

Abstract
Rationale
The current understanding of human lung development derives mostly from animal studies. Although transcript-level studies have analyzed human donor tissue to identify genes expressed during normal human lung development, protein-level analysis that would enable the generation of new hypotheses on the processes involved in pulmonary development are lacking.
Objectives
To define the temporal dynamic of protein expression during human lung development.
Methods
We performed proteomics analysis of human lungs at 10 distinct times from birth to 8 years to identify the molecular networks mediating postnatal lung maturation.
Measurements and Main Results
We identified 8,938 proteins providing a comprehensive view of the developing human lung proteome. The analysis of the data supports the existence of distinct molecular substages of alveolar development and predicted the age of independent human lung samples, and extensive remodeling of the lung proteome occurred during postnatal development. Evidence of post-transcriptional control was identified in early postnatal development. An extensive extracellular matrix remodeling was supported by changes in the proteome during alveologenesis. The concept of maturation of the immune system as an inherent part of normal lung development was substantiated by flow cytometry and transcriptomics.
Conclusions
This study provides the first in-depth characterization of the human lung proteome during development, providing a unique proteomic resource freely accessible at Lungmap.net. The data support the extensive remodeling of the lung proteome during development, the existence of molecular substages of alveologenesis, and evidence of post-transcriptional control in early postnatal development.
Keywords: proteomics, lung, development, alveolarization
At a Glance Commentary
Scientific Knowledge on the Subject
Proteins execute the bulk of cellular processes. However, proteome-level analysis of human lung development is lacking. In-depth characterization of the human lung proteome during normal development provides a molecular framework of understanding to address disrupted development in premature infants and lung injury repair in adults.
What This Study Adds to the Field
We hope this article will serve as a resource for pulmonary clinicians and biologists working in pediatrics, development, and regenerative medicine.
Human lung morphogenesis is parsed into five morphologically distinct stages: embryonic, pseudoglandular, canalicular, saccular, and alveolar stages (1). Much of what has been learned about lung development has been gleaned from murine models. However, there are important differences between murine and human lung development (2). In humans, the transition from saccular to alveolar stage occurs in utero, and at term, the human lungs are in the early alveolar stage. In contrast, at birth the murine lungs are at the saccular stage, and the alveolar stage initiates postnatally (2). Thus, there is a need to molecularly understand lung development using human tissue, especially for the alveolar stage, during which physiological differences exist between murine and human lung structure and function (3).
There is a paucity of data using state-of-the-art, unbiased omics approaches to understand the molecular mechanism(s) driving normal human lung development. Kho and colleagues (4) used RNA sequencing (RNA-seq) to characterize global gene expression during the pseudoglandular and canalicular phases of lung development. More recently, RNA-seq was used to characterize global gene expression changes within alveolar parenchyma isolated by laser capture microdissection (5). Although RNA transcripts provide an important view of biological systems, post-translational biological processes (e.g., protein turnover, degradation, or secretion) may result in discrepancies between RNA and protein concentrations.
Ding and colleagues (5) reported the first global proteomics data from the developing human lung during the postnatal alveolar stage. These data provided limited coverage of the proteome, requiring pooling of tissue from four time segments to reduce noise in the analysis. Herein we performed proteomics profiling to a depth of 8,938 proteins on whole-lung tissue from 10 distinct times from 11 donors of age 1 day to 8 years to identify molecular networks associated with postnatal lung maturation. The datasets generated were substantiated with selected validation experiments to demonstrate the accuracy of proteome measurements. This study is aimed to be an invaluable hypothesis generation resource for pediatric clinicians, researchers, or tissue engineers attempting to rebuild pulmonary tissues. Some of the results presented in the current manuscript have been previously reported in the form of abstracts (6, 7).
Methods
Human Lung Samples
Postmortem human lung samples were provided by the LungMAP Human Tissue Core (University of Rochester Medical Center). Sample collection and processing and handling of the samples are described on protocols.io (8, 9).
Protein Sample Preparation and Analysis by Liquid Chromatography–Tandem Mass Spectrometry
Proteomic sample preparation and liquid chromatography–tandem mass spectrometry analysis using an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) was performed as described previously (10, 11) and detailed in Supplementary Material and Methods in the online supplement.
Bulk RNA-Seq
Genome-wide transcriptomics was performed on 10 donor lungs of similar age range as the proteomics. The methods were previously described (12) and are detailed in Supplementary Material and Methods.
Proteomics Data Analysis
Raw mass spectrometry data were converted to peak lists using the DeconMSn (version 2.3.1.2) (13) and searched with MSGF+ (14) against the Homo sapiens UniProt/SwissProt database (15), bovine trypsin, and human keratin sequences. Spectra were filtered based on their MSGF+ scores, and only the proteins with two proteospecific peptides were conserved (false discovery rate <1%). For the quantitative analysis, the tandem mass tag (TMT) reporter ion intensities were extracted with MASIC (16). Peptides observed in at least two samples and proteins with two or more peptides were retained. These criteria resulted in 106,551 peptides with quantitative information attributed to human proteins (Dataset E2). Missing values were replaced with a value of NA, and data were log2 transformed. A median normalization (17, 18) was applied to the data, and peptide-level data were quantified to the protein level using a reference-based rollup approach (19).
Trend Analysis
Individual abundance trend analyses were performed using regression models to identify the proteins or transcripts that exhibited significant changes in normalized abundances across time. A linear and quadratic model of the normalized protein abundances were fit for each analyte. The two types of models employed are detailed in Supplementary Material and Methods. For each quantified biomolecule, the model with the smallest fitting P value was retained to describe its abundance trend during postnatal development.
Age Prediction Analysis
For age prediction, we first fit an elastic net regression model to the 11-donor isobaric labeled TMT proteomics data (Dataset E3). To estimate the age of independent human postnatal alveolar samples, we generated a 28-donor label-free proteomics dataset (Dataset E4). Subsequently, the model fitted using the TMT dataset was then used to predict the age of donors in the independent label-free dataset as described in Supplementary Material and Methods.
Immunofluorescence
Tissue sections (7 μm) were stained with primary antibodies, including NKX2.1, SFTPC, PECAM1, AGER, and ACTA2, as described in Supplementary Material and Methods, and sections were mounted using ProLong Gold (Thermo-Fisher) mounting medium and coverslipped.
Flow Cytometry
Tissue digests were used for analysis. Digestion and staining were previously described (12), where the list of antibodies employed is detailed.
Proteasome Validation Experiments
Western blots were performed to verify the modulation of proteasomal β5 and immunoproteasomal iβ5 subunits. Proteolytic activities were evaluated using the Proteasome-Glo 3 Substrate System (Promega). Details are provided in Supplementary Material and Methods.
Data Availability
Proteomics data are deposited and freely available at ProteomeXchange data repository, ProteomeXchangeID: PXD020798, and MassIVE data repository, MassIVE ID: MSV000085929. All the code used to generate the data was deposited on GitHub (https://github.com/GeremyClair/Proteomics_analysis_of_human_lung_development/).
Results and Discussion
LC–MS–based Identification of Proteins in Normal Developing Human Lung
We profiled the proteome of whole-lung tissue from 11 human donors at 10 time points from Postnatal Day 1 to 8 years of age using LC–MS–based proteomics. Donor lungs were selected based on normal tissue morphology and absence of pulmonary pathology as assessed by a pediatric pathologist; clinical information associated with each donor is provided in Dataset E1. We identified 8,938 proteins (false discovery rate < 1%) (Dataset E3), of which 6,281 proteins were used for quantification (i.e., detected with at least two peptides per protein and detected in at least 2/10 time points) (Dataset E3). A prior proteomics study of normal human lung development identified 2,254 proteins in tissues from pooled samples from four time segments (5). Although peptide amounts per LC–MS run were similar in both studies, the present study used a two-dimensional LC–MS approach with a state-of-the-art Lumos Orbitrap mass spectrometer, differences that may account for the almost fourfold increase in proteome coverage reported herein.
Temporal Dynamics of the Human Lung Proteome during Development
We performed individual protein trend analyses using regression models to identify the proteins modulated with age. Data were fit to both linear and quadratic models. Likelihood ratios tests were conducted to identify the proteins changing with significant linear or quadratic temporal trends. Only the trend with the best fitting P value was retained for each protein. Of the 6,281 quantified proteins, 53.7% were modulated following either a linear or quadratic trend (P < 0.05). The observed extensive remodeling of the proteome during the alveolar stage is consistent with previous findings in a murine model (11) in which approximately 50% of proteome was modulated from the canalicular to the late alveolar stage. Of the 3,373 proteins modulated over time, 2,418 were linearly decreasing; 608 were linearly increasing; 204 fit a quadratic concave model (i.e., increasing to an apex and then decreasing); and 143 fit a quadratic convex model (i.e., decreasing to a minimum and then increasing). The observation that more than 70% of all modulated proteins decreased over time suggests a global move toward cellular homeostasis as the lung matures. As expected, alveolar cell-specific marker proteins (Figures 1A and 1B) were increased during postnatal alveolar septation (20) (linear fitting P < 0.05) (Figures 1C–1F). Markers comprise the AT1-specific receptor for advanced glycation end products (RAGE_HUMAN), the AT2-specific surfactant protein C (PSPC_HUMAN) and ABCA3 (ABCA3_HUMAN), and actin α-2 (ACTA2_HUMAN) highly expressed in myofibroblasts. The increase in the abundance of RAGE/AGER was validated by Western blot (t test P = 0.0125; <30 d old n = 5 vs. >30 d old n = 6, Figures 1G and 1H).
Figure 1.

Immunofluorescence staining and relative protein abundance of the alveolar cell marker proteins in the tandem mass tag (TMT) proteomics dataset proteins during postnatal development. (A) Confocal immunofluorescence staining of AGER (green), PECAM1 (red), NKX2.1 (white), and DAPI (blue) for a 4-year-old donor. (B) Confocal immunofluorescence staining of SFTPC (green), ABCA3 (red), ACTA2 (white), and DAPI (blue) for a 3-year-old donor. (C–F) Relative abundance of the alveolar cell marker proteins measured by proteomics: AGER_HUMAN (C), ABCA3_HUMAN (D), ACTA_HUMAN (E), and PSPC_HUMAN (F). Likelihood ratio tests were conducted to identify the proteins with evidence of significant linear increases with age (*P < 0.05 and **P <0.01). (G) Western blot performed for RAGE. (H) Quantitative assessment made on the Western blots normalized by the abundance of GAPDH (*Student’s t test P = 0.0131; bars represent the mean values, and error bars are SE). Scale bars, 100 μm.
Silhouette Analysis Identifies Molecular Substages of Postnatal Alveolarization
Prior transcriptomic analyses of lung development support substages within the traditionally defined morphological phases of lung development using principal component analysis (PCA). Kho and colleagues suggested a previously unappreciated molecular subphase of early human lung development within the pseudoglandular stage (21). Using RNA-seq to characterize murine lung development, Beauchemin and colleagues identified four molecularly distinct stages of alveolar development between Postnatal Day 0 and Postnatal Day 18 (22). Principal component analysis was applied to our human lung proteome dataset to further evaluate the potential partitioning of developmental substages (Figure 2A). Silhouette analysis revealed that the proteome profiles of the donors were partitioned into two main groups; K-means clustering identified two distinct groups corresponding to one with ages from 1 to 24 days and a second from 3 months to 8 years. Using the same methods, two further subgroups were identified, resulting in four groups: 1) 1 day; 2) 5 days to 24 days; 3) 3 months to 2 years; and 4) 4 years to 8 years, similar to the four alveolarization substages identified by Beauchemin and colleagues in the murine lung (22).
Figure 2.

Proteomics profiling identifies substages of alveologenesis and predicts age of an independent cohort. (A) Principal component analysis showing the sample partitioning based on their tandem mass tag (TMT) proteomics profile. The four age subgroups depicted were identified using silhouette analysis. The labels indicate the donor identifiers and their age in days. (B) Age prediction from elastic net regression on the TMT proteomics dataset (blue dots). The model was fitted using the leave one out cross-validation. A 28-donor label-free proteomics dataset composed of the profile of the 11 donors previously profiled using the TMT dataset (red dots) and of 16 independent donors (green dots). The age model fitting yielded an R2 value of 0.75 when compared with the true ages of the 16 donors. LF = label free; PC = principal component.
Proteome Profiles Predict the Developmental Maturity of the Lung
We investigated the robustness of our proteomics data by testing the ability to estimate the ages of independent human postnatal lung samples. To do this, we used an elastic net regression model (23) to predict donor age. Elastic net models enable fitting a predictive model while conducting feature selection. The normalized log-transformed protein abundance profile for each donor was used as a set of explanatory variables. The log-transformed age was used as the dependent variable. We estimated the age of an independent human postnatal lung sample cohort (16 donors). Predictions of the ages of donors used in model fitting, as expected, yielded high R2 values (Figure 2B). Importantly, the elastic net also predicted donor ages for the independent validation set of 16 donors with an R2 value of 0.75 when compared with true donor age. The ability of the elastic net to predict the ages of donors based on an independent validation set demonstrates that the major trends in protein expression behave consistently across the examined datasets, and the proteins selected by the elastic net (Dataset E5) have strong predictive power related to donor age.
Biological Processes Modulated during Normal Human Lung Postnatal Development
A heat map representing the 3,373 differentially expressed proteins during normal lung development is depicted in Figure 3. To identify biological processes modulated within distinct temporal trends (i.e., linear increasing, linear decreasing, quadratic concave, and quadratic convex), we performed functional enrichment analysis using DAVID. Biological Process Gene Ontology terms (GO_BPs) enriched for the proteins fitting to each temporal trend are reported in Figure 3 (left panel), Figure E1, and Dataset E6. Telomerase activity correlates well with the differentiation state of cells, with diminished activity in mature terminally differentiated cells and enhanced activity in less mature progenitor and progenitor-like cells (24). Our proteomics data revealed telomerase-related GO_BPs were enriched in the group of proteins decreasing over time. These proteins included WRAP53/TCAB1 (telomerase Cajal body protein 1), coilin, the MRN complex protein RAD50, and multiple proteins member of the chaperone TCP-1 ring complex that mediates TCAB1 folding (25), consistent with prior studies in the murine lung, which showed that telomerase expression and activity peaked at birth and declined to undetectable concentrations by Postnatal Day 9 (26). Taken together, our data support the concept that proliferative progenitor cells continue to expand into distinct lineages beyond birth in humans. For the group of proteins fitting a linearly decreasing trend, various terms associated with post-transcriptional and post-translational modifications were enriched, including proteasome, RNA splicing, and multiple post-translational modifications (e.g., sumoylation, phosphorylation, and ubiquitination). These findings suggest a significant contribution of nontranscriptional regulation of postnatal lung growth and development. Ontology terms related to immunity, including “inflammation,” “immune regulation,” “platelets,” “phagocytosis,” and “defense to bacteria, viruses, and fungi” were enriched in the subset of proteins changing with linearly increasing trend, consistent with a recent report by single-cell RNA analysis in the murine lung (27). Other functional enrichments of proteins following a linearly increasing trend included “lipid metabolism,” likely associated with increased surfactant production (28), and “extracellular matrix,” necessary to provide support for alveolar structures (29).
Figure 3.

Biological Process Gene Ontology (GO_BP) terms and transcription factors associated with different temporal models. The heat map depicts the proteins fitted to linear decreasing (blue), linear increasing (red), quadratic concave (yellow), and quadratic convex (purple) models (fitting P < 0.05). The boxes on the left panel represent the GO_BP enrichments generated using DAVID. All GO_PB terms enriched for the proteins belonging to either of the quadratic trends are depicted in the right panel. For the linear trends, to reduce GO_BP redundancy, the number of “GO classes” was reduced using the cytoscape plugin EnrichmentMap (a detailed version of these enrichment maps is provided as Figure E1). The left panel lists the transcription regulators fitted to each of the trends. In bold red are the transcription regulators previously described as important for pulmonary development (see Datasets E7 and E9).
Transcriptional Control of Human Postnatal Lung Development
Although RNA-seq studies provide comprehensive information related to gene expression patterns of transcription factors during normal human lung development, comprehensive information at the protein level is limited. The deep proteome coverage achieved enabled the identification of transcription factors modulated in abundance at the protein level during normal human lung development. A total of 271/444 proteins annotated as transcription factor in the Ingenuity Pathway Database were modulated in abundance. Using a literature mining strategy detailed in Supplementary Material and Methods, we found that several of these transcription regulators were previously identified to be essential for pre- or postnatal lung development (Figure 3, right panel). For example, Hoxa5 (30, 31), Hop (32), and JunB (33) followed the same abundance trend as the ones described in the literature. We speculate that additional members in this subset may play unappreciated roles in postnatal lung formation and function.
Modulation of the Proteasome Abundance and Activities in the Postnatal Lung
Proteins associated with post-transcriptional mechanisms were more abundant during early postnatal development. For example, proteasome-related Gene Ontology (GO) terms were enriched for the proteins following a decreasing trend (Figure 3). The proteasome is the primary protein degradation system within human lungs (20). The caspase-, trypsin-, and chymotrypsin-like activities of the standard proteasome are mediated though three subunits: β1 (PSB1), β2 (PSB2), and β5 (PSB5), respectively. Most proteins of the 20S core and the 19S regulatory particle of the standard proteasome followed a decreasing trend (23/32 subunits including β1, β2, and β5) (Figure 4A and Dataset E9). In the immunoproteasome, the proteolytic subunits are exchanged with their inducible counterparts, iβ1 (PSB9), iβ2 (PSB10), and iβ5 (PSB8), and the regulatory particle is replaced by the PA28-ɑβ particle (34). All three inducible subunits followed an increasing trend, and the PA28-ɑβ seemed to increase as well (Figure 4B). We performed Western blots for β5/PSB5 and iβ5/PSB8, the subunits contributing to the chymotrypsin-like activity (Figures 4C–4F). In agreement with our proteome profiling, β5 was significantly more abundant in younger donors (<30 d) than older donors, whereas iβ5 followed an opposite trend. Although caspase- and trypsin-like activities were not significantly modulated, chymotrypsin-like activity increased in the lung homogenate of donors older than 30 days (Figure 4I). Collectively, these results suggest that chymotrypsin-like activity increases with the increased expression of the immunoproteasome subunit iβ5/PSB8. The caspase-like and the trypsin-like activities may remain equivalent in lung homogenates owing to the compensation of the decreased abundance of the standard proteasome subunits (β1/PSB1 and β2/PSB2) by the increased abundance of their immunoproteasome counterpart (iβ1/PSB9 and iβ2/PSB10).
Figure 4.
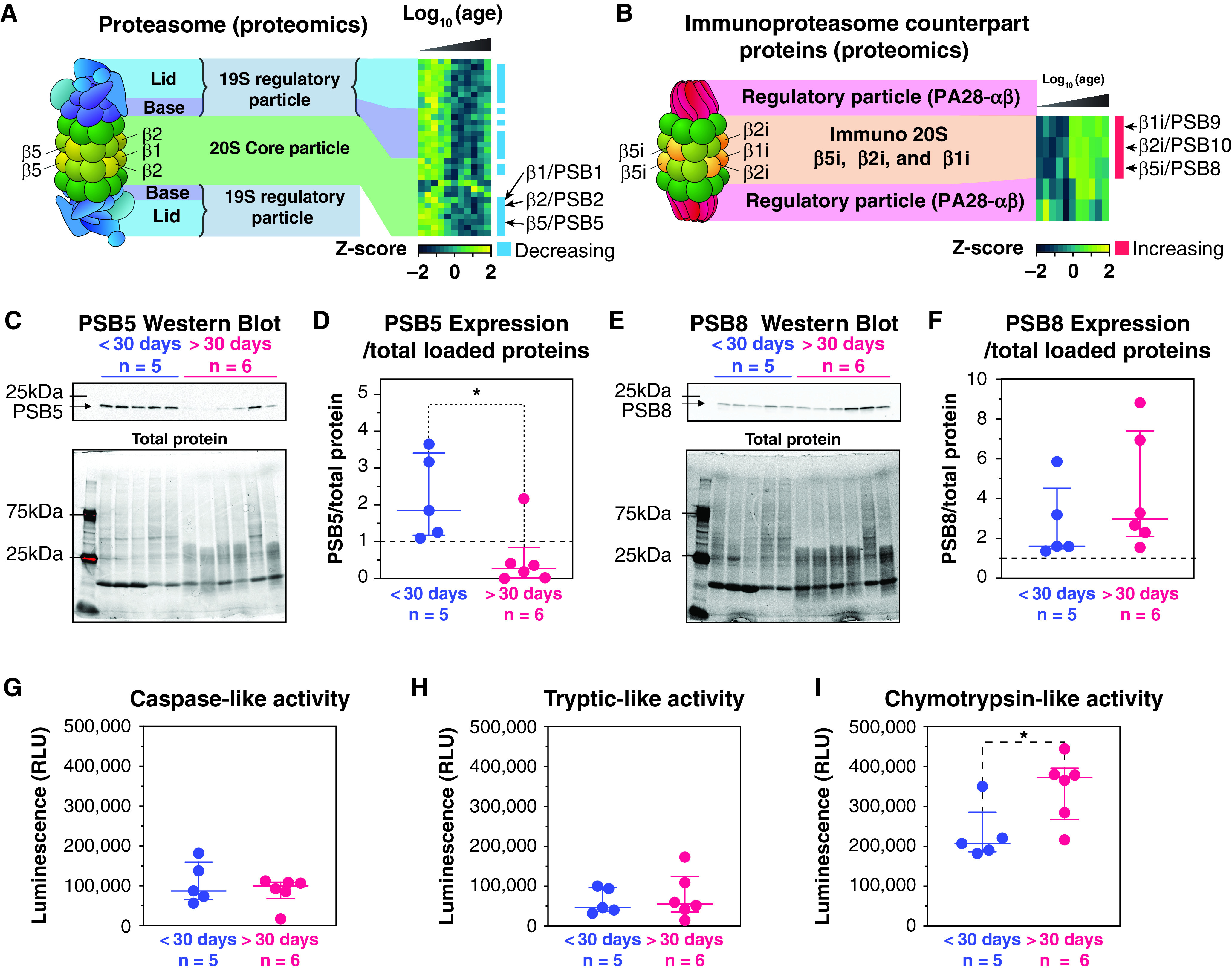
Proteasomal proteins are downregulated during postnatal lung development. (A) Heat map depicting the abundance of the standard proteasome subunits and of the 19S regulatory particle. (B) Heat map depicting the abundance of the immune-proteasome subunits and of the PA28-ɑβ regulatory particle. (C) Western blot of the PSB5/β5 subunit of the proteasome (top panel) and the total protein abundance (bottom panel). (D) PSB5 abundance normalized by the total loaded protein intensity from Western blotting. (E) Western blot of the PSB8/iβ5 subunit of the proteasome (top panel) and the total protein abundance (bottom panel). (F) PSB8 abundance normalized by the total loaded protein intensity from Western blotting. (G–I) Measured proteasome caspase-like (G), tryptic-like (H), and chymotrypsin-like (I) activities expressed in relative light units (RLU). *P < 0.05.
Comparison of the Proteome and Transcriptome during Postnatal Development
To verify how proteomics and transcriptomics datasets compared, a transcriptomics dataset generated on 10 donors with age ranging between Postnatal Day 1 to 3 years in age was used. Noteworthily, for 5/10 of these donors, proteomics data was also generated in this study. An identical trend analysis as the one performed for the proteomics was applied on the transcriptomics results. We identified 5,773 protein/transcript pairs with quantitative values for both RNA and proteins. 652 pairs were fitted to one of the above-mentioned trends for both the protein and the transcript (Figure 5A and Dataset E10). A moderate correlation was found between the transcripts and proteins with 344/652 pairs following identical trend (52.8%). A total of 82 had opposite linear trends, and 7 had opposite quadratic trends. DAVID enrichments revealed that matching decreasing protein/transcript pairs were enriched, for example, in GO terms related to RNA splicing, chromatin remodeling, or cellular adhesion (Figure 5B and Dataset E11). Noteworthily, 85/116 members of the “spliceosomal” protein were fitted to a linearly decreasing model (P < 0.05), suggesting an important role of splicing right after birth (Figure E2 and Dataset E12). Unfortunately, the deepness of coverage of the transcriptomics analysis did not allow identification of splicing variants differentially expressed. Matching linearly increasing pairs were enriched in GO terms related to oxidoreduction processes, immune function (e.g., innate immune response, positive regulation of T cell, or B-cell receptor signaling pathway). Interestingly, pairs with transcript linearly decreasing but protein linearly increasing were enriched in GO terms related to the organization of the extracellular matrix. Collagen IVa, IVb, VIa, and VIb all increased at the protein level while matching mRNA abundance decreased with age. This anticorrelation for collagens may result from the fact that collagen deposition and turnover are slow (35).
Figure 5.
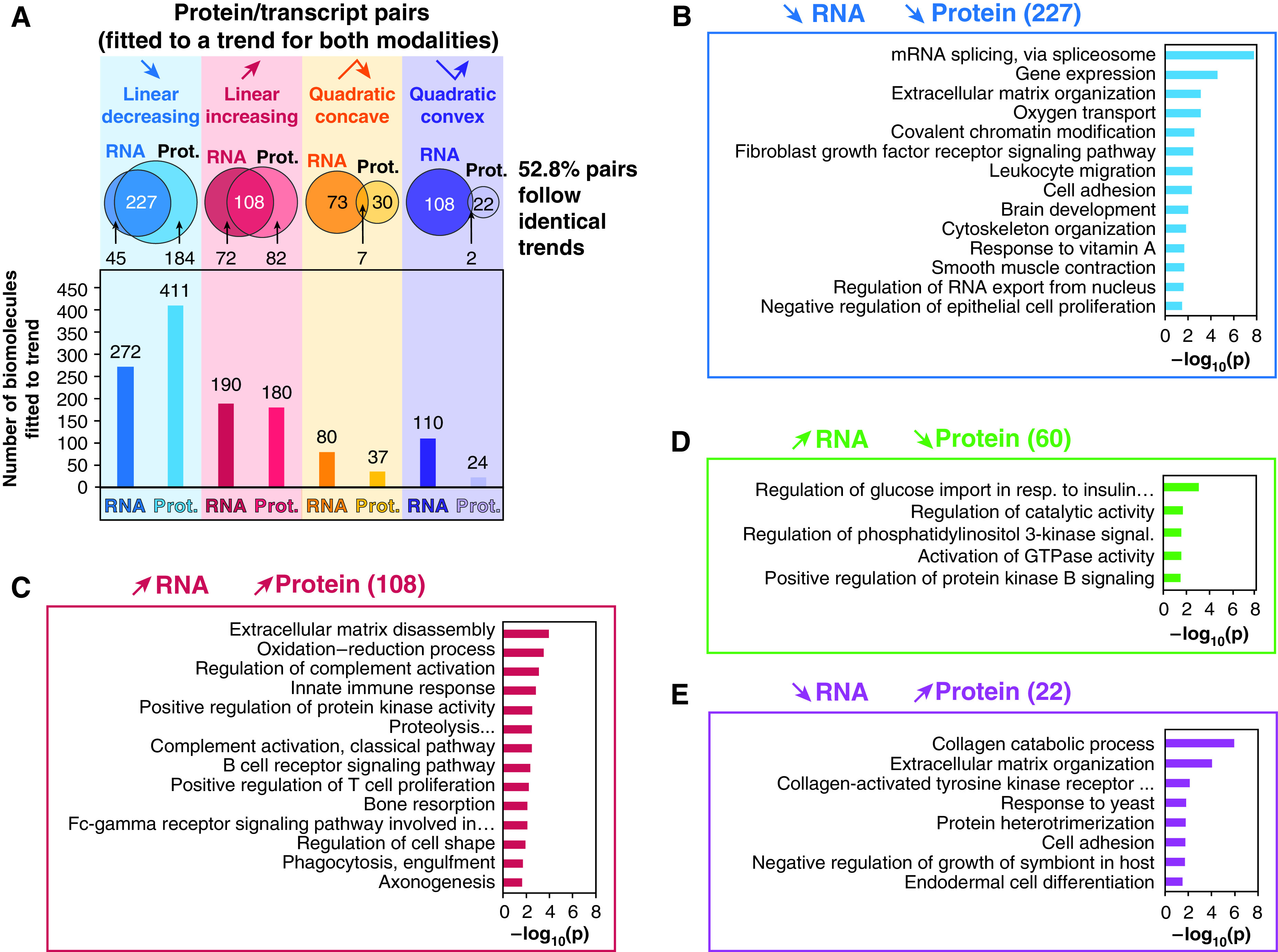
Trend and enrichment analysis for the RNA/protein pairs. Only the 652 pairs of RNA/proteins both fitted to one of the four described trends were used to generate this figure. (A) The bar plots show the count of RNA/proteins fitted to each one of the four trends, and the Venn diagram indicates the number of matching and nonmatching protein/RNA pairs for each trend. (B–E) Representative DAVID GO_BP enrichment analysis identified enriched pathways associated to linear matching (B and C) or opposite trends (D and E) for RNA/protein pairs. Prot. = protein; resp. = response.
Extracellular Matrix Remodeling during Human Postnatal Development
Our proteome dataset contained 118 core matrisome and 138 matrisome-associated proteins. Of the 256 extracellular matrix-related proteins detected in the present study, more than 50% (135 proteins) fit one of the four temporal trends previously described (Figure 6 and Dataset E13), suggesting an extensive remodeling of the extracellular matrix during alveologenesis. During alveolar development, the basement membrane of the alveolar epithelium comes into close apposition to endothelial cell membranes of the capillary microcirculation facilitating gas exchange (36). Laminins and collagens are the major protein families forming the basement membranes, and both groups of proteins are required for normal lung morphogenesis (37, 38). Although the relative abundance of various laminins (e.g., laminins subunit α-1, subunit α-4, and subunit β-1) decreased during postnatal development, the relative abundance of various collagen proteins (e.g., subunits of collagen IV, VI, and VIII) increased in our data. A recent study showed progressive thinning and simplification of the laminin network within the alveolar septal wall after birth (39), consistent with the decrease in abundance observed in our proteomics results.
Figure 6.

Extracellular matrix remodeling during lung development. The heat map depicts the 135/262 core matrisome (brown rectangles) or matrisome-associated (green rectangles) proteins fitted to one of the four trends described (P < 0.05). Examples of proteins following a linearly increasing trend (collagen IV and VI subunits) and linearly decreasing trend (laminins and tropoelastin) are in the boxes on the right panel of the figure. A full list of proteins is provided in Dataset E13.
Immune Cells Are Increasing in Number and Diversity during Postnatal Development
We identified several proteins that were increased with age in the proteomics relating to at least seven immune response pathways, including adaptive immune responses, neutrophil chemotaxis, macrophage-related proteins, complement activation, Type I IFN signaling, B-cell receptor signaling, and T-cell proliferation (Figure 7A). Importantly, the hematopoietic marker PTPRC/CD45 linearly increased with age, suggesting that the immune-related functions were not just an acute response to birth and adaptation to the ambient environment. From this result, we hypothesized that the amount and nature of the immune cells present in pulmonary tissue was modulated as the immune system maturated. We validated the age-related increase of CD45+ cells by flow cytometry using cells isolated from digested lung tissue from 34 donors (Figure 7B). In the same cohort, nonhematopoietic cells decreased in abundance (CD45−) (P < 0.0001) (Figure 7C). More specifically, endothelial cells decreased (CD45−/PECAM+, P < 0.001), epithelial cells did not change significantly (CD45−/EPCAM+), and other cell populations slightly decreased (CD45−/EPCAM−/PECAM−) (P < 0.05) as shown in Figure E3. It is important to note that some of the proteomics and transcriptomics changes presented in this study may be driven by cell population shift. Sorted-population omics analyses across ages would be required to identify cell population-specific patterns.
Figure 7.
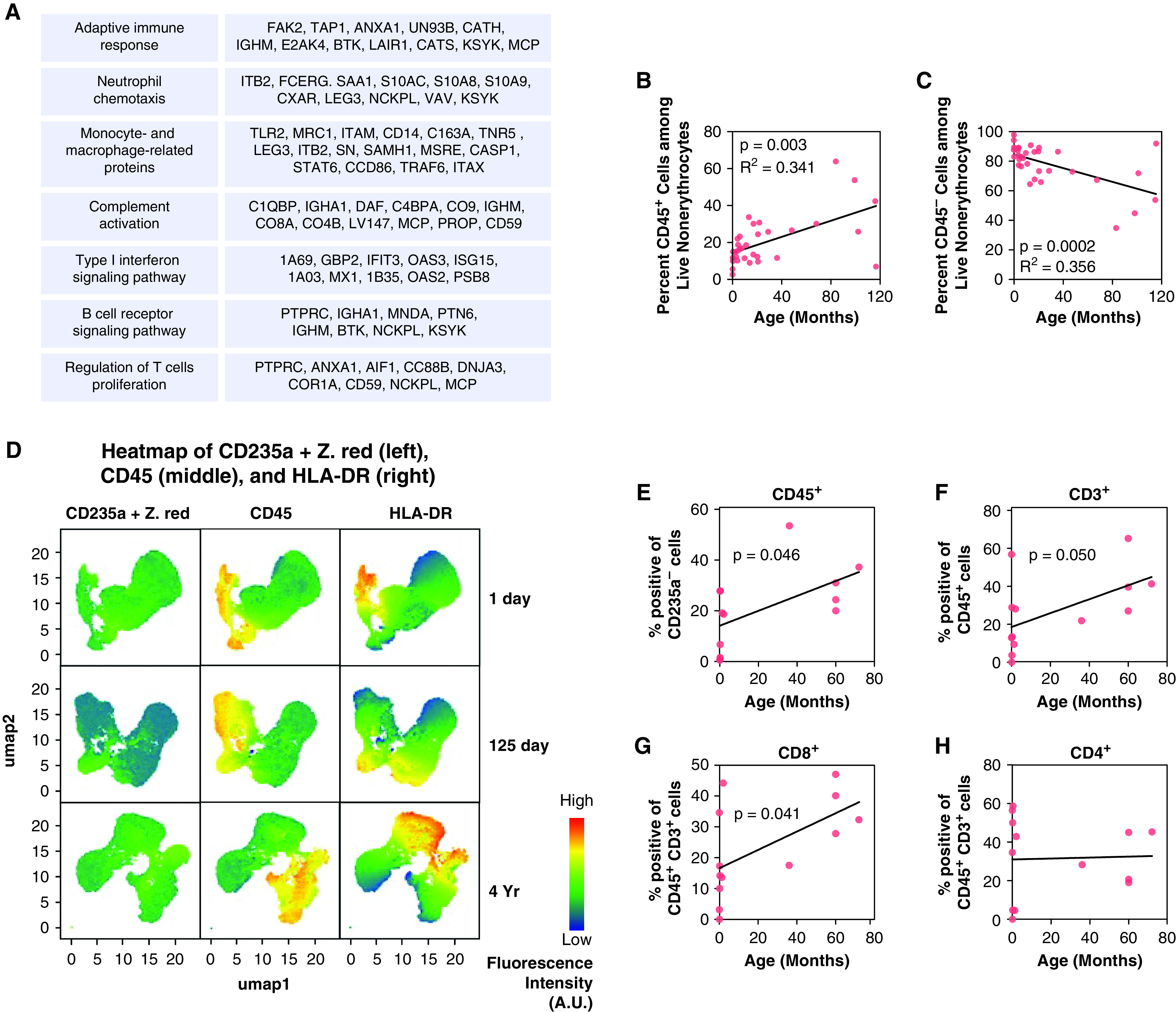
Dynamics of immune cell population markers during lung development. (A) Lists of immune-related proteins belonging to seven immune pathways following a linearly increasing trend. (B) Flow cytometry count of nonerythrocyte CD45+ immune cells showing an increase of the number of immune cells within the lungs of 35 donors of age ranging from 0 to 120 months. (C) Flow cytometry count of nonerythrocyte CD45− cells showing a decrease of nonimmune, nonerythrocyte cells within the lungs of 35 donors of age ranging from 0 to 120 months. (D) Heat map from three donors depicting the age-dependent increase in expression of HLA-DR on cells that originated from the hematopoietic system (CD45+) or other nonhematopoietic lineages (CD235a−). Dead cells and erythrocytes were filtered out using the viability dye Zombie Red (Z. red) and CD235a PE-Cy5. The figure shows that the proportion of nonleukocytes (CD45−) expressing HLA-DR increases with age. Fluorescence intensity is expressed in arbitrary units (A.U.). (E–H) Immune cells were identified by flow cytometry as expressing CD45 but not the red blood cell marker CD235a (E). T cells were identified by CD3 expression (F), which were further subdivided into CD4 (G) and CD8 (H) cells. A linear regression analysis was performed, and P values are displayed.
One of the hallmarks of immune activation is the upregulation of the antigen-presenting protein, HLA-DR. We demonstrated that there was an age-dependent increase in expression of HLA-DR on cells of that originated from the hematopoietic system (CD45+ ) or from other nonhematopoietic lineages (CD45−) (Figure 7D). For example, cultured human endothelial cells have been demonstrated to express low amounts of MHC II, and that amount increased log-fold with IFN-γ treatment (40). Expression of MHC II by lung nonleukocytes shows their critical role in triggering immune response by presenting antigen. We found neonatal lung nonleukocytes did not express MHC II (by assessing membrane HLA-DR) but a fraction of infant and older pediatric lung CD45 negative cell population expressed MHC II. This finding validates that developing pediatric lungs become more capable of triggering pathogen-specific immune response.
We then looked at obtaining specific details on changes occurring in T cells. CD3 is a protein complex and T-cell coreceptor that is involved in activating both the cytotoxic T cell (CD8+ naive T cells) and T helper cells (CD4+ naive T cells). The overall frequency of T cells (CD3+) increased in pulmonary tissue with age; interestingly, although the frequency of CD8+ T cells increased with age, it was not the case for CD4+ T cells (Figures 7E–7H). In addition, transcript-protein pairs with a matching increasing abundance were enriched in many immune-related GO terms (Dataset E11). Taken together, our orthogonal analysis of immune cell populations sampled using flow cytometry from birth to late childhood/early adulthood and the transcriptomics results substantiated our hypothesis generated from the proteomics dataset.
Conclusions
In this study, we examined the human lung proteome to elucidate the processes underlying human lung development during the postnatal alveolar stage. A limitation of the present study is the relatively small number of time points (10 time points) examined; however, we note that it is the most detailed study of its type yet available. Analysis of more time points provides a higher-resolution view of the temporal dynamics of protein expression, potentially revealing the presence of additional molecular substages beyond the four reported substages that the current data provide. In addition, as the present study used whole tissue proteomic analyses, the roles of specific cell types and specific cells in lung formation and function remain to be identified. Nevertheless, our study identified important biological processes associated with postnatal maturation of the human lung at the level of translation and provides a unique hypothesis-generating resource that the lung biology community can mine to develop novel hypotheses to further understand human lung formation and function.
Footnotes
Supported by NHLBI grants HL134745 and HL148856 (J.A.W.), HL148861 (G.S.P.), and HL148860 (C.A.). The opinions expressed in this article are the authors’ own and do not reflect the view of the NIH, the Department of Health and Human Services, or the U.S. government. Parts of this work were performed in the Environmental Molecular Science Laboratory, a U.S. Department of Energy national scientific user facility at Pacific Northwest National Laboratory in Richland, Washington.
Author Contributions: Conception of study: G.C., G.S.P., and C.A. Acquisition of study data: G.C., R.M., J.A.K., S.B., H.L.H., M.D.M., and G.B. Analysis of study data: G.C., L.M.B., R.M., M.D.M., G.H.D., S.B., S.F., V.G.D., H.B., and C.A. Drafting of manuscript: G.C., L.M.B., M.D.M., G.B., R.M., J.N.A., and C.A. Revision of manuscript: G.C., L.M.B., R.M., M.D.M., J.A.K., G.H.D., T.J.M., J.P.C., J.A.W., G.S.P., and J.N.A.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202008-3303OC on November 9, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Schittny JC. Development of the lung. Cell Tissue Res . 2017;367:427–444. doi: 10.1007/s00441-016-2545-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Warburton D. Overview of lung development in the newborn human. Neonatology . 2017;111:398–401. doi: 10.1159/000458465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, et al. Lung organogenesis. Curr Top Dev Biol . 2010;90:73–158. doi: 10.1016/S0070-2153(10)90003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kho AT, Bhattacharya S, Tantisira KG, Carey VJ, Gaedigk R, Leeder JS, et al. Transcriptomic analysis of human lung development. Am J Respir Crit Care Med . 2010;181:54–63. doi: 10.1164/rccm.200907-1063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ding J, Ahangari F, Espinoza CR, Chhabra D, Nicola T, Yan X, et al. Integrating multiomics longitudinal data to reconstruct networks underlying lung development. Am J Physiol Lung Cell Mol Physiol . 2019;317:L556–L568. doi: 10.1152/ajplung.00554.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clair G, Bramer L, Kyle J, Zink E, Pryhuber GS, Misra R, et al. Proteomics and lipidomics analysis of the developing post-natal human lung. Am J Resp Crit Care Med . 2020;201:A5979. [Google Scholar]
- 7. Clair G, Kyle J, Piehowski P, Zhu Y, Zink E, Bramer L, et al. The proteomics and lipidomics landscape of the developing human lung during the post-natal alveolarization process. Am J Resp Crit Care Med . 2019;199:A6082. [Google Scholar]
- 8. Pryhuber GS. URMC HTC Non-inflated fresh-frozen embedded lung and associated tissue. https://www.protocols.io/view/615-1-urmc-htc-non-inflated-fresh-frozen-embedded-bjtnknme [Google Scholar]
- 9. Pryhuber GS, Huyck H, Rogers L, Poole C. Donor acceptance criteria for URMC HTC HuBMAP and LungMAP inclusion. https://www.protocols.io/view/602-2-donor-acceptance-criteria-for-urmc-htc-hubma-bjuxknxn [Google Scholar]
- 10. Dautel SE, Kyle JE, Clair G, Sontag RL, Weitz KK, Shukla AK, et al. Lipidomics reveals dramatic lipid compositional changes in the maturing postnatal lung. Sci Rep . 2017;7:40555. doi: 10.1038/srep40555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moghieb A, Clair G, Mitchell HD, Kitzmiller J, Zink EM, Kim YM, et al. Time-resolved proteome profiling of normal lung development. Am J Physiol Lung Cell Mol Physiol . 2018;315:L11–L24. doi: 10.1152/ajplung.00316.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bandyopadhyay G, Huyck HL, Misra RS, Bhattacharya S, Wang Q, Mereness J, et al. Dissociation, cellular isolation, and initial molecular characterization of neonatal and pediatric human lung tissues. Am J Physiol Lung Cell Mol Physiol . 2018;315:L576–L583. doi: 10.1152/ajplung.00041.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mayampurath AM, Jaitly N, Purvine SO, Monroe ME, Auberry KJ, Adkins JN, et al. DeconMSn: a software tool for accurate parent ion monoisotopic mass determination for tandem mass spectra. Bioinformatics . 2008;24:1021–1023. doi: 10.1093/bioinformatics/btn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim S, Pevzner PA. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun . 2014;5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The UniProt Consortium. UniProt [accessed 2017 Apr 12]. Available from: https://www.uniprot.org. [Google Scholar]
- 16. Monroe ME, Shaw JL, Daly DS, Adkins JN, Smith RD. MASIC: a software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput Biol Chem . 2008;32:215–217. doi: 10.1016/j.compbiolchem.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webb-Robertson BJ, Matzke MM, Jacobs JM, Pounds JG, Waters KM. A statistical selection strategy for normalization procedures in LC-MS proteomics experiments through dataset-dependent ranking of normalization scaling factors. Proteomics. 2011;11:4736–4741. doi: 10.1002/pmic.201100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stratton KG, Webb-Robertson BM, McCue LA, Stanfill B, Claborne D, Godinez I, et al. pmartR: quality control and statistics for mass spectrometry-based biological data. J Proteome Res . 2019;18:1418–1425. doi: 10.1021/acs.jproteome.8b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Polpitiya AD, Qian WJ, Jaitly N, Petyuk VA, Adkins JN, Camp DG, II, et al. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics . 2008;24:1556–1558. doi: 10.1093/bioinformatics/btn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature . 2020;587:619–625. doi: 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kho AT, Bhattacharya S, Mecham BH, Hong J, Kohane IS, Mariani TJ. Expression profiles of the mouse lung identify a molecular signature of time-to-birth. Am J Respir Cell Mol Biol. 2009;40:47–57. doi: 10.1165/rcmb.2008-0048OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beauchemin KJ, Wells JM, Kho AT, Philip VM, Kamir D, Kohane IS, et al. Temporal dynamics of the developing lung transcriptome in three common inbred strains of laboratory mice reveals multiple stages of postnatal alveolar development. PeerJ . 2016;4:e2318. doi: 10.7717/peerj.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Simon N, Friedman J, Hastie T, Tibshirani R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J Stat Softw . 2011;39:1–13. doi: 10.18637/jss.v039.i05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mattson MP, Zhang P, Fu W. Madame Curie Bioscience Database. Austin, Texas: Landes Bioscience; 2000. Roles for TERT and telomerase in cell differentiation and apoptosis. [Google Scholar]
- 25. Freund A, Zhong FL, Venteicher AS, Meng Z, Veenstra TD, Frydman J, et al. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell . 2014;159:1389–1403. doi: 10.1016/j.cell.2014.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Driscoll B, Buckley S, Bui KC, Anderson KD, Warburton D. Telomerase in alveolar epithelial development and repair. Am J Physiol Lung Cell Mol Physiol . 2000;279:L1191–L1198. doi: 10.1152/ajplung.2000.279.6.L1191. [DOI] [PubMed] [Google Scholar]
- 27. Guo M, Du Y, Gokey JJ, Ray S, Bell SM, Adam M, et al. Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat Commun . 2019;10:37. doi: 10.1038/s41467-018-07770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Agassandian M, Mallampalli RK. Surfactant phospholipid metabolism. Biochim Biophys Acta . 2013;1831:612–625. doi: 10.1016/j.bbalip.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y, Horowitz JC, Naba A, Ambalavanan N, Atabai K, Balestrini J, et al. Extracellular matrix in lung development, homeostasis and disease. Matrix Biol . 2018;73:77–104. doi: 10.1016/j.matbio.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jeannotte L, Lemieux M, Charron J, Poirier F, Robertson EJ. Specification of axial identity in the mouse: role of the Hoxa-5 (Hox1.3) gene. Genes Dev . 1993;7:2085–2096. doi: 10.1101/gad.7.11.2085. [DOI] [PubMed] [Google Scholar]
- 31. Aubin J, Lemieux M, Tremblay M, Bérard J, Jeannotte L. Early postnatal lethality in Hoxa-5 mutant mice is attributable to respiratory tract defects. Dev Biol . 1997;192:432–445. doi: 10.1006/dbio.1997.8746. [DOI] [PubMed] [Google Scholar]
- 32. Yin Z, Gonzales L, Kolla V, Rath N, Zhang Y, Lu MM, et al. Hop functions downstream of Nkx2.1 and GATA6 to mediate HDAC-dependent negative regulation of pulmonary gene expression. Am J Physiol Lung Cell Mol Physiol . 2006;291:L191–L199. doi: 10.1152/ajplung.00385.2005. [DOI] [PubMed] [Google Scholar]
- 33. Wilkinson DG, Bhatt S, Ryseck RP, Bravo R. Tissue-specific expression of c-jun and junB during organogenesis in the mouse. Development . 1989;106:465–471. doi: 10.1242/dev.106.3.465. [DOI] [PubMed] [Google Scholar]
- 34. Kammerl IE, Dann A, Mossina A, Brech D, Lukas C, Vosyka O, et al. Impairment of immunoproteasome function by cigarette smoke and in chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2016;193:1230–1241. doi: 10.1164/rccm.201506-1122OC. [DOI] [PubMed] [Google Scholar]
- 35. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J . 2017;50:1601805. doi: 10.1183/13993003.01805-2016. [DOI] [PubMed] [Google Scholar]
- 36. Morrissey MA, Sherwood DR. An active role for basement membrane assembly and modification in tissue sculpting. J Cell Sci . 2015;128:1661–1668. doi: 10.1242/jcs.168021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen NM, Senior RM. Laminin isoforms and lung development: all isoforms are not equal. Dev Biol . 2006;294:271–279. doi: 10.1016/j.ydbio.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 38. Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development . 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 39. Luo Y, Li N, Chen H, Fernandez GE, Warburton D, Moats R, et al. Spatial and temporal changes in extracellular elastin and laminin distribution during lung alveolar development. Sci Rep . 2018;8:8334. doi: 10.1038/s41598-018-26673-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cunningham AC, Zhang JG, Moy JV, Ali S, Kirby JA. A comparison of the antigen-presenting capabilities of class II MHC-expressing human lung epithelial and endothelial cells. Immunology . 1997;91:458–463. doi: 10.1046/j.1365-2567.1997.d01-2249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Proteomics data are deposited and freely available at ProteomeXchange data repository, ProteomeXchangeID: PXD020798, and MassIVE data repository, MassIVE ID: MSV000085929. All the code used to generate the data was deposited on GitHub (https://github.com/GeremyClair/Proteomics_analysis_of_human_lung_development/).