

Abstract
Background:
Secondary spontaneous pneumothorax (SSP) in interstitial lung disease (ILD) may influence prognosis of any ILD, and SSP onset predicts poor outcome in idiopathic pulmonary fibrosis (IPF). Recently, progressive fibrosing ILD (PF-ILD) has rapidly acquired importance.
Objective:
We hypothesized that PF-ILD would strongly influence the prognosis of patients with any ILD complicated with SSP.
Methods:
We retrospectively surveyed and collected data from patients hospitalized for SSP from January 2016 to June 2020. PF-ILD was defined as the following occurring within 24 months before SSP develops: relative decline in %forced vital capacity (FVC) ≥10% or two of the following: relative decline in %FVC between 5% and 10%, worsening respiratory symptoms, or increased extent of fibrosis on high-resolution computed tomography.
Results:
We analyzed 32 patients hospitalized for SSP in ILD. This study comprised 18 patients with PF-ILD and 14 patients with non-PF-ILD. PF-ILD patients had lower body mass index (BMI) and %FVC. No significant differences in survival regarding follow-up period from the time of ILD diagnosis and hospitalization for SSP were observed between the PF-ILD and non-PF-ILD patients. Older age and lower BMI were significant predictors of mortality by multivariate Cox regression analysis. ROC analysis showed BMI ≤17.8 kg/m2 to reliably predict poor prognosis.
Conclusions:
Regardless of whether patients have PF-ILD, older age and lower BMI in patients with ILD places them at higher risk of developing SSP, and prognosis is poor if SSP develops. Therefore, clinical management of physique is important to improve the prognosis of ILD patients.
Keywords: Pneumothorax, Progressive fibrosing interstitial lung disease, Prognosis
Introduction
Pneumothorax is classified as primary spontaneous pneumothorax or secondary spontaneous pneumothorax (SSP) (1). The most common etiology of SSP is pulmonary emphysema, followed by interstitial lung disease (ILD), lung cancer, pulmonary infection, catamenial, and others (2). Nishimoto et al. reported that among these disorders, SSP in patients with idiopathic pulmonary fibrosis (IPF) could lead to a poor prognosis (3). These authors also recently reported that SSP was a negative prognostic factor in connective tissue disease (CTD)-associated ILD (CTD-ILD) (4). Thus, SSP might influence prognosis for other ILDs, although there are very few reports on the impact of SSP in ILDs other than IPF and CTD-ILD.
A recent approach based on patterns of disease behavior is useful for managing various ILDs (5,6). Thus, the concept of progressive fibrosing interstitial lung disease (PF-ILD) has rapidly acquired importance in the management of patients with non-IPF progressive ILD based on the results of the INBUILD trial (7). This trial defined PF-ILD as progression due to a combination of a decline in forced vital capacity (FVC), increased extent of fibrosis on high-resolution computed tomography (HRCT), and worsening of respiratory symptoms. It was expected that nintedanib as an anti-fibrotic agent could be effective in various progressive ILDs as well as IPF (7). Also in clinical practice, we often encounter patients with non-IPF progressive ILD. Therefore, we hypothesized that the prognostic influence of PF-ILD may be strongly affected by any ILD in patients complicated with SSP.
Materials and methods
Study sample
This study received approval from the institutional review board of Saitama Red Cross Hospital (approval no. 19-D). Because of the retrospective nature of the study, the review board waived the need for written informed consent from the patients. We retrospectively surveyed the patients hospitalized for SSP from January 2016 to June 2020. We then collected data from each patient’s medical records that included characteristics, laboratory data, and chest CT findings at the time of SSP diagnosis on admission to our hospital. In addition, we collected baseline pulmonary function test results within 6 months of their hospitalization for SSP. Survival was defined as the time from the date of either the ILD diagnosis or hospitalization for SSP to the date of death or censoring. A part of this patient cohort was already the subject of a previous study focusing on SSP in patients with ILD and pulmonary emphysema (8).
Data collection
In this study, PF-ILD was defined on the basis of the INBUILD trial as the presence of one of the following within 24 months before SPP develops: a relative decline in %FVC ≥10% or any two of the following: a relative decline in %FVC between 5% and 10%, worsening respiratory symptoms, or an increase in the extent of fibrosis on HRCT imaging (7). Worsening of respiratory symptoms was judged as positive when the patient complained of apparent increasing dyspnea before SSP occurred based on the medical record. On the basis of a comparison of the HRCT images obtained farthest before and closest to the onset of SSP within a 24-month period, increasing evidence of the extent of fibrosis on HRCT was defined as an apparent increase in reticular patterns and/or honeycombing with architectural distortion and vessel-related structures by two pulmonologists (S. Sato, H. Matsushima) and one radiologist (H. Sasaki) (9). HRCT patterns on the image obtained closest to the time of onset of SSP were classified as recent according to the guidelines for IPF (10). The ILD diagnosis of each patient was based on multidisciplinary discussions with pulmonary physicians, a radiologist, and pathologist, who made clinical diagnoses based on the clinical, radiographic, and/or pathological findings of each ILD case (10,11). In patients with combined pulmonary fibrosis with emphysema (CPFE), positive findings of emphysema were visually defined as the presence of an area of low attenuation indicating the lack of a distinct alveolar wall threshold over 10% (12). We also reviewed treatments for ILD such as the use of anti-inflammatory agents (prednisolone, cyclosporine, and tacrolimus) and antifibrotic agents (pirfenidone and nintedanib) during the hospitalization for SSP and which were not a part of previous treatments administered during the observation period.
Statistical methods
In the statistical analysis, Fisher’s exact test, unpaired t-test, or Mann-Whitney U test was used as appropriate to detect differences between groups. We investigated potential risk factors of mortality with each variable chosen for entry into univariate Cox regression analysis. Thereafter, the variables that achieved a modest level of statistical significance (P < 0.05 on univariate analysis) based on the forced entry method were assessed in a multivariate analysis. We considered P < 0.05 to represent statistical significance. All data were analyzed with SPSS version 22.0 (IBM Japan, Tokyo, Japan).
Results
Overall patient characteristics
The study cohort included 88 patients hospitalized for SSP. After excluding the patients with causes of SSP that included pulmonary emphysema (n = 43), catamenial (n = 7), lung cancer (n = 3), nontuberculous mycobacterial pulmonary disease (n = 2), or Birt-Hogg-Dubé syndrome (n = 1), we finally identified 32 patients with SSP in ILD (Table 1).
Table 1.
Baseline characteristics of the study patients
| Total | PF-ILD | Non-PF-ILD | P Value | |
| No. of patients | 32 | 18 | 14 | |
| Male [N, (%)] | 27 (84.4%) | 15 (83.3%) | 12 (85.7%) | >0.999 |
| Age, mean ± SD | 74.1 ± 7.6 | 73.2 ± 6.3 | 75.4 ± 9.1 | 0.425 |
| Current or ex-smoker [N, (%)] | 24 (75.0%) | 12 (66.7%) | 12 (85.7%) | 0.412 |
| BMI (kg/m2), mean ± SD | 17.8 ± 3.7 | 16.6 ± 2.6 | 19.4 ± 4.4 | 0.028 |
| Albumin (g/dL), mean ± SD | 3.5 ± 0.6 | 3.4 ± 0.5 | 3.7 ± 0.6 | 0.110 |
| KL-6 (U/mL), mean ± SD | 921.4 ± 755.6 | 873.9 ± 445.5 | 982.6 ± 1046.2 | 0.693 |
| HRCT pattern | 0.511 | |||
| UIP | 9 (28.1%) | 7 (38.9%) | 2 (14.3%) | |
| Probable UIP | 12(37.5%) | 6 (33.3%) | 6 (42.9%) | |
| Indeterminate for UIP | 6 (18.8%) | 3 (16.7%) | 3 (21.4%) | |
| Alternative | 5 (15.6%) | 2 (11.1%) | 3 (21.4%) | |
| CPFE [N, (%)] | 12 (37.5%) | 4 (22.2%) | 8 (57.1%) | 0.068 |
| IPF [N, (%)] | 11 (34.4%) | 11 (61.1%) | 0 (0.0%) | – |
| CTD-ILD [N, (%)] | 4 (12.5%) | 2 (11.1%) | 2 (14.3%) | >0.999 |
| Upper lobe-dominant pulmonary fibrosis [N, (%)] | 4 (12.5%) | 3 (16.7%) | 1 (7.1%) | 0.613 |
| %FVC, mean ± SD (available N = 23) | 65.4 ± 20.9 | 57.4 ± 14.8 | 83.7 ± 22.1 | 0.003 |
| %DLCO, mean ± SD (available N = 19) | 51.3 ± 23.4 | 49.3 ± 20.2 | 55.6 ± 30.9 | 0.597 |
| Comorbidity | ||||
| Malignancy [N, (%)] | 10 (41.7%) | 3 (16.7%) | 7 (50.0%) | 0.062 |
| Chronic pulmonary aspergillosis [N, (%)] | 3 (9.4%) | 2 (11.1%) | 1 (7.1%) | >0.999 |
| Cerebro-cardiovascular disease [N, (%)] | 4 (12.5%) | 0 (0.0%) | 4 (28.6%) | 0.028 |
| Receive HOT [N, (%)] | 19 (59.4%) | 12 (66.7%) | 7 (50.0%) | 0.473 |
| Treatment for ILD at hospitalization for SSP | ||||
| Anti-inflammatory agents | 7 (21.9%)* | 5 (27.8%)* | 2 (14.3%)* | 0.426 |
| Anti-fibrotic agents (nintedanib or pirfenidone) | 8 (25.0%) | 7 (38.9%) | 1 (7.1%) | 0.053 |
| Treatment for SSP (including duplication) | ||||
| Chest tube drainage [N, (%)] | 27 (84.4%) | 14 (77.8%) | 13 (92.9%) | 0.355 |
| Surgery [N, (%)] | 5 (15.6%) | 2 (11.1%) | 3 (21.4%) | 0.631 |
| Pleurodesis [N, (%)] | 13 (40.6%) | 8 (44.4%) | 5 (35.7%) | 0.725 |
| EWS [N, (%)] | 4 (12.5%) | 2 (11.1%) | 2 (14.3%) | >0.999 |
| Recurrence of pneumothorax [N, (%)] | 14 (43.8%) | 9 (50.0%) | 5 (35.7%) | 0.490 |
| Observation period from hospitalization for SSP (days) | 162.0 (50.3-351.5) | 179.5 (50.5-407.8) | 152.0 (47.5-269.3) | 0.569 |
| Deaths (during follow-up), [N, (%)] | 22 (68.8) | 14 (77.8) | 8 (57.1) | 0.267 |
Data are presented as the mean or median (interquartile ranges). PF-ILD, progressive fibrosing interstitial lung disease; BMI, body mass index; KL-6, Krebs von den Lungen-6; HRCT, high-resolution computed tomography; UIP, usual interstitial pneumonia; CPFE, combined pulmonary fibrosis with emphysema; IPF, idiopathic pulmonary fibrosis; CTD: connective tissue disease; FVC: forced vital capacity; DLCO, diffusing capacity of the lung for carbon monoxide; HOT: home oxygen therapy; SSP, secondary spontaneous pneumothorax; EWS, endobronchial Watanabe spigot. *All patients received prednisolone, and 1 patient also received cyclosporine, and 1 patient also received tacrolimus.
Among these patients, we classified 18 patients as having PF-ILD (56.3%) and 14 (43.8%) as not having PF-ILD. Among the PF-ILD patients, two thirds (n = 12) had a relative decline in %FVC ≥10%, 2 patients had both worsening dyspnea and an increased extent of fibrosis on HRCT without a relative decline in %FVC ≥5%, and one patient had a relative decline in %FVC of 7.3% and progressive respiratory symptoms with extension of fibrosis on HRCT. In contrast, 14 patients had a lack of %FVC data for comparison, and thus, 3 patients met the PF-ILD criteria for worsening respiratory symptoms and extent of fibrosis on HRCT (Table 2). All IPF patients (n = 11) were classified as having PF-ILD. Among the patients with CTD-ILD, 2 were PF-ILD patients and 2 were non-PF-ILD patients. Among those with upper lobe-dominant pulmonary fibrosis, 3 were PF-ILD patients and 1 was a non-PF-ILD patient (Table 1). The most frequent comorbidities were malignancy (41.7%), cerebro-cardiovascular disease (12.5%), and chronic pulmonary aspergillosis (9.4%). Of the patients, 43.8% had a history of recurrence of pneumothorax (PF-ILD patients: 50.0% and non-PF-ILD patients: 35.7%). The median follow-up period for analyzing the change of %FVC within 24 months was 13.3 (range, 4–20) months, and that for analyzing disease extent by comparing the closest and farthest HRCT images was 16.1 (range, 3–23) months.
Table 2.
Study patients meeting the criteria for PF-ILD
| Relative decline in %FVC | ≥10% | 5% to <10% | <5% | Lack of data for comparison | |
| n = 12 | n = 1 | n = 5 | n = 14 | ||
| Increased extent of fibrosis on HRCT | Yes | 11 (92%) | 1 (100%) | 2 (40%) | 5 (36%) |
| No | 1 (8%) | 0 (0%) | 3 (60%) | 9 (64%) | |
| Worsening respiratory symptoms | Yes | 10 (83%) | 1 (100%) | 2 (40%) | 5 (36%) |
| No | 2 (17%) | 0 (0%) | 3 (60%) | 9 (64%) | |
| Met the criteria for PF-ILD | 12 (100%) | 1 (100%) | 2 (40%) | 3 (21%) | |
PF-ILD, progressive fibrosing interstitial lung disease; FVC, forced vital capacity; HRCT, high-resolution computed tomography.
As shown in Table 1, characteristics of the PF-ILD patients at hospitalization for SSP were lower body mass index (BMI) and %FVC than those in the non-PF-ILD patients. As treatment for ILD during hospitalization for SSP, 5 PF-ILD patients (27.8%) and 2 non-PF-ILD patients (14.3%) received anti-inflammatory agents although the difference was not significant. All of the patients received prednisolone, with 1 patient also receiving cyclosporine and 1 patient also receiving tacrolimus. All of the patients with IPF were classified as having PF-ILD. Just under 40% of the PF-ILD patients received antifibrotic agents, and the tendency for their use was higher than that in the non-PF-ILD patients (P = 0.053).
Survival
Death from any cause occurred in 22 patients (68.8%) during a median follow-up period from hospitalization of 162 days. The cause of death was ILD progression or SSP in 17 patients (77.3%), pneumonia in 3 patients (17.6%), and other causes in 2 patients (9.1%).
Kaplan-Meier survival curves and log-rank test showed that the IPF patients had significantly poorer survival than the non-IPF patients from the time of ILD diagnosis (Fig. 1A) (P = 0.047; mean survival time: IPF, 45.6 months and non-IPF, 76.4 months). However, in view of the follow-up period after hospitalization for SSP, there was no significant difference between the IPF and non-IPF patients (P = 0.193; mean survival time: IPF, 8.7 months and non-IPF, 22.5 months) (Fig. 1B). The PF-ILD patients showed no significant difference in survival compared with the non-PF-ILD patients from the viewpoints of the follow-up period from ILD diagnosis and that from hospitalization for SSP (Fig. 1C, D). Mean survival time from ILD diagnosis was 60.7 months in the PF-ILD patients and 64.4 months in the non-PF-ILD patients (P = 0.551), and that from the date of hospitalization for SSP was 15.6 months for the PF-ILD patients versus 20.2 months for the non-PF-ILD patients (P = 0.728).
Figure 1.
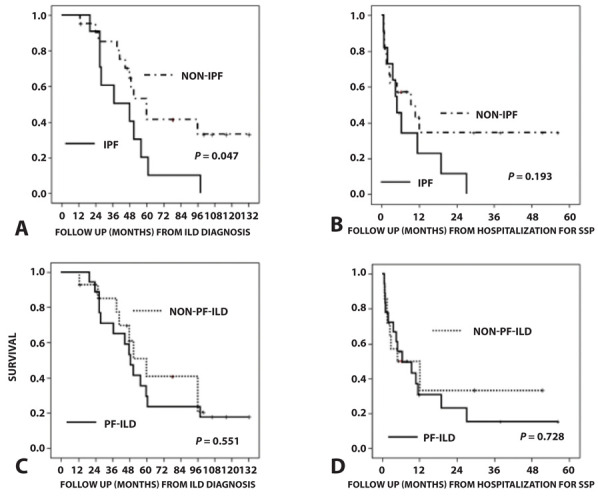
Kaplan-Meier survival curves of all-cause mortality at ILD diagnosis and after the start of hospitalization for SSP. (A) IPF patients had significantly poorer survival than non-IPF patients in terms of time from ILD diagnosis (P = 0.047; mean survival time: IPF: 45.6 months, non-IPF: 76.4 months). (B) There was no significant difference in survival after the start of hospitalization for SSP between IPF and non-IPF patients (P = 0.193; mean survival time: IPF: 8.7 months, non-IPF: 22.5 months). (C) PF-ILD patients showed no significant difference in survival time compared with non-PF-ILD patients (P = 0.551; mean survival time: PF-ILD: 60.7 months, non-PF-ILD: 64.4 months). (D) There was also no significant difference in survival after the start of hospitalization for SSP between PF-ILD and non-PF-ILD patients (P = 0.165; mean survival time: PF-ILD: 15.6 months, non-PF-ILD: 20.2 months). ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; PF-ILD, progressive fibrosing interstitial lung disease; SSP, secondary spontaneous pneumothorax.
Older age and lower BMI at hospitalization for SSP were significant predictors of mortality by univariate and multivariate Cox regression analyses (Table 3). We next conducted ROC analysis to determine the optimal BMI cutoff level that represents increased poor prognosis in the ILD patients with hospitalization for SSP by using the Youden index. A BMI of ≤17.8 kg/m2 was shown to reliably predict poor prognosis (Fig. 2).
Table 3.
Analysis of predictors of mortality from hospitalization for SSP
| Univariate Cox regression | Multivariate Cox regression | |||||||
| HR | 95% CI | P value | HR | 95% CI | P value | |||
| Male | 1.163 | 0.344, 3.938 | 0.808 | – | ||||
| Age (per 1 year) | 1.145 | 1.048, 1.251 | 0.003 | 1.144 | 1.050,1.247 | 0.002 | ||
| Current/ex-smoker | 0.812 | 0.317, 2.082 | 0.665 | – | ||||
| BMI (per 1 kg/m2) | 0.746 | 0.612, 0.909 | 0.004 | 0.716 | 0.574,0.893 | 0.003 | ||
| Albumin (per 1 g/dL) | 0.793 | 0.366, 1.720 | 0.558 | – | ||||
| KL-6 (per 1 U/mL) | 0.999 | 0.998, 1.000 | 0.285 | – | ||||
| Receive HOT | 1.799 | 0.716, 4.524 | 0.212 | – | ||||
| IPF (vs. non-IPF) | 1.739 | 0.748, 4.039 | 0.198 | – | ||||
| CTD-ILD | 0.998 | 0.294, 3.389 | 0.998 | – | ||||
| Upper lobe-dominant pulmonary fibrosis | 3.084 | 0.991, 9.595 | 0.052 | – | ||||
| HRCT pattern | – | |||||||
| UIP | 1.000 | ref | ||||||
| Probable UIP | 0.701 | 0.255, 1.926 | 0.491 | |||||
| Indeterminate for UIP | 0.464 | 0.116, 1.853 | 0.277 | |||||
| Alternative | 0.887 | 0.226, 3.475 | 0.863 | |||||
| CPFE | 0.450 | 0.179, 1.131 | 0.090 | – | ||||
| %FVC (per 1%) | 0.971 | 0.940, 1.004 | 0.081 | – | ||||
| %DLCO (per 1%) | 0.999 | 0.973, 1.025 | 0.919 | – | ||||
| PF-ILD (vs. Non-PF-ILD) | 1.167 | 0.487, 2.796 | 0.729 | – | ||||
| Comorbidity | ||||||||
| Malignancy | 1.193 | 0.473, 3.008 | 0.708 | – | ||||
| Chronic pulmonary aspergillosis | 1.062 | 0.245, 4.612 | 0.936 | – | ||||
| Cerebro-cardiovascular disease | 0.635 | 0.148, 2.724 | 0.541 | – | ||||
| Treatment for SSP | ||||||||
| Chest tube drainage | 0.594 | 0.193, 1.825 | 0.363 | – | ||||
| Surgery | 0.358 | 0.083, 1.545 | 0.169 | – | ||||
| Pleurodesis | 0.765 | 0.325, 1.797 | 0.539 | – | ||||
| EWS | 0.968 | 0.280, 3.344 | 0.959 | – | ||||
| Recurrence of pneumothorax | 0.846 | 0.362, 1.981 | 0.701 | – | ||||
| Treatment for ILD at hospitalization for SSP | ||||||||
| Anti-inflammatory agents | 0.584 | 0.195, 1.750 | 0.337 | – | ||||
| Anti-fibrotic agents | 0.462 | 0.154, 1.382 | 0.167 | – | ||||
SSP, secondary spontaneous pneumothorax; HR, hazard ratio; CI, confidence interval; BMI, body mass index; KL-6, Krebs von den Lungen-6; HOT, home oxygen therapy; IPF, idiopathic pulmonary fibrosis; CTD-ILD: connective tissue disease-interstitial lung disease; HRCT, high-resolution computed tomography; UIP, usual interstitial pneumonia; CPFE, combined pulmonary fibrosis with emphysema; FVC: forced vital capacity; DLCO, diffusing capacity of the lung for carbon monoxide; PF-ILD: progressive fibrosing interstitial lung disease; EWS, endobronchial Watanabe spigot.
Figure 2.
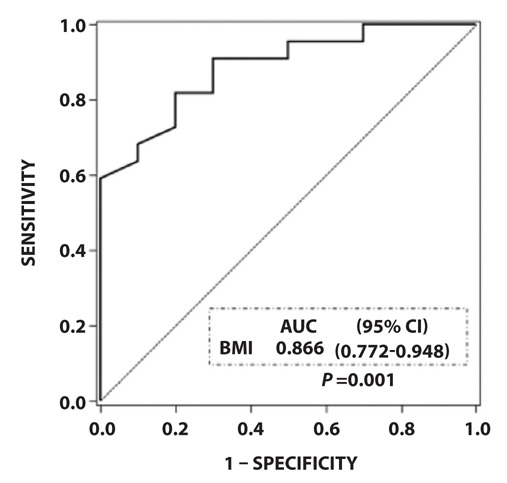
Reciever-operating characterictic curve analysis to determine the optimal cutoff value for BMI that represents increased poor prognosis in the ILD patients hospitalized for SSP. A BMI level of ≤17.8 kg/m2 was shown to reliably predict poor prognosis. AUC, area under the curve; BMI, body mass index; CI, confidence interval; ILD, interstitial lung disease; SSP, secondary spontaneous pneumothorax.
Discussion
We investigated whether PF-ILD could influence the prognosis of patients with ILDs who required hospitalization for SSP. Our findings revealed that older age and lower BMI were significant prognostic factors in these patients. Unexpectedly, however, PF-ILD had no significant impact on prognosis of the patients with ILDs complicated with SSP. The present study resulted in three important findings.
First, both lower BMI and older age were significant predictors of prognosis in the ILD patients hospitalized for SSP. In general, and not limited to ILD, pneumothorax itself occurs significantly more frequently in patients with a lower BMI, and these patients require extended hospitalization and vigilant treatment (13). In IPF, lower BMI was not only a significant predictor of SSP but also of poor prognosis (4). Therefore, the negative impact of SSP can effect some ILD patients with lower BMI, and not just IPF patients.
Second, there was no significant difference in the prognosis of SSP regardless of whether the patient had PF-ILD, even though the patients with PF-ILD accounted for more than half of the population of ILD patients requiring hospitalization for SSP. Iwasawa et al. reported that the initial SSP in IPF occurred at a mean of 9.1 months before death (14). This survival time was similar to that in the present study (mean survival time in the IPF patients was 8.7 months). However, PF-ILD was not a significant factor in the prognosis. Among the patients with CTD-ILD as the IPF type, the mortality rate was significantly higher in the patients with SSP than in those without SSP (3,4). Taken together, SSP itself that requires hospitalization may lead to a poor prognosis for any ILD patient including both non-PF-ILD and PF-ILD patients. The pathophysiology of the progression of ILD is believed to be based on recurrent epithelial cell injury and abnormal wound repair responses, including aberrant fibroblast activity, and although the cause of epithelial injury remains unknown, chronic viral infections, exposure to wood and dust particles, gastroesophageal reflux, micro-aspiration, and drug toxicity might be associated with the progression of ILD (15). Pneumothorax causes a collapsed lung due to injury to the lung and may be trigger of the progression of ILD; therefore, the prognosis of any patient with ILD could be poor after the onset of SSP.
Third, the PF-ILD patients in the present study had lower BMI and %FVC than the non-PF-ILD patients. The association between low %FVC and PF-ILD was obviously based on the definition of PF-ILD. Notably, many patients with PF-ILD had the poor prognostic factor of lower BMI at hospitalization for SSP. A recent study reported that weight loss is a common finding among patients with ILD and appears to be a marker of disease progression in those with any ILD type (16). Although we could not analyze the change of BMI between ILD diagnosis and the onset of SSP, we encounter body weight loss particularly in the ILD patients with progression of fibrosis in clinical practice. Moreover, in the INPULSIS and ASCEND trials, weight loss was reported in 9.7% and 12.6% of participants, respectively, which required careful management when using antifibrotic agents (pirfenidone and nintedanib) (17,18). Therefore, considering that SSP is a poor prognostic factor, we believe that nutrition and rehabilitation therapy to prevent weight loss and SSP are very important in the management of ILD patients. The present study showed that an optimal cutoff level for BMI of ≤17.8 kg/m2 may be a useful marker in daily clinical practice to manage the body weight of each ILD patient.
Our study has several limitations. First, it is a single-center, retrospective study with a modest number of cases. Second, this study restricted patients to those hospitalized, which could potentially cause bias because hospitalization was left to the judgment of each clinician. However, we thought the present study was meaningful because it was based more on real-world clinical practice. Third, the follow-up period and time intervals of the FVC and CT examinations were different for each patient, which may have caused some uncertainty when dividing the patients into the PF-ILD or non-PF-ILD group. For example, half of the non-PF-ILD patients received home oxygen therapy. Therefore, it is possible that some of the non-PF-ILD patients in fact converted to PF-ILD. Fourth, differences in the underlying ILD and comorbidities (e.g., malignancy) might have affected the results of our study indicating that PF-ILD was not a factor in survival following hospitalization for SSP. Fifth, we could not determine whether the clinical diagnosis of ILD impacted treatment decisions and as such, the natural course of the disease. Sixth, the study subjects did not receive uniform therapy because a treatment regimen has not been well established for ILD-associated SSP.
We conclude that despite these limitations, our findings revealed that older age and lower BMI were negative prognostic factors for ILD-associated SSP. In other words, in patients with any type of ILD regardless of whether it is PF-ILD, older age and/or lower BMI can place them at higher risk of developing SSP and of having a poor prognosis after SSP. Thus, clinicians should be aware of the importance of nutrition and rehabilitation therapy in the management of patients with ILD to prevent weight loss and potential SSP.
Acknowledgments:
We sincerely thank Yosuke Sasaki of Satista Co., Ltd. for his advice on statistical analysis. The authors also thank Rise Japan LLC for the professional English language review.
Statements:
All work was performed at the Saitama Red Cross Hospital.
Author contributions:
H.Y. takes responsibility for the completeness of the data presented herein. H.Y., Y.T., S.S. and H.M. performed the primary data analysis. K.K. and H.S. had the idea for the study. S.S., H.S. and H.M. reviewed the chest radiographs and HRCT scans. H.Y. wrote the manuscript. S.S., H.O., G.K., T. Na, T.Ni, R.K., T.O., K.A., M.A., K.K., H.S., T.T. and H.M. reviewed the manuscript. All authors approved the final version of the manuscript.
Conflicts of interest:
Each author declares that he or she has no commercial associations (e.g. consultancies, stock ownership, equity interest, patent/licensing arrangement etc.) that might pose a conflict of interest in connection with the submitted article.
References
- 1.Bobbio A, Dechartres A, Bouam S, et al. Epidemiology of spontaneous pneumothorax: gender-related differences. Thorax. 2015;70:653–658. doi: 10.1136/thoraxjnl-2014-206577. [DOI] [PubMed] [Google Scholar]
- 2.Maskell N. British Thoracic Society Pleural Disease Guideline Group. British Thoracic Society Pleural Disease Guidelines--2010 update. Thorax. 2010;65:667–669. doi: 10.1136/thx.2010.140236. [DOI] [PubMed] [Google Scholar]
- 3.Nishimoto K, Fujisawa T, Yoshimura K, et al. The prognostic significance of pneumothorax in patients with idiopathic pulmonary fibrosis. Respirology. 2018;23:519–525. doi: 10.1111/resp.13219. [DOI] [PubMed] [Google Scholar]
- 4.Nishimoto K, Fujisawa T, Yoshimura K, et al. Pneumothorax in connective tissue disease-associated interstitial lung disease. PLoS One. 2020;15:e0235624. doi: 10.1371/journal.pone.0235624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamakawa H, Kitamura H, Takemura T, et al. Prognostic factors and disease behaviour of pathologically proven fibrotic non-specific interstitial pneumonia. Respirology. 2018;23:1032–1040. doi: 10.1111/resp.13313. [DOI] [PubMed] [Google Scholar]
- 7.Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381:1718–1727. doi: 10.1056/NEJMoa1908681. [DOI] [PubMed] [Google Scholar]
- 8.Tsukahara Y, Yamakawa H, Tsumiyama E, et al. Clinical features and prognosis of secondary pneumothorax in pulmonary emphysema, interstitial pneumonia, and combined pulmonary fibrosis and emphysema. Nihon Kokyuki Gakkai Zasshi. 2020;9:160–165. [Google Scholar]
- 9.Wuyts WA, Wijsenbeek M, Bondue B, et al. Idiopathic pulmonary fibrosis: best practice in monitoring and managing a relentless fibrotic disease. Respiration. 2020;99:73–82. doi: 10.1159/000504763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198:e44–68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 11.Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6:138–153. doi: 10.1016/S2213-2600(17)30433-2. [DOI] [PubMed] [Google Scholar]
- 12.Ryerson CJ, Hartman T, Elicker BM, et al. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest. 2013;144:234–240. doi: 10.1378/chest.12-2403. [DOI] [PubMed] [Google Scholar]
- 13.Huang TW, Cheng YL, Tzao C, et al. Factors related to primary bilateral spontaneous pneumothorax. Thorac Cardiovasc Surg. 2007;55:310–312. doi: 10.1055/s-2007-964902. [DOI] [PubMed] [Google Scholar]
- 14.Iwasawa T, Ogura T, Takahashi H, et al. Pneumothorax and idiopathic pulmonary fibrosis. Jpn J Radiol. 2010;28:672–679. doi: 10.1007/s11604-010-0494-1. [DOI] [PubMed] [Google Scholar]
- 15.Lee JS, Collard HR, Raghu G, et al. Does chronic microaspiration cause idiopathic pulmonary fibrosis? Am J Med. 2010;123:304–311. doi: 10.1016/j.amjmed.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pugashetti J, Graham J, Boctor N, et al. Weight loss as a predictor of mortality in patients with interstitial lung disease. Eur Respir J. 2018;52:1801289. doi: 10.1183/13993003.01289-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King TE, Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 18.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]