

ABSTRACT
The opportunistic pathogen Pseudomonas aeruginosa relies upon type IV pili (Tfp) for host colonization and virulence. Tfp are retractile surface appendages that promote adherence to host tissue and mediate twitching motility, a form of surface-associated translocation. Tfp are composed of a major structural pilin protein (PilA), several less abundant, fiber-associated pilin-like proteins (FimU, PilV, PilW, PilX, and PilE), and a pilus-associated tip adhesin and surface sensor (PilY1). Several proteins critical for Tfp biogenesis and surface sensing are encoded by the fimU-pilVWXY1Y2E operon. Tfp biogenesis is regulated by the global transcription factor Vfr and its allosteric effector, cyclic AMP (cAMP). Our investigation into the basis for reduced Tfp production in cAMP/vfr mutants revealed a defect in the expression of the fimU operon. We found that cAMP/Vfr activation of the fimU operon occurs via direct binding of Vfr to a specific fimU promoter sequence. We also refined the role of the AlgZ/AlgR two-component system in fimU regulation by demonstrating that phosphorylation of the response regulator AlgR is required for maximal binding to the fimU promoter region in vitro. Vfr also regulates expression of the algZR operon, revealing an indirect regulatory loop affecting fimU operon transcription. Overall, these results demonstrate that two linked but independent regulatory systems couple the expression of Tfp biogenesis and surface sensing genes and highlight the regulatory complexity governing expression of P. aeruginosa virulence factors.
KEYWORDS: AlgR, Pseudomonas aeruginosa, Vfr, cAMP, type IV pili
INTRODUCTION
Pseudomonas aeruginosa is an opportunistic pathogen that causes a wide variety of diseases ranging from acute and disseminating tissue infections to debilitating chronic lung infections in individuals with muco-obstructive lung disease (1–3). Acute P. aeruginosa infections are characterized by an intense neutrophil-dominated inflammatory response, extensive tissue damage, and sepsis. In contrast, chronic infections of the cystic fibrosis (CF) lung are noninvasive and cause progressive damage and respiratory decline over time. P. aeruginosa virulence relies on the expression of many surface-exposed and secreted factors (4, 5), and individual virulence factors are largely associated with either the acute or chronic mode of infection (6). Acute infection is partially mediated by direct injection of exotoxins into host cells by a type III secretion system (T3SS) and a variety of toxins, degradative proteases, and lipases secreted by a type II secretion system (T2SS) (7). Acute virulence is also mediated by type IV pili (Tfp), which are filamentous surface appendages that promote interactions with host cells (8). Chronic infection is typified by a biofilm-like mode of growth, where bacteria exist as microcolonies encased in one of several exopolysaccharides (9). Chronic CF isolates commonly exhibit a mucoid colony phenotype, resulting from overproduction of the exopolysaccharide alginate (10).
Despite the association of specific P. aeruginosa factors with either acute or chronic infection lifestyles, Tfp have the potential to contribute to both infection modes. In addition to host cell attachment (8), Tfp also mediate a form of surface-associated motility (twitching motility) that promotes bacterial dissemination within infected tissue (11). Tfp are also important for the initiation of biofilm formation (12) and play a role in maintaining biofilm structure (13). The various functions of Tfp fibers are mediated by rapid, repeated cycles of extension and retraction (14). The Tfp fiber is primarily composed of pilin (or PilA) subunits, which are produced as prepilins containing a short amino-terminal leader sequence that is cleaved prior to assembly (15). Assembly and disassembly of Tfp occur in an energy-dependent manner at the inner membrane via the ATPases PilB and PilT, which respectively facilitate extension and retraction of the pilus fiber through an outer membrane pore (16). In addition to pilin, several less abundant minor pilins (FimU, PilV, PilW, PilX, and PilE), are required for pilus assembly, function, and surface stability (17–22). The minor pilins are present in the mature pilus fiber (23) and believed to form a complex at regular intervals along the length of the fiber or at the fiber tip. PilY1 is a multifunctional pilus-associated protein that antagonizes pilus retraction (22) and acts as an adhesin for abiotic surfaces and integrins located at the basolateral membrane of human epithelial cells (8, 24, 25). PilY1 also functions as a mechanosensor, linking attachment with surface-associated virulence behaviors and fiber retraction (25–28). Upon contact with a surface, PilY1 regulates virulence by modulating the production of alkyl quinolones through regulation of the AlgZR two-component system (29, 30). In addition, PilY1 sensing is associated with repression of swarming motility and implicated in other surface-associated behaviors, such as biofilm formation, through the regulation of cyclic di-GMP (26, 31, 32).
Multiple P. aeruginosa signal transduction pathways participate in the reciprocal regulation of virulence factors associated with acute and chronic infection (6). The AlgZR two-component system (TCS) regulates multiple virulence systems. AlgZ (also referred to as FimS) and AlgR are both required for twitching motility and Tfp production, suggesting that AlgR phosphorylation by AlgZ is required for activity (33–35). AlgR controls transcription of the fimU-pilVWXY1Y2E operon (fimU) (17–20, 35). Proteins encoded within the fimU operon negatively regulate AlgR activity through a poorly understood mechanism that appears to involve surface sensing by PilY1 (27, 28, 30, 32). AlgR also mediates the conversion to mucoidy (alginate overproduction) that occurs during chronic infection in the CF lung (36–39).
The global transcriptional regulator Vfr coordinately controls the expression of over 200 genes, many of which are involved in P. aeruginosa virulence (40–42). Vfr activity is regulated by the second messenger cyclic AMP (cAMP), which is synthesized by two adenylate cyclases (CyaA and CyaB) (42–45). Vfr was identified in a screen for twitching motility-defective mutants (43), and Tfp production requires both Vfr and cAMP (41, 46). Furthermore, spontaneous mucA mutation, a common adaptation found in chronic-CF isolates, results in downregulation of invasive virulence factors via inhibition of vfr expression, an effect that involves both AlgU and AlgR (41). Thus, when overexpressed due to mucA mutation, AlgR not only mediates the switch to a chronic infection phenotype via activation of alginate production but also represses acute virulence factors through inhibition of cAMP/Vfr signaling.
Although cAMP/Vfr signaling is implicated in Tfp biogenesis and function, the exact mechanism of this regulation is unknown. In this study, we sought to identify the specific defect in cAMP/Vfr signaling mutants that accounts for loss of Tfp biogenesis and determine how the cAMP/Vfr system is integrated within the complex Tfp biogenesis regulatory network.
RESULTS
Vfr/cAMP and AlgZR control surface abundance of Tfp.
Although both Vfr and cAMP are required for Tfp production and function, a mechanism for this requirement has not been determined (41, 43, 46). To characterize the Tfp defect in strains lacking Vfr or cAMP production, we evaluated the abundance and morphology of Tfp surface fibers by transmission electron microscopy (TEM). TEM images of the wild-type strain show both flagella (thick fibers) and abundant Tfp (thin fibers and fiber bundles) extending from the bacterial surface (Fig. 1A). In contrast, the vfr mutant and a cyaA cyaB (cyaAB) double mutant, which lacks the two adenylate cyclases necessary for cAMP synthesis, showed a substantial reduction in surface Tfp. The length, diameter (relative to flagella), and morphology of surface Tfp produced by the vfr and cyaAB mutants were indistinguishable from those of Tfp produced by the wild-type strain. Both mutants displayed approximately 1 to 2 Tfp fibers per cell, as assessed by evaluating bacteria in multiple TEM images (data not shown). Despite the dramatic reduction in Tfp, both the vfr and cyaAB mutants could be distinguished from a nonpiliated pilA mutant lacking the major pilus structural subunit (47).
FIG 1.

P. aeruginosa adenylate cyclases and Vfr are required for Tfp production. (A) Visualization of Tfp (thin fibers and fiber bundles) produced by wild-type (wt) and mutant P. aeruginosa strains by TEM. Thin Tfp fibers and fiber bundles are indicated by arrowheads and an asterisk, respectively. Flagella are indicated by arrows. Bar = 250 nm. (B) Tfp abundance on wild-type and mutant P. aeruginosa strains by IF microscopy. P. aeruginosa cells (green) were identified with Hoechst stain, and Tfp (red) were detected with pilin-specific antiserum and Alexa Red-conjugated goat anti-rabbit IgG (Molecular Probes). Representative IF micrographs are shown at a magnification of ×100. (C) (Top) Coomassie blue-stained SDS-polyacrylamide gel showing surface pilin from purified pilus fractions isolated from the wild-type (WT) and indicated mutant strains. (Middle and bottom) Immunoblots of whole-cell lysates (WC) probed with pilin-specific monoclonal antibody or PilW-specific antiserum. An uncropped blot showing equal protein loading is shown in Fig. S1A. (D) Activity of the fimU promoter as measured by β-galactosidase assay in the indicated strains harboring a chromosomal PfimU-lacZ transcriptional reporter. Values are means and standard errors of the means (SEM) (n ≥ 4). When compared pairwise, values for all mutants were significantly different (*, P < 0.05; ****, P < 0.0001) from those for the wild-type strain as determined by one-way analysis of variance (ANOVA) with multiple comparisons using Bonferroni’s correction.
Uncropped α-PilW Western blots. α-PilW blots for Fig. 1C (A), Fig. 5B and C (B), and Fig. 9B (C). Blots for PilW and PilA were run on the same samples, which were generated as described in Materials and Method. Protein was normalized based on total protein levels as determined by a Bradford assay. Download FIG S1, TIF file, 0.5 MB (501.5KB, tif) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To confirm the TEM results, wild-type and mutant P. aeruginosa strains were labeled with pilin-specific antibodies and imaged by immunofluorescence (IF) microscopy (Fig. 1B). This technique allows the direct visualization of Tfp abundance on the surface of live bacteria (8, 48). When examined by IF, the wild-type strain displayed an extensive network of Tfp fibers, and the pilA mutant lacked any appreciable Tfp staining. The cyaAB and vfr mutants both displayed considerably less Tfp staining than the wild type, indicating that the reduction in Tfp seen by TEM was not due to Tfp shedding but rather reflects a reduction in Tfp expression and/or elaboration on the surface.
As pilin is the major structural component of Tfp, we also evaluated the relative amounts of pilin recovered in sheared pilus preparations, which correlates with surface Tfp abundance. As previously demonstrated (41, 43, 46), the vfr and cyaAB mutants displayed dramatically reduced levels of surface pilin compared to the wild-type strain (Fig. 1C, top). With the exception of the pilA mutant, similar levels of pilin were detected in whole-cell lysates of all strains by immunoblotting (Fig. 1C, middle), confirming that the reduction in surface Tfp seen in the vfr and cyaAB mutants is due to a defect in fiber assembly rather than a consequence of reduced pilin synthesis.
The AlgZR two-component system controls Tfp production by directly regulating the fimU operon (35, 49). To determine whether the AlgZR and cAMP/Vfr pathways play a similar regulatory role in Tfp production, we assessed the relative abundance of surface and total cellular pilin in mutants with disruption of either algZ and algR individually or in combination (algZR). All three mutants lacked detectable surface Tfp (Fig. 1C, top) but retained the capacity to synthesize pilin (Fig. 1C, middle). Next, we assessed the impact of vfr, cyaAB, algZ, and algR mutations on fimU operon expression. As a readout for fimU operon expression, we initially evaluated the level of PilW, a protein encoded within the fimU operon. Each of the mutants produced substantially less PilW than the wild-type strain, as determined by immunoblotting of whole-cell lysates (Fig. 1C, bottom). Raw immunoblotting data, demonstrating equal sample loading, are available in Fig. S1 in the supplemental material.
As an alternative readout for the presence of functional pilin, we performed a phage susceptibility assay using the pilus-specific lytic phage PO4 (50). PO4 infection requires both surface-accessible pili and pilus retraction for attachment and entry, respectively (16). Despite possessing dramatically reduced surface pili, the vfr, cyaAB, and algZR mutants were indistinguishable from the wild type and exhibited complete sensitivity to PO4 (Fig. S2). In contrast, an isogenic pilA mutant was resistant to lysis even at the highest phage concentration. This result suggests that, despite the lack of appreciable surface fibers, the vfr, cyaAB, and algZR mutants can assemble pilus structures that are sufficient to support phage attachment and entry. Given that proteins encoded in the fimU operon are required for pilus stability (22), we hypothesize that the regulatory mutants may produce dysfunctional pili that can extend only to the bacterial cell surface or outer membrane prior to retraction, such that they can support phage attachment and entry but do not form conspicuous surface fibers. Regardless of the specific defect, these findings support the notion that cAMP/Vfr and AlgZR both control a functionally related aspect of pilus biogenesis.
vfr, cyaAB, and algZR mutants exhibit sensitivity to the pilin-specific phage PO4. Phage susceptibility was determined using a double-agar layer as described in Materials and Methods. Phage was serially diluted from 108 PFU/mL to 100 PFU/mL. Ten microliters of each phage dilution, as well as broth control, was spotted onto double agar containing the indicated strains and incubated overnight before imaging. Phage concentrations above 105 PFU/mL also resulted in complete lysis. The assay was performed in duplicate. Download FIG S2, TIF file, 0.9 MB (891.8KB, tif) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To determine if altered transcription of the fimU operon accounts for reduced PilW in the cAMP/Vfr and algZR mutants, we constructed a transcriptional reporter by fusing the putative fimU promoter region (PfimU) to the β-galactosidase-encoding lacZ gene and introduced the construct onto the chromosomes of wild-type and mutant strains. Each of the mutants showed a significant reduction in PfimU-lacZ reporter activity compared to that of the wild-type strain (Fig. 1D). Wild-type levels of PfimU-lacZ reporter activity were restored in each of the mutants by plasmid-based expression of the corresponding genes in trans (Fig. S3). These results demonstrate that cAMP/Vfr and AlgZR are required for optimal expression of the fimU operon and confirm previous reports (42, 43).
Restoration of PfimU-lacZ reporter activity in mutants. The wild-type (WT) or indicated mutant strains containing either vector, pPa-cyaB, pPa-vfr, pPa-algZ, pPa-algR, or pPa-algZR expression plasmids were grown in LB broth containing 30 μg/mL carbenicillin (Cb) and 50 μM IPTG. Values are means and SEM (n ≥ 3). Download FIG S3, TIF file, 0.3 MB (286KB, tif) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
AlgR affinity for the fimU promoter is dramatically enhanced by the phosphodonor phosphoamidate.
The fimU promoter region contains two putative AlgR binding sites (designated ABS1 and ABS2) (Fig. 2A) (35). Our finding that both algZ and algR are required for activation of the fimU promoter (Fig. 1D) suggested that AlgZ-dependent phosphorylation of AlgR is required for PfimU activation. This hypothesis is consistent with the finding that an algZ mutant is defective for Tfp-dependent function (34). Furthermore, an AlgR mutant lacking the phospho-accepting aspartate residue (AlgRD54N) is defective for Tfp production and unable to bind the fimU promoter in vitro (34, 35). Despite these findings, direct evidence that phosphorylation increases AlgR DNA binding activity is lacking. To confirm AlgR binding to the fimU promoter and to assess the impact of phosphorylation, we purified full-length AlgR with an amino-terminal histidine fusion tag and assessed binding to a PfimU promoter probe in the presence and absence of small-molecule phosphodonor compounds. In the absence of a phosphodonor, binding to the PfimU probe was observed only when high concentrations of AlgR (351 nM) were used, and less than 50% of the probe was shifted (Fig. 2B). AlgR bound to a nonspecific probe devoid of identifiable AlgR binding sites with similar kinetics, suggesting that binding to PfimU in the absence of AlgR phosphorylation is not biologically relevant (Fig. 2B). In contrast, addition of the phosphodonor phosphoamidate (PAM) significantly enhanced AlgR binding to the PfimU promoter probe (Fig. 2C). We also observed an increase in the intensity of the nonspecific probe shift; however, specific binding was enhanced to a greater degree. Although the exact increase in affinity could not be calculated due to the poor binding activity of unphosphorylated AlgR, we estimate that affinity of AlgR for the PfimU promoter probe increased by at least 1 order of magnitude in the presence of PAM. These results confirm that AlgR binds the fimU promoter and that phosphorylation is required for specific high-affinity binding and support the hypothesis that AlgZ functions as the cognate sensor kinase responsible for AlgR phosphorylation. Although Belete et al. (35) reported two shifted products, our results show a single shift product (Fig. 2C). While two putative AlgR binding sites have been identified (35), our results suggest that only a single site is occupied or that AlgR binding is cooperative such that both sites become occupied simultaneously. Further studies are required to determine the individual contributions of the two putative AlgR binding sites.
FIG 2.

AlgR requires phosphorylation for fimU promoter recognition. (A) Diagram of the fimU promoter region. Numbering (in base pairs) is relative to the fimU translational start site; the ATG is indicated in bold type. The partial coding sequence of fimU is highlighted in gray. Locations of oligonucleotides used to generate the PfimU reporter (5′PfimU rep. and 3′PfimU rep.) and PfimU EMSA probes (5′EMSA probe and 3′ EMSA probe) are indicated. The boxed region represents the sequence protected by cAMP-Vfr in DNase I footprinting assays (Fig. S4) with the identified VBS (centered at −130 and −115 bp) is indicated in bold and underlined. The previously identified AlgR binding sites (ABS1 and ABS2) are indicated. (B and C) Specific (PfimU, lanes 1 and 3 to 8) and nonspecific (Non-Sp, lane 2) probes (0.25 nM) were incubated in the absence (lane 1) or presence (lanes 2 to 8) of various concentrations of AlgR (11 to 351 nM) for 15 min, followed by electrophoresis and phosphorimaging. Reactions were performed in the absence (B) or presence (C) of the small molecule phosphodonor PAM (50 mM). The nonspecific probe shift is indicated by an asterisk.
Vfr protected region of the fimU operon. DNase I footprinting of the fimU promoter region. Samples contained a DNA fragment (0.4 nM) corresponding to ∼−300 bp relative to the translational start site. The top strand of the DNA probe was radiolabeled on one end and was incubated in the absence or presence of cAMP-Vfr (114, 13, or 1 nM) prior to DNase I treatment. DNase I-generated fragments were separated by electrophoresis, and Maxam-Gilbert sequencing ladders were made using the same DNA. Sequence corresponding region protected by Vfr binding is bracketed, with the predicted half-sites underlined and in bold. Download FIG S4, TIF file, 0.6 MB (604KB, tif) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Vfr directly controls algZ expression.
Based on the evidence that AlgZR positively regulates the fimU operon and that algZ expression was reduced in a vfr mutant (42), we hypothesized that decreased algZR expression in the cAMP and vfr mutants could account for the defect in Tfp production observed. AlgZR are coexpressed from two promoters (designated ZT1 and ZT2). Expression of the proximal ZT1 promoter (Fig. 3A) is entirely Vfr dependent, whereas the far-upstream ZT2 promoter is Vfr independent (51). To examine the role of the Vfr-dependent algZ promoter, we engineered an algZ transcriptional reporter (PalgZ-lacZ) by fusing the putative Vfr-dependent ZT1 promoter to lacZ. PalgZ-lacZ reporter activity was significantly reduced in the vfr and cyaAB mutants, and the defect was complemented by plasmid-based expression of vfr (pPa-vfr) or cyaB (pPa-cyaB), respectively (Fig. 3B).
FIG 3.

Vfr is required for algZ promoter expression. (A) Diagram of the algZ promoter region. Numbering (in base pairs) is relative to the algZ translational start site. Also depicted is the divergently transcribed gene argH. The translational start codons are indicated in bold type. Partial coding sequences of algZ and argH are highlighted in gray. The proposed Vfr-binding sites VBS1 (centered between −166 and −151 bp), VBS2 (centered between -109 and −94 bp), and VBS3 (centered between −89 and −74) are indicated in bold and underlined. Locations of the oligonucleotides (5′PalgZ rep. and 3′PalgZ rep.) used to generate the PalgZ-lacZ reporter fragment and PalgZ EMSA probe used for the lower panel are indicated. The Vfr dependent PZT1 is located 23 bp upstream of algZ, whereas the Vfr-independent PZT2 (not shown) is located 1,044 bp upstream of algZ. (B) Activity of the algZ promoter as measured by β-galactosidase assay in the indicated strains harboring a chromosomal PalgZ-lacZ transcriptional reporter. The wild-type (WT) or indicated mutant strains contained vector, pPa-cyaB, or pPa-vfr expression plasmids. Strains containing expression vectors were grown in LB broth containing 30 μg/mL carbenicillin (Cb) and 50 μM IPTG. Values are means and SEM (n ≥ 3). Values for the cyaAB and vfr mutants containing empty vector were significantly different from that of WT (P ≤ 0.0001), as determined by one-way ANOVA with multiple comparisons using Bonferroni’s correction. (C) Specific probe (lanes 1 and 3 to 8) and nonspecific probe (lane 2) (0.25 nM) were incubated in the absence (lane 1) or presence (lanes 2 to 8) of various concentrations of cAMP-Vfr (9 to 271 nM) for 15 min followed by electrophoresis and phosphorimaging. Bands representing shifting of the nonspecific probe (∼200 bp) are indicated by an asterisk. Arrows indicate the two cAMP-Vfr-dependent shift complexes (Shift 1 and Shift 2). (D) Apo-Vfr (271 nM) incubated with specific (PalgZ, lanes 1 and 3 to 8) and nonspecific (Non-Sp, lane 2) probes (0.25 nM) in the absence (lane 2) or presence (lanes 3 to 9) of increasing concentrations of cAMP (8 to 500 nM) for 15 min followed by electrophoresis and phosphorimaging.
Kanack et al. previously demonstrated that Vfr bound a DNA probe encompassing the algZR promoter region by electrophoretic mobility shift assay (EMSA) (52). Based on visual inspection, the authors identified a putative Vfr-binding site (5′-TTTTTTGCGGC:AGATGGCATTT [boldface indicates the conserved half-sites to which Vfr is predicted to bind, and a colon represents the axis of symmetry within the binding site]), although direct binding to this site was not confirmed (52). Based on the Vfr-binding consensus sequence (VCS, 5′-ANWWTGNGAWNY:AGWTCACAT), we identified two additional candidate Vfr-binding sites, 5′-GAACGAGCGCT:GTTTCGCGGTT and 5′-AAGTGCGACGA:TGATAGCAGGC in the algZ promoter region (44). We designated these putative binding sites VBS1, VBS2, and VBS3 based on their arrangement within the algZ promoter region (Fig. 3A). We first confirmed that purified Vfr binds to a PalgZ promoter probe encompassing all three putative Vfr binding sites by EMSA. Two shift products were produced with increasing concentrations of cAMP-saturated Vfr, consistent with Vfr binding to at least two distinct sites (Fig. 3C, lane 8). The alternative possibility that the second shift is due to Vfr oligomerization at a single site is addressed below (Fig. 4). Vfr binding to an unrelated DNA probe was observed only at the highest Vfr concentrations tested, demonstrating that binding to the algZ promoter probe is highly specific (Fig. 3C, lanes 7 and 8). To determine the role of cAMP in Vfr binding, we generated cAMP-free Vfr (apo-Vfr) as previously described (44). Apo-Vfr did not shift the PalgZ probe (Fig. 3D, lane 2), but the two shift products were restored by the addition of cAMP to the binding reactions (Fig. 3D, lanes 3 to 9), demonstrating that Vfr requires cAMP for binding to the PalgZ probe.
FIG 4.

VBS 2 and 3 are required for full activity of Vfr-dependent algZ promoter expression. (A) Putative Vfr binding sites (VBS). Mutations in different VBS were generated by site-directed mutagenesis. (B) Promoter activity in strains containing either the wild-type or mutant PalgZ-lacZ reporter as measured by β-galactosidase assay. Values are the means and SEM (n ≥ 3). Values for VBS2 and VBS3 mutants were significantly different when compared pairwise to that of the wild-type strain (**, P < 0.01; ****, P < 0.0001). Combinatorial VBS1-2 and VBS2-3 mutations produced additive reporter activity losses compared to individual VBS2 (#, P < 0.05) and VBS3 (&, P < 0.0001) mutations, respectively. Statistical analysis was performed using one-way ANOVA with multiple comparisons using Bonferroni’s correction. (C) EMSA analysis of PalgZ probes containing individual or combinatorial mutations of putative Vfr binding sites using the EMSA probes indicated in Fig. 3A. A double shift of the wild-type promoter probe is significantly abrogated in a triple VBS1-2-3 PalgZ probe. (D) Promoter activity in strains containing either the wild-type or mutant PalgZ-VBS1-2-3 reporter as measured by β-galactosidase assay. Reporter constructs were in either a wild-type (WT) or vfr mutant strain background. ****, P < 0.0001 compared to WT + vector; #, P < 0.0001 compared to vfr + pPa-vfr. Statistical analysis was performed using one-way ANOVA with multiple comparisons using Bonferroni’s correction.
To determine which of the putative binding sites contribute to algZ transcription and Vfr binding, we disrupted the sites individually and in combination (Fig. 4A). While PalgZ-lacZ reporter activity in the VBS1 mutant was identical to wild-type, mutations in VBS2 and VBS3 significantly lowered activity (P < 0.01 and P < 0.0001, respectively) (Fig. 4B). In contrast to the reporter assays, mutation of the VBS1 site resulted in loss of the second shift product, while probes carrying mutations in VBS2 or VBS3 retained the capacity to generate two Vfr-dependent shift products (Fig. 4C). Combinatorial VBS1-2 and VBS2-3 mutations produced small yet additive losses in PalgZ-lacZ reporter activity compared to the VBS2 and VBS3 mutations, respectively (Fig. 4B). Reporter activity of the VBS13 mutant was indistinguishable from that of the single VBS3 mutant. All three double-mutant probes retained the capacity to generate a single Vfr-dependent shift product (Fig. 4C). While PalgZ-lacZ activity was identical in VBS2-3 and VBS1-2-3 mutants (Fig. 4B), all three mutations were required to eliminate Vfr binding by EMSA (Fig. 4C). Activity of the wild-type PalgZ-lacZ reporter in a vfr mutant was indistinguishable from that of a wild-type strain carrying the VBS1-2-3 mutant PalgZ-lacZ reporter (Fig. 4D). Furthermore, deletion of vfr in the VBS1-2-3 mutant PalgZ-lacZ reporter strain caused no further loss in reporter activity and activity could not be restored by plasmid based vfr expression (pPa-vfr) (Fig. 4D).
Based on the shift patterns, it appears that Vfr binding to VBS2 and that to VBS3 are mutually exclusive (i.e., binding to one excludes binding to the other), with VBS2 being preferentially occupied when both intact sites are available (Fig. 4C, lanes 2 and 3 versus 4 and 5). Binding to VBS2 or VBS3 was noncompetitive with VBS1. Surprisingly, mutation of VBS3 results in a shift pattern most similar to that of the wild-type probe, but mutation of VBS3 showed the greatest reduction of PalgZ reporter activity in vivo (Fig. 4B and C). In general, the EMSA results suggest that all three binding sites are functional; however, the in vitro binding patterns are not consistent with promoter activity, indicating that additional factors may contribute to promoter activity in vivo. Taken together, these results indicate that all three candidate sites support cAMP-dependent Vfr binding within the algZ promoter region and that VBS3 is the primary site involved in Vfr regulation of PalgZ promoter activity. While VBS1 appears to have little or no influence on algZ promoter activity, full activity requires both VBS2 and VBS3.
Both Vfr and AlgZR are required for expression of the fimU operon.
Based on the findings above, we hypothesized that cAMP and Vfr may indirectly regulate expression of the fimU operon by controlling algZ expression. This was tested by conducting a series of epistasis experiments. Plasmid-based expression of algZR (pPa-algZR) was sufficient to restore PfimU-lacZ reporter activity to wild-type levels in an algZR mutant background but not in an isogenic vfr algZR triple mutant (Fig. 5A). Similarly, plasmid-expressed vfr restored PfimU-lacZ activity in a vfr mutant but not in the vfr algZR mutant (Fig. 5A). Consistent with these results, plasmid-based expression of either vfr or algZR in the vfr algZR mutant failed to restore PilW production (Fig. 5C) or Tfp production, as measured by the recovery of pilin in sheared surface fractions (Fig. 5B, top), despite similar levels of pilin in whole-cell lysates (Fig. 5B, bottom). These results indicate that the cAMP/Vfr and AlgZR systems do not act in a strictly linear pathway to control fimU operon expression. Overall, these results suggest that cAMP/Vfr and AlgZR are both required for maximal control of fimU promoter activity.
FIG 5.

Both AlgR and cAMP/Vfr are required for fimU transcription and Tfp production. (A) The wild-type (WT) or indicated mutant strains containing either vector, pPa-vfr, or pPa-algZR expression plasmids were grown as previously described. Values are means and SEM (n ≥ 3). The value for the algZR/vfr mutant containing any expression plasmid was statistically different from that of the WT strain (****, P ≤ 0.0001) as determined by one-way ANOVA with multiple comparisons using Bonferroni’s correction. (B) Coomassie blue-stained SDS-PAGE gel showing pilin from purified pilus fractions isolated from the WT and indicated mutant strains (top) and immunoblot of whole-cell lysates probed with α-PilA antibody (bottom). (C) Immunoblot of whole-cell lysates probed with PilW-specific antiserum. An uncropped blot showing equal protein loading is shown in Fig. S1B.
cAMP/Vfr directly regulates transcription of the fimU operon.
As expression of the fimU operon was dependent on both AlgZR and Vfr, we hypothesized that Vfr may also have a direct role in regulating transcription of the fimU operon. To test this hypothesis, we assessed Vfr binding to the fimU promoter region (Fig. 6A) by EMSA. Shifting of the PfimU probe was observed with increasing concentrations of cAMP-saturated Vfr (Fig. 6A, lanes 3 to 8). In contrast, apo-Vfr did not shift the PfimU probe (Fig. 6B, lane 2), but shifting was restored by the addition of cAMP to the binding reaction mixture (Fig. 6B, lanes 3 to 9), thus demonstrating that Vfr requires cAMP for binding to the PfimU probe.
FIG 6.
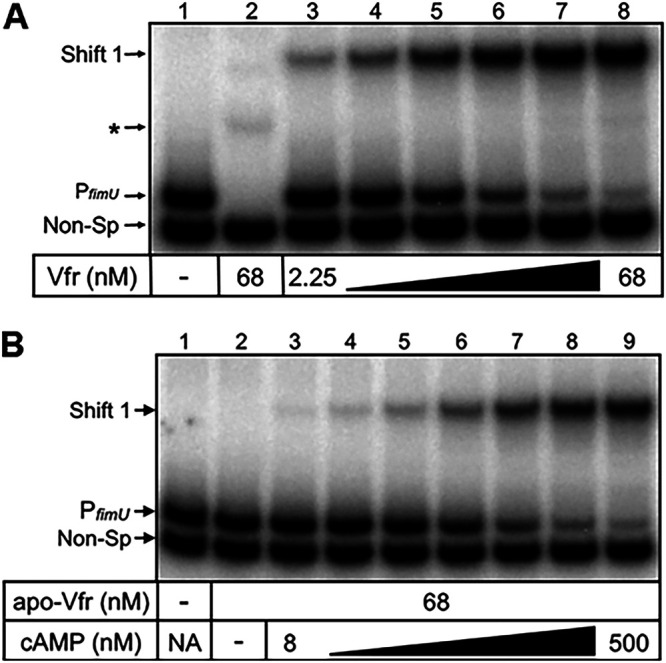
cAMP is required for optimal Vfr binding to PfimU. (A) Specific (PfimU, lanes 1 and 3 to 8) and nonspecific (Non-Sp, lane 2) probes (0.25 nM) were incubated in the absence (lane 1) or presence (lanes 2 to 8) of various concentrations of cAMP-Vfr (2.25 to 68 nM) for 15 min followed by electrophoresis and phosphorimaging. The nonspecific probe shift is indicated by an asterisk. (B) Apo-Vfr (68 nM) incubated with specific (PfimU) and nonspecific (Non-Sp) probes (0.25 nM) in the absence (lane 2) or presence (lanes 3 to 9) of increasing concentrations of cAMP for 15 min followed by electrophoresis and phosphorimaging.
To identify the Vfr binding site within the fimU promoter region, we performed DNase I footprinting. cAMP-saturated Vfr altered the DNase I cleavage pattern of a 41-bp sequence within the PfimU promoter probe (Fig. S4), corresponding to nucleotides −142 to −102 relative to the fimU translational start codon. Enhanced cleavage sites were detected at nucleotides −127 and −117 (Fig. S4). The spacing of DNase I hypersensitivity sites within PfimU is consistent with the previously reported Vfr footprints of the PfleQ, PlasR, PtoxR, PregA, PcpdA, and Pvfr promoters (44, 52–56).
To confirm the Vfr binding site within the fimU promoter, we performed site-directed mutagenesis to alter the predicted half-sites (Fig. 7A). Initially, we mutated the half-sites in the PfimU transcriptional reporter to more closely mirror the Vfr consensus half-sites (TGAGC→TGAGA) and (GCACT→TCACT). The resulting reporter (PfimU M1-lacZ) resulted in a slight but significant increase in reporter activity relative to the wild-type PfimU-lacZ reporter (Fig. 7B). Mutating the PfimU Vfr half-sites such that they were more divergent from the consensus half-sites (TGAGC→TCAGC and GCACT→GGACT) in the PfimU M2-lacZ reporter (Fig. 7A) resulted in a significant decrease in reporter activity compared to PfimU-lacZ (Fig. 7B). The PfimU M2 mutation had the same effect on promoter activity as a vfr mutant (Fig. 7B).
FIG 7.

Mutation of the putative Vfr binding site influences fimU transcription. (A) Site-directed mutagenesis of the putative Vfr binding half-sites within the fimU promoter region. The base changes used to create two mutated versions of the fimU promoter (fimUM1 and fimUM2) are indicated in bold type. PfimU M1 contains mutations to make the sequence more closely resemble the VCS reported by Fuchs et al. (44). PfimU M2 contains mutations predicted to disrupt Vfr binding. (B) Promoter activity in strains containing either the wild-type fimU promoter reporter (PfimU) or one of the two mutated fimU promoter reporters (PfimUM1 or PfimUM2), as measured by β-galactosidase assay. Reporter constructs were in either a wild-type (WT) or vfr mutant strain background. Values are means and SEM) (n ≥ 3). Values for all of the mutants were significantly different (P ≤ 0.0001) when compared pairwise to that of the wild-type strain using one-way ANOVA with multiple comparisons using Bonferroni’s correction. (C and D) EMSA analysis of PfimU probes containing mutations indicated in Fig. 7A. The probes PfimUM1 (C) and PfimUM2 (D) were incubated in the absence (lane 1) or presence (lanes 2 to 8) of various concentrations of cAMP-Vfr (2.25 to 68 nM) for 15 min followed by electrophoresis and phosphorimaging. The nonspecific probe shift is indicated by an asterisk.
To determine whether mutations in the predicted Vfr binding site of PfimU altered DNA recognition by Vfr, we generated DNA probes corresponding to PfimU M1 and PfimU M2 (Fig. 7A) and assessed Vfr binding by EMSA. As expected, cAMP-saturated Vfr shifted the PfimU M1 probe with no apparent change in binding affinity compared to the wild-type probe (Fig. 6A and 7C). In contrast, cAMP-Vfr failed to bind PfimU M2 (Fig. 7D). Taken together, these results identify the Vfr binding site within the fimU promoter and support the finding that cAMP-bound Vfr binds directly to the fimU promoter region to activate transcription.
Vfr and AlgR simultaneously bind the fimU promoter.
AlgR and Vfr are both necessary for activation of the fimU promoter (Fig. 5) and capable of independently binding to the fimU promoter region (Fig. 2 and 6). To determine whether AlgR and Vfr simultaneously bind the fimU promoter and whether binding is cooperative, we assessed the binding kinetics by EMSA. The PfimU probe was incubated with a saturating concentration of cAMP-saturated Vfr and increasing concentrations of AlgR in the presence of PAM (Fig. 8A). Addition of cAMP-saturated Vfr resulted in the expected PfimU shift product (Fig. 8A, lane 3). The further addition of AlgR resulted in the formation of a second discrete concentration-dependent supershift product (Fig. 8A, lanes 4 to 9), indicating that both proteins simultaneously bound the PfimU probe. The concentration of AlgR required to shift the PfimU probe in the presence of Vfr (supershift; Fig. 8A) was similar to the concentration required to shift the probe in the absence of Vfr (Fig. 2C), indicating that Vfr did not significantly influence AlgR binding.
FIG 8.

AlgR and Vfr bind to the PfimU WT probe. (A) PfimU WT probe incubated in the absence (lane 1) or presence (lanes 2 to 9) of AlgR and Vfr. Lanes 2 and 3 show the concentrations of AlgR and Vfr, respectively resulting in ∼90% shifting of the PfimU WT probe. Lanes 4 to 9 depict incubation of the PfimU WT probe and Vfr (68 nM) with increasing concentrations of AlgR (11 to 351 nM). Reactions were conducted in the presence of 50 mM PAM. (B) PfimU WT probe incubated as for panel A, except that the AlgR concentration was held constant (176 nM) while cAMP-saturated Vfr was added at increasing concentrations (2.25 to 68 nM). Shifted complexes consisting of fimU with each of the individual proteins (Vfr shift and AlgR shift) or both proteins (AlgR/Vfr shift) are indicated. Reactions were conducted in the presence of 50 mM PAM.
To determine the impact of AlgR binding on Vfr recruitment, the PfimU probe was incubated with a saturating amount of AlgR (in the presence of PAM) and increasing concentrations of cAMP-saturated Vfr (Fig. 8B). AlgR formed the expected shift product (Fig. 8B, lane 3), and addition of Vfr resulted in formation of a second discrete shift product. Overall, these results demonstrate that both AlgR and Vfr can simultaneously bind the fimU promoter, and binding of either protein appears to be largely independent of the other. However, simultaneous recognition of the fimU promoter by AlgR and Vfr has a positive and combined effect on fimU transcription (Fig. 5A).
fimU operon complementation does not restore the Tfp defect of vfr mutants.
To determine whether the Tfp defect of algZR and vfr mutants is attributed solely to the lack of fimU transcription, we complemented the algZR and vfr mutants with plasmid-based expression of the entire fimU operon (pPa-fimU-pilE). When expressed under conditions that restored Tfp production in a fimU-pilE mutant (Fig. 9A, lane 3), pPa-fimU-pilE was sufficient to restore Tfp production in the algZR mutant (Fig. 9A, lane 9) but not in the vfr mutant (Fig. 9A, lane 6). As a further control, we confirmed that plasmid-based expression of vfr (pPa-vfr) and algZR (pPa-algZR) was sufficient to restore Tfp production in the vfr and algZR mutants, respectively (Fig. 9A, lanes 5 and 8). pPa-fimU-pilE complementation also restored PilW expression to both the vfr and algZR mutants (Fig. 9B). Taken together, these results suggest that the Tfp defect in the algZR mutant can be attributed solely to AlgZR regulation of the fimU operon, whereas the Tfp defect of vfr mutants involves regulation of other factors in addition to the fimU operon.
FIG 9.

The fimU operon is not sufficient to restore the Tfp defect of a vfr mutant. (A) Tfp production as determined by Coomassie blue-stained SDS-PAGE gels of purified pilus fractions from WT and indicated mutant strains. (B) Immunoblots from whole-cell lysates of WT and indicated mutant strains probed with PilW-specific antiserum. An uncropped blot showing equal protein loading is shown in Fig. S1C.
DISCUSSION
The regulatory complexity of P. aeruginosa Tfp production and function is not surprising given the number of components involved, their various functions, and their genetic arrangement. There are distinct mechanisms governing the structural components of Tfp as well as the factors involved in processing and assembly, all of which may be regulated at the level of transcription and/or function (e.g., fiber extension/retraction and surface sensing). While this study builds upon the work of multiple investigators (33, 35, 52, 57), we present several novel results that contribute to the understanding of Tfp regulation. We identified two independent, but linked, regulatory systems involving the transcription factors AlgR and Vfr that control expression of the fimU operon. These findings suggest a mechanistic explanation for the Tfp biogenesis and twitching motility defects of algZR and vfr mutants. However, as shown in Fig. 9, the Tfp defect in the vfr mutant is not solely attributed to fimU operon control, as complementation of fimU does not restore Tfp expression. The possibility that Vfr controls additional factors necessary for Tfp biogenesis and expression is also supported by microarray analysis, which identified several Tfp biogenesis-related genes with altered expression in a vfr mutant (42). Several categories of Tfp biogenesis-related genes displayed altered expression levels in the microarray study, including (i) Chp chemosensory system genes (pilG, pilJ, and pilK), which regulates twitching motility and cAMP production; (ii) essential pilus biogenesis machinery components (pilNOPQ, pilZ, and pilU); (iii) additional regulators of Tfp biogenesis and/or function (fimV and pilS); and (iv) pilD, the prepilin peptidase. Altered expression of most of these genes (except pilS) does not affect intracellular pilin levels but instead controls Tfp production at the level of assembly and/or function and could contribute to the Tfp biogenesis and twitching motility defect of vfr mutants.
A previous bioinformatic analysis predicted a Vfr binding site in the algZ promoter and showed that Vfr binds to a PalgZ probe by EMSA, suggesting that Vfr regulates fimU indirectly through the AlgZR system (52). We demonstrated that cAMP/Vfr positively regulates both the fimU and algZ promoters, revealing both direct and indirect modes of fimU transcriptional control. Although Kanack et al. (52) observed a single Vfr-PalgZ shift product via EMSA, we visually identified two additional Vfr binding sites within the algZ promoter and observed two shift products in our in vitro analysis. We showed that mutation of all three sites is required for maximal loss of PalgZ-lacZ activity and Vfr binding to a PalgZ via EMSA. Although individual mutations in VBS2 and VBS3 but not VBS1 resulted in reduced PalgZ-lacZ activity, combinatorial VBS1-2 and VBS2-3 mutations exhibited additive reporter activity decreases. Our results are consistent with the microarray analysis of a vfr mutant, which initially indicated Vfr-dependent regulation of algZ expression (42).
Despite the fact that the fimU promoter lacked a predicted Vfr binding site, our study provides the following evidence for Vfr-dependent regulation of fimU transcription: (i) PfimU-lacZ reporter activity was significantly decreased in both cyaAB and vfr mutants, (ii) Vfr shifted a PfimU probe in a concentration- and cAMP-dependent manner in vitro, and (iii) mutation to the Vfr half-sites identified by DNase I footprinting diminished PfimU-lacZ reporter activity and abolished binding of Vfr in vitro. In fact, the VBS (5′-AACGTGAGCTAT:GCAGCACTT) we identified within the fimU promoter region shares reasonable consensus with the previously identified VCS (5′-ANWWTGNGAWNY:AGWTCACAT). The upstream Vfr half-site (TGAGC) shares considerable conservation with the upstream VCS half-site (TGNGA), while the downstream fimU Vfr half-site (GCACT) deviates significantly from the downstream VCS half-site (TCACA). Given the results of previous studies that have characterized the Vfr binding sites for several Vfr-dependent targets (44, 52), it is clear that Vfr binding sites display significant variability, and thus, it is not surprising that several Vfr-dependent genes are directly regulated by Vfr activity that were not previously identified via bioinformatics.
Activation of alginate production by AlgR is AlgZ independent and therefore likely phosphorylation independent (36). However, since both AlgZ and AlgR control Tfp function, albeit by different assays in different strains, it suggests that AlgR phosphorylation by AlgZ is required for Tfp formation (34, 35, 58). AlgR phosphorylation impacts regulation of the iron-scavenging molecules pyocyanin and pyoverdine, further highlighting the role of different AlgR phosphorylation states (59).
Previous work demonstrated that AlgR positively regulates the fimU operon and identified two AlgR binding sites within the fimU promoter region (35). We extended these findings by analyzing the Tfp phenotype and PfimU-lacZ reporter activity of mutants in which either algZ or algR was inactivated. Both algZ and algR single mutants were defective for Tfp production and displayed PfimU-lacZ reporter activity equivalent to an algZR double mutant, suggesting that AlgZ activity is required for AlgR regulation of fimU. Although previous studies found that AlgR can be phosphorylated in vitro using the Escherichia coli CheA kinase as a phosphodonor (34, 60), these studies did not directly address the role of AlgR phosphorylation in activation of the fimU operon. Our fimU promoter EMSA studies using PAM as a phosphodonor resulted in a significant increase in AlgR binding affinity. Taken together, these results provide strong evidence that AlgR phosphorylation is necessary for regulation of the fimU operon. Due to the absence of conserved kinase motifs (D/F and G boxes) required for ATP binding, it has been suggested that AlgZ does not function as a sensor kinase but may instead function as a phosphatase (36). However, experimental results supporting either function have been limited to the characterization of algZ mutant phenotypes.
Based on response regulators homologous to AlgR, we predicted that phosphorylation would affect DNA binding (61). In our study, unphosphorylated AlgR bound the PfimU probe at high concentrations, and phosphorylation of AlgR dramatically reduced the amount of AlgR required to shift the PfimU probe. This result suggests that, regardless of phosphorylation status, AlgR recognizes the fimU promoter, but phosphorylation serves to enhance DNA affinity. Interestingly, promoters known to be regulated by unphosphorylated AlgR are associated with chronic infection phenotypes (algC and algD) (62), whereas phosphorylated AlgR regulates genes more often associated with acute phenotypes (fimU operon and hcnA) (35, 63). Thus, AlgR phosphorylation may serve as a signal responsible for the commitment to a particular virulence phenotype.
Given the requirement of both transcriptional regulators Vfr and AlgR for activation of the fimU promoter, we attempted to determine whether the binding of either required the presence of the other. We determined that phosphorylated AlgR and cAMP-saturated Vfr are capable of binding the PfimU probe simultaneously. While binding of either Vfr or AlgR to the fimU promoter was largely independent of the presence of the other, there is a combined effect of AlgR and Vfr on activation of the fimU promoter. Collectively, the results of our study demonstrate that AlgR activates the fimU operon in a phosphorylation-dependent manner, presumably via phosphorylation by the cognate putative sensor kinase AlgZ. Furthermore, we extend previous phenotypic observations implicating Vfr in Tfp expression by demonstrating that Vfr directly regulates the fimU operon in a cAMP-dependent manner.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and plasmids.
The bacterial strains and plasmids used in this study are described in Table S1. Bacteria (E. coli and P. aeruginosa) were routinely grown at 37°C in LB medium. pMMB-based expression plasmids were maintained in P. aeruginosa with 150 μg/mL carbenicillin (Cb), except where indicated. Bacterial growth in liquid culture was assessed by optical density at 600 nm (OD600). Assays for β-galactosidase activity were performed as previously described (56).
Strains and plasmids used in this study. Download Table S1, DOCX file, 0.05 MB (56.8KB, docx) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
With the exception of algZ, all gene deletion alleles were constructed, introduced onto the chromosomes of the appropriate P. aeruginosa strains, and confirmed as previously described (42). The algZ gene was disrupted by converting the translational start codon into a stop codon (ATG→TGA), using splice PCR, to avoid ablating a previously identified algR promoter within the 3′ region of the algZ coding sequence (51). All other point mutations were generated by site-directed mutagenesis using a commercially available kit (QuikChange II; Agilent Technologies). See Table S2 for primer sequences.
Primers used in this study. Mutated sites are indicated by italics. EcoRI and BamHI restriction sites are underlined, and attB1 and attB2 tails are in boldface. Download Table S2, DOCX file, 0.05 MB (54.4KB, docx) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PalgZ and PfimU transcriptional reporters were constructed by PCR amplifying the fimU promoter region (−311 to +153 bp relative to the fimU transcriptional start site) and the algZ promoter region (−283 to +107 bp relative to the algZ transcriptional start site) from P. aeruginosa strain K (PAK) chromosomal DNA with EcoRI and BamHI restriction sites and attB1 and attB2 tails for cloning into pDONR201 via Gateway cloning (see Table S2 for primers). Promoter fragments were digested from pDONR201 using EcoRI and BamHI, ligated into mini-CTX-lacZ and introduced into P. aeruginosa as previously described (44, 64).
Phage susceptibility assay.
Susceptibility to the pilin-specific phage PO4 was assayed using the double-agar method as previously described (50). Briefly, overnight cultures of each strain were mixed with molten LB with 0.75% agar (top agar) and poured over standard LB agar plates. Phage were serially diluted from an initial phage stock of 108 PFU/mL. After the top agar solidified, 10 μL of each phage dilution, as well as a broth control, was spotted onto the top agar. Plates were incubated at 37°C overnight and imaged. A mutant of pilA and its complement were included as controls. IPTG (isopropyl-β-d-thiogalactopyranoside; 250 μM) was added for the pilA complement (pPa-pilA).
Electron and immunofluorescence microscopy.
Transmission electron microscopy (TEM) was performed as described previously (65), with the exception that grids were placed on a drop of bacterial suspension at 22°C for 10 min and the samples were stained with an aqueous 0.5% ammonium molybdate solution for 10 min before being viewed in a Philips CM100 transmission electron microscope. Immunofluorescence (IF) microscopy was performed as described previously (48), except that the P. aeruginosa strains were grown to an OD600 of 0.2 prior to incubating the bacteria on poly-l-lysine-coated glass coverslips. P. aeruginosa pilin-specific antiserum (gift from E. C. Gotschlich, Rockefeller University) was used as a primary antibody for Tfp labeling, followed by an Alexa Red 594-conjugated goat anti-rabbit IgG (Molecular Probes). P. aeruginosa cells were stained with 4′-6-diamidino-2-phenylindole (DAPI) at 1 μg/mL in Mowiol mounting medium (Sigma) containing 2% 1,4-diazabicyclo(2)octane (DABCO) prior to viewing with a Nikon Eclipse C400 fluorescence microscope.
Isolation of surface Tfp.
Surface pilin was isolated as previously described (8). Briefly, P. aeruginosa was grown on LB agar plates until confluent. Bacteria were collected and suspended in 1 mL 0.15 M NaCl, 0.2% formaldehyde and vortexed vigorously for 1 min to release surface Tfp. Bacterial cells were removed by centrifugation at 12,000 × g for 5 min, and the total protein content of the bacterial pellet was determined by a Bradford assay. Supernatants were transferred to new tubes, adjusted to 0.1 M MgCl2, and incubated at 4°C for 12 h. Following centrifugation at 12,000 × g for 5 min, the resulting Tfp pellets were suspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer, normalized based on total protein in the bacterial pellet fraction, and separated by SDS-PAGE (18% polyacrylamide), and pilin was visualized by Coomassie blue staining.
Immunoblotting.
Whole-cell lysates were prepared from bacteria grown in LB broth to mid-exponential growth phase (OD600 = 1). Strains harboring plasmids were grown in the presence of 30 μg/mL Cb and the indicated amount of IPTG. Bacteria were collected by centrifugation, suspended in 100 μl of SDS-PAGE sample buffer, incubated at 95°C for 10 min, and normalized based on total protein. The lysate was diluted ∼1:10, separated on SDS-polyacrylamide gels (18% for pilin, 7.5% for PilW), and transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes, respectively. Membranes were probed with PKL1 anti-pilin mouse monoclonal antibody (66) (1:30,000 dilution; gift from Randall Irvin, University of Alberta) or anti-PilW rabbit serum (23) (1:8,000 dilution). Horseradish peroxidase-conjugated secondary antibodies were used for the detection of specific antibody-antigen complexes. Blots were developed with chemiluminescence reagents (Millipore) and visualized via autoradiography.
EMSA and DNase I footprinting.
DNA promoter probes were generated by PCR using the indicated oligonucleotides and end labeled using 10 μCi of [γ-32]ATP (GE Healthcare) and 10 U of T4 polynucleotide kinase (New England Biolabs). EMSAs were performed as previously described (67). Briefly, probes (0.25 nM each) were incubated in binding buffer (10 mM Tris [pH 7.5], 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol [DTT], 5% glycerol, and 100 μg/mL bovine serum albumin) containing 5 μg/mL poly(dI-dC) for 5 min at 25°C. cAMP-saturated Vfr, apo-Vfr, or His6-AlgR protein were added as indicated in the figure legends to a final volume of 20 μL and incubated for an additional 15 min at 25°C. Where indicated (in the figure legends), 50 mM PAM and 1 mM MgCl2 was incubated with His6-AlgR. Samples were subjected to electrophoresis on a 5% polyacrylamide glycine gel (10 mM Tris [pH 7.5], 380 mM glycine, 1 mM EDTA) at 4°C. Imaging and data analyses were performed using an FLA-700 Phosphorimager (Fujifilm) and MultiGauge v3.0 software (Fujifilm). EMSAs were repeated a minimum of two times, and representative gels are shown.
Single end-labeled probe for DNase I footprinting was generated by PCR and labeled with 32P as previously described (56). Labeled probe (10 fmol) was incubated with 5 μg/mL poly(dI-dC) (Sigma) in DNase I reaction buffer (10 mM Tris [pH 8.0], 50 mM KCl, 2 mM MgCl2, 0.5 mM DTT, 100 μg/mL bovine serum albumin, 10% glycerol with purified Vfr as indicated in a final reaction volume of 25 μl. Reaction mixtures were incubated for 15 min at 25°C, fractionated on denaturing sequencing gel, and analyzed using a FLA-700 Phosphorimager.
Protein purification.
Apo-Vfr and cAMP saturated Vfr were generated as previously described (42, 54). The hexa-histidine tagged AlgR expression plasmid was constructed as follows. The algR coding sequence was amplified from PAK genomic DNA using primers. The resulting PCR fragment was gel purified (Qiagen), digested with NdeI and BamHI, and ligated into the corresponding sites in pET28a. After confirmation via sequencing, the resulting plasmid was transformed into E. coli BL21(DE3) strains for protein overexpression.
An overnight culture of E. coli strain BL21 carrying the pET28a-AlgR overexpression construct was subcultured (1:100) into 1 L LB containing 25 μg/mL kanamycin and grown at 37°C to an OD600 of ∼0.8. The flask was then chilled to room temperature (RT), induced with 0.5 mM IPTG, and cultured at 23°C overnight on a rotary shaker (∼30 rpm). Cells were collected and washed twice via resuspension in 40 mL of binding buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 20 mM imidazole). Cells were harvested, washed twice with binding buffer, and suspended in a 20-mL binding buffer. Following a 30-min incubation with 0.5 mg/mL lysozyme on ice, the cells were lysed by sonication and centrifuged at 10,000 × g for 30 min to remove cellular debris. The soluble fraction was then incubated with 3 mL of Ni2+-charged resin at 4°C for 2 h with gentle rotation. The resin was loaded onto a 20-mL Bio-Rad disposable column, washed twice with wash buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 40 mM imidazole), and then eluted with elution buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 100 mM imidazole). Fractions containing significant amounts of 6His-AlgR, as determined by SDS-PAGE analysis, were concentrated using a Centriprep concentrator and subsequently loaded on a Sephadex 75 size elution column. Fractions from a peak corresponding to AlgR (27 kDa) were concentrated to ∼3 mL, loaded into a Slide-a-Lyzer (Pierce), dialyzed twice against 2 L of buffer (20 mM Tris [pH 8.0], 500 mM NaCl, 1 mM DTT, 0.5% Tween 20), and then dialyzed into storage buffer (20 mM Tris [pH 8.0], 500 mM NaCl, 1 mM DTT, 0.5% Tween 20, 50% glycerol). Protein concentration was determined by Bradford assay.
ACKNOWLEDGMENTS
We thank E. C. Gotschlich (Rockefeller University, New York, NY) and Randall Irvin (University of Alberta) for the gifts of PilA-specific antibodies.
This study was supported (to H.C.W.-L. and M.K.) by funds from the Research Council of Norway Functional Genomics initiative (FUGE) directed through The Consortium of Advanced Microbial Sciences and Technologies (CAMST). Work in the Wolfgang and Yahr laboratories was supported by the National Institutes of Health (AI069116 [to M.C.W.], AI055042 [to T.L.Y.], and AI097264 [to M.C.W. and T.L.Y.]).
Footnotes
This article is a direct contribution from Timothy L. Yahr, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Daniel Wozniak, The Ohio State University, and Matthew Wargo, University of Vermont.
Contributor Information
Matthew C. Wolfgang, Email: matthew_wolfgang@med.unc.edu.
Matthew Parsek, University of Washington.
REFERENCES
- 1.Deretic V, Schurr MJ, Yu H. 1995. Pseudomonas aeruginosa, mucoidy and the chronic infection phenotype in cystic fibrosis. Trends Microbiol 3:351–356. doi: 10.1016/S0966-842X(00)88974-X. [DOI] [PubMed] [Google Scholar]
- 2.Lam J, Chan R, Lam K, Costerton JW. 1980. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect Immun 28:546–556. doi: 10.1128/iai.28.2.546-556.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Govan JR, Deretic V. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 5.Rahme LG, Ausubel FM, Cao H, Drenkard E, Goumnerov BC, Lau GW, Mahajan-Miklos S, Plotnikova J, Tan MW, Tsongalis J, Walendziewicz CL, Tompkins RG. 2000. Plants and animals share functionally common bacterial virulence factors. Proc Natl Acad Sci USA 97:8815–8821. doi: 10.1073/pnas.97.16.8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coggan KA, Wolfgang MC. 2012. Global regulatory pathways and cross-talk control Pseudomonas aeruginosa environmental lifestyle and virulence phenotype. Curr Issues Mol Biol 14:47–70. [PubMed] [Google Scholar]
- 7.Jurado-Martín I, Sainz-Mejías M, McClean S. 2021. Pseudomonas aeruginosa: an audacious pathogen with an adaptable arsenal of virulence factors. Int J Mol Sci 22:3128. doi: 10.3390/ijms22063128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heiniger RW, Winther-Larsen HC, Pickles RJ, Koomey M, Wolfgang MC. 2010. Infection of human mucosal tissue by Pseudomonas aeruginosa requires sequential and mutually dependent virulence factors and a novel pilus-associated adhesin. Cell Microbiol 12:1158–1173. doi: 10.1111/j.1462-5822.2010.01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faure E, Kwong K, Nguyen D. 2018. Pseudomonas aeruginosa in chronic lung infections: how to adapt within the host? Front Immunol 9:2416. doi: 10.3389/fimmu.2018.02416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedersen SS. 1992. Lung infection with alginate-producing, mucoid Pseudomonas aeruginosa in cystic fibrosis. APMIS Suppl 28:1–79. [PubMed] [Google Scholar]
- 11.Comolli JC, Hauser AR, Waite L, Whitchurch CB, Mattick JS, Engel JN. 1999. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect Immun 67:3625–3630. doi: 10.1128/IAI.67.7.3625-3630.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giltner CL, van Schaik EJ, Audette GF, Kao D, Hodges RS, Hassett DJ, Irvin RT. 2006. The Pseudomonas aeruginosa type IV pilin receptor binding domain functions as an adhesin for both biotic and abiotic surfaces. Mol Microbiol 59:1083–1096. doi: 10.1111/j.1365-2958.2005.05002.x. [DOI] [PubMed] [Google Scholar]
- 13.Mikkelsen H, Sivaneson M, Filloux A. 2011. Key two-component regulatory systems that control biofilm formation in Pseudomonas aeruginosa. Environ Microbiol 13:1666–1681. doi: 10.1111/j.1462-2920.2011.02495.x. [DOI] [PubMed] [Google Scholar]
- 14.Leighton TL, Buensuceso RNC, Howell PL, Burrows LL. 2015. Biogenesis of Pseudomonas aeruginosa type IV pili and regulation of their function. Environ Microbiol 17:4148–4163. doi: 10.1111/1462-2920.12849. [DOI] [PubMed] [Google Scholar]
- 15.Strom MS, Lory S. 1991. Amino acid substitutions in pilin of Pseudomonas aeruginosa. Effect on leader peptide cleavage, amino-terminal methylation, and pilus assembly. J Biol Chem 266:1656–1664. doi: 10.1016/S0021-9258(18)52345-0. [DOI] [PubMed] [Google Scholar]
- 16.Mattick JS. 2002. Type IV pili and twitching motility. Annu Rev Microbiol 56:289–314. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- 17.Alm RA, Mattick JS. 1996. Identification of two genes with prepilin-like leader sequences involved in type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J Bacteriol 178:3809–3817. doi: 10.1128/jb.178.13.3809-3817.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alm RA, Hallinan JP, Watson AA, Mattick JS. 1996. Fimbrial biogenesis genes of Pseudomonas aeruginosa: pilW and pilX increase the similarity of type 4 fimbriae to the GSP protein-secretion systems and pilY1 encodes a gonococcal PilC homologue. Mol Microbiol 22:161–173. doi: 10.1111/j.1365-2958.1996.tb02665.x. [DOI] [PubMed] [Google Scholar]
- 19.Alm RA, Mattick JS. 1995. Identification of a gene, pilV, required for type 4 fimbrial biogenesis in Pseudomonas aeruginosa, whose product possesses a pre-pilin-like leader sequence. Mol Microbiol 16:485–496. doi: 10.1111/j.1365-2958.1995.tb02413.x. [DOI] [PubMed] [Google Scholar]
- 20.Russell MA, Darzins A. 1994. The pilE gene product of Pseudomonas aeruginosa, required for pilus biogenesis, shares amino acid sequence identity with the N-termini of type 4 prepilin proteins. Mol Microbiol 13:973–985. doi: 10.1111/j.1365-2958.1994.tb00489.x. [DOI] [PubMed] [Google Scholar]
- 21.Jacobsen T, Bardiaux B, Francetic O, Izadi-Pruneyre N, Nilges M. 2020. Structure and function of minor pilins of type IV pili. Med Microbiol Immunol 209:301–308. doi: 10.1007/s00430-019-00642-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen Y, Sugiman-Marangos S, Harvey H, Bell SD, Charlton CL, Junop MS, Burrows LL. 2015. Pseudomonas aeruginosa minor pilins prime type IVa pilus assembly and promote surface display of the PilY1 adhesin. J Biol Chem 290:601–611. doi: 10.1074/jbc.M114.616904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giltner CL, Habash M, Burrows LL. 2010. Pseudomonas aeruginosa minor pilins are incorporated into type IV pili. J Mol Biol 398:444–461. doi: 10.1016/j.jmb.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 24.Orans J, Johnson MDL, Coggan KA, Sperlazza JR, Heiniger RW, Wolfgang MC, Redinbo MR. 2010. Crystal structure analysis reveals Pseudomonas PilY1 as an essential calcium-dependent regulator of bacterial surface motility. Proc Natl Acad Sci USA 107:1065–1070. doi: 10.1073/pnas.0911616107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webster SS, Mathelié-Guinlet M, Verissimo AF, Schultz D, Viljoen A, Lee CK, Schmidt C, Wong GCL, Dufrene YF, O’Toole GA. 2021. Force-induced changes of PilY1 drive surface sensing by Pseudomonas aeruginosa. bioRxiv doi: 10.1101/2021.08.24.457478. [DOI] [PMC free article] [PubMed]
- 26.Rodesney CA, Roman B, Dhamani N, Cooley BJ, Katira P, Touhami A, Gordon VD. 2017. Mechanosensing of shear by Pseudomonas aeruginosa leads to increased levels of the cyclic-di-GMP signal initiating biofilm development. Proc Natl Acad Sci USA 114:5906–5911. doi: 10.1073/pnas.1703255114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siryaporn A, Kuchma SL, O'Toole GA, Gitai Z. 2014. Surface attachment induces Pseudomonas aeruginosa virulence. Proc Natl Acad Sci USA 111:16860–16865. doi: 10.1073/pnas.1415712111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Persat A, Inclan YF, Engel JN, Stone HA, Gitai Z. 2015. Type IV pili mechanochemically regulate virulence factors in Pseudomonas aeruginosa. Proc Natl Acad Sci USA 112:7563–7568. doi: 10.1073/pnas.1502025112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vrla GD, Esposito M, Zhang C, Kang Y, Seyedsayamdost MR, Gitai Z. 2020. Cytotoxic alkyl-quinolones mediate surface-induced virulence in Pseudomonas aeruginosa. PLoS Pathog 16:e1008867. doi: 10.1371/journal.ppat.1008867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marko VA, Kilmury SLN, MacNeil LT, Burrows LL. 2018. Pseudomonas aeruginosa type IV minor pilins and PilY1 regulate virulence by modulating FimS-AlgR activity. PLoS Pathog 14:e1007074. doi: 10.1371/journal.ppat.1007074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuchma SL, Ballok AE, Merritt JH, Hammond JH, Lu W, Rabinowitz JD, O'Toole GA. 2010. Cyclic-di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa: the pilY1 gene and its impact on surface-associated behaviors. J Bacteriol 192:2950–2964. doi: 10.1128/JB.01642-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo Y, Zhao K, Baker AE, Kuchma SL, Coggan KA, Wolfgang MC, Wong GCL, O’Toole GA. 2015. A hierarchical cascade of second messengers regulates Pseudomonas aeruginosa surface behaviors. mBio 6:e02456-14. doi: 10.1128/mBio.02456-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitchurch CB, Alm RA, Mattick JS. 1996. The alginate regulator AlgR and an associated sensor FimS are required for twitching motility in Pseudomonas aeruginosa. Proc Natl Acad Sci USA 93:9839–9843. doi: 10.1073/pnas.93.18.9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitchurch CB, Erova TE, Emery JA, Sargent JL, Harris JM, Semmler ABT, Young MD, Mattick JS, Wozniak DJ. 2002. Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J Bacteriol 184:4544–4554. doi: 10.1128/JB.184.16.4544-4554.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belete B, Lu H, Wozniak DJ. 2008. Pseudomonas aeruginosa AlgR regulates type IV pilus biosynthesis by activating transcription of the fimU-pilVWXY1Y2E operon. J Bacteriol 190:2023–2030. doi: 10.1128/JB.01623-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu H, Mudd M, Boucher JC, Schurr MJ, Deretic V. 1997. Identification of the algZ gene upstream of the response regulator algR and its participation in control of alginate production in Pseudomonas aeruginosa. J Bacteriol 179:187–193. doi: 10.1128/jb.179.1.187-193.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schurr MJ, Yu H, Martinez-Salazar JM, Boucher JC, Deretic V. 1996. Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J Bacteriol 178:4997–5004. doi: 10.1128/jb.178.16.4997-5004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohr CD, Leveau JH, Krieg DP, Hibler NS, Deretic V. 1992. AlgR-binding sites within the algD promoter make up a set of inverted repeats separated by a large intervening segment of DNA. J Bacteriol 174:6624–6633. doi: 10.1128/jb.174.20.6624-6633.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohr CD, Hibler NS, Deretic V. 1991. AlgR, a response regulator controlling mucoidy in Pseudomonas aeruginosa, binds to the FUS sites of the algD promoter located unusually far upstream from the mRNA start site. J Bacteriol 173:5136–5143. doi: 10.1128/jb.173.16.5136-5143.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suh S-J, Runyen-Janecky LJ, Maleniak TC, Hager P, MacGregor CH, Zielinski-Mozny NA, Phibbs PV, West SEH. 2002. Effect of vfr mutation on global gene expression and catabolite repression control of Pseudomonas aeruginosa. Microbiology (Reading) 148:1561–1569. doi: 10.1099/00221287-148-5-1561. [DOI] [PubMed] [Google Scholar]
- 41.Jones AK, Fulcher NB, Balzer GJ, Urbanowski ML, Pritchett CL, Schurr MJ, Yahr TL, Wolfgang MC. 2010. Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. J Bacteriol 192:5709–5717. doi: 10.1128/JB.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolfgang MC, Lee VT, Gilmore ME, Lory S. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell 4:253–263. doi: 10.1016/S1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 43.Beatson SA, Whitchurch CB, Sargent JL, Levesque RC, Mattick JS. 2002. Differential regulation of twitching motility and elastase production by Vfr in Pseudomonas aeruginosa. J Bacteriol 184:3605–3613. doi: 10.1128/JB.184.13.3605-3613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuchs EL, Brutinel ED, Jones AK, Fulcher NB, Urbanowski ML, Yahr TL, Wolfgang MC. 2010. The Pseudomonas aeruginosa Vfr regulator controls global virulence factor expression through cyclic AMP-dependent and -independent mechanisms. J Bacteriol 192:3553–3564. doi: 10.1128/JB.00363-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.West SE, Sample AK, Runyen-Janecky LJ. 1994. The vfr gene product, required for Pseudomonas aeruginosa exotoxin A and protease production, belongs to the cyclic AMP receptor protein family. J Bacteriol 176:7532–7542. doi: 10.1128/jb.176.24.7532-7542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fulcher NB, Holliday PM, Klem E, Cann MJ, Wolfgang MC. 2010. The Pseudomonas aeruginosa Chp chemosensory system regulates intracellular cAMP levels by modulating adenylate cyclase activity. Mol Microbiol 76:889–904. doi: 10.1111/j.1365-2958.2010.07135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saiman L, Ishimoto K, Lory S, Prince A. 1990. The effect of piliation and exoproduct expression on the adherence of Pseudomonas aeruginosa to respiratory epithelial monolayers. J Infect Dis 161:541–548. doi: 10.1093/infdis/161.3.541. [DOI] [PubMed] [Google Scholar]
- 48.Winther-Larsen HC, Wolfgang M, Dunham S, van Putten JPM, Dorward D, Løvold C, Aas FE, Koomey M. 2005. A conserved set of pilin-like molecules controls type IV pilus dynamics and organelle-associated functions in Neisseria gonorrhoeae. Mol Microbiol 56:903–917. doi: 10.1111/j.1365-2958.2005.04591.x. [DOI] [PubMed] [Google Scholar]
- 49.Lizewski SE, Schurr JR, Jackson DW, Frisk A, Carterson AJ, Schurr MJ. 2004. Identification of AlgR-regulated genes in Pseudomonas aeruginosa by use of microarray analysis. J Bacteriol 186:5672–5684. doi: 10.1128/JB.186.17.5672-5684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bradley DE. 1973. Basic characterization of a Pseudomonas aeruginosa pilus-dependent bacteriophage with a long noncontractile tail. J Virol 12:1139–1148. doi: 10.1128/JVI.12.5.1139-1148.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pritchett CL, Little AS, Okkotsu Y, Frisk A, Cody WL, Covey CR, Schurr MJ. 2015. Expression analysis of the Pseudomonas aeruginosa AlgZR two-component regulatory system. J Bacteriol 197:736–748. doi: 10.1128/JB.02290-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanack KJ, Runyen-Janecky LJ, Ferrell EP, Suh S-J, West SEH. 2006. Characterization of DNA-binding specificity and analysis of binding sites of the Pseudomonas aeruginosa global regulator, Vfr, a homologue of the Escherichia coli cAMP receptor protein. Microbiology (Reading) 152:3485–3496. doi: 10.1099/mic.0.29008-0. [DOI] [PubMed] [Google Scholar]
- 53.Albus AM, Pesci EC, Runyen-Janecky LJ, West SE, Iglewski BH. 1997. Vfr controls quorum sensing in Pseudomonas aeruginosa. J Bacteriol 179:3928–3935. doi: 10.1128/jb.179.12.3928-3935.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dasgupta N, Ferrell EP, Kanack KJ, West SEH, Ramphal R. 2002. Ramphal R. fleQ, the gene encoding the major flagellar regulator of Pseudomonas aeruginosa, is sigma70 dependent and is downregulated by Vfr, a homolog of Escherichia coli cyclic AMP receptor protein. J Bacteriol 184:5240–5250. doi: 10.1128/JB.184.19.5240-5250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferrell E, Carty NL, Colmer-Hamood JA, Hamood AN, West SEH. 2008. Regulation of Pseudomonas aeruginosa ptxR by Vfr. Microbiology (Reading) 154:431–439. doi: 10.1099/mic.0.2007/011577-0. [DOI] [PubMed] [Google Scholar]
- 56.Fuchs EL, Brutinel ED, Klem ER, Fehr AR, Yahr TL, Wolfgang MC. 2010. In vitro and in vivo characterization of the Pseudomonas aeruginosa cyclic AMP (cAMP) phosphodiesterase CpdA, required for cAMP homeostasis and virulence factor regulation. J Bacteriol 192:2779–2790. doi: 10.1128/JB.00168-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whitchurch CB, Hobbs M, Livingston SP, Krishnapillai V, Mattick JS. 1991. Characterisation of a Pseudomonas aeruginosa twitching motility gene and evidence for a specialised protein export system widespread in eubacteria. Gene 101:33–44. doi: 10.1016/0378-1119(91)90221-V. [DOI] [PubMed] [Google Scholar]
- 58.Whitchurch CB, Leech AJ, Young MD, Kennedy D, Sargent JL, Bertrand JJ, Semmler ABT, Mellick AS, Martin PR, Alm RA, Hobbs M, Beatson SA, Huang B, Nguyen L, Commolli JC, Engel JN, Darzins A, Mattick JS. 2004. Characterization of a complex chemosensory signal transduction system which controls twitching motility in Pseudomonas aeruginosa. Mol Microbiol 52:873–893. doi: 10.1111/j.1365-2958.2004.04026.x. [DOI] [PubMed] [Google Scholar]
- 59.Little AS, Okkotsu Y, Reinhart AA, Damron FH, Barbier M, Barrett B, Oglesby-Sherrouse AG, Goldberg JB, Cody WL, Schurr MJ, Vasil ML, Schurr MJ. 2018. Pseudomonas aeruginosa AlgR phosphorylation status differentially regulates pyocyanin and pyoverdine production. mBio 9:e02318-17. doi: 10.1128/mBio.02318-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deretic V, Leveau JH, Mohr CD, Hibler NS. 1992. In vitro phosphorylation of AlgR, a regulator of mucoidy in Pseudomonas aeruginosa, by a histidine protein kinase and effects of small phospho-donor molecules. Mol Microbiol 6:2761–2767. doi: 10.1111/j.1365-2958.1992.tb01455.x. [DOI] [PubMed] [Google Scholar]
- 61.Nikolskaya AN, Mulkidjanian AY, Beech IB, Galperin MY. 2003. MASE1 and MASE2: two novel integral membrane sensory domains. J Mol Microbiol Biotechnol 5:11–16. doi: 10.1159/000068720. [DOI] [PubMed] [Google Scholar]
- 62.Ma S, Selvaraj U, Ohman DE, Quarless R, Hassett DJ, Wozniak DJ. 1998. Phosphorylation-independent activity of the response regulators AlgB and AlgR in promoting alginate biosynthesis in mucoid Pseudomonas aeruginosa. J Bacteriol 180:956–968. doi: 10.1128/JB.180.4.956-968.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cody WL, Pritchett CL, Jones AK, Carterson AJ, Jackson D, Frisk A, Wolfgang MC, Schurr MJ. 2009. Pseudomonas aeruginosa AlgR controls cyanide production in an AlgZ-dependent manner. J Bacteriol 191:2993–3002. doi: 10.1128/JB.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 65.Winther-Larsen HC, Wolfgang MC, van Putten JPM, Roos N, Aas FE, Egge-Jacobsen WM, Maier B, Koomey M. 2007. Pseudomonas aeruginosa Type IV pilus expression in Neisseria gonorrhoeae: effects of pilin subunit composition on function and organelle dynamics. J Bacteriol 189:6676–6685. doi: 10.1128/JB.00407-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu L, Lee KK, Hodges RS, Paranchych W, Irvin RT. 1994. Adherence of Pseudomonas aeruginosa and Candida albicans to glycosphingolipid (asialo-GM1) receptors is achieved by a conserved receptor-binding domain present on their adhesins. Infect Immun 62:5213–5219. doi: 10.1128/iai.62.12.5213-5219.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brutinel ED, Vakulskas CA, Brady KM, Yahr TL. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol 68:657–671. doi: 10.1111/j.1365-2958.2008.06179.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Uncropped α-PilW Western blots. α-PilW blots for Fig. 1C (A), Fig. 5B and C (B), and Fig. 9B (C). Blots for PilW and PilA were run on the same samples, which were generated as described in Materials and Method. Protein was normalized based on total protein levels as determined by a Bradford assay. Download FIG S1, TIF file, 0.5 MB (501.5KB, tif) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
vfr, cyaAB, and algZR mutants exhibit sensitivity to the pilin-specific phage PO4. Phage susceptibility was determined using a double-agar layer as described in Materials and Methods. Phage was serially diluted from 108 PFU/mL to 100 PFU/mL. Ten microliters of each phage dilution, as well as broth control, was spotted onto double agar containing the indicated strains and incubated overnight before imaging. Phage concentrations above 105 PFU/mL also resulted in complete lysis. The assay was performed in duplicate. Download FIG S2, TIF file, 0.9 MB (891.8KB, tif) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Restoration of PfimU-lacZ reporter activity in mutants. The wild-type (WT) or indicated mutant strains containing either vector, pPa-cyaB, pPa-vfr, pPa-algZ, pPa-algR, or pPa-algZR expression plasmids were grown in LB broth containing 30 μg/mL carbenicillin (Cb) and 50 μM IPTG. Values are means and SEM (n ≥ 3). Download FIG S3, TIF file, 0.3 MB (286KB, tif) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Vfr protected region of the fimU operon. DNase I footprinting of the fimU promoter region. Samples contained a DNA fragment (0.4 nM) corresponding to ∼−300 bp relative to the translational start site. The top strand of the DNA probe was radiolabeled on one end and was incubated in the absence or presence of cAMP-Vfr (114, 13, or 1 nM) prior to DNase I treatment. DNase I-generated fragments were separated by electrophoresis, and Maxam-Gilbert sequencing ladders were made using the same DNA. Sequence corresponding region protected by Vfr binding is bracketed, with the predicted half-sites underlined and in bold. Download FIG S4, TIF file, 0.6 MB (604KB, tif) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains and plasmids used in this study. Download Table S1, DOCX file, 0.05 MB (56.8KB, docx) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Mutated sites are indicated by italics. EcoRI and BamHI restriction sites are underlined, and attB1 and attB2 tails are in boldface. Download Table S2, DOCX file, 0.05 MB (54.4KB, docx) .
Copyright © 2022 Coggan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.