelski K, et al. Adjuvant chemotherapy with gemcitabine and long‐term outcomes among patients with resected pancreatic cancer: the CONKO‐001 randomized trial. JAMA. 2013;310(14):1473–81. [DOI] [PubMed] [Google Scholar]
- 12. Neoptolemos JP, Moore MJ, Cox TF, Valle JW, Palmer DH, McDonald AC, et al. Effect of adjuvant chemotherapy with fluorouracil plus folinic acid or gemcitabine vs observation on survival in patients with resected periampullary adenocarcinoma: the ESPAC‐3 periampullary cancer randomized trial. JAMA. 2012;308(2):147–56. [DOI] [PubMed] [Google Scholar]
- 13. Uesaka K, Boku N, Fukutomi A, Okamura Y, Konishi M, Matsumoto I, et al. Adjuvant chemotherapy of S‐1 versus gemcitabine for resected pancreatic cancer: a phase 3, open‐label, randomised, non‐inferiority trial (JASPAC 01). Lancet. 2016;388(10041):248–57. [DOI] [PubMed] [Google Scholar]
- 14. Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul J‐L, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379(25):2395–406. [DOI] [PubMed] [Google Scholar]
- 15. Goldstein D, El‐Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, et al. nab‐Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long‐term survival from a phase III trial. J Natl Cancer Inst. 2015;107(2):dju413. [DOI] [PubMed] [Google Scholar]
- 16. Tempero MA, Reni M, Riess H, Pelzer U, O'Reilly EM, Winter JM, et al. APACT: phase III, multicenter, international, open‐label, randomized trial of adjuvant nab‐paclitaxel plus gemcitabine (nab‐P/G) vs gemcitabine (G) for surgically resected pancreatic adenocarcinoma. J Clin Oncol. 2019;37(15_suppl):4000. [Google Scholar]
- 17. Shrinivas SA, Shanta SH, Prajakta BB. <DNA repair 2017.pdf>. Glob J Pharmaceu Sci. 2017;3(2):555613. [Google Scholar]
- 18. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–82. [DOI] [PubMed] [Google Scholar]
- 19. Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. [DOI] [PubMed] [Google Scholar]
- 20. Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med. 2006;7(3):165–72. [DOI] [PubMed] [Google Scholar]
- 21. Kaufman B, Shapira‐Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lowery MA, Kelsen DP, Capanu M, Smith SC, Lee JW, Stadler ZK, et al. Phase II trial of veliparib in patients with previously treated BRCA‐mutated pancreas ductal adenocarcinoma. Eur J Cancer. 2018;89:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Reilly EM, Lee JW, Lowery MA, Capanu M, Stadler ZK, Moore MJ, et al. Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: germline BRCA mutation carriers and wild‐type BRCA pancreatic ductal adenocarcinoma. Cancer. 2018;124(7):1374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, et al. Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation. JCO Precis Oncol. 2018;2:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Golan T, Hammel P, Reni M, Cutsem EV, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA‐mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol. 2020;38(13):1378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pu N, Lou W, Yu J. PD‐1 immunotherapy in pancreatic cancer: current status. J Pancreatol. 2019;2(1):6–10. [Google Scholar]
- 28. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker‐defined disease. J Immunother Cancer. 2018;6(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahmad‐Nielsen SA, Bruun Nielsen MF, Mortensen MB, Detlefsen S. Frequency of mismatch repair deficiency in pancreatic ductal adenocarcinoma. Pathol Res Pract. 2020;216(6):152985. [DOI] [PubMed] [Google Scholar]
- 30. Kaur J, Singh P, Enzler T, Sahai V. Emerging antibody therapies for pancreatic adenocarcinoma: a review of recent phase 2 trials. Expert Opin Emerg Drugs. 2021;26(2):103–29. [DOI] [PubMed] [Google Scholar]
- 31. Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy M, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2(1):41–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;6(2):166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17(7):569–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Couch FJ, Johnson MR, Rabe KG, Brune K, de Andrade M, Goggins M, et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2007;16(2):342–6. [DOI] [PubMed] [Google Scholar]
- 35. Hahn SA, Greenhalf B, Ellis I, Sina‐Frey M, Rieder H, Korte B, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003;95(3):214–21. [DOI] [PubMed] [Google Scholar]
- 36. Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor‐Das A, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol. 2017;35(30):3382–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. 2015;148(3):556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018;319(23):2401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brand R, Borazanci E, Speare V, Dudley B, Karloski E, Peters MLB, et al. Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer. 2018;124(17):3520–7. [DOI] [PubMed] [Google Scholar]
- 40. Yurgelun MB, Chittenden AB, Morales‐Oyarvide V, Rubinson DA, Dunne RF, Kozak MM, et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet Med. 2019;21(1):213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rapposelli IG, Zampiga V, Cangini I, Arcangeli V, Ravegnani M, Valgiusti M, et al. Comprehensive analysis of DNA damage repair genes reveals pathogenic variants beyond BRCA and suggests the need for extensive genetic testing in pancreatic cancer. BMC Cancer. 2021;21(1):611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. (NCCN) NCCN NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) Pancreatic Adenocarcinoma; Version 2.2021.
- 43. Pishvaian MJ, Bender RJ, Halverson D, Rahib L, Hendifar AE, Mikhail S, et al. Molecular profiling of patients with pancreatic cancer: initial results from the know your tumor initiative. Clin Cancer Res. 2018;24(20):5018–27. [DOI] [PubMed] [Google Scholar]
- 44. Hu H, Zhu Y, Pu N, Burkhart RA, Burns W, Laheru D, et al. Association of germline variants in human DNA damage repair genes and response to adjuvant chemotherapy in resected pancreatic ductal adenocarcinoma. J Am Coll Surg. 2020;231(5):527–35.e514. [DOI] [PubMed] [Google Scholar]
- 45. Fountzilas E, Eliades A, Koliou GA, Achilleos A, Loizides C, Tsangaras K, et al. Clinical significance of germline cancer predisposing variants in unselected patients with pancreatic adenocarcinoma. Cancers (Basel). 2021;13(2):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cho A, Park JY, Lee SW, Kim D‐Y, Suh D‐S, Kim J‐H, et al. Real‐world experience of olaparib as maintenance therapy in BRCA‐mutated recurrent ovarian cancer. Arch Gynecol Obstet. 2021;304(4):1055–63. [DOI] [PubMed] [Google Scholar]
- 47. Zheng F, Zhang Y, Chen S, Weng X, Rao Y, Fang H. Mechanism and current progress of Poly ADP‐ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed Pharmacother. 2020;123:109661. [DOI] [PubMed] [Google Scholar]
- 48. Javle M, Shacham‐Shmueli E, Xiao L, Varadhacnary G, Halpern N, Fogelman D, et al. Olaparib monotherapy for previously treated pancreatic cancer with DNA damage repair genetic alterations other than germline BRCA variants: findings from 2 phase 2 nonrandomized clinical trials. JAMA Oncol. 2021;7(5):693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pishvaian MJ, Wang H, He AR, Hwang JJ, Smaglo BG, Kim SS, et al. A phase I/II study of veliparib (ABT‐888) in combination with 5‐fluorouracil and oxaliplatin in patients with metastatic pancreatic cancer. Clin Cancer Res. 2020;26(19):5092–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reiss KA, Mick R, O'Hara MH, Teitelbaum U, Karasic TB, Schneider C, et al. Phase II study of maintenance rucaparib in patients with platinum‐sensitive advanced pancreatic cancer and a pathogenic germline or somatic variant in BRCA1, BRCA2, or PALB2. J Clin Oncol. 2021;39(22):2497–505. [DOI] [PubMed] [Google Scholar]
- 51. Blair AB, Groot VP, Gemenetzis G, Wei J, Cameron JL, Weiss MJ, et al. BRCA1/BRCA2 germline mutation carriers and sporadic pancreatic ductal adenocarcinoma. J Am Coll Surg. 2018;226(4):630–7.e631. [DOI] [PMC free article] [PubMed] [Google Scholar]