

Abstract
Rationale
The ability of peripheral blood biomarkers to assess chronic obstructive pulmonary disease (COPD) risk and progression is unknown. Genetics and gene expression may capture important aspects of COPD-related biology that predict disease activity.
Objectives
Develop a transcriptional risk score (TRS) for COPD and assess the contribution of the TRS and a polygenic risk score (PRS) for disease susceptibility and progression.
Methods
We randomly split 2,569 COPDGene (Genetic Epidemiology of COPD) participants with whole-blood RNA sequencing into training (n = 1,945) and testing (n = 624) samples and used 468 ECLIPSE (Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points) COPD cases with microarray data for replication. We developed a TRS using penalized regression (least absolute shrinkage and selection operator) to model FEV1/FVC and studied the predictive value of TRS for COPD (Global Initiative for Chronic Obstructive Lung Disease 2–4), prospective FEV1 change (ml/yr), and additional COPD-related traits. We adjusted for potential confounders, including age and smoking. We evaluated the predictive performance of the TRS in the context of a previously derived PRS and clinical factors.
Measurements and Main Results
The TRS included 147 transcripts and was associated with COPD (odds ratio, 3.3; 95% confidence interval [CI], 2.4–4.5; P < 0.001), FEV1 change (β, −17 ml/yr; 95% CI, −28 to −6.6; P = 0.002), and other COPD-related traits. In ECLIPSE cases, we replicated the association with FEV1 change (β, −8.2; 95% CI, −15 to −1; P = 0.025) and the majority of other COPD-related traits. Models including PRS, TRS, and clinical factors were more predictive of COPD (area under the receiver operator characteristic curve, 0.84) and annualized FEV1 change compared with models with one risk score or clinical factors alone.
Conclusions
Blood transcriptomics can improve prediction of COPD and lung function decline when added to a PRS and clinical risk factors.
Keywords: COPD, transcriptomic, polygenic risk score, risk prediction, FEV1 decline
At A Glance Commentary
Current Scientific Knowledge on the Subject
Genetics and gene expression may capture important aspects of chronic obstructive pulmonary disease (COPD)–related biology that predict disease activity. Whether a blood-based gene expression or transcriptional risk score (TRS) for COPD adds value to a polygenic risk score for predicting disease susceptibility and progression is unknown.
What This Study Adds to the Field
A TRS was predictive of COPD, COPD-related traits, and prospective FEV1 decline in two cohorts of smokers. Models including polygenic risk score, TRS, and clinical factors were more predictive of COPD and annualized FEV1 change than models with one risk score or clinical factors alone. These results demonstrate that blood transcriptomics can improve prediction of COPD and lung function decline.
Chronic obstructive pulmonary disease (COPD) is characterized by airflow obstruction and primarily develops in the setting of cigarette smoking exposure (1). Only a minority of smokers develop the disease (2), and only certain individuals with the disease experience rapidly progressive lung function decline (3), exacerbations (4), and higher mortality (5). Identifying individuals at high risk of COPD and COPD progression is crucial for focusing public health interventions and drug development.
Genetic studies have identified hundreds of variants associated with COPD and low lung function (6). Although genome-wide significant variants are important in COPD pathobiology, individual variants account for a small amount of phenotypic variability and are poor for risk prediction. By contrast, aggregating genome-wide significant variants into a risk score or, even more powerfully, millions of variants including those not reaching genome-wide significance, into a polygenic risk score (PRS) can identify those at high risk for COPD (7); yet the variants most important for prediction are not necessarily the most biologically relevant variants. Despite these advances in genetics-based risk prediction, a majority of COPD risk is nongenetic (6, 7), and an individual’s genetic makeup does not change in response to environmental stimuli. Other omics data, such as gene expression, can change in response to environmental stimuli (e.g., cigarette smoking), and may correlate with the onset and timing of disease progression.
Gene expression, or transcriptome-based, risk scores have shown promise in complex diseases (8–10). A transcriptome-based risk score was more predictive of disease progression than a traditional PRS in inflammatory bowel diseases (8). Serial sampling of blood transcriptomic profiles was used to develop a risk score predictive of lung function decline in idiopathic pulmonary fibrosis (10). In COPD, transcriptomic profiles from peripheral blood have been associated with COPD susceptibility (11) and exacerbations (12, 13), although with limited capacity to predict lung function decline. A lung gene expression signature for IL-17A was associated with FEV1 decline in a subset of patients on inhaled glucocorticoids (14). These results support the value of gene expression data in predicting COPD and COPD progression.
We hypothesized that a peripheral blood transcriptional risk score (TRS) trained on spirometry baseline FEV1/FVC would be complementary to our previously published PRS (15) and be predictive of COPD in the COPDGene (Genetic Epidemiology of COPD) (16) study and FEV1 decline in COPDGene and COPD cases from the ECLIPSE (Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points) study (17). We used FEV1/FVC as it represents the main diagnostic criteria for COPD and has been associated with worse outcomes (18). In addition to studying subjects with COPD, we also included smokers without airflow obstruction because this group has been shown to have emphysema and airway pathology on computed tomography (CT) scans, respiratory symptoms, exacerbations, FEV1 decline, and rapid progression (19). Identifying individuals without substantial airflow obstruction who are likely to experience accelerated lung function decline may allow for early intervention. The TRS was developed using data from more than 2,500 individuals from COPDGene (16) with RNA sequencing (RNA-seq) data, nearly 1,000 of whom also had 5-year follow-up spirometry data. We sought replication of our results in microarray data from the ECLIPSE study (17).
Methods
Study Populations
Written informed consent was obtained from all study participants, and institutional review board approval was obtained at all study centers. We included non-Hispanic White and African American participants with whole-blood RNA-seq data from the 5-year follow-up of the COPDGene study (16). We also included participants with whole-blood microarray data and at least two FEV1 measurements from the ECLIPSE study (17). Additional details regarding study populations are in the Supplementary Methods in the online supplement.
Preparation of Gene Expression Data
RNA-seq data
Details regarding generation of RNA-seq data were previously published (20). Details regarding RNA processing and preparation of count data are in the Supplementary Methods. Counts were adjusted for library depth, and batch effects were removed using the limma removeBatchEffects function (21).
Microarray data
Details regarding microarray processing were previously published (22) and are further described in the Supplementary Methods. Batch effects were removed using the limma removeBatchEffects function (21).
After the above processing steps were performed, we took two additional steps to facilitate transportability across RNA-seq and microarray data platforms. First, we limited transcripts to those present in both data sets based on HUGO Gene Nomenclature Committee symbols. Second, we scaled and centered the RNA-seq count and microarray gene expression data.
Statistical Analyses
Overview of study design
We randomly split COPDGene participants into training (75%) and testing (25%) samples (Figure 1). Model development was performed exclusively within the training sample. We tested associations with cross-sectional and longitudinal outcomes in the testing sample and replicated our results in ECLIPSE.
Figure 1.

Schematic of study design. COPD = chronic obstructive pulmonary disease; COPDGene = Genetic Epidemiology of COPD; CPM = counts per million; ECLIPSE = Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points; LASSO = least absolute shrinkage and selection operator; RNAseq = RNA sequencing.
Derivation of a TRS
To develop a TRS, we applied a penalized regression framework using the glmnet R package (23) to construct a transcriptome-based regression model for FEV1/FVC. We trained our model to FEV1/FVC because it is a lung function measurement used to define COPD, reflects emphysema and airway wall pathology, has been associated with worse outcomes (18), is easily obtainable, and was available in all participants. Furthermore, prediction on a continuous variable offers greater power than that on dichotomized variables and baseline lung function is a major predictor of lung function decline (24).
For model training, we used least absolute shrinkage and selection operator (25), which shrinks coefficients toward zero and provides automated feature selection. The model was tuned within the training sample using 10-fold cross-validation, minimizing the mean squared error (MSE) on the left-out fold. The regression model with the minimum MSE was used to calculate a TRS in participants in the testing sample and ECLIPSE replication sample. The TRS was then centered and scaled to facilitate statistical analyses. We also examined the association of TRS tertiles with primary outcomes; tertiles were chosen to represent low-, medium-, and high-risk categories and to mirror categories of FEV1 decline (see Outcomes). We trained an additional risk score in the subset of participants from the COPDGene training sample with 5-year follow-up spirometry data optimized to the outcome of prospective FEV1 change in milliliters per year (TRSdeltaFEV1). We tested the predictive performance of this score and the TRS for annualized FEV1 decline using MSE in the COPDGene testing sample. Gene enrichment analyses with respect to Reactome (26) pathways were performed with the sigora R package (27).
Model specifications and performance evaluations
We adjusted all linear and logistic regression models for age, sex, race, height, pack-years of cigarette smoking, and current smoking status. For annualized change in FEV1, we additionally adjusted models for baseline FEV1. Additional adjustments based on outcomes are detailed in the Supplementary Methods. We examined the association of the TRS with exacerbation frequency and severe exacerbations using negative binomial regression models (pscl R package (28, 29)), adjusted for age, sex, race, smoking pack-years, current smoking status, and ± baseline FEV1. We performed stratified analyses in COPD cases and controls to examine whether the TRS has differential effects in these strata.
We compared models with clinical risk factors alone (see Predictors), PRS alone, TRS alone, and all three predictors together. We assessed predictive performances of models for binary outcomes using area under the receiver operator characteristic curve (AUC) metrics and compared AUCs with the DeLong test (30) using the pROC R package (31). Further details regarding performance metrics are in the Supplementary Methods.
Predictors
We evaluated the associations and predictive performances of clinical risk factors, the TRS, and a previously published PRS (15) with respect to each outcome. “Clinical” risk factors included age, sex, race, height, pack-years of cigarette smoking, and current smoking status. For FEV1 change between Phases 2 and 3 (see Outcomes), baseline FEV1 (i.e., at phase 2) was also included as a clinical risk factor. Further details regarding the PRS are in the Supplementary Methods.
Outcomes
We examined two primary outcomes: 1) moderate to very severe COPD (Global Initiative for Chronic Obstructive Lung Disease [GOLD] spirometry grades 2–4: post-bronchodilator FEV1/FVC <0.7 and FEV1% predicted <80%) compared with smokers with normal spirometry (post-bronchodilator FEV1/FVC ⩾0.7 and FEV1% predicted ⩾80% [formerly known as GOLD 0]), as previously reported (15), and 2) prospective annualized change in FEV1 in milliliters per year. In addition to examining the continuous measure of decline, we also examined a rapid decline phenotype by comparing the highest versus the lowest tertiles of FEV1 decline (3). Secondary outcomes are described in the Supplementary Methods.
Differential gene expression analyses
We grouped participants from the overall COPDGene sample based on TRS tertiles (chosen to mirror the groupings of annualized FEV1 decline [3] above) and examined differential gene expression between the top versus middle and the bottom versus middle TRS tertiles using limma (21). After performing differential expression analyses, we used mitch (32) to perform multicontrast gene set enrichment analyses. We also used limma to perform differential gene expression analyses (adjusting for age, sex, race, pack-years of smoking, and current smoking status) in the COPDGene training sample according to previously described methods (33) and determined which transcripts overlapped with TRS transcripts.
Results
Characteristics of Study Populations
Characteristics of the study populations are shown in Table 1. In total, we included 3,037 (2,569 COPDGene; 468 ECLIPSE) participants. Anthropometric, spirometric, and CT imaging measures are evenly distributed among COPDGene training (n = 1,945) and testing (n = 624) samples (all P > 0.05). Characteristics of the subset of individuals with FEV1 measurements collected prospectively at the 10-year follow-up of the COPDGene study (i.e., phase 3) are shown in Table E1 in the online supplement. In total, 937 participants had 5-year follow up spirometry measures, 209 of which were in the testing sample.
Table 1.
Characteristics of Participants Included in This Study
| Characteristic | COPDGene |
ECLIPSE (n = 468) | P Value | |
|---|---|---|---|---|
| Training Sample (n = 1,945) | Testing Sample (n = 624) | |||
| Age, yr, mean (SD) | 65.31 (8.70) | 66.05 (8.28) | 64.43 (6.09) | 0.006 |
| Sex, F, n (%) | 942 (48.4) | 299 (47.9) | 156 (33.3) | <0.001 |
| Race, n (%) | 486 (25.0) | 156 (25.0) | 0 (0.0) | <0.001 |
| Height, cm, mean (SD) | 169.56 (9.56) | 169.46 (9.49) | 169.31 (8.61) | 0.874 |
| BMI, kg/m2, mean (SD) | 28.83 (6.19) | 29.25 (6.69) | 27.11 (5.46) | <0.001 |
| Pack-years of smoking, mean (SD) | 43.92 (23.71) | 44.27 (24.04) | 49.33 (26.87) | <0.001 |
| Current smoking, n (%) | 700 (36.0) | 216 (34.6) | 70 (15.0) | <0.001 |
| FEV1% predicted, mean (SD) | 78.84 (24.25) | 77.79 (24.08) | 44.22 (14.64) | <0.001 |
| FEV1/FVC, mean (SD) | 0.68 (0.15) | 0.68 (0.15) | 0.44 (0.11) | <0.001 |
| % LAA <−950 HU, median (IQR)* | 1.63 (0.51–5.20) | 1.51 (0.45–5.31) | 17.66 (9.98–26.74) | <0.001 |
| Perc15, mean (SD)* | −916.40 (29.14) | −914.72 (31.03) | −959.03 (49.24) | <0.001 |
| Pi10, mean (SD) | 2.25 (0.57) | 2.26 (0.58) | 4.40 (0.20) | <0.001 |
| WA%, mean (SD) | 49.85 (8.24) | 50.10 (8.56) | 65.37 (3.76) | <0.001 |
| Primary outcomes | ||||
| Moderate to severe COPD, n (%) | 658 (33.8) | 209 (33.5) | 468 (100.0) | <0.001 |
| FEV1 decline, ml/yr, mean (SD) | −42.09 (55.30) | −42.52 (59.08) | −30.58 (71.82) | 0.004 |
Definition of abbreviations: % LAA = % low-attenuation area; BMI = body mass index; COPD = chronic obstructive pulmonary disease; COPDGene = Genetic Epidemiology of COPD; ECLIPSE = Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points; HU = Hounsfield units; IQR = interquartile range; Perc15 = 15th percentile of the lung density histogram on inspiratory scans; Pi10 = square root of wall area of a hypothetical airway with internal perimeter of 10 mm; WA% = wall area percent.
P values indicate comparisons across COPDGene samples and ECLIPSE.
% LAA <−950 HU and Perc15 were calculated using Thirona at phase 2 (5-yr follow-up) in COPDGene and Slicer in ECLIPSE.
Development and Characterization of a TRS
A schematic of the study design is shown in Figure 1. Using the COPDGene training sample, we trained a least absolute shrinkage and selection operator model using FEV1/FVC and used 10-fold cross-validation to identify the lambda (λ = 0.00485) that minimized the MSE (Figure E1). This approach identified 147 transcripts (Table E2). Density plots in COPD cases and controls for the transcripts most negatively (GPR15) and positively (PTPN13) associated with FEV1/FVC are shown in Figure E2. In Reactome (26) gene enrichment analyses, these 147 transcripts implicate immunoregulatory interactions between lymphoid and nonlymphoid cells, PPAR-α (peroxisome proliferator-activated receptor α)–activated gene expression, and chondroitin sulfate biosynthesis (Table E3). The final regression model was used to calculate a TRS for each individual. The TRS was weakly correlated with our previously developed PRS; this correlation reached statistical significance in the training (r = 0.06, P = 0.01) but not the testing (r = 0.07, P = 0.094) sample of COPDGene (Figure E3).
A TRS Is Associated with COPD and Is a Predictor of Prospective Loss of Lung Function
Our primary goal was to develop a blood-based TRS to predict COPD and accelerated FEV1 decline in individuals who smoke. The association of the TRS with moderate to very severe COPD and FEV1 decline (milliliters per year [ml/yr]) is demonstrated in Figure 2. Compared with the bottom tertile, being in the top tertile of the TRS was associated with odds ratios of 11.0 (95% confidence interval [CI], 5.4–21.0) and 2.0 (95% CI, 1.1–3.4) for COPD and rapid FEV1 decline (i.e., top vs. bottom tertile of decline), respectively. Clinical characteristics of COPDGene participants grouped by TRS tertile are shown in Table E4. Participants in higher tertiles were more likely to be older, male, and non-Hispanic White, have more pack-years of smoking, lower baseline spirometry measures, more emphysema, and thicker airways, and experience greater FEV1 decline.
Figure 2.

(A) A box plot with interquartile ranges demonstrating the association of the TRS with moderate to very severe COPD. The P value represents the results of a Student’s t test. (B) Participants were grouped into tertiles of the TRS, and the effects of being in the middle and top tertiles compared with the bottom tertile of the TRS on COPD affection status are reported as odds ratios and 95% confidence intervals. Models were adjusted for age, sex, race, pack-years of smoking, and current smoking status. (C) The TRS was linearly associated with FEV1 decline. Pearson correlation coefficient and associated P value are displayed above the plot. (D) Participants were grouped into tertiles of the TRS, and the effects of being in the middle and top tertiles compared with the bottom tertile of the TRS on rapid FEV1 decline (defined as top vs. bottom tertile of FEV1 decline) are reported as odds ratios and 95% confidence intervals. Models were adjusted for age, sex, race, pack-years of smoking, current smoking status, and baseline FEV1. COPD = chronic obstructive pulmonary disease; OR = odds ratio; TRS = transcriptional risk score.
For moderate to very severe COPD, the results of multivariable analyses of the TRS in the COPDGene testing sample is shown in Table 2. In the COPDGene testing sample, one SD increment in the TRS was associated with COPD (odds ratio, 3.4; 95% CI, 2.5–4.5; P < 0.001). In ECLIPSE, both microarray and follow-up FEV1 data were not available in spirometrically normal (formerly GOLD 0) subjects, so we could not evaluate the association of the TRS with moderate to very severe COPD.
Table 2.
Associations of the TRS with Moderate to Severe COPD and Outcomes Related to COPD Heterogeneity
| Outcome | COPDGene Testing Sample |
ECLIPSE |
||
|---|---|---|---|---|
| TRS | P Value | TRS | P Value | |
| Moderate to severe COPD, OR (95% CI) | 3.35 (2.48–4.53) | <0.0001 | N/A | N/A |
| SGRQ total score | 0.4 (0.053) | <0.0001 | 3.2 (0.85) | 0.00021 |
| 6-min-walk distance | −140 (19) | <0.0001 | −61 (19) | 0.0015 |
| % LAA <−950 HU | 0.53 (0.078) | <0.0001 | 0.12 (0.04) | 0.0033 |
| Perc15 | −7.7 (1.2) | <0.0001 | −5.4 (2.5) | 0.033 |
| Pi10 | 0.19 (0.026) | <0.0001 | 0.0023 (0.0092) | 0.8 |
| WA% | 2.1 (0.37) | <0.0001 | 0.58 (0.18) | 0.0018 |
Definition of abbreviations: % LAA = % low-attenuation area; CI = confidence interval; COPD = chronic obstructive pulmonary disease; COPDGene = Genetic Epidemiology of COPD; CT = computed tomography; ECLIPSE = Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points; HU = Hounsfield units; N/A = not applicable; OR = odds ratio; Perc15 = 15th percentile of the lung density histogram on inspiratory scans; Pi10 = square root of wall area of a hypothetical airway with internal perimeter of 10 mm; TRS = transcriptional risk score; SGRQ = St. George’s Respiratory Questionnaire; WA% = wall area percent.
Data are shown as β (SE) unless otherwise indicated. Multivariable models were constructed in the testing sample of COPDGene and ECLIPSE. Covariates included age, sex, race, pack-years of smoking, and current smoking status. The model for 6-minute-walk distance was additionally adjusted for height and weight. Models for CT imaging variables (% LAA <−950 HU, Perc15, Pi10, and WA%) were additionally adjusted for CT scanner and body mass index. Bonferroni-adjusted significance level is P < 0.05/7 outcomes = 0.007. ECLIPSE included 468 individuals with microarray and follow-up FEV1 data; all of these participants had moderate to severe COPD, so replication of this outcome could not be performed.
We also examined the association of the TRS with related cross-sectional outcomes in the COPDGene testing sample and ECLIPSE (Table 2). In the COPDGene testing sample, the TRS was associated with higher St. George’s Respiratory Questionnaire scores (β = 0.4; SE, 0.05; P < 0.0001), lower 6-minute-walk distance (β = −140; SE, 19; P < 0.0001), two measures of emphysema (% low attenuation area < −950 HU and 15th percentile of the lung density histogram on inspiratory scans; both P < 0.0001), and two measures of airway pathology (square root of wall area of a hypothetical airway with internal perimeter of 10 mm [Pi10] and wall area percent; both P < 0.0001). In ECLIPSE, we were able to replicate associations of the TRS with a majority of COPD-related outcomes. Compared with COPDGene, we observed concordant directions of effects and P values for all outcomes in ECLIPSE, except for Pi10. By comparison, the PRS was associated with COPD, CT imaging traits, and St. George’s Respiratory Questionnaire total score (all P < 0.05; Table E5), although with smaller effect sizes than the TRS. The PRS was not associated with 6-minute-walk distance.
Having demonstrated the association between the TRS and COPD-related cross-sectional measures, we then tested whether the TRS is associated with prospective changes in lung function. In multivariable analyses, including the PRS and other potential confounders (Table 3), the TRS was significantly associated with change in FEV1 in the COPDGene testing sample (β = −17 ml/yr; 95% CI, −28 to −6.6; P = 0.002) and ECLIPSE (β = −8.2 ml/yr; 95% CI, −15 to −1; P = 0.025). In stratified analyses, the TRS was associated with prospective annualized FEV1 decline in participants with COPD (β = −26 ml/yr; 95% CI, −48 to −3.1; P = 0.031). In control participants, the association of the TRS with annualized FEV1 decline trended toward significance (β = −17 ml/yr; 95% CI, −33 to −0.24; P = 0.051). Adjusting for cell counts (Table E6) demonstrated similar results in COPDGene; the association of the TRS with FEV1 decline was attenuated and did not reach statistical significance in ECLIPSE after adjusting for cell counts. A risk score trained to FEV1 decline (TRSdeltaFEV1) had lower predictive performance for annualized FEV1 decline (MSE = 3,474.19) than the TRS (MSE = 3,408.51) (Table E7). The TRS was associated with exacerbation frequency and severe exacerbations in the COPDGene testing sample, but these associations were attenuated after adjusting for baseline FEV1 and did not replicate in ECLIPSE (Table E8).
Table 3.
Multivariable Models Including Clinical Factors, PRS, and TRS for Moderate-to-Severe COPD and Change in FEV1
| Variable | COPDGene Testing Sample |
ECLIPSE |
||||
|---|---|---|---|---|---|---|
| Moderate to Severe COPD |
Change in FEV1 (ml/yr) |
Change in FEV1 (ml/yr) |
||||
| OR (95% CI) | P Value | β (95% CI) | P Value | β (95% CI) | P Value | |
| PRS | 1.67 (1.31 to 2.13) | <0.0001 | −4.2 (−14 to 5.1) | 0.37 | 1.3 (−5.3 to 8) | 0.69 |
| TRS | 3.27 (2.38 to 4.5) | <0.0001 | −17 (−28 to −6.6) | 0.002 | −8.2 (−15 to −1) | 0.025 |
| Baseline FEV1, L | N/A | N/A | −24 (−38 to −9.9) | 0.0011 | −34 (−50 to −19) | 1.90 × 10−5 |
Definition of abbreviations: CI = confidence interval; COPD = chronic obstructive pulmonary disease; COPDGene = Genetic Epidemiology of COPD; ECLIPSE = Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points; N/A = not applicable; OR = odds ratio; PRS = polygenic risk score; TRS = transcriptional risk score.
Multivariable models including clinical factors (age, sex, race, pack-years of smoking, current smoking status, and baseline FEV1 [for change in FEV1]), PRS, and TRS for moderate to severe COPD and change in FEV1 (ml/yr) were constructed in the testing sample of COPDGene and replicated in ECLIPSE. Models were also adjusted for principal components of genetic ancestry. Bonferroni-adjusted significance level is 0.05/3 (2 outcomes in COPDGene, 1 outcome in ECLIPSE) = 0.017. The COPDGene testing data set included 624 individuals, of whom 209 had 5-year follow-up spirometric data. ECLIPSE included 468 individuals with microarray and follow-up FEV1 data; all of these participants had moderate to severe COPD, so replication of this outcome could not be performed.
We then studied the relative utility of the PRS, TRS, and clinical risk factors for predicting COPD and annualized change in FEV1. We constructed a series of models to predict COPD or FEV1 decline using the PRS, TRS, and/or clinical factors (age, sex, race, height, pack-years of cigarette smoking, current smoking status, ± baseline FEV1 [as appropriate]). In the COPDGene training sample, the TRS predicted moderate to very severe COPD with an AUC of 0.80. Figure 3 shows that in the COPDGene testing sample, the TRS (AUC 0.79) performed significantly better than both the PRS (AUC 0.64) and clinical factors (AUC 0.72) for predicting COPD (P [TRS vs. PRS] < 0.0001; P [TRS vs. clinical factors] = 0.0058). Adding the PRS and clinical factors into a model with the TRS did not significantly improve the AUC (AUC 0.84; P [PRS + TRS + clinical factors vs. TRS] = 0.08). For annualized change in FEV1, a model with all predictors again had the best performance based on MSE in the COPDGene testing sample and ECLIPSE (Figure E4). There was no significant evidence of miscalibration (Figure E5, Hosmer-Lemeshow P values >0.05). Performance metrics for COPD and FEV1 decline are shown in Table E9.
Figure 3.

Receiver operator characteristic curves for prediction of moderate to severe chronic obstructive pulmonary disease in the testing sample of COPDGene. The clinical model contained age, sex, race, pack-years of smoking, and current smoking status. AUC = area under the curve; COPD = chronic obstructive pulmonary disease; COPDGene = Genetic Epidemiology of COPD; PRS = polygenic risk score; TRS = transcriptional risk score.
Transcriptomic Characterization of TRS-defined Risk Groups
To gain biological insight into why TRS-defined subgroups have distinct disease-related characteristics, we performed multicontrast pathway enrichment analyses comparing the top versus middle and bottom versus middle tertiles of the TRS (Figure 4). The top tertile group demonstrated upregulation of IL-6/JAK/STAT3, TNF-α [tumor necrosis factor α] via NFKB (nuclear factor-κB), peroxisome, complement, and IFN-α and -γ signaling. Contrast ranks for IFN-α signaling are shown in Figure 4B. In the bottom tertile, we observed upregulation of signaling in MYC Targets, G2M Checkpoint, IL-2/STAT5, and Wnt/β-catenin. Contrast ranks for Wnt/β-catenin signaling are shown in Figure 4C.
Figure 4.
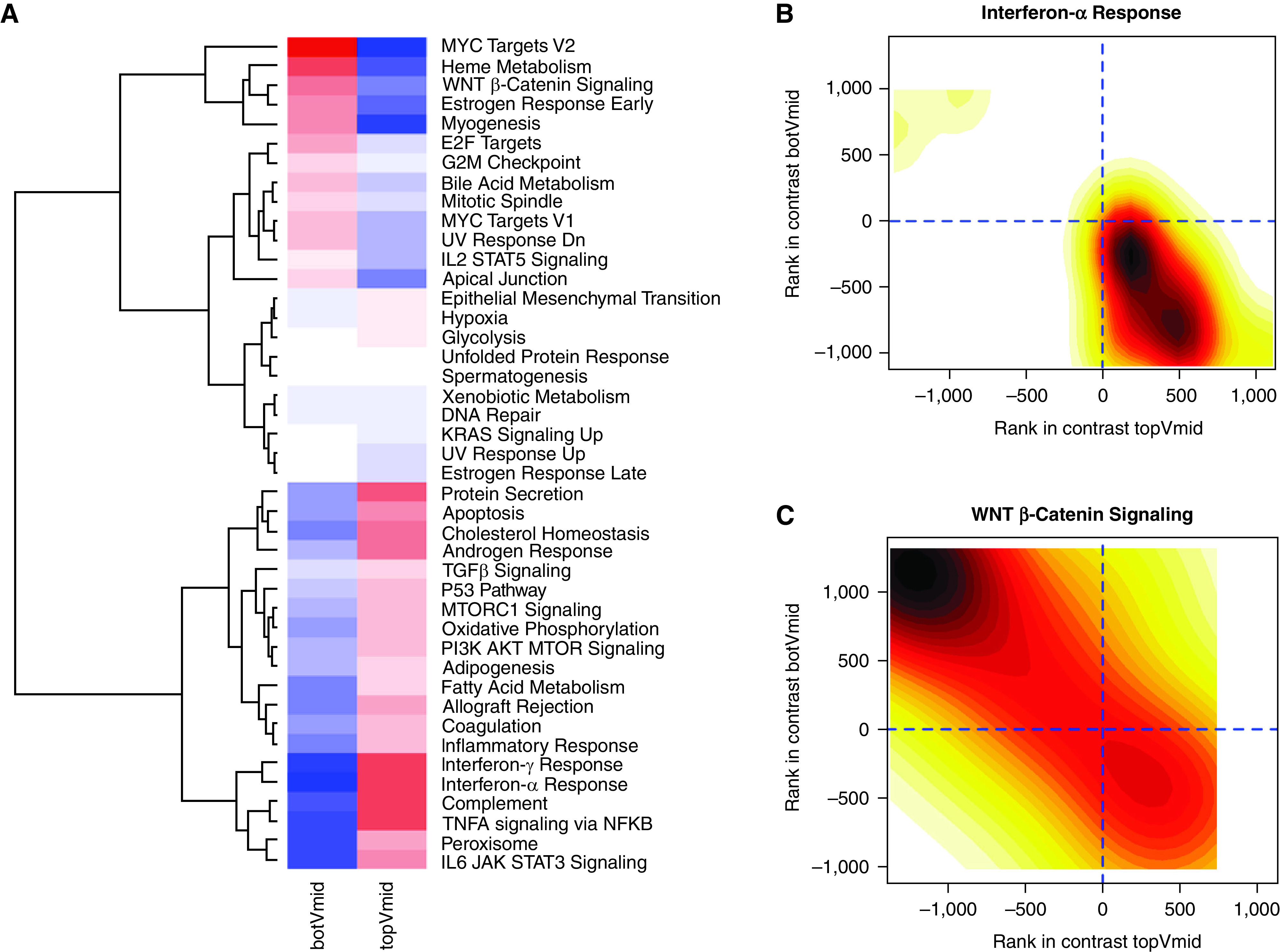
Gene expression of participants in the overall COPDGene sample (n = 2,569) in the top tertile of the transcriptional risk score (TRS) were compared with those in the middle tertile, and those in the bottom tertile were compared with the middle tertile. (A) A heatmap of enrichment scores for gene expression contrasts between these two comparisons with hierarchical clustering. Cells in red denote a positive enrichment score, whereas cells in blue denote a negative enrichment score. (B) Density plot of gene ranks in the top (x-axis) and bottom (y-axis) tertiles of the TRS are shown for IFN-α. (C) Density plot of gene ranks in the top (x-axis) and bottom (y-axis) tertiles of the TRS are shown for WNT β-catenin signaling. For density plots, the color gradients indicate numbers of genes at the given rank coordinates, with darker shades indicating higher numbers and lighter shades indicating lower numbers of genes. botVmid = bottom versus middle tertile; COPD = chronic obstructive pulmonary disease; COPDGene = Genetic Epidemiology of COPD; topVmid = top versus middle tertile.
In differential gene expression analyses for FEV1/FVC in the COPDGene training sample, 43 of the 147 TRS transcripts had a false discovery rate [FDR] P value <0.05 (Table E10), and 216 transcripts were associated with FEV1/FVC (FDR P < 0.05) (Table E11).
Discussion
In this study of more than 3,000 smokers in two cohorts, we demonstrate that a TRS is associated with COPD and can predict lung function decline, is complementary to a PRS and clinical predictors, and is enriched for specific biologic pathways that may be relevant for COPD pathogenesis. These results suggest that blood-based omics risk scores can risk-stratify individuals with cigarette smoking exposure.
The TRS and PRS were only weakly correlated, yet both risk scores were significantly associated with and predictive of COPD when included in a single model. These data suggest that the two blood-based omics risk scores may reflect different aspects of COPD pathobiology. PRS variants may have more representation of biological pathways important for lung growth and development (15, 34, 35), and the TRS may indicate activation of specific inflammatory pathways (discussed below). In addition, the two risk scores were differentially associated with COPD-related outcomes. The TRS was associated with 6-minute-walk distance, CT imaging traits, and FEV1 decline. The PRS has demonstrated robust associations with COPD affection status across multiple general population and case–control cohorts (15), but it had smaller associations with CT imaging traits and was not associated with 6-minute walk distance or FEV1 decline. Thus, the PRS and TRS appear to offer different, yet complementary, predictive value for COPD-related traits and outcomes and may be useful for risk stratification and subtyping. All CT imaging traits replicated in ECLIPSE, except for Pi10. The reason for this lack of replication may be accounted for by differences in imaging techniques; significant differences in CT measures between COPDGene and ECLIPSE have been reported despite matching clinical characteristics (36).
The TRS also improved upon clinical risk factors and the PRS for prediction of COPD and FEV1 decline. We were interested in identifying individuals at high risk of accelerated lung function decline across a range of baseline spirometric values, as certain smokers with normal lung function may rapidly progress to COPD (19). Whereas individuals with higher baseline FEV1 tend to have a greater rate of decline (3, 37–39), individuals in the top TRS tertile had greater FEV1 decline despite lower baseline FEV1, suggesting that the TRS can identify subgroups particularly prone to lung function loss. For every SD increase in the TRS, an additional ∼8–17 ml of FEV1 is lost per year. Although the minimally clinically importance difference in FEV1 has been debated (40), a change in FEV1 of ∼100 ml is generally perceivable by patients (41). Thus, the TRS appears to be associated with a clinically meaningful FEV1 decline over a 5- to 10-year period. These results demonstrate the potential utility of transcriptomics in predicting COPD progression.
We observed that the TRS, which was trained to the outcome of FEV1/FVC, predicted FEV1 decline as well as or better than one trained to FEV1 decline. There are several plausible explanations for this result, such as the smaller sample of participants with 5-year follow-up spirometry data and the inherent noisiness of repeated measure spirometry data over time. Regardless, it appears that, at least in our data sets, a transcriptional model built on cross-sectional lung function may actually predict longitudinal decline as well as or better than one trained to longitudinal change in lung function.
Compared with prior studies, our results corroborate that transcriptome-based risk scores can improve prediction of complex disease susceptibility and progression (8–10, 14, 42). Christenson and colleagues developed an airway-derived IL-17A gene expression risk score from 238 smokers with and without COPD and demonstrated an association of this gene signature with FEV1 decline in 79 patients receiving inhaled glucocorticoids (14). By contrast, we developed a TRS with 1,945 COPDGene participants using cross-sectional lung function data, applied the risk score to an independent test set of 624 COPDGene participants, and replicated our results in 468 individuals from the ECLIPSE study. We applied the TRS to risk stratify a heterogeneous group of smokers and identified those at high risk for severe disease and progression. We then compared groups at a molecular level, identifying multiple previously described and several novel pathways that require further validation.
Becker and colleagues (43) recently used microarray data from bronchial brushings to identify 171 differentially expressed genes that were associated with FEV1 decline. These genes were involved in mucin production and unfolded protein endoplasmic reticulum responses. Gene expression profiles derived from the lung compartment are appealing because the lung is the site of injury (i.e., the site of cigarette smoking exposure), yet obtaining lung tissue is challenging in clinical practice. By contrast, our TRS appears to reflect systemic inflammatory and immune system responses, and blood is routinely obtained in clinical care. Comparing these two gene expression profiles, only one transcript, CCDC170, was present in both scores and had opposite directions of effects. Therefore, a systematic integration of gene expression profiles from lung and blood is needed to facilitate clinical use and disentangle the local and systemic gene expression changes associated with COPD.
Using gene expression data to construct the TRS can also identify biological pathways that are activated or repressed in the peripheral blood and predictive of FEV1/FVC. We note that lasso selects features to optimize prediction, but these are not necessarily the most biologically important transcripts, so biological mechanisms must be interpreted with caution. Out of 147 TRS transcripts, 43 had FDR-significant P values in traditional gene expression analyses. This result reinforces the notion that the transcripts most important for risk prediction are not necessarily the same transcripts identified by differential gene expression. The 147 TRS transcripts are enriched for genes involved in immunoregulatory interactions between lymphoid and nonlymphoid cells and PPAR-α signaling. B-cell–activating factor expression has been associated with COPD severity and expansion of pulmonary lymphoid follicles (44), and lung gene expression suggests that B-cell activation is increased in emphysema (45). PPAR-α signaling may be important for antiinflammatory processes in the airways (46) and has been suggested as a therapeutic target for airway diseases (47).
To gain insight into the molecular differences between those in the highest and lowest risk groups defined by the TRS, we performed multicontrast enrichment analyses, using the middle tertile as the reference group. Individuals in the top tertile of TRS risk had higher activation of pathways implicated in COPD pathogenesis, such as IL-6 (48–50), TNF-α (51, 52), transforming growth factor β (53, 54), and IFNs (55). Those in the top tertile had lower activation of WNT-β-catenin signaling. This finding is consistent with murine and human studies that suggest that COPD genome-wide association study–identified variants in FAM13A can decrease WNT-β-catenin signaling and increase emphysema risk (56, 57). Other pathways identified by multicontrast enrichment analyses offer avenues for follow-up functional studies.
Strengths of this study include that we leverage a large, deeply phenotyped cohort with extensive genetic and transcriptomic data, which allowed us to develop a TRS in a training sample and evaluate performance in a held-out testing sample. We replicated many findings in ECLIPSE, which represents an external cross-technology replication (i.e., RNA-seq to microarray). The TRS was derived using automated feature selection and added information to known clinical risk factors and spirometric measures. We were able to identify those at high risk for FEV1 decline, despite two heterogeneous cohorts. The extent to which technical issues in transcriptome analysis (e.g., use of microarray vs. globin depleted RNA-seq) may have attenuated our ability to identify a stronger replication signal is unknown. ECLIPSE also did not have control subjects with microarray data and follow-up FEV1 measures, so the association of the TRS with COPD could not be assessed. A second measurement of gene expression may improve stable prediction of longitudinal outcomes (10). As low lung function may reflect lung structural abnormalities, the association of a lung function–based genetic risk score with FEV1/FVC was attenuated by CT imaging measures of emphysema and airway pathology (58). Whether the associations of the TRS with COPD and FEV1 decline are mediated through CT imaging traits is unknown. The TRS was not associated with exacerbations after adjusting for baseline FEV1, suggesting that shared gene expression profiles exist between these outcomes. Investigating an optimal TRS for exacerbations that adds predictive value above FEV1 measures can be undertaken in future studies.
In conclusion, we demonstrate that blood-based genetic and transcriptional risk scores combined with clinical factors can predict COPD, CT imaging traits, and FEV1 decline in two cohorts. These and other omics could be extended to predict additional measures of COPD progression, such as exacerbations, increased emphysema, and death. This approach can lend insight into biological mechanisms and offers the potential for development of personalized therapies.
Footnotes
Supported by T32HL007427 (M.M. and A.J.G.); NHLBI grant K08HL136928 (B.D.H.); and NIH grants U01 HL089856 (B.D.H.); K08HL141601 (A.B.); K08HL146972 (J.H.Y.); R01HL130512 and R01HL125583 (C.P.H.); R01HL137927, R01HL135142, HL147148, and HL089856 (M.H.C.); R01HL124233 and R01HL147326 (P.J.C.); and R01 HL137927, R01 HL147148, U01 HL089856, R01 HL133135, P01 HL132825, and P01 HL114501 (E.K.S.). The COPDGene project described was supported by NHLBI Award Number U01 HL089897 and Award Number U01 HL089856. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH. The ECLIPSE study (NCT00292552; GSK code SCO104960) was funded by GlaxoSmithKline.
Author Contributions: Study design: M.M., M.H.C., and P.J.C. Acquisition, analysis, or interpretation of the data: M.M., B.D.H., M.H.C., E.K.S., A.B., A.J.G., A.S., S.L., Z.X., C.P.H., D.D.S., R.T.-S., and P.J.C. Statistical analysis: M.M., B.D.H., A.B., A.J.G., A.S., Z.X., M.H.C., and P.J.C. Obtained funding: P.J.C., E.K.S., and M.H.C. Critical revision of the manuscript for important intellectual content: all authors.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202107-1584OC on November 5, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
Contributor Information
on behalf of the HAPIN Investigators:
James D. Crapo, Edwin K. Silverman, Barry J. Make, Elizabeth A. Regan, Terri Beaty, Ferdouse Begum, Peter J. Castaldi, Michael Cho, Dawn L. DeMeo, Adel R. Boueiz, Marilyn G. Foreman, Eitan Halper-Stromberg, Lystra P. Hayden, Craig P. Hersh, Jacqueline Hetmanski, Brian D. Hobbs, John E. Hokanson, Nan Laird, Christoph Lange, Sharon M. Lutz, Merry-Lynn McDonald, Margaret M. Parker, Dmitry Prokopenko, Dandi Qiao, Phuwanat Sakornsakolpat, Edwin K. Silverman, Emily S. Wan, Juan Pablo Centeno, Jean-Paul Charbonnier, Harvey O. Coxson, Craig J. Galban, MeiLan K. Han, Eric A. Hoffman, Stephen Humphries, Francine L. Jacobson, Philip F. Judy, Ella A. Kazerooni, Alex Kluiber, David A. Lynch, Pietro Nardelli, John D. Newell, Jr., Aleena Notary, Andrea Oh, James C. Ross, Raul San Jose Estepar, Joyce Schroeder, Jered Sieren, Berend C. Stoel, Juerg Tschirren, Edwin Van Beek, Bram van Ginneken, Eva van Rikxoort, Gonzalo Vegas Sanchez-Ferrero, Lucas Veitel, George R. Washko, Carla G. Wilson, Robert Jensen, Douglas Everett, Jim Crooks, Katherine Pratte, Matt Strand, Gregory Kinney, Sharon M. Lutz, Kendra A. Young, Surya P. Bhatt, Jessica Bon, Alejandro A. Diaz, Susan Murray, Xavier Soler, Russell P. Bowler, Katerina Kechris, and Farnoush Banaei-Kashani
References
- 1. Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, Bourbeau J, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 report. GOLD Executive Summary. Am J Respir Crit Care Med . 2017;195:557–582. doi: 10.1164/rccm.201701-0218PP. [DOI] [PubMed] [Google Scholar]
- 2. Rennard SI, Vestbo J. COPD: the dangerous underestimate of 15% Lancet . 2006;367:1216–1219. doi: 10.1016/S0140-6736(06)68516-4. [DOI] [PubMed] [Google Scholar]
- 3. Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, et al. ECLIPSE Investigators Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med . 2011;365:1184–1192. doi: 10.1056/NEJMoa1105482. [DOI] [PubMed] [Google Scholar]
- 4. Hurst JR, Vestbo J, Anzueto A, Locantore N, Müllerova H, Tal-Singer R, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med . 2010;363:1128–1138. doi: 10.1056/NEJMoa0909883. [DOI] [PubMed] [Google Scholar]
- 5. Celli BR, Cote CG, Marin JM, Casanova C, Montes de Oca M, Mendez RA, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med . 2004;350:1005–1012. doi: 10.1056/NEJMoa021322. [DOI] [PubMed] [Google Scholar]
- 6. Palmer LJ, Knuiman MW, Divitini ML, Burton PR, James AL, Bartholomew HC, et al. Familial aggregation and heritability of adult lung function: results from the Busselton Health Study. Eur Respir J . 2001;17:696–702. doi: 10.1183/09031936.01.17406960. [DOI] [PubMed] [Google Scholar]
- 7. Zhou JJ, Cho MH, Castaldi PJ, Hersh CP, Silverman EK, Laird NM. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am J Respir Crit Care Med . 2013;188:941–947. doi: 10.1164/rccm.201302-0263OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marigorta UM, Denson LA, Hyams JS, Mondal K, Prince J, Walters TD, et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat Genet . 2017;49:1517–1521. doi: 10.1038/ng.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herazo-Maya JD, Sun J, Molyneaux PL, Li Q, Villalba JA, Tzouvelekis A, et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: an international, multicentre, cohort study. Lancet Respir Med . 2017;5:857–868. doi: 10.1016/S2213-2600(17)30349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang Y, Oldham JM, Ma S-F, Unterman A, Liao S-Y, Barros AJ, et al. Blood transcriptomic predicts progression of pulmonary fibrosis and associates natural killer cells. Am J Respir Crit Care Med . 2021;204:197–208. doi: 10.1164/rccm.202008-3093OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ling Song SH. Predicting COPD status with a random generalized linear model. Syst Biomed . 2013;1:261–267. [Google Scholar]
- 12. Singh D, Fox SM, Tal-Singer R, Bates S, Riley JH, Celli B. Altered gene expression in blood and sputum in COPD frequent exacerbators in the ECLIPSE cohort. PLoS One . 2014;9:e107381. doi: 10.1371/journal.pone.0107381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bertrams W, Griss K, Han M, Seidel K, Klemmer A, Sittka-Stark A, et al. Transcriptional analysis identifies potential biomarkers and molecular regulators in pneumonia and COPD exacerbation. Sci Rep . 2020;10:241. doi: 10.1038/s41598-019-57108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Christenson SA, van den Berge M, Faiz A, Inkamp K, Bhakta N, Bonser LR, et al. An airway epithelial IL-17A response signature identifies a steroid-unresponsive COPD patient subgroup. J Clin Invest . 2019;129:169–181. doi: 10.1172/JCI121087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moll M, Sakornsakolpat P, Shrine N, Hobbs BD, DeMeo DL, John C, et al. International COPD Genetics Consortium SpiroMeta Consortium. Chronic obstructive pulmonary disease and related phenotypes: polygenic risk scores in population-based and case-control cohorts. Lancet Respir Med . 2020;8:696–708. doi: 10.1016/S2213-2600(20)30101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, et al. Genetic epidemiology of COPD (COPDGene) study design. COPD . 2010;7:32–43. doi: 10.3109/15412550903499522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, Edwards L, et al. ECLIPSE Investigators Evaluation of COPD longitudinally to identify predictive surrogate end-points (ECLIPSE) Eur Respir J . 2008;31:869–873. doi: 10.1183/09031936.00111707. [DOI] [PubMed] [Google Scholar]
- 18. Bhatt SP, Balte PP, Schwartz JE, Cassano PA, Couper D, Jacobs DR, Jr, et al. Discriminative accuracy of FEV1:FVC thresholds for COPD-related hospitalization and mortality. JAMA . 2019;321:2438–2447. doi: 10.1001/jama.2019.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lowe KE, Regan EA, Anzueto A, Austin E, Austin JHM, Beaty TH, et al. COPDGene® 2019: redefining the diagnosis of chronic obstructive pulmonary disease Chronic Obstr Pulm Dis (Miami) 2019. 6 384 399 31710793 [Google Scholar]
- 20. Parker MM, Chase RP, Lamb A, Reyes A, Saferali A, Yun JH, et al. RNA sequencing identifies novel non-coding RNA and exon-specific effects associated with cigarette smoking. BMC Med Genomics . 2017;10:58. doi: 10.1186/s12920-017-0295-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res . 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Obeidat M, Nie Y, Chen V, Shannon CP, Andiappan AK, Lee B, et al. Network-based analysis reveals novel gene signatures in peripheral blood of patients with chronic obstructive pulmonary disease. Respir Res . 2017;18:72. doi: 10.1186/s12931-017-0558-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw . 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 24. Baarnes CB, Thuesen BH, Linneberg A, Ustrup AS, Pedersen SK, Ulrik CS. Predictors of accelerated FEV1 decline in adults with airflow limitation: findings from the Health2006 cohort. Chron Respir Dis . 2019;16:1479973119838278. doi: 10.1177/1479973119838278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc Series B Stat Methodol . 1996;58:267–288. [Google Scholar]
- 26. Fabregat A, Sidiropoulos K, Viteri G, Forner O, Marin-Garcia P, Arnau V, et al. Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics . 2017;18:142. doi: 10.1186/s12859-017-1559-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Foroushani ABK, Brinkman FSL, Lynn DJ. Pathway-GPS and SIGORA: identifying relevant pathways based on the over-representation of their gene-pair signatures. PeerJ . 2013;1:e229. doi: 10.7717/peerj.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.S J. pscl: Classes and methods for R Developed in the Political Science Computational Laboratory. United States Stud. Centre, Univ. Sydney, Sydney, New South Wales, Australia. R Package version 1.5.5 2020https://github.com/atahk/pscl/.
- 29. Zeileis A, Kleiber C, Jackman S. Regression models for count data in R. J Stat Softw . 2008;27 doi: 10.18637/jss.v027.i08. [DOI] [Google Scholar]
- 30. DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics . 1988;44:837–845. [PubMed] [Google Scholar]
- 31.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves BMC Bioinformatics 20111277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaspi A, Ziemann M. mitch: multi-contrast pathway enrichment for multi-omics and single-cell profiling data. BMC Genomics . 2020;21:447. doi: 10.1186/s12864-020-06856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghosh AJ, Saferali A, Lee S, Chase R, Moll M, Morrow J, et al. Blood RNA sequencing shows overlapping gene expression across COPD phenotype domains. Thorax . 2021 doi: 10.1136/thoraxjnl-2020-216401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sakornsakolpat P, Prokopenko D, Lamontagne M, Reeve NF, Guyatt AL, Jackson VE, et al. SpiroMeta Consortium International COPD Genetics Consortium. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet . 2019;51:494–505. doi: 10.1038/s41588-018-0342-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shrine N, Guyatt AL, Erzurumluoglu AM, Jackson VE, Hobbs BD, Melbourne CA, et al. Understanding Society Scientific Group New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet . 2019;51:481–493. doi: 10.1038/s41588-018-0321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moll M, Qiao D, Regan EA, Hunninghake GM, Make BJ, Tal-Singer R, et al. Machine learning and prediction of all-cause mortality in COPD. Chest . 2020;158:952–964. doi: 10.1016/j.chest.2020.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fletcher C, Peto R, Tinker CSF. The natural history of chronic bronchitis and emphysema: an eight-year study of early chronic obstructive lung disease in working men in London. Oxford, England: Oxford University Press; 1976. [Google Scholar]
- 38. Bhatt SP, Soler X, Wang X, Murray S, Anzueto AR, Beaty TH, et al. COPDGene Investigators Association between functional small airway disease and FEV1 decline in chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2016;194:178–184. doi: 10.1164/rccm.201511-2219OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oelsner EC, Balte PP, Bhatt SP, Cassano PA, Couper D, Folsom AR, et al. Lung function decline in former smokers and low-intensity current smokers: a secondary data analysis of the NHLBI Pooled Cohorts Study. Lancet Respir Med . 2020;8:34–44. doi: 10.1016/S2213-2600(19)30276-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jones PW, Beeh KM, Chapman KR, Decramer M, Mahler DA, Wedzicha JA. Minimal clinically important differences in pharmacological trials. Am J Respir Crit Care Med . 2014;189:250–255. doi: 10.1164/rccm.201310-1863PP. [DOI] [PubMed] [Google Scholar]
- 41. Donohue JF. Minimal clinically important differences in COPD lung function. COPD . 2005;2:111–124. doi: 10.1081/copd-200053377. [DOI] [PubMed] [Google Scholar]
- 42. Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma SF, Tseng GC, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med . 2013;5:205ra136. doi: 10.1126/scitranslmed.3005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Becker EJ, Faiz A, van den Berge M, Timens W, Hiemstra PS, Clark K, et al. Bronchial gene expression signature associated with rate of subsequent FEV1 decline in individuals with and at risk of COPD. Thorax . 2021 doi: 10.1136/thoraxjnl-2019-214476. [DOI] [PubMed] [Google Scholar]
- 44. Polverino F, Cosio BG, Pons J, Laucho-Contreras M, Tejera P, Iglesias A, et al. B cell-activating factor. An orchestrator of lymphoid follicles in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2015;192:695–705. doi: 10.1164/rccm.201501-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Faner R, Cruz T, Casserras T, López-Giraldo A, Noell G, Coca I, et al. Network analysis of lung transcriptomics reveals a distinct B-cell signature in emphysema. Am J Respir Crit Care Med . 2016;193:1242–1253. doi: 10.1164/rccm.201507-1311OC. [DOI] [PubMed] [Google Scholar]
- 46. Belvisi MG, Mitchell JA. Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. Br J Pharmacol . 2009;158:994–1003. doi: 10.1111/j.1476-5381.2009.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Remels AH, Gosker HR, Schrauwen P, Langen RC, Schols AM. Peroxisome proliferator-activated receptors: a therapeutic target in COPD? Eur Respir J . 2008;31:502–508. doi: 10.1183/09031936.00068207. [DOI] [PubMed] [Google Scholar]
- 48. Chang Y, Glass K, Liu Y-Y, Silverman EK, Crapo JD, Tal-Singer R, et al. COPD subtypes identified by network-based clustering of blood gene expression. Genomics . 2016;107:51–58. doi: 10.1016/j.ygeno.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. He J-Q, Foreman MG, Shumansky K, Zhang X, Akhabir L, Sin DD, et al. Associations of IL6 polymorphisms with lung function decline and COPD. Thorax . 2009;64:698–704. doi: 10.1136/thx.2008.111278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pérez-Rubio G, Silva-Zolezzi I, Fernández-López JC, Camarena Á, Velázquez-Uncal M, Morales-Mandujano F, et al. Genetic variants in IL6R and ADAM19 are associated with COPD severity in a Mexican mestizo population. COPD . 2016;13:610–615. doi: 10.3109/15412555.2016.1161017. [DOI] [PubMed] [Google Scholar]
- 51. Gingo MR, Silveira LJ, Miller YE, Friedlander AL, Cosgrove GP, Chan ED, et al. Tumour necrosis factor gene polymorphisms are associated with COPD. Eur Respir J . 2008;31:1005–1012. doi: 10.1183/09031936.00100307. [DOI] [PubMed] [Google Scholar]
- 52. Yao Y, Zhou J, Diao X, Wang S. Association between tumor necrosis factor-α and chronic obstructive pulmonary disease: a systematic review and meta-analysis. Ther Adv Respir Dis . 2019;13 doi: 10.1177/1753466619866096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Verhamme FM, Bracke KR, Joos GF, Brusselle GG. Transforming growth factor-β superfamily in obstructive lung diseases. more suspects than TGF-β alone. Am J Respir Cell Mol Biol . 2015;52:653–662. doi: 10.1165/rcmb.2014-0282RT. [DOI] [PubMed] [Google Scholar]
- 54. Hersh CP, Hansel NN, Barnes KC, Lomas DA, Pillai SG, Coxson HO, et al. ICGN Investigators Transforming growth factor-beta receptor-3 is associated with pulmonary emphysema. Am J Respir Cell Mol Biol . 2009;41:324–331. doi: 10.1165/rcmb.2008-0427OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Johnston SL. IFN therapy in airway disease: is prophylaxis a new approach in exacerbation prevention? Am J Respir Crit Care Med . 2020;201:9–11. doi: 10.1164/rccm.201909-1850ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jiang Z, Lao T, Qiu W, Polverino F, Gupta K, Guo F, et al. A chronic obstructive pulmonary disease susceptibility gene, FAM13A, regulates protein stability of β-catenin. Am J Respir Crit Care Med . 2016;194:185–197. doi: 10.1164/rccm.201505-0999OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Castaldi PJ, Guo F, Qiao D, Du F, Naing ZZC, Li Y, et al. Identification of functional variants in the FAM13A chronic obstructive pulmonary disease genome-wide association study locus by massively parallel reporter assays. Am J Respir Crit Care Med . 2019;199:52–61. doi: 10.1164/rccm.201802-0337OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oelsner EC, Ortega VE, Smith BM, Nguyen JN, Manichaikul AW, Hoffman EA, et al. A genetic risk score associated with COPD susceptibility and lung structure on computed tomography. Am J Respir Crit Care Med . 2019;200:721–731. doi: 10.1164/rccm.201812-2355OC. [DOI] [PMC free article] [PubMed] [Google Scholar]