

ABSTRACT
Mycobacterium abscessus (Mab) infections are a growing menace to the health of many patients, especially those suffering from structural lung disease and cystic fibrosis. With multidrug resistance a common feature and a growing understanding of peptidoglycan synthesis in Mab, it is advantageous to identify potent β-lactam and β-lactamase inhibitor combinations that can effectively disrupt cell wall synthesis. To improve existing therapeutic regimens to address serious Mab infections, we evaluated the ability of durlobactam (DUR), a novel diazobicyclooctane β-lactamase inhibitor to restore in vitro susceptibilities in combination with β-lactams and provide a biochemical rationale for the activity of this compound. In cell-based assays, susceptibility of Mab subsp. abscessus isolates to amoxicillin (AMOX), imipenem (IMI), and cefuroxime (CXM) was significantly enhanced with the addition of DUR. The triple drug combinations of CXM-DUR-AMOX and IMI-DUR-AMOX were most potent, with MIC ranges of ≤0.06 to 1 μg/mL and an MIC50/MIC90 of ≤0.06/0.25 μg/mL, respectively. We propose a model by which this enhancement may occur, DUR potently inhibited the β-lactamase BlaMab with a relative Michaelis constant (Ki app) of 4 × 10−3 ± 0.8 × 10−3 μM and acylation rate (k2/K) of 1 × 107 M−1 s−1. Timed mass spectrometry captured stable formation of carbamoyl-enzyme complexes between DUR and LdtMab2-4 and Mab d,d-carboxypeptidase, potentially contributing to the intrinsic activity of DUR. Molecular modeling showed unique and favorable interactions of DUR as a BlaMab inhibitor. Similarly, modeling showed how DUR might form stable Michaelis-Menten complexes with LdtMab2-4 and Mab d,d-carboxypeptidase. The ability of DUR combined with amoxicillin or cefuroxime and imipenem to inactivate multiple targets such as d,d-carboxypeptidase and LdtMab2,4 supports new therapeutic approaches using β-lactams in eradicating Mab.
KEYWORDS: antibiotic resistance, bacteria, inhibitor, antibiotics, durlobactam, β-lactams, Mycobacterium abscessus, β-lactamase inhibitor, diazabicyclooctane, Mycobacterium
INTRODUCTION
Mycobacterium abscessus (Mab) is a multidrug-resistant (MDR) nontuberculous mycobacterium (NTM) that causes invasive pulmonary and disseminated infections, particularly in patients with structural lung disease, cystic fibrosis, or compromised immunity (1). Recently updated guidelines by four international societies, the American Thoracic Society (ATS), European Respiratory Society (ERS), European Society of Clinical Microbiology and Infectious Diseases (ESCMID), and Infectious Diseases Society of America (IDSA) published in July 2020 recommend a multidrug regimen that contains at least three active antimicrobials (2). However, treatment outcomes remain poor compared to other mycobacterial diseases (3), and the optimal combination and duration of therapy remain unknown. Furthermore, the presence of acquired or inducible macrolide resistance [23S rRNA mutations or erm(41) induction, respectively] is associated with a significant rate of treatment failure (2, 4, 5). In focal pulmonary infections, surgical resection of bronchiectatic lung lobes is often recommended, reminiscent of the dramatic approach to drug-susceptible pulmonary tuberculosis in the era before chemotherapy. Novel therapeutic strategies with sustainable efficacy for the treatment of Mab are needed.
β-Lactams and β-lactamase inhibitors (BLIs) are agents that may provide promise for Mab infections. Within the current guidelines, imipenem (IMI) and cefoxitin (FOX) are the only two β-lactam antibiotics recommended for inclusion in a multidrug regimen (1). Unfortunately, IMI- and FOX-resistant isolates are reported (5). Nevertheless, insights into the intrinsic mechanisms of β-lactam resistance provide opportunities to expand the use of this drug class. One such important mechanism of resistance is BlaMab, a class A β-lactamase encoded on a Mab gene. BlaMab demonstrates catalytic activity against penicillins, some cephalosporins, and to a lesser extent, carbapenems; BlaMab hydrolyzes FOX and IMI, albeit at a comparatively lower rate (low k2/K) than other β-lactams (6). Unlike BlaC, the β-lactamase of Mycobacterium tuberculosis, BlaMab is poorly inhibited by clavulanate (6).
Another important source of β-lactam resistance in Mab are the l,d-transpeptidases (Ldts). The better-studied d,d-transpeptidases (Ddts; also referred to as high-molecular-weight penicillin-binding proteins or HMW PBPs) form 4,3-cross-links between adjacent peptidoglycan stem peptides. In contrast, Ldts form 3,3-cross-links, and in Mab, this linkage predominates (7). Ldts in Mab are resistant to penicillins but are inhibited by carbapenems and some cephalosporins (8, 9). Important to the function of Ldts are the d,d-carboxypeptidases (d,d-c), a group of low-molecular-weight penicillin-binding proteins (LMW PBPs) that convert pentapeptides of peptidoglycan subunits to tetrapeptides that can subsequently be used by Ldts; these enzymes also appear to be inhibited by β-lactams (10).
Recently, it was hypothesized that the synergy exhibited by dual β-lactams in susceptibility studies may be driven by inhibition of multiple enzymes in the peptidoglycan synthesis pathway (i.e., “target redundancy”) (8, 9). This “target redundancy” may lead to an unanticipated synergistic effect using β-lactam combinations. This hypothesis is supported by the finding that combinations of certain β-lactams and β-lactamase inhibitors inhibit more than one critical enzyme in the cell wall synthesis process. In addition to inhibition of Ldts, select cephalosporins and carbapenems are known to inhibit the traditionally recognized targets of β-lactams (Ddts). Sayed et al. demonstrated that PBPs of Mab (PonA1, PonA2, and PbpA) are inhibited by ceftriaxone, cefotaxime, and carbapenems at relatively low concentrations (11). Interest in combinations of β-lactams with and without β-lactamase inhibitors is growing as laboratory findings demonstrate improvement of in vitro susceptibilities in Mab isolates (9, 12–15). The application of these findings awaits in vivo studies.
Diazabicyclooctanes (DBOs) are a recently developed class of β-lactamase inhibitors that may provide an opportunity to address the challenges of BlaMab and Ldts and expand the use of β-lactams in the treatment of Mab. Avibactam, nacubactam, and zidebactam restore susceptibility to β-lactams and inhibit growth of Mab in vitro (16–20). The DBO avibactam is a potent inhibitor of BlaMab (21). DBOs also participate in the inhibition of peptidoglycan transpeptidases. Avibactam has been shown to inhibit l,d transpeptidation of M. tuberculosis (Mtb) and form carbamoyl-enzyme complexes with some Ldts of Mtb and Mab (9, 22). DBO inhibition of Ldts may be important not only for ensuring broad inhibition of multiple Ldts, Ddts, and d,d-c but also for exploiting the notion of target redundancy that may underlie the synergy between some drug combinations.
Durlobactam (DUR, previously designated ETX2514) is a novel DBO with the unique features of a double bond within the DBO ring between C-3 and C-4 and a methyl group at C-3 (Fig. 1); these features enhance reactivity and binding to β-lactamases (23). As such, DUR is a potent inhibitor of class A, C, and D serine β-lactamases. It is currently in clinical development in combination with sulbactam for the treatment of carbapenem-resistant Acinetobacter baumannii infections. To date, DUR activity against BlaMab and LdtMab of Mab has not been investigated. As part of a concerted effort to improve existing therapeutic regimens to address serious Mab infections, we sought to evaluate the ability of DUR to restore in vitro susceptibilities in combination with β-lactams and provide a biochemical rationale for activity. We hypothesized that DUR would act as a “dual function” compound and enhance the activity of β-lactam partners through both protection from BlaMab and inhibition of peptidoglycan transpeptidation. We also propose a model by which this enhancement may occur.
FIG 1.

Structures of the DBOs, avibactam, and DUR (ETX2514).
RESULTS
Susceptibility of Mycobacterium abscessus subsp. abscessus isolates to amoxicillin, imipenem, or cefuroxime is enhanced by the addition of DUR.
An initial assessment of antibacterial activity for amoxicillin (AMOX), IMI, and DUR alone plus each of the β-lactams in a 1:1 combination with DUR was determined for M. abscessus (Mab) ATCC 19977 in Middlebrook 7H9 broth supplemented with 10% (vol/vol) oleic albumin dextrose catalase and 0.05% (vol/vol) Tween 80. As anticipated, IMI had better activity than AMOX alone with MIC values ranging from 0.5 to 4 μg/mL. Unlike avibactam and relebactam (9), DUR demonstrates antimicrobial activity with MIC values ranging from 2 to 8 μg/mL. The MIC values for AMOX-DUR combination were one twofold dilution lower than those for DUR alone, and the MIC values for the IMI-DUR combination were generally one twofold dilution lower than those for IMI alone (Table 1 and Fig. 2).
TABLE 1.
Summary of activity of DUR, AMOX, IMI, or CXM alone or in various combinations against 101 clinical isolates of Maba
| Antibiotic | Test method | MIC (μg/mL) |
||
|---|---|---|---|---|
| Range | MIC50 | MIC90 | ||
| DUR | Alone | 2−8 | 4 | 8 |
| AMOX | Alone | 128 to >256 | >256 | >256 |
| IMI | Alone | 1−4 | 4 | 4 |
| IMI-AMOX | Titrate IMI + AMOX fixed at 8 μg/mL | 0.25−8 | 1 | 2 |
| AMOX-DUR | Fixed 1:1 ratio | 1−4 | 2 | 4 |
| IMI-DUR | Fixed 1:1 ratio | 0.5−4 | 2 | 2 |
| IMI-DUR-AMOX | Titrate IMI-DUR 1:1 + AMOX fixed at 8 μg/mL | ≤0.06−2 | ≤0.06 | 0.25 |
| CXM | Alone | 4−128 | 8 | 16 |
| CXM-AMOX | Titrate CXM + AMOX fixed at 8 μg/mL | 0.5 to >64 | 4 | 8 |
| CXM-DUR | Fixed 1:1 ratio | 0.5−4 | 2 | 2 |
| CXM-DUR-AMOX | Titrate CXM-DUR 1:1 + AMOX fixed at 8 μg/mL | ≤0.06−1 | ≤0.06 | 0.25 |
DUR, durlobactam; AMOX, amoxicillin; IMI, imipenem; CXM, cefuroxime. CLSI antimicrobial agent and susceptibility breakpoints for testing rapidly growing mycobacteria are available only for imipenem and are as follows: susceptible (≤8 μg/mL), intermediate (16 μg/mL), and resistant (≥32 μg/mL).
FIG 2.

MIC distribution against 101 clinical isolates of Mycobacterium abscessus (Mab) subsp. abscessus. The drugs used were cefuroxime (CXM), durlobactam (DUR), amoxicillin (AMOX), and imipenem (IMI). When more than two drugs were combined, AMOX was added at a fixed concentration of 8 μg/mL to serial dilutions of CXM-DUR or IMI-DUR. Mab isolates were incubated with test agents at 30°C for 48 h, and MIC was defined as lowest antibiotic concentration that prevented visible bacterial growth.
Further exploration of the antibacterial activity of combinations of β-lactams and DUR in our entire set of previously characterized isolates of Mab subsp. abscessus revealed specific susceptibilities to the combinations of AMOX, IMI, and DUR. Cefuroxime (CXM) alone and in combination with DUR or AMOX, IMI-DUR, and triple combinations of IMI-DUR-AMOX and CXM-DUR-AMOX were also tested against our clinical isolates (Table 1 and Fig. 2).
The intrinsic antibacterial activity of DUR for this larger collection of Mab isolates was similar to that observed against Escherichia coli and other Enterobacterales spp. (23), with an MIC50/MIC90 of 4/8 μg/mL. IMI also showed similar activity against Mab as observed in the previous study (9), with an MIC50/MIC90 of 4/4 μg/mL. Addition of DUR to IMI in a 1:1 fixed ratio improved the MIC50/MIC90 value by one dilution (to 2/2 μg/mL). AMOX added at a fixed concentration of 8 μg/mL to IMI had an even greater effect, lowering the MIC50/MIC90 to 1/2 μg/mL. Addition of DUR to AMOX in a 1:1 fixed ratio also improved the MIC50/MIC90 (to 2/4 μg/mL). The triple combination (IMI-DUR 1:1 in the presence of AMOX fixed at 8 μg/mL) had the greatest activity, with an MIC range of ≤0.06 to 2 μg/mL and an MIC50/MIC90 of ≤0.06/0.25 μg/mL (Table 1 and Fig. 2).
Cefuroxime (CXM) alone had less activity than IMI, with an MIC50/MIC90 of 8/16 μg/mL against this collection of isolates. Addition of AMOX at a fixed concentration of 8 μg/mL to CXM improved the MIC50/MIC90 by one dilution (to 4/8 μg/mL). DUR added to CXM in a fixed 1:1 ratio had a greater effect, lowering the MIC50/MIC90 to 2/2 μg/mL. The triple combination (CXM-DUR titrated 1:1 in the presence of 8 μg/mL AMOX) was very potent and similar to the triple combination of IMI-DUR-AMOX, with an MIC range of ≤0.06 to 1 μg/mL and an MIC50/MIC90 of ≤0.06/0.25 μg/mL (Table 1 and Fig. 2).
Kinetics of BlaMab inactivation by DUR is superior to AVI and REL.
We next sought to determine the biochemical parameters of Mab inactivation by DUR under steady-state conditions using a reporter substrate, nitrocefin. We previously reported apparent Ki (Ki app) values for the inhibition of BlaMab by avibactam (AVI) and relebactam (REL) (0.30 ± 0.03 μM and 136 ± 14 μM, respectively) (9). For DUR, we determined a Ki app (4.0 × 10−3 ± 0.8 × 10−3 μM) nearly 75-fold lower than that of avibactam (Table 2). The k2/K or “on rate” constant for DUR with BlaMab was 1.0 × 107 ± 0.3 × 107 M−1 s−1, nearly 30-fold and 17,000-fold higher than the values with avibactam and relebactam, respectively. The koff and half-life (t1/2) with DUR were similar to those with avibactam and DUR had a slightly higher turnover number (kcat/kinact). We thus calculated the dissociation constant (Kd) to be 0.2 ± 0.1 nM, which again is comparatively lower than that of avibactam and relebactam, Taken together, these kinetic data show that DUR is a highly potent inhibitor of BlaMab.
TABLE 2.
Kinetic parameters of BlaMab inhibition by DUR, relebactam, and avibactama
| BlaMab | Ki app (μM) | k2/K (M−1s−1) | Koff (s−1) | t1/2 (min) | Kd (nM) | Kcat/kinact |
|---|---|---|---|---|---|---|
| Durlobactam | (4.0 ± 0.8) × 10−3 | (1.0 ± 0.3) × 107 | (2.0 ± 0.2) × 10−3 | 6.0 ± 0.6 | 0.2 ± 0.1 | 4 |
| Avibactam | 0.30 ± 0.03 | (3.4 ± 0.4) × 105 | (2.0 ± 0.2) × 10−3 | 6.0 ± 0.6 | 6.0 ± 0.6 | 1 |
| Relebactam | 140 ± 14 | (6.0 ± 0.6) × 102 | (1.0 ± 0.1) × 10−1 | 12 ± 1.2 | (1.7 ± 0.2) × 103 | 3 |
Relebactam and avibactam values were previously determined (9). t1/2 = ln2/koff and Kd = koff/(k2/K).
Timed electrospray ionization mass spectrometry. (i) Capturing DUR carbamoyl-enzyme complex formation with Mab BlaMab.
In order to confirm DUR inhibition of BlaMab by covalent binding, we next aimed to capture carbamoyl-enzyme complexes between DUR and BlaMab with timed electrospray ionization mass spectrometry. The compound was incubated in excess with BlaMab for 2 h. Carbamoyl-enzyme complexes of BlaMab with avibactam and relebactam were previously reported (9). As expected, a peak corresponding to the mass of a BlaMab-DUR carbamoyl-enzyme complex was seen (Fig. 3A).
FIG 3.

Timed mass spectrometry measuring durlobactam acyl-enzyme complex formation between Mab BlaMab (A), d,d-c (B), and l,d-transpeptidases LdtMab2 (C), LdtMab3 (D), and LdtMab4 (E).
(ii) DUR binds to LdtMab2, LdtMab3, LdtMab4, and d,d-c.
On the basis of DUR intrinsic antimicrobial activity, we explored whether there was formation of stable acyl-enzyme complexes with DUR and several Ldts and the d,d-c of Mab. We previously captured carbamoyl-enzyme complexes between avibactam and relebactam with these enzymes (9). Again, excess DUR was incubated with enzyme for 2 h. DUR formed carbamoyl-enzyme complexes with d,d-c (Fig. 3B), LdtMab2, and LdtMab4 (Fig. 3C and E). A complex was not captured with LdtMab3 (Fig. 3D and Table 3). These binding patterns are reminiscent of those with avibactam and relebactam except that binding was not observed between relebactam and LdtMab4.
TABLE 3.
Mass spectrometry analyses of BlaMab, LdtMab 2,3,4, and d,d-c alone and incubated with durlobactam
| Protein (nominal MWa [Da]) |
Compound (nominal MW [Da]) |
Observed MW (±5 Da) |
Changes in MWb |
|---|---|---|---|
| BlaMab (28,432) | None | 28,432 | |
| Durlobactam (276) | 28,709 | +277 | |
| LdtMab2 (39,214) | None | 39,214 | |
| Durlobactam (276) | 39,492 | +278 | |
| Amoxicillin (365) | 39, 214 | +0 | |
| LdtMab3 (44,791) | None | 44,791 | |
| Durlobactam (276) | 44,791 | +0 | |
| Amoxicillin (365) | 44,791 | +0 | |
| LdtMab4 (32,565) | None | 32,565 | |
| Durlobactam (276) | 32,841 | +276 | |
| Amoxicillin (365) | 32,841 | +0 | |
| Mab d,d-carboxypeptidase (26,789) | None | 26,789 | |
| Durlobactam (276) | 27,066 | +277 | |
| Amoxicillin (365) | 27156 | +367 | |
MW, molecular weight.
Mass accuracy, ±5 Da.
(iii) AMOX binds to d,d-c but not to LdtMab2, LdtMab3, and LdtMab4.
In order to further explore a biochemical rationale supporting the observation that the addition of AMOX further lowered MICs when added to IMI-DUR and CXM-DUR, we sought to investigate whether AMOX formed acyl-enzyme complexes with LdtMab2, LdtMab3, LdtMab4, and d,d-c. In these investigations, AMOX was incubated in excess with enzymes for 2 h. Consistent with previous studies examining the activity of penicillins and Mab Ldts, acyl-enzyme complexes of AMOX with Ldts were not captured. In contrast, an acyl-enzyme complex with AMOX and d,d-c was captured, demonstrating that it is a potential target of the drug (Table 3).
Molecular modeling of DUR in the active site of BlaMab.
To better describe the potential molecular basis of DUR inhibition of BlaMab, we performed molecular modeling of DUR in the active site of BlaMab. We previously described the positioning of avibactam and relebactam in BlaMab using molecular modeling (9). Starting with the Michaelis-Menten complex, we show that DUR is well positioned in the active site of the enzyme (Fig. 4A) and forms potential hydrogen bonds with catalytic site residues N132, S130, and K232. The DBO ring may be stabilized by W105 and by hydrophobic interactions between F237 with the C-3 methyl group of DUR (Fig. 4B). While W105 and F237 had a favorable interaction with DUR, in our previous molecular docking of BlaMab, we hypothesized these residues were a source of steric hindrance for the piperidine ring of relebactam (9).
FIG 4.

Molecular docking of DUR into the active site of BlaMab as Michaelis-Menten complex. (A) DUR is perfectly positioned into the active site of the enzyme and makes H-bond interactions with catalytic residues N132, S130, and K232. (B) The DBO ring is stabilized by W105 and by hydrophobic interactions of F237 with the methyl group of DUR. (C and D) The carbamoyl-enzyme complex between BlaMab and DUR is formed (C), and productive interactions with the enzyme are created (D). The carbamoyl is positioned into the oxyanion hole (H-bond interactions with S70:NH and G235:NH), the steric interactions between DBO and W105 are preserved, and H-bonds with K234, S130, N132 and K73 are formed.
When the carbamoyl-enzyme complex between BlaMab and DUR is formed (Fig. 4C), the carbamoyl group is positioned into the oxyanion hole, forming stabilizing hydrogen bond interactions with (S70:NH, G235:NH); there are also steric interactions between the DBO ring and W105 and hydrogen bonds (H-bonds) with K232, S130, N132, and K73. All these interactions support the finding that DUR is a potent and efficient inhibitor of BlaMab.
Molecular modeling of DUR in the active site of d,d-c and LdtMab2.
To assess the potential interactions between d,d-c and DUR, the molecular docking of DUR into the active site of d,d-c was performed and analyzed. The docking of intact DUR generated the formation of Michaelis-Menten complex (Fig. 5A) with the carbamoyl group at C-7 perfectly positioned into the oxyanion hole, close to nucleophilic S70 and F230:NH. When the carbamoyl-enzyme complex (Fig. 5B) between d,d-c and DUR is formed, the carbamoyl is maintaining the position into the oxyanion hole (H-bond interactions with S70:NH and F230:NH) and the sulfate group makes H-bonds with Q213 and S123.
FIG 5.

Molecular docking of DUR into the active site of D,D-c, and the formation of Michaelis-Menten (A) and carbamoyl enzyme complexes (B) between d,d-carboxypeptidases and DUR. The carbamoyl is positioned into the oxyanion hole (H-bond interactions with S70:NH and F230:NH), and the SO3− group makes H-bonds with Q213 and S123.
Using the data from mass spectrometry, which show the formation of stable complex between DUR and LdtMab2 after 2 h incubation (Fig. 3C), and previous molecular studies (9), the molecular docking of DUR was performed into the “open” active site of LdtMab2. In Ldts, C351 is the catalytic residue (24). When intact DUR is docked into the active site of LdtMab2 (see Fig. 8A), the carbamoyl is positioned into the proposed catalytic site with H-bond formation with C351:NH and G351:NH. Based on the molecular dynamic (MD) simulation of LdtMab2 and DUR Michaelis-Menten complex (Fig. 6A), the distance between C351:SH and the H333:ND2 imidazole ring is less than 2 Å. This makes it possible for the activation (deprotonation) of the thiol group of C351 by H333 imidazole ring and provides assistance with the formation of the thio ester complex (Fig. 6B). The first step is deprotonation of a thiol (C351) in the enzyme’s active site by H333 (which is part of the catalytic dyad, C351-H333); this is followed by nucleophilic attack by the deprotonated cysteine's anionic sulfur on the carbonyl carbon (C-7) (Fig. 7). When the thioester was formed, the results of the docking generated several possible conformations (Fig. 6C, with carbamoyl “out” of oxianion hole, and Fig. 6D, with carbamoyl toward oxianion hole). These results and the fact that the complex between LdtMab2 and DUR was stable for more than 2 h (previous data with avibactam show strong interaction for more than 8 h, and up to 4 weeks) (24) suggest that DUR may form a long-lived complex in the active site of LdtMab2, without undergoing deacylation.
FIG 8.

d,d-Carboxypeptidase and amoxicillin Michaelis-Menten (A) and acyl enzyme (B) complexes. (Flexible docking does not result in productive conformations for acylation and covalent bond formation [figure not shown]). Manual docking, followed by minimization and bond formation for acyl enzyme complex, resulted in conformations with the amoxicillin carboxyl group positioned in the oxyanion hole, and the formation of H-bond networking with active site cavity residues.
FIG 6.

Intact durlobactam (DUR) was docked into the open active site of LdtMab2 (A). The molecular dynamics of the Michelis-Menten complex suggest that His333 activates (deprotonates) the thiol group of Cys351 and assists the acyl-transfer step. The nucleophilic Cys351 attacks C6 of DUR and forms the acyl-enzyme complex.
FIG 7.

His333 activates (deprotonates) the thiol group of C351 and assists the acyl-transfer step (22, 31). The nucleophilic C351 attacks C-6 of DUR and forms the acyl-enzyme complex. As for the mechanism, the first step is deprotonation of a thiol (C351) in the enzyme's active site by a histidine (H333, not H349) residue. The next step is nucleophilic attack by the deprotonated cysteine’s anionic sulfur on the peptide carbonyl carbon (C-6).
Molecular modeling of AMOX in the active site of d,d-c.
The formation of the acyl complex of AMOX with d,d-c captured by the timed mass spectroscopy was examined using molecular docking. Flexible docking does not result in productive conformations for acylation and covalent bond formation (figure not shown). Manual docking, followed by minimization and bond formation for acyl-enzyme complex, resulted in conformations with the AMOX carboxyl group positioned in the oxionion hole (S70:NH and F230:NH), for Michaelis-Menten (Fig. 8A) and acyl-enzyme (Fig. 8B) complexes. In both complexes, amoxicillin is making H-bond networks with active site cavity residues (S70, F230, K73, and K227).
DISCUSSION
DUR enhances susceptibility of Mab to IMI, CXM, and AMOX.
The ability of dual β-lactam combinations (carbapenems and cephalosporin) to inactivate multiple Ddt and Ldt targets, the relative stability of carbapenems against hydrolysis by BlaMab, and the use of novel β-lactamase inhibitors all together support the notion that dual β-lactam therapy may be a potential candidate for evaluation against Mab. Combining two β-lactams in several in vitro studies revealed promising results (9, 13, 25, 26). Here, the results observed in our cell-based assays for dual β-lactam combinations present a more complex picture. First, DUR had significant intrinsic antimicrobial activity on its own, which is supported by its ability to form stable complexes with the cell wall synthesis proteins LdtMab2, LdtMab4, and Mab d,d-c. Second, while DUR had a profound effect on the MIC of AMOX, only a modest impact on the activity of IMI or CXM was observed. Third, the combination of AMOX with IMI or CXM revealed only modest improvement. Last, the addition of DUR to the combination of AMOX with either IMI or CXM resulted in profound reductions in MICs into submicromolar range (Table 4). This hitherto unappreciated synergy seen by inhibition of d,d-c was very revealing.
TABLE 4.
Cumulative MIC distribution of CXM, DUR, AMOX, and IMI for clinical isolates of Mab and ATCC 19977 control straina
| Antibiotic(s) | No. (cumulative %) of isolates inhibited at the following MIC (μg/mL): |
MIC50 (μg/mL) |
MIC90 (μg/mL) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ≤0.06 | 0.12 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 32 | 64 | ≥128 | |||
| DUR | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 10 (9.9) | 74 (84.1) | 17 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 | 8 |
| Amoxicillin | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 101 (100) | >256 | >256 |
| AMOX-DUR | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (1.9) | 83 (84) | 16 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 | 4 |
| IMI | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 7 (7) | 40 (46.6) | 54 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 | 4 |
| IMI-AMOX | 0 (0) | 0 (0) | 12 (11.8) | 21 (32.5) | 36 (68.1) | 25 (92.8) | 6 (98.7) | 1 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 | 2 |
| IMI-DUR | 0 (0) | 0 (0) | 0 (0) | 2 (1.9) | 30 (31.6) | 68 (98.9) | 1 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 | 2 |
| IMI-DUR-AMOX | 87 (86.1) | 1 (87) | 3 (89.9) | 7 (96.8) | 2 (98.7) | 1 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | ≤0.06 | 0.25 |
| CXM | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (1.9) | 69 (70.2) | 21 (90.9) | 8 (98.8) | 0 (0) | 1 (100) | 8 | 16 |
| CXM-AMOX | 0 (0) | 0 (0) | 0 (0) | 3 (2.9) | 9 (11.8) | 32 (43.4) | 41 (83.9) | 12 (95.7) | 3 (98.6) | 0 (0) | 0 (0) | 1 (100) | 4 | 8 |
| CXM-DUR | 0 (0) | 0 (0) | 0 (0) | 1 (0.9) | 42 (42.4) | 57 (98.8) | 1 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 | 2 |
| CXM-DUR-AMOX | 83 (82.1) | 5 (87) | 5 (91.9) | 6 (97.8) | 2 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | ≤0.06 | 0.25 |
DUR, durlobactam; AMOX, amoxicillin; IMI, imipenem; CXM, cefuroxime. When more than two drugs were combined, AMOX was added at fixed concentration of 8 μg/mL to serial dilutions of 1:1 CXM-DUR or IMI-DUR.
In this context, we advance the idea that DUR leads to effective protection of AMOX, allowing for synergy with IMI or CXM, respectively. A recent in vitro report noted that the addition of AMOX at a fixed concentration of 32 μg/mL lowered MICs when combined with IMI and relebactam against Mab. The lower concentration of 8 μg/mL used in our study proved to be effective and is attainable in patients using 500-mg three times daily dose regimens (15). In vitro data for the activity of new DBOs and prior clinically approved BLIs were previously reported (15, 17, 27). None of these drugs inhibited BlaMab as potently as DUR and MIC values were at least fourfold higher. Further in vitro studies are needed, but durlobactam in combination with β-lactam partners holds potential as an addition to the relatively limited array of drugs available for Mab infections.
DUR is a potent and efficient inhibitor of BlaMab.
As a β-lactamase with a broad substrate profile and relative resistance to previous BLIs, BlaMab remains a challenge to be surmounted for the use of β-lactams in the treatment of Mab infections (6). The DBOs avibactam and relebactam exhibit potent inhibitory activity against BlaMab and restore susceptibility to AMOX (15, 21) and ceftaroline (9, 13). Relebactam combined with IMI-cilastatin and AMOX showed minimal enhancement of IMI bactericidal activity against Mab isolates with declines in MIC ranges (15). We anticipated DUR would also be an effective inhibitor of BlaMab. Our kinetics establish DUR as an effective inhibitor to BlaMab and also demonstrate that it is more potent and efficient than avibactam with Ki app 75-fold lower and k2/K 30-fold higher.
Molecular modeling reveals unique interactions with the C-3 methyl group of DUR and the hydrophobic W105 residue of BlaMab.
While other class A β-lactamases have a polar tyrosine at position 105 (Y105), BlaMab has a hydrophobic tryptophan (W105). BlaMab also has a phenylalanine at position 237, which is usually occupied by alanine or serine in other class A β-lactamases. We had previously shown that these residues were a steric hindrance for the piperidine side group of relebactam into the active site of BlaMab (9). In contrast, whereas W105 and F237 were obstacles for the interaction of relebactam into the active site, they were favorable for DUR binding. In particular, the C-3 methyl group of DUR has hydrophobic interactions with these residues in the Michaelis-Menten complex. There is also a stabilizing interaction between the DUR DBO ring and W105. These unique, favorable interactions likely underlie the enhanced efficacy of DUR as a BlaMab inhibitor compared to relebactam.
Blockage of LdtMab2 by IMI and DUR may prevent Ldts from catalyzing transpeptidation.
Penicillins and cephalosporins tend to be poor Ldt inhibitors, while carbapenems and penems are typically the most potent (8). LdtMab2 is an ortholog of LdtMt2 with significant sequence homology (25). Mutant M. tuberculosis strains that are deficient in LdtMt2 were more susceptible to AMOX and clavulanic acid compared to the wild type, and colonies were smaller and smooth (28). We hypothesize that inhibition of the LdtMab2 by IMI and DUR interferes with peptidoglycan transpeptidation and sensitizes Mab to AMOX. In addition, disruption of peptidoglycan metabolism may potentially alter bacterial β-lactamase production (21, 29). For AMOX to exhibit activity against Mab, it must have a critical target to inhibit and must be protected against hydrolysis by BlaMab. We showed that AMOX formed stable acyl-enzyme complexes with d,d-c and demonstrated potent inhibition of BlaMab activity by DUR. Figure 9 summarizes the interactions between AMOX, IMI, and DUR with Ldt2,4, d,d-c, and BlaMab.
FIG 9.
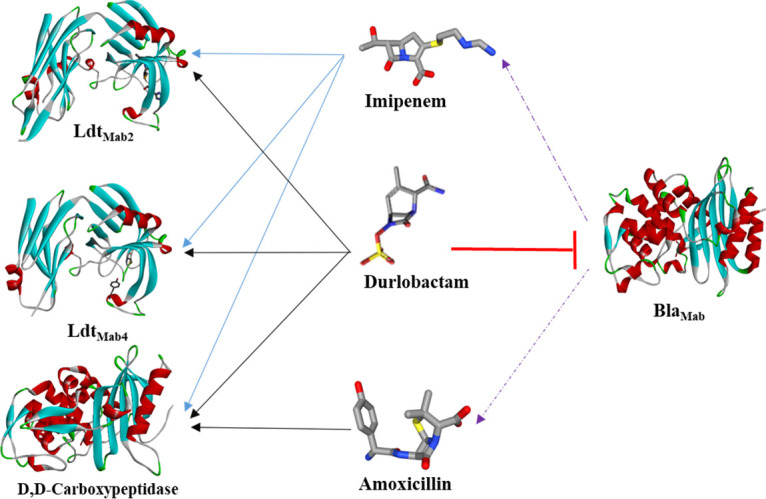
Interactions between Mab BlaMab, d,d-c, and l,d-transpeptidases LdtMab2 and LdtMab4 with IMI, AMOX, and DUR. DUR is a potent inhibitor of BlaMab, provides protection of AMOX and IMI against hydrolysis. Like IMI, DUR has intrinsic activity forming stable acyl enzyme complexes with LdtMab2 and LdtMab4. AMOX has no activity against LdtMab2 and LdtMab4 but stably binds to d,d-c. The ability of DUR to protect amoxicillin and imipenem against BlaMab and its intrinsic activity along with the dual β-lactam target redundancy can explain the rationale behind potent activity of this combination.
Conclusions.
In this study, we have demonstrated that DUR, a novel DBO BLI, is an active agent with potent intrinsic activity against BlaMab, LdtMab2,4, and d,d-c with MIC ranges comparable to those for IMI. The triple combination of DUR with AMOX and either IMI or CXM is highly potent. We hypothesize that DUR improves the β-lactam’s activity by protecting AMOX against the hydrolytic activity of BlaMab and by targeting multiple steps in peptidoglycans synthesis. Data on use of dual β-lactams and BLI for treatment of Mab are emerging and DUR may have potential as an active agent against Mab, adding to the growing evidence of the potential clinical benefit of utilizing dual β-lactams with a BLI against serious Mab infections. The use of highly active β-lactam/β-lactamase inhibitor combinations may prove to be effective therapy, reduce toxicity in Mab treatment regimens, and lead to improved treatment outcomes by overcoming resistance. Further studies are required to define the role of dual novel β-lactams and BLI in the treatment of Mab infections.
MATERIALS AND METHODS
Clinical strains, chemical reagents, and antibiotics.
One hundred clinical respiratory isolates were received at University Hospitals Cleveland Medical Center and MetroHealth Hospital in 2018 and from the National Jewish Hospital, Denver, CO. Control M. abscessus type strain, ATCC 19977, was obtained from the American Type Culture Collection (ATCC). Durlobactam (DUR) was a gift from Entasis Therapeutics. Cefuroxime (CXM) salts (active ingredient), imipenem (IMI), and amoxicillin (AMOX) were purchased from AchemBlock.
In vitro susceptibility testing.
The MICs of CXM, IMI, and AMOX with or without DUR were determined using a serial microdilution method. Approximately 5 × 105 CFU/mL were inoculated into Middlebrook 7H9 broth supplemented with 10% (vol/vol) oleic albumin dextrose catalase and 0.05% (vol/vol) Tween 80. When more than two drugs were combined, AMOX was added at a fixed concentration of 8 μg/mL to serial dilutions of CXM-DUR or IMI-DUR. Mab isolates were incubated with test agents at 30°C for 48 h, and MIC was defined as the lowest antibiotic concentration that prevented visible bacterial growth.
Cloning and purification of BlaMab.
A truncated sequence of Mab blaMab (Δ1–30 blaMab) was generated by Celtek Biosciences (Franklin, TN), cloned into the pET28(a)+ vector, and transformed into Escherichia coli BL21(DE3). Cells were grown to an optical density at 600 nm (OD600) of 0.8, and protein expression was induced with 1 mM isopropyl-d-1-thiogalactopyranoside (IPTG). After incubation for 18 h at 16°C, cells were harvested and lysed using a QIA express nickel-nitrilotriacetic acid (Ni-NTA) fast-start kit, followed by nickel column purification of the His-tagged protein according to the manufacturer’s protocol (Qiagen, Inc., Valencia, CA). Briefly, the His tag was removed from the protein by adding thrombin (Novagen, Madison, WI) overnight at 4°C (1.6 U/mg protein). The cleaved protein was separated from the His-tagged peptides by size exclusion chromatography using a HiLoad 16/60 Superdex 75 column (GE Healthcare Life Science, Uppsala, Sweden). The predicted mass of the BlaMab enzyme (28,433 Da) was confirmed by mass spectrometry.
Cloning and purification of LdtMab2,3,4 and d,d-c.
Cloning and purification of LdtMab2,3,4 and d,d-c were previously described (9, 25). Briefly, truncated sequences of LdtMab 2,3,4 and d,d-c (Δ1–41) were generated by Celtek Biosciences and cloned into the pET28(a)+ vector with a TEV (tobacco etch virus) protease cleavage site prior to the start codon of the target protein sequences. Clones were transformed into E. coli BL2(DE3) and grown at an OD600 of 0.8, and protein expression was induced with 0.25 mM IPTG. After incubation for 18 h at 18°C, cells were harvested and stored at −20°C overnight. LdtMab2,3,4 and d,d-c cell pellets were resuspended in buffer containing 50 mM Tris (pH 8.0), 400 mM sodium chloride, and 1 mM Tris (2-carboxyethyl) phosphine hydrochloride (TCEP), followed by sonication and centrifugation. The supernatant was passed through a His Prep FF 16/10 column (GE Healthcare) and washed with 25 column volumes of buffer, and bound protein was eluted with a gradient of 1 to 500 mM imidazole. Eluted protein was subjected to dialysis overnight at 4°C in buffer containing 50 mM Tris (pH 8.0), 150 mM sodium chloride, and 0.5 mM TCEP in the presence of His-tagged TEV protease (ratio of TEV protease to LdtMab2,3,4 and d,d-c, 1:30). To remove the His tag, uncleaved fusion protein, and His-tagged TEV protease, passage over the His Prep FF 16/10 column was performed. Fractions containing LdtMab2,3,4 and d,d-c were pooled, concentrated, and stored in 20% glycerol at −20°C.
Mass spectrometry analyses.
Ten micrograms of BlaMab, LdtMab2,3,4, and Mab d,d-c was incubated at room temperature with substrate (DUR) at a molar ratio of 1:20 for 2 h in 50 mM Tris-HCl (pH 7.5) and 300 mM sodium chloride for a total reaction volume of 20 μl. Reactions were quenched with 10 μl acetonitrile, and the mixtures were added to 1 ml of 0.1% formic acid in water. Samples were analyzed using a quadrupole time-of-flight (Q-TOF) Waters Synapt-G2-Si electrospray ionization mass spectrometer (ESI-MS) and Waters Acquity H class ultraperformance liquid chromatography (UPLC) with a BEH C18 1.7-μm column (2.1 by 50 mm). The Synapt G2-Si spectrometer was calibrated with sodium iodide with an m/z mass range of 50 to 2,000. MassLynx V4.1 was used to deconvolute protein peaks. The tune settings for each sample were as follows: capillary voltage at 3 kV, sampling cone at 35 V, source offset at 35, source temperature of 100°C, desolvation temperature of 500°C, cone gas at 100 L/h, desolvation gas at 800 L/h, and 6.0 nebulizer. Mobile phase A was 0.1% formic acid (FA) in water. Mobile phase B was 0.1% FA in acetonitrile. The mass accuracy for this system is ±5 Da.
Inactivation kinetics.
The kinetic parameters of BlaMab with avibactam and relebactam were previously determined (9). DUR inhibition kinetics were performed with purified BlaMab enzyme as previously described (9, 30). In brief, the reaction scheme is represented in equation 1:
| (1) |
where E is the enzyme and I is the inhibitor. Kinetic parameters were determined using an Agilent 8453 diode array spectrophotometer (Agilent Technologies, Inc., Santa Clara, CA), and reactions were conducted in 100 mM morpholino ethanesulfonic acid (MES) (pH 6.4) at 20°C. A direct competition assay was performed to approximate the relative Michaelis constant (Ki app) of the inhibitor which was the inhibitor concentration leading to reduction of the velocity by 50% as measured by nitrocefin hydrolysis. A final concentration of 5 × Km of nitrocefin (NCF) (KmNCF = 22 μM) was used as the indicator substrate in the presence of 7.0 nM BlaMab. Initial velocities were measured over the first 10 s of the reaction in the presence of durlobactam concentrations ranging from 0 to 0.05 μM. The Ki app was determined by dividing the y intercept over the slope of the simple linear regression of 1/velocity versus the concentration of DUR. This value was then corrected to account for the affinity of nitrocefin for BlaMab according to equation 2:
| (2) |
To determine the acylation rate (k2/K), which is the second-order rate constant for enzyme and inhibitor complex inactivation with K = k1/k−1, assays were performed using fixed concentrations of enzyme and NCF and increasing concentrations of DUR. The progress curves to obtain observed k (kobs) values for inactivation were fit graphically using equation 3:
| (3) |
where Vf is the final velocity, V0 is the initial velocity, and A0 is the initial absorbance at 482 nm. Fitting to equation 3 was performed in Origin 8.1 (Origin Lab, Northampton, MA). The values for kobs versus DUR concentration were plotted. The acylation rate constant was determined by correcting the slope of the line to account for the affinity of nitrocefin (KmNCF) using equation 4:
| (4) |
The off-rate constant (koff) was determined by incubating BlaMab with DUR at 500 × Kiapp for 5 min. The mixture was diluted 1:1,000, and 100 μM nitrocefin was added. Progress curves of nitrocefin hydrolysis were measured and fitted to single exponential decay equation to obtain koff. Reaction mixtures containing BlaMab alone and DUR alone were used as controls. The residence half-life (t1/2) of the drug-enzyme complex was (ln2/koff). Partition ratios (kcat/kinact) were obtained by incubating BlaMab with increasing ratios of DUR for 24 h. The ratio of inhibitor to enzyme required to inhibit the hydrolysis of nitrocefin by >90% is kcat/kinact.
Molecular modeling.
The crystal structure of class A β-lactamase from M. abscessus (PDB accession no. 4YFM) was used for simulation and molecular docking. Discovery Studio 2020 Client (BIOVIA, Dassault Systémes, 2020, San Diego) molecular modeling software was used as previously described (9). The structure was minimized using a conjugate gradient method, with a root mean square (RMS) gradient of 0.001 kcal/(mol × Å). Generalized Born with a simple Switching (GBSW) solvation model was used, and long-range electrostatics were treated using a Particle Mesh Ewald method with periodic boundary condition. The SHAKE algorithm was applied.
The intact and acyl DUR was built and minimized. The CDOCKER protocol was used to dock the DUR into the active site of BlaMab. Similarly, d,d carboxypeptidases crystal structure (PDB accession no. 4RYE) and LdtMab2 (PDB accession no. 5UWV) were used for docking DUR into the active site. The protocol uses a CHARMm-based molecular dynamics (MD) scheme to dock ligands into a receptor binding site. Random ligand conformations were generated using high-temperature MD. The conformations are then translated into the binding site. Candidate poses are created using random rigid-body rotations, followed by simulated annealing. A final minimization is used to refine the ligand poses. The generated poses were analyzed, and the best ranked poses were used to create the Michaelis-Menten and acyl-enzyme complexes and were further minimized.
ACKNOWLEDGMENTS
R.A.B. receives support from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) under award numbers R01AI100560, R01AI063517, and R01AI072219. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, award number 1I01BX001974 to R.A.B. from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development, and the Geriatric Research Education and Clinical Center VISN 10. R.A.B. also received research support provided by Entasis Therapeutics.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Department of Veterans Affairs.
REFERENCES
- 1.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother 67:810–818. doi: 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- 2.Daley CL, Iaccarino JM, Lange C, Cambau E, Wallace RJ, Jr, Andrejak C, Bottger EC, Brozek J, Griffith DE, Guglielmetti L, Huitt GA, Knight SL, Leitman P, Marras TK, Olivier KN, Santin M, Stout JE, Tortoli E, van Ingen J, Wagner D, Winthrop KL. 2020. Treatment of nontuberculous mycobacterial pulmonary disease: an official ATS/ERS/ESCMID/IDSA clinical practice guideline. Eur Respir J 56:2000535. doi: 10.1183/13993003.00535-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pasipanodya JG, Ogbonna D, Ferro BE, Magombedze G, Srivastava S, Deshpande D, Gumbo T. 2017. Systematic review and meta-analyses of the effect of chemotherapy on pulmonary Mycobacterium abscessus outcomes and disease recurrence. Antimicrob Agents Chemother 61:e01206-17. doi: 10.1128/AAC.01206-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nash KA, Brown-Elliott BA, Wallace RJ, Jr.. 2009. A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother 53:1367–1376. doi: 10.1128/AAC.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maurer FP, Ruegger V, Ritter C, Bloemberg GV, Bottger EC. 2012. Acquisition of clarithromycin resistance mutations in the 23S rRNA gene of Mycobacterium abscessus in the presence of inducible erm(41). J Antimicrob Chemother 67:2606–2611. doi: 10.1093/jac/dks279. [DOI] [PubMed] [Google Scholar]
- 6.Soroka D, Dubee V, Soulier-Escrihuela O, Cuinet G, Hugonnet JE, Gutmann L, Mainardi JL, Arthur M. 2014. Characterization of broad-spectrum Mycobacterium abscessus class A beta-lactamase. J Antimicrob Chemother 69:691–696. doi: 10.1093/jac/dkt410. [DOI] [PubMed] [Google Scholar]
- 7.Lavollay M, Fourgeaud M, Herrmann JL, Dubost L, Marie A, Gutmann L, Arthur M, Mainardi JL. 2011. The peptidoglycan of Mycobacterium abscessus is predominantly cross-linked by l,d-transpeptidases. J Bacteriol 193:778–782. doi: 10.1128/JB.00606-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Story-Roller E, Maggioncalda EC, Cohen KA, Lamichhane G. 2018. Mycobacterium abscessus and beta-lactams: emerging insights and potential opportunities. Front Microbiol 9:2273. doi: 10.3389/fmicb.2018.02273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dousa KM, Kurz SG, Taracila MA, Bonfield T, Bethel CR, Barnes MD, Selvaraju S, Abdelhamed AM, Kreiswirth BN, Boom WH, Kasperbauer SH, Daley CL, Bonomo RA. 2020. Insights into the l,d-transpeptidases and D, D-carboxypeptidase of Mycobacterium abscessus: ceftaroline, imipenem, and novel diazabicyclooctane inhibitors. Antimicrob Agents Chemother 64: e00098-20. doi: 10.1128/AAC.00098-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandey SD, Pal S, Kumar NG, Bansal A, Mallick S, Ghosh AS. 2018. Two dd-carboxypeptidases from Mycobacterium smegmatis affect cell surface properties through regulation of peptidoglycan cross-linking and glycopeptidolipids. J Bacteriol 200:e00760-17. doi: 10.1128/JB.00760-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sayed ARM, Shah NR, Basso KB, Kamat M, Jiao Y, Moya B, Sutaria DS, Lang Y, Tao X, Liu W, Shin E, Zhou J, Werkman C, Louie A, Drusano GL, Bulitta JB. 2020. First penicillin-binding protein occupancy patterns for 15 beta-lactams and beta-lactamase inhibitors in Mycobacterium abscessus. Antimicrob Agents Chemother 65:e01956-20. doi: 10.1128/AAC.01956-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Story-Roller E, Maggioncalda EC, Lamichhane G. 2019. Select beta-lactam combinations exhibit synergy against Mycobacterium abscessus in vitro. Antimicrob Agents Chemother 63:02613-18. doi: 10.1128/AAC.02613-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pandey R, Chen L, Manca C, Jenkins S, Glaser L, Vinnard C, Stone G, Lee J, Mathema B, Nuermberger EL, Bonomo RA, Kreiswirth BN. 2019. Dual beta-lactam combinations highly active against Mycobacterium abscessus complex in vitro. mBio 10:e02895-18. doi: 10.1128/mBio.02895-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar P, Kaushik A, Lloyd EP, Li SG, Mattoo R, Ammerman NC, Bell DT, Perryman AL, Zandi TA, Ekins S, Ginell SL, Townsend CA, Freundlich JS, Lamichhane G. 2017. Non-classical transpeptidases yield insight into new antibacterials. Nat Chem Biol 13:54–61. doi: 10.1038/nchembio.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopeman RC, Harrison J, Rathbone DL, Desai M, Lambert PA, Cox JAG. 2020. Effect of amoxicillin in combination with imipenem-relebactam against Mycobacterium abscessus. Sci Rep 10:928. doi: 10.1038/s41598-020-57844-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meir M, Bifani P, Barkan D. 2018. The addition of avibactam renders piperacillin an effective treatment for Mycobacterium abscessus infection in an in vivo model. Antimicrob Resist Infect Control 7:151. doi: 10.1186/s13756-018-0448-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lefebvre AL, Le Moigne V, Bernut A, Veckerle C, Compain F, Herrmann JL, Kremer L, Arthur M, Mainardi JL. 2017. Inhibition of the beta-lactamase BlaMab by avibactam improves the in vitro and in vivo efficacy of imipenem against Mycobacterium abscessus. Antimicrob Agents Chemother 61:e02440-16. doi: 10.1128/AAC.02440-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubee V, Soroka D, Cortes M, Lefebvre AL, Gutmann L, Hugonnet JE, Arthur M, Mainardi JL. 2015. Impact of beta-lactamase inhibition on the activity of ceftaroline against Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob Agents Chemother 59:2938–2941. doi: 10.1128/AAC.05080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Run E, Arthur M, Mainardi JL. 2019. In vitro and intracellular activity of imipenem combined with tedizolid, rifabutin, and avibactam against Mycobacterium abscessus. Antimicrob Agents Chemother 63:e01915-18. doi: 10.1128/AAC.01915-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaushik A, Ammerman NC, Parrish NM, Nuermberger EL. 2019. New beta-lactamase inhibitors nacubactam and zidebactam improve the in vitro activity of beta-lactam antibiotics against Mycobacterium abscessus complex clinical isolates. Antimicrob Agents Chemother 63:e00733-19. doi: 10.1128/AAC.00733-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubee V, Bernut A, Cortes M, Lesne T, Dorchene D, Lefebvre AL, Hugonnet JE, Gutmann L, Mainardi JL, Herrmann JL, Gaillard JL, Kremer L, Arthur M. 2015. Beta-lactamase inhibition by avibactam in Mycobacterium abscessus. J Antimicrob Chemother 70:1051–1058. doi: 10.1093/jac/dku510. [DOI] [PubMed] [Google Scholar]
- 22.Edoo Z, Iannazzo L, Compain F, Li de la Sierra Gallay I, van Tilbeurgh H, Fonvielle M, Bouchet F, Le Run E, Mainardi JL, Arthur M, Etheve-Quelquejeu M, Hugonnet JE. 2018. Synthesis of avibactam derivatives and activity on beta-lactamases and peptidoglycan biosynthesis enzymes of mycobacteria. Chemistry 24:8081–8086. doi: 10.1002/chem.201800923. [DOI] [PubMed] [Google Scholar]
- 23.Durand-Reville TF, Guler S, Comita-Prevoir J, Chen B, Bifulco N, Huynh H, Lahiri S, Shapiro AB, McLeod SM, Carter NM, Moussa SH, Velez-Vega C, Olivier NB, McLaughlin R, Gao N, Thresher J, Palmer T, Andrews B, Giacobbe RA, Newman JV, Ehmann DE, de Jonge B, O’Donnell J, Mueller JP, Tommasi RA, Miller AA. 2017. ETX2514 is a broad-spectrum beta-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat Microbiol 2:17104. doi: 10.1038/nmicrobiol.2017.104. [DOI] [PubMed] [Google Scholar]
- 24.Steiner EM, Schneider G, Schnell R. 2017. Binding and processing of beta-lactam antibiotics by the transpeptidase LdtMt2 from Mycobacterium tuberculosis. FEBS J 284:725–741. doi: 10.1111/febs.14010. [DOI] [PubMed] [Google Scholar]
- 25.Kumar P, Chauhan V, Silva JRA, Lameira J, d’Andrea FB, Li SG, Ginell SL, Freundlich JS, Alves CN, Bailey S, Cohen KA, Lamichhane G. 2017. Mycobacterium abscessus l,d-transpeptidases are susceptible to inactivation by carbapenems and cephalosporins but not penicillins. Antimicrob Agents Chemother 61:e00866-17. doi: 10.1128/AAC.00866-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Story-Roller E, Galanis C, Lamichhane G. 2021. Beta-lactam combinations that exhibit synergy against Mycobacteroides abscessus clinical isolates. Antimicrob Agents Chemother 65:e02545-20. doi: 10.1128/AAC.02545-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaushik A, Ammerman NC, Lee J, Martins O, Kreiswirth BN, Lamichhane G, Parrish NM, Nuermberger EL. 2019. In vitro activity of the new beta-lactamase inhibitors relebactam and vaborbactam in combination with beta-lactams against Mycobacterium abscessus complex clinical isolates. Antimicrob Agents Chemother 63:e02623-18. doi: 10.1128/AAC.02623-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta R, Lavollay M, Mainardi JL, Arthur M, Bishai WR, Lamichhane G. 2010. The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat Med 16:466–469. doi: 10.1038/nm.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacoby GA. 2009. AmpC beta-lactamases. Clin Microbiol Rev 22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papp-Wallace KM, Nguyen NQ, Jacobs MR, Bethel CR, Barnes MD, Kumar V, Bajaksouzian S, Rudin SD, Rather PN, Bhavsar S, Ravikumar T, Deshpande PK, Patil V, Yeole R, Bhagwat SS, Patel MV, van den Akker F, Bonomo RA. 2018. Strategic approaches to overcome resistance against Gram-negative pathogens using beta-lactamase inhibitors and beta-lactam enhancers: activity of three novel diazabicyclooctanes WCK 5153, zidebactam (WCK 5107), and WCK 4234. J Med Chem 61:4067–4086. doi: 10.1021/acs.jmedchem.8b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lohans CT, Chan HTH, Malla TR, Kumar K, Kamps J, McArdle DJB, van Groesen E, de Munnik M, Tooke CL, Spencer J, Paton RS, Brem J, Schofield CJ. 2019. Non-hydrolytic beta-lactam antibiotic fragmentation by l,d-transpeptidases and serine beta-lactamase cysteine variants. Angew Chem Int Ed Engl 58:1990–1994. doi: 10.1002/anie.201809424. [DOI] [PMC free article] [PubMed] [Google Scholar]