

Abstract
Aim:
We investigated the effect of ataluren plus standard of care (SoC) on age at loss of ambulation (LoA) and respiratory decline in patients with nonsense mutation Duchenne muscular dystrophy (nmDMD) versus patients with DMD on SoC alone.
Patients & methods:
Study 019 was a long-term Phase III study of ataluren safety in nmDMD patients with a history of ataluren exposure. Propensity score matching identified Study 019 and CINRG DNHS patients similar in disease progression predictors.
Results & conclusion:
Ataluren plus SoC was associated with a 2.2-year delay in age at LoA (p = 0.0006), and a 3.0-year delay in decline of predicted forced vital capacity to <60% in nonambulatory patients (p = 0.0004), versus SoC. Ataluren plus SoC delays disease progression and benefits ambulatory and nonambulatory patients with nmDMD. ClinicalTrials.gov: NCT01557400.
Keywords: : ataluren, dystrophin, efficacy, loss of ambulation, nonsense mutation Duchenne muscular dystrophy, respiratory function, Study 019
Duchenne muscular dystrophy (DMD) is a rare, X-linked, progressive, debilitating and ultimately fatal disease resulting from the loss of function of dystrophin protein due to mutations in the DMD gene. The disease occurs in approximately 1 in every 3600–6000 live male births [1,2].
The DMD disease process starts before birth; consequences of reduced or absent dystrophin levels are already evident in muscle biopsies of affected newborns and infants, and serum CK is grossly elevated in patients with DMD at birth [3]. These patients have measurable deficits in gross and fine motor function before the age of 3 years, resulting in delays in developmental motor milestones such as attainment of independent walking. Deterioration of ambulation occurs in the first decade of life, leading to wheelchair dependency, which typically occurs in the early teens [1,4]. Weakness eventually develops in the upper limb, respiratory and cardiac muscles, resulting in respiratory or cardiac failure and death in the second to fourth decades of life [5–7].
Loss of ambulation (LoA) is a particularly challenging milestone for patients and families of patients affected by DMD; patients who have lost ambulation require increased assistance with the activities of daily living as they age [8]. Moreover, sequential loss of early developmental milestones, such as getting up from the floor, predicts subsequent disease progression. Similarly, age at LoA is predictive of age at respiratory decline [9].
Respiratory function in patients with DMD typically increases with increasing age and growth until just before a patient loses ambulation. After reaching a peak, forced vital capacity (FVC) values start to decline over time [10]. Patients with DMD then require increasing levels of respiratory intervention starting at nighttime, and ultimately need continuous assisted ventilation. Disease milestones indicative of increasing deterioration in respiratory function and disease progression are percentage predicted FVC of <60%, <50% and <30%, and absolute FVC of <1 l [9–13]. Predicted FVC of <60% is indicative of the first need for intervention using lung volume recruitment, when patients require mechanical ventilation (through a manual ventilation bag or an insufflation–exsufflation device) to preserve lung function [10]. Predicted FVC of <50% is indicative of the need for assisted coughing techniques and nocturnal-assisted ventilation; noninvasive ventilation is strongly recommended [10]. Once patients with DMD have declined to a predicted FVC of <30% they are considered to have severe respiratory insufficiency, for which noninvasive ventilation is necessary [9,11]. Absolute FVC decline to <1 l is a threshold that is strongly predictive of mortality within 3 years and is associated with a fourfold increased risk of death [12,13]. Given the clear association between respiratory function and disease progression, including mortality, FVC is an important assessment, especially for patients in the nonambulatory stage of the disease.
Approximately 10–15% of patients with DMD have the disease owing to a nonsense mutation in the DMD gene [14,15]. A nonsense mutation results in a premature stop codon in the protein-coding region of the mRNA [16]. When a premature stop codon is present, ribosomal translation of mRNA is interrupted before a full-length functional protein is generated. The resulting truncated protein is unstable and rapidly degraded, leading to the typical absence of dystrophin in patient muscle biopsies. Genetic testing is used to determine the presence of such a mutation in the DMD gene and thereby diagnose patients with nonsense mutation DMD (nmDMD) [17].
Ataluren is an oral therapy for ambulatory patients with nmDMD [17]. It is a small molecule that is designed to enable the ribosome to read through a premature stop codon in the mRNA so that translation continues as normal, although with reduced efficiency, resulting in the production of a full-length dystrophin protein [18]. By restoring the production of dystrophin, treatment with ataluren helps to preserve muscle mass and prolong function in patients with nmDMD. Ataluren 40 mg/kg/day is indicated for the treatment of nmDMD in ambulatory patients aged at least 2 years in member states of the European Union, Belarus, Brazil, Great Britain, Northern Ireland, Iceland, Israel, Kazakhstan, Liechtenstein, Norway, the Republic of Korea and Russia, or aged at least 5 years in Chile and Ukraine (under special state registration in Ukraine) [17]. In Brazil, the indication is restricted to pediatric male patients.
The efficacy of ataluren has previously been investigated in two randomized, placebo-controlled studies: PTC124-GD-007-DMD (Study 007; ClinicalTrials.gov identifier: NCT00592553) and PTC124-GD-020-DMD (Study 020; ClinicalTrials.gov identifier: NCT01826487) [19,20]. Even though these two studies did not meet their primary end points, evaluation of the totality of the data obtained indicates that ataluren dosed at 40 mg/kg/day (10 mg/kg in the morning, 10 mg/kg at midday and 20 mg/kg in the evening) can delay disease progression compared with placebo as measured by a range of motor function end points over a 48-week period [19–22]. Furthermore, ataluren is well tolerated in patients with nmDMD [19,20].
The safety and effectiveness of ataluren have also been demonstrated by data from the STRIDE (Strategic Targeting of Registries and International Database of Excellence) Registry, an ongoing, multicenter registry providing real-world data on ataluren use in patients with nmDMD in clinical practice [23,24]. Safety outcomes in the STRIDE Registry were consistent with the known safety profile of ataluren. Propensity-score matched analyses demonstrated that ataluren 40 mg/kg/day plus standard of care (SoC) significantly delayed the age at LoA in patients with nmDMD in the STRIDE Registry compared with patients with DMD receiving SoC alone in an external natural history cohort, the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS; ClinicalTrials.gov identifier: NCT00468832) [23]. SoC refers to various mutation nonspecific therapies recommended for the management of DMD, including corticosteroid treatment regimens [10,25,26].
Study PTC124-GD-019-DMD (019; ClinicalTrials.gov identifier: NCT01557400) was a long-term safety study that included patients from prior ataluren clinical studies who had been treated at sites outside the USA. Several efficacy assessments were also collected in this study, including age at LoA and age at decline in respiratory function as measured by FVC end points. Herein, we describe the effect of ataluren on these key clinically meaningful milestones of disease progression by comparing patients with nmDMD treated with ataluren plus SoC in Study 019 with patients with DMD treated with SoC alone in the CINRG DNHS.
Patients & methods
Study design & methodology
Study 019 was a Phase III, long-term, multicenter, international, open-label study designed to assess the safety and tolerability of ataluren in patients with nmDMD who had received ataluren in prior PTC Therapeutics-sponsored studies at investigational sites outside USA. The study was conducted at 21 sites in ten countries. Ataluren was manufactured by PTC Therapeutics International Limited (Dublin, Ireland) and was supplied by PTC Therapeutics at all study sites. The primary objective of this study was to assess the long-term safety and tolerability of ataluren at a dose of 40 mg/kg/day (10 mg/kg in the morning, 10 mg/kg at midday and 20 mg/kg in the evening) in patients with nmDMD. Secondary objectives explored the long-term efficacy of ataluren and included measuring the ages at LoA and decline in respiratory function (i.e., to defined FVC events). Respiratory function was only evaluated in nonambulatory patients (i.e., throughout the study for those who were nonambulatory upon entering the study, or once the patient had lost ambulation for those who were ambulatory at study start and transitioned to a nonambulatory status during the study). Of note, patients who were nonambulatory at study entry had previously received ataluren in a clinical trial during which they were ambulatory.
Study design and data collection methodology for the CINRG DNHS have been described in full previously [27–29]. The study enrolled patients with DMD (i.e., various DMD genotypes; not only those with nmDMD) aged 2–28 years at 20 centers in nine countries between 2006 and 2016. Because Study 019 did not contain a placebo control arm, data from patients receiving SoC in the CINRG DNHS are used in the present analysis as a control to provide context for assessing the effects of ataluren plus SoC in Study 019 patients. Here, SoC refers to corticosteroid and palliative therapies.
Patient eligibility & enrollment
Study 019
Eligible patients were males with nmDMD who had a history of exposure to ataluren in a prior PTC Therapeutics-sponsored study in nmDMD. The largest recruitment pool for Study 019 consisted of patients who had completed the prior randomized, controlled, Phase IIb study of 48 weeks' duration (Study 007) and the subsequent open-label extension study (Study 007e; ClinicalTrials.gov identifier: NCT00847379). Patients who had participated in the Phase IIa study (Study 004; ClinicalTrials.gov identifier: NCT00264888) and the subsequent open-label extension study (Study 004e; ClinicalTrials.gov identifier: NCT00759876) were also included in the trial. One patient did not have previous exposure to ataluren and entered Study 019 through an institutional review board (IRB)- and US FDA-approved special exemption.
CINRG DNHS
Patient eligibility for the CINRG DNHS has been described previously [28,29]. Briefly, patients aged 2–4 years were required to have a diagnosis of DMD confirmed by one or more of the following: dystrophin immunofluorescence, dystrophin immunoblot, an out-of-frame deletion or complete DMD gene sequencing in the proband or sibling. Patients aged 5–28 years were required to meet the same criteria or to have documented clinical symptoms of DMD. Direct support of the diagnosis by either a positive DNA analysis, a muscle biopsy showing abnormal dystrophin, or a combination of increased CK levels and an X-linked pedigree was also required. In total, 440 patients were enrolled in the CINRG DNHS. Patients were excluded from the present analyses if they received investigational drugs in previous clinical trials.
Statistical analyses
Study populations
The as-treated population in Study 019 included all patients who received at least one dose of ataluren. Study 019 patients were considered ambulatory if they had a 10-m walk/run time of 30 s or less at screening and nonambulatory if their 10-m walk/run time was more than 30 s at screening. Nonambulatory patients from the CINRG DNHS were defined as such when they self-reported full-time wheelchair use. Data for continuous and categorical variables are presented descriptively.
Study 019 safety & clinical laboratory data
Adverse events (AEs) were classified using the Medical Dictionary for Regulatory Activities (version 20.1) classification system. AE severity was graded by the investigator according to the Common Terminology Criteria for Adverse Events (CTCAE), Version 3.0, whenever possible. A treatment emergent AE (TEAE) was defined as an AE that occurred or worsened in the period extending from the day of a patient's first ataluren dose in this study to 6 weeks after the last ataluren dose in this study.
Clinical laboratory analysis methodology is described in the Supplementary Materials.
Study 019 & CINRG DNHS data comparisons
To remove bias and allow for a robust comparison between Study 019 and CINRG DNHS, propensity score matching (one-to-one) was used to identify patients in Study 019 who were similar to those in the CINRG DNHS according to established predictors of disease progression [23,30]. The propensity score was created using a logistic regression model with the following four covariates, which are known predictors of disease progression: age at first clinical symptoms, age at initiation of corticosteroids, duration of deflazacort use (<1 month, ≥1 month to <12 months and ≥12 months) and duration of other corticosteroid use (<1 month, ≥1 month to <12 months and ≥12 months) [28,31,32]. Mutation type was not used as a covariate for propensity score matching because there were only 26/440 (5.9%) patients in the CINRG DNHS with nmDMD, meaning if mutation type had been a covariate, this would have substantially reduced the number of patients available for a statistically meaningful analysis.
It should be stated that in Study 019, the age at first symptoms was not recorded, which makes it unavailable for use as a covariate for propensity score matching with patients with DMD from the CINRG DNHS, as was the case with analyses of the STRIDE ataluren real-world registry data [23]. As an alternative for this assessment, we decided to use age at diagnosis since those data were collected in Study 019. However, to confirm that the use of age at diagnosis as a proxy for the age at first symptoms covariate did not result in inappropriately matched study cohorts, sensitivity analyses in which the age at diagnosis was used as the fourth covariate in both Study 019 and CINRG DNHS populations were conducted for completeness.
Following the calculation of a propensity score for each eligible patient in Study 019 and the CINRG DNHS, a matching control patient from the CINRG DNHS to each Study 019 patient was identified using a local optimal (greedy) algorithm based on nearest neighbor approaches without replacement [33]. Based on this approach, patients from Study 019 and CINRG DNHS were first randomly sorted and the first patient from Study 019 was selected to find the closest matching patient from CINRG DNHS. The closest match was based on the absolute value of the difference between their propensity scores (predicted probability from the logistic regression model). Once a patient from CINRG DNHS was chosen as the closest match, this patient was not available for further matching. The procedure was repeated for all Study 019 patients, resulting in a one-to-one match.
To be eligible for the propensity-score matched analysis of age at LoA, patients must have had available data for age at LoA and the four covariates used for matching. To be eligible for the propensity-score matched analysis of age at decline in respiratory function, patients must have been nonambulatory and had available data for age at LoA and the three respiratory end points described below and the four covariates used for matching, and must not have experienced a decline to below one of the FVC end points listed below before Study 019 entry. The age at LoA was defined as the age at which disease progression to LoA was reported as an event or the age at the first of two consecutive visits at which the patient consistently took more than 30 s to run/walk 10 m, whichever occurred earlier, for patients in Study 019. For CINRG DNHS patients, age at LoA was defined as the age at self-reported full-time wheelchair use.
Kaplan–Meier analysis and Cox regression were used to estimate the distribution of age at LoA and age at which FVC declined to <60% predicted, <50% predicted, <30% predicted and <1 l (absolute). For each time-to-event variable, the median age at the event was determined for the Study 019 and CINRG DNHS populations separately, and the 95% confidence intervals (CIs) were determined by log–log transformation. Distribution of the variables was compared between Study 019 and CINRG DNHS populations using a log-rank test stratified by the duration of deflazacort use and the duration of other corticosteroid use (<1 month, ≥1 month to <12 months and ≥12 months). The corticosteroid durations were calculated up to the time of LoA or the latest time that the patient was still known to be ambulatory (for analyses of LoA), or to the time at which the respiratory function end point (predicted FVC <60%, predicted FVC <50%, predicted FVC <30% or FVC <1 l) was reached for respiratory function analyses. Gaps between corticosteroid use were excluded and overlaps of different corticosteroid use were not double counted.
Results
Patient disposition, demographics & baseline characteristics
The duration of Study 019 was 240 weeks (~4.5 years), or up to 336 weeks in Canada. In total, 94 male patients with nmDMD were enrolled in the study, of whom 90 had participated in both the Phase IIb pivotal study (Study 007) [19] and the extension study (Study 007e), three had participated in both the Phase IIa study (Study 004) [34] and extension study (Study 004e), and one had not had prior ataluren exposure and had petitioned to be allowed into the study (Figure 1).
Figure 1. . The different study populations used in the analyses presented herein.

aThe as-treated population included patients with nmDMD who received at least one dose of ataluren during Study 019.
bThis patient had not participated in a previous trial involving ataluren and was included in Study 019 due to a successful petition.
cThe four covariates used for propensity score matching were: age at first clinical symptoms; age at initiation of corticosteroids; duration of deflazacort use (<1 month, ≥1 month to <12 months, and ≥12 months); and duration of other corticosteroid use (<1 month, ≥1 month to <12 months and ≥12 months).
dThe four respiratory outcome measures assessed were: predicted FVC <60%; predicted FVC <50%; predicted FVC <30%; and absolute FVC <1 l. These respiratory outcome measures were only performed in nonambulatory patients.
eStudy 007; Study 007e; Study 004; Study 004e.
fThe 33 patients comprised patients who (a) received placebo in Study 007 and ataluren 80 mg/kg/day in Study 007e, (b) received ataluren 80 mg/kg/day in Study 007 and ataluren 80 mg/kg/day in Study 007e, or (c) received ataluren 80 mg/kg/day in Study 004 and ataluren 80 mg/kg/day in Study 004e.
FVC: Forced vital capacity; LoA: Loss of ambulation; nmDMD: Nonsense mutation Duchenne muscular dystrophy; SoC: Standard of care.
The mean (standard deviation [SD]) treatment duration in Study 019 was 988 (590) days (equivalent to 2.7 years) before LoA. Not all patients immediately entered Study 019 from a prior ataluren study; the mean (SD) treatment gap between the prior studies and Study 019 for the 93 patients who had participated in previous trials was 2.9 (0.5) years. At study entry, 50 patients were ambulatory and 44 patients were nonambulatory (Table 1). The mean (SD) age of patients enrolled in Study 019 at baseline was 12.8 (2.4) years. Patients who were nonambulatory were heavier and had a higher body mass index, as expected and likely owing to decreased physical activity and caloric expenditure relative to ambulatory patients. Of the 50 ambulatory patients and 44 nonambulatory patients in Study 019, 47 (94%) and 37 (84%) were receiving corticosteroids, respectively.
Table 1. . Baseline demographics and characteristics for all patients with nonsense mutation Duchenne muscular dystrophy receiving ataluren 40 mg/kg/day plus standard of care in Study 019 (as-treated population).
| Parameter, n (%) | Ambulatory (n = 50) | Nonambulatory (n = 44) | Overall (n = 94) |
|---|---|---|---|
| Age (years) | 12.1 (2.1) | 13.7 (2.5) | 12.8 (2.4) |
| Age groups, n (%) 6 to ≤11 years 12 to ≤17 years ≥18 years |
18 (36.0) 31 (62.0) 1 (2.0) |
6 (13.6) 34 (77.3) 4 (9.1) |
24 (25.5) 65 (69.1) 5 (5.3) |
| Race, n (%) Caucasian Asian Other |
46 (92.0) 3 (6.0) 0 |
41 (93.2) 1 (2.3) 2 (4.5) |
87 (92.6) 4 (4.3) 2 (2.1) |
| Weight (kg) | 39.5 (9.5) | 53.1 (15.0) | 45.8 (14.0) |
| Height (cm)† | 131.6 (11.1) | 135.0 (7.0) | 132.1 (10.7) |
| BMI (kg/m2) | 22.8 (4.6) | 26.7 (4.8) | 23.3 (4.8) |
| Corticosteroid use, n (%)‡ Prednisone/prednisolone Deflazacort |
47 (94.0) 14 (28.0) 35 (70.0) |
37 (84.1) 21 (47.7) 16 (36.4) |
84 (89.4) 35 (37.2) 51 (54.3) |
| Time to walk/run 10 m, s | 8.4 (4.7) | 37.0 (N/A)§ | 8.9 (6.1) |
| FVC (l)¶,# | N/A | 1.9 (0.5) | NA |
| % predicted FVC¶,# | N/A | 72.7 (20.6) | NA |
Baseline values were the last non-missing numeric value on or before the first dose of study medication. Data are mean (SD) unless indicated otherwise.
Height values for some nonambulatory patients were not collected.
Patients could be treated with more than one corticosteroid.
Data for one of the 44 patients were available for the time to walk/run 10 m assessment before the first dose of study treatment was administered. Despite this one patient being defined as nonambulatory at Study 019 entry as per the definition of taking >30 s to run/walk 10 m, he completed baseline assessments intended for ambulatory patients (including time to walk/run 10 m, s).
Baseline respiratory function assessments were only performed for nonambulatory patients.
One of the 44 nonambulatory patients had missing baseline FVC and percentage predicted FVC values.
FVC: Forced vital capacity; NA: Not applicable; SD: Standard deviation.
Of 440 patients enrolled in the CINRG DNHS, 22 had participated in previous clinical trials of ataluren or had received eteplirsen, drisapersen or tadalafil and were thus excluded before propensity score matching (n = 418) [23]. Before propensity score matching, differences were observed between the Study 019 and CINRG DNHS control cohorts with respect to non-deflazacort corticosteroid use and time to stand from supine (P-values <0.05) (Table 2). A propensity score was used to match patients with similar disease characteristics in Study 019 with those in the CINRG DNHS (see Patients & Methods for additional detail).
Table 2. . Baseline demographics and disease characteristics for all patients with nonsense mutation Duchenne muscular dystrophy receiving ataluren 40 mg/kg/day plus standard of care in Study 019 (as-treated population) and patients with Duchenne muscular dystrophy receiving standard of care alone in the CINRG DNHS, before propensity score matching.
| Assessment | Study 019 (n = 94) | CINRG DNHS (n = 418) | p-value |
|---|---|---|---|
| Age at first symptoms, years† | 0.0634‡ | ||
| n | 405 | ||
| Mean (SD) | NA | 3.2 (1.7) | |
| Age at diagnosis, years | |||
| n | 93 | 417 | |
| Mean (SD) | 3.6 (1.9) | 4.4 (2.1) | |
| Age at corticosteroid initiation, years§ | |||
| n | 94 | 417 | |
| Mean (SD) | 13.0 (9.5) | 11.5 (9.7) | 0.1828 |
| Deflazacort duration, n (%)¶ | |||
| <1 month | 48 (51.1) | 249 (59.6) | 0.0800 |
| ≥1 to <12 months | 2 (2.1) | 21 (5.0) | |
| ≥12 months | 44 (46.8) | 148 (35.4) | |
| Other corticosteroid duration, n (%)¶ | |||
| <1 month | 66 (70.2) | 216 (51.7) | 0.0046 |
| ≥1 to <12 months | 4 (4.3) | 35 (8.4) | |
| ≥12 months | 24 (25.5) | 167 (40.0) | |
| Baseline 6MWD, m | |||
| n | 90 | 134 | |
| Mean (SD) | 358.3 (99.1) | 350.1 (123.6) | 0.5808 |
| Time to climb four stairs at first assessment, s# | |||
| n | 92 | 250 | |
| Mean (SD) | 6.6 (6.9) | 6.5 (5.4) | 0.9118 |
| Time to walk/run 10 m at first assessment, s# | |||
| n | 92 | 261 | |
| Mean (SD) | 7.4 (4.6) | 7.5 (3.9) | 0.8131 |
| Time to stand from supine at first assessment, s# | |||
| n | 92 | 230 | |
| Mean (SD) | 10.5 (10.3) | 6.9 (4.8) | 0.0019 |
P-values were calculated based on a two-sample t-test for continuous variables or a χ2 test for categorical variables.
Data are mean (SD) unless indicated otherwise.
The patients' age at first symptoms was not captured in Study 019.
P-value is for the comparison between the age at diagnosis for Study 019 patients and age at first symptoms for CINRG DNHS patients.
Age at initiation of corticosteroid use for steroid-naive patients (patients who had never used steroids or used steroids after loss of ambulation) in Study 019 was set to 30 years.
Corticosteroid duration is calculated from starting use of corticosteroid to loss of ambulation/censored date.
Time to climb four stairs, walk/run 10 m, and stand from supine at first assessment were determined using baseline values from the prior ataluren studies that the patients were enrolled in, i.e., Study 007/007e or Study 004/004e.
6MWD: 6-minute walking distance; NA: Not available; SD: Standard deviation.
Age at LoA
To evaluate the impact of ataluren therapy on the median age at LoA, Kaplan–Meier analyses were performed to address whether patients who received the 40 mg/kg/day dose of ataluren plus SoC in Study 019 lose ambulation at a later age than matched CINRG DNHS patients who received SoC alone.
The various study populations used for the analyses presented here are shown in Figure 1. In total, 85 patients in Study 019 had a date for LoA and data for the four covariates used for propensity score matching and were therefore eligible for propensity score matching. Of these 85 patients, 60 were receiving ataluren 40 mg/kg/day plus SoC at the point of LoA or censoring (i.e., when the final data collection was completed for that patient). This group was used for a comparative analysis of the age at LoA in patients receiving ataluren 40 mg/kg/day relative to matched patients receiving SoC in the CINRG DNHS. The 60 patients in this analysis were comprised of two subgroups of patients. The first subgroup consisted of 27 patients who received ataluren 40 mg/kg/day plus SoC in prior clinical trials (Studies 004 and 004e or 007 and 007e) and Study 019, and were either ambulatory (16 patients) or nonambulatory (11 patients) at entry to Study 019. The second subgroup consisted of 33 patients who received placebo and then ataluren 80 mg/kg/day (20 mg/kg in the morning, 20 mg/kg at midday and 40 mg/kg in the evening) or only ataluren 80 mg/kg/day plus SoC during prior clinical trials and began treatment with ataluren 40 mg/kg/day plus SoC upon entry to Study 019, and were ambulatory at entry to Study 019.
The 60-patient ataluren treatment group had similar baseline demographics and disease characteristics to propensity-score matched CINRG DNHS controls (Table 3). This was also the case for the 59-patient cohorts matched using the age at diagnosis covariate in both populations for the corresponding sensitivity analysis (Supplementary Table 1).
Table 3. . Baseline demographics and disease characteristics for Study 019 patients with nonsense mutation Duchenne muscular dystrophy who received ataluren (40 mg/kg/day) plus standard of care in at least Study 019 (n = 60) and for propensity-score matched patients with Duchenne muscular dystrophy receiving standard of care alone in the CINRG DNHS (n = 60), for the evaluation of loss of ambulation.
| Study 019 (n = 60) | CINRG DNHS (n = 60) | p-value | |
|---|---|---|---|
| Age at first symptoms, years† | |||
| Mean (SD) | NA | 3.9 (1.7) | 0.3859‡ |
| Age at diagnosis, years | |||
| Mean (SD) | 3.6 (2.0) | 4.9 (2.3) | |
| Age at corticosteroid initiation, years§ | |||
| Mean (SD) | 10.9 (8.1) | 10.1 (8.1) | 0.6182 |
| Deflazacort duration, n (%)¶ | |||
| <1 month | 24 (40.0) | 27 (45.0) | 0.6865 |
| ≥1 to <12 months | 1 (1.7) | 2 (3.3) | |
| ≥12 months | 35 (58.3) | 31 (51.7) | |
| Other corticosteroid duration, n (%)¶ | |||
| <1 month | 37 (61.7) | 37 (61.7) | 0.6816 |
| ≥1 to <12 months | 4 (6.7) | 2 (3.3) | |
| ≥12 months | 19 (31.7) | 21 (35.0) | |
| Time to climb four stairs at first assessment, s# | |||
| n | 60 | 31 | |
| Mean (SD) | 5.3 (5.9) | 6.9 (6.5) | 0.2247 |
| Time to walk/run 10 m at first assessment, s# | |||
| n | 60 | 33 | |
| Mean (SD) | 6.6 (4.2) | 8.2 (4.5) | 0.0851 |
| Time to stand from supine at first assessment, s# | |||
| n | 60 | 26 | |
| Mean (SD) | 7.8 (8.5) | 7.2 (5.9) | 0.7296 |
P-values were calculated based on a two-sample t-test for continuous variables or a χ2 test for categorical variables.
The patients' age at first symptoms was not captured in Study 019.
P-value is for the comparison between the age at diagnosis for Study 019 patients and age at first symptoms for CINRG DNHS patients.
Age at initiation of corticosteroid use for steroid-naive patients (patients who had never used steroids or used steroids after loss of ambulation) in Study 019 was set to 30 years.
Corticosteroid duration is calculated from starting use of corticosteroid to loss of ambulation/censored date.
Time to climb four stairs, walk/run 10 m, and stand from supine at first assessment were determined using baseline values from the prior ataluren studies that the patients were enrolled in, i.e., Study 007/007e or Study 004/004e.
NA: Not available; SD: Standard deviation.
Ataluren treatment at 40 mg/kg/day plus SoC was associated with a significant delay of approximately 2.2 years in age at LoA compared with SoC alone in the CINRG DNHS study. The median age at LoA for the 60 patients treated with ataluren 40 mg/kg/day plus SoC in Study 019 was 15.5 years, compared with 13.3 years in the matched CINRG DNHS cohort (p = 0.0006 [each n = 60]) (Figure 2). Data generated from a sensitivity analysis, in which age at diagnosis was used as the fourth covariate for propensity-score matching for both Study 019 and CINRG DNHS patient populations, yielded similar results: the median age at LoA was 15.5 and 12.0 years for Study 019 and CINRG DNHS cohorts, respectively (p = 0.0343 [each n = 59]) (Supplementary Figure 1).
Figure 2. . Age at loss of ambulation for Study 019 patients with nonsense mutation Duchenne muscular dystrophy who received ataluren 40 mg/kg/day plus standard of care in at least Study 019† compared with propensity-score matched patients with Duchenne muscular dystrophy receiving standard of care alone in the CINRG DNHS (each n = 60).
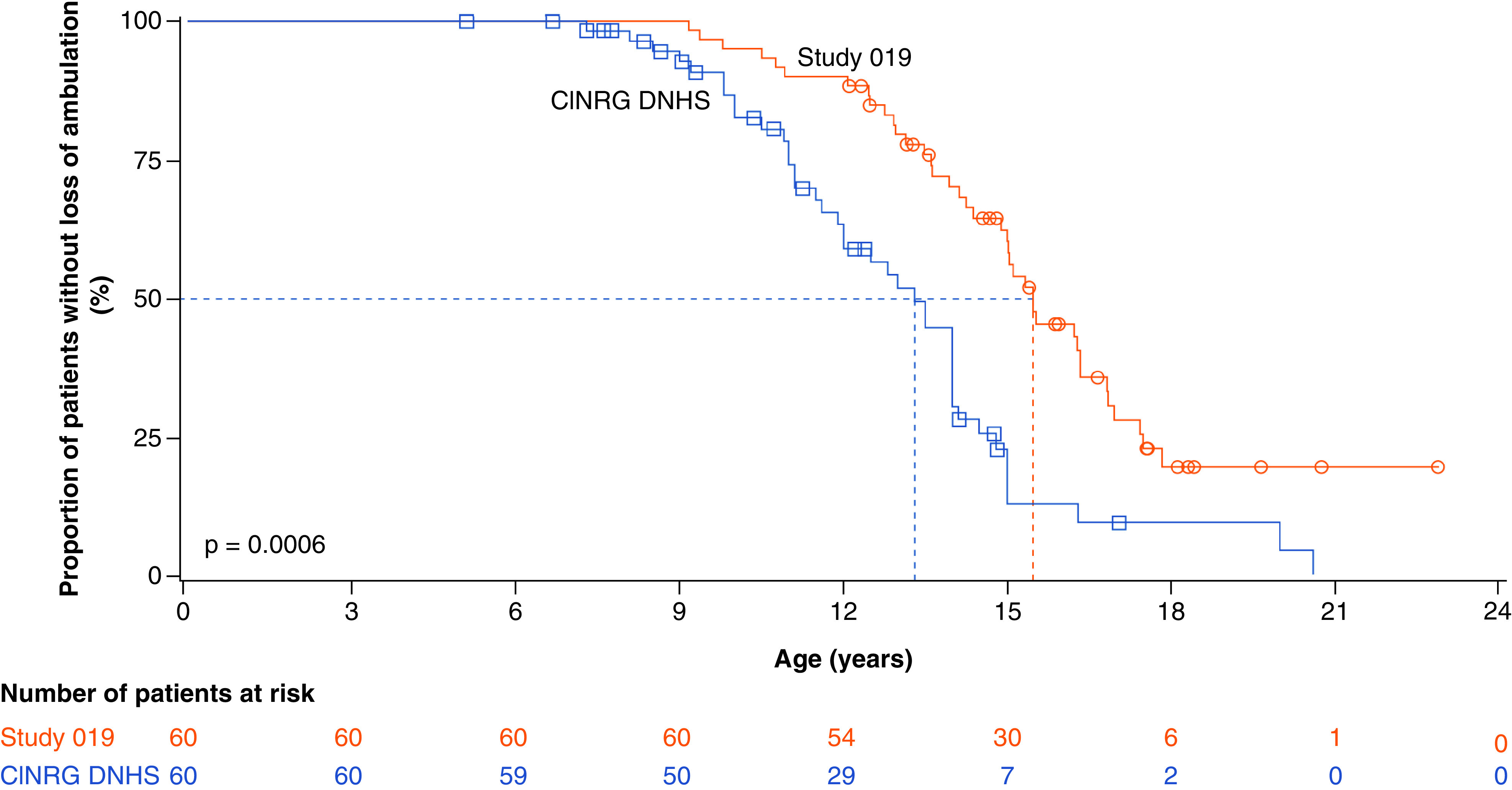
†The 60 patients in this analysis are comprised of 27 patients who received 40 mg/kg/day ataluren plus standard of care (SoC) in prior ataluren studies and 33 patients who received placebo and then ataluren 80 mg/kg/day or only ataluren 80 mg/kg/day plus SoC in prior clinical trials, and began treatment with ataluren 40 mg/kg/day plus SoC upon entry to Study 019, and were ambulatory at entry to Study 019.
Age at decline in respiratory function
Kaplan–Meier analyses were also used to compare the decline in respiratory function between nonambulatory patients treated with ataluren 40 mg/kg/day plus SoC in at least Study 019 and those receiving SoC alone in the CINRG DNHS. The analysis of respiratory function included 45 of the 85 Study 019 patients (eligible for propensity score matching, with available data for age at LoA and the four covariates used for propensity score matching), who received ataluren 40 mg/kg/day plus SoC in Study 019 and any of placebo, ataluren 40 mg/kg/day or ataluren 80 mg/kg/day in prior clinical trials; had data for the three respiratory outcome measures assessed below; who did not experience a decline to one of these FVC milestones before Study 019 entry; and who were nonambulatory at study data cut-off. At Study 019 entry, 18 patients were ambulatory and 27 patients were nonambulatory. As this analysis used FVC data gathered throughout Study 019 for patients who were nonambulatory at study entry or data only gathered once patients lost ambulation in Study 019 for patients who were ambulatory at study entry, only nonambulatory patients are included in this analysis. As for the analysis of age at LoA, Study 019 and CINRG DNHS patients were matched using propensity scoring as described previously. The resulting matched populations were similar, with no significant differences across a range of important baseline demographic and disease state characteristics (Table 4). This was also the case for the 45-patient cohorts matched using the age at diagnosis covariate in both populations for the corresponding sensitivity analysis (Supplementary Table 2).
Table 4. . Baseline demographics and disease characteristics for nonambulatory Study 019 patients with nonsense mutation Duchenne muscular dystrophy who received ataluren 40 mg/kg/day plus standard of care in at least this study (n = 45) and propensity-score matched patients with Duchenne muscular dystrophy who received standard of care alone in the CINRG DNHS (n = 45), for evaluation of respiratory function.
| Study 019 (n = 45) | CINRG DNHS (n = 45) | p-value | |
|---|---|---|---|
| Age at first symptoms, years† | 0.4938‡ | ||
| Mean (SD) | NA | 3.5 (1.5) | |
| Age at diagnosis, years | |||
| Mean (SD) | 3.8 (1.8) | 4.8 (2.2) | |
| Age at initiation of corticosteroids, years§ | |||
| Mean (SD) | 10.6 (6.9) | 10.2 (7.4) | 0.8066 |
| Deflazacort duration, n (%)¶ | |||
| <1 month | 22 (48.9) | 20 (44.4) | 0.5722 |
| ≥1 to <12 months | 0 (0.0) | 1 (2.2) | |
| ≥12 months | 23 (51.1) | 24 (53.3) | |
| Other corticosteroid duration, n (%)¶ | 0.8385 | ||
| <1 month | 26 (57.8) | 27 (60.0) | |
| ≥1 to <12 months | 2 (4.4) | 1 (2.2) | |
| ≥12 months | 17 (37.8) | 17 (37.8) | |
| Time to climb four stairs at first assessment, s# | |||
| n | 45 | 23 | |
| Mean (SD) | 7.1 (7.7) | 10.8 (8.5) | 0.0777 |
| Time to walk/run 10 m at first assessment, s# | |||
| n | 45 | 30 | |
| Mean (SD) | 8.2 (5.7) | 9.3 (4.8) | 0.3769 |
| Time to stand from supine at first assessment, s# | |||
| n | 45 | 17 | |
| Mean (SD) | 12.4 (11.1) | 9.2 (5.9) | 0.1488 |
P-values were calculated based on a two-sample t-test for continuous variables or a χ2 test for categorical variables.
The patients' age at first symptoms was not captured in Study 019.
P-value is for the comparison between the age at diagnosis for Study 019 patients and age at first symptoms for CINRG DNHS patients.
Age at initiation of corticosteroid use for steroid-naive patients (patients who had never used steroids or used steroids after loss of ambulation) in Study 019 was set to 30 years.
Corticosteroid duration is calculated from starting use of corticosteroid to loss of ambulation/censored date.
Time to climb four stairs, walk/run 10 m, and stand from supine at first assessment were determined using baseline values from the prior ataluren studies that the patients were enrolled in, i.e., Study 007/007e or Study 004/004e.
NA: Not available; SD: Standard deviation.
Ataluren treatment (40 mg/kg/day) plus SoC resulted in a 3.0-year delay in patients' predicted FVC declining to <60% compared with SoC alone. In the matched populations, 23 of 45 (51.1%) patients in Study 019 and 32 of 45 (71.1%) patients in the CINRG DNHS experienced a decline to below the 60% predicted FVC threshold. The median age for this milestone was 18.1 years in Study 019 and 15.1 years in the CINRG DNHS (p = 0.0004) (Figure 3A).
Figure 3. . Age at (A) predicted forced vital capacity <60%, (B) predicted forced vital capacity <50%, (C) predicted forced vital capacity <30% and (D) forced vital capacity <1 l (absolute), and (E) the percentage predicted forced vital capacity since loss of ambulation, for patients with nonsense mutation Duchenne muscular dystrophy who received ataluren 40 mg/kg/day plus standard of care in at least Study 019 (all n = 45), compared with propensity-score matched patients with Duchenne muscular dystrophy who received SoC alone in the CINRG DNHS (n = 45).
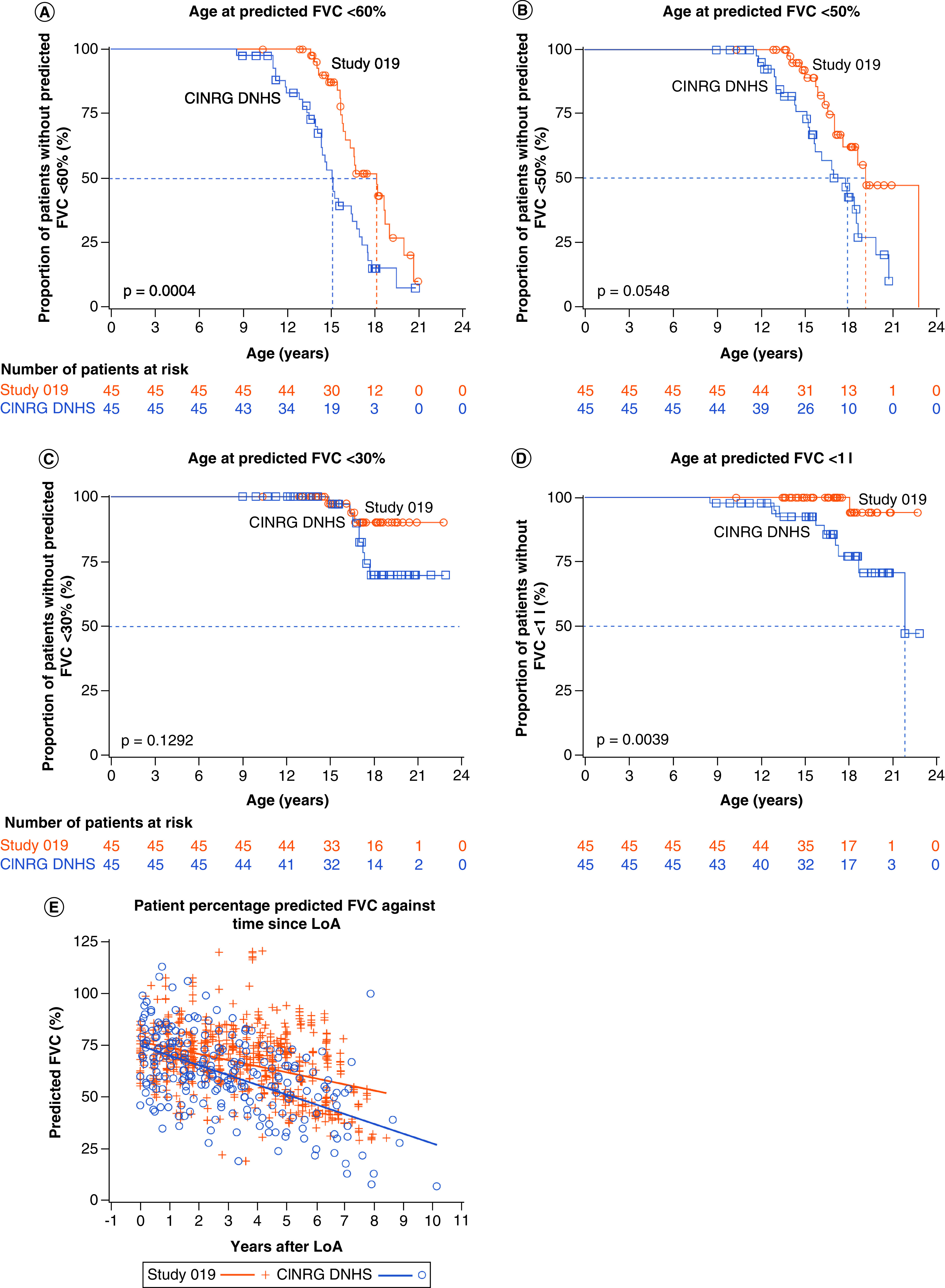
FVC: Forced vital capacity.
To evaluate whether the delay in decline to predicted FVC of <60% is not simply reflective of the 2.2-year delay in LoA, the time from LoA to when patients reached predicted FVC <60% was compared for propensity-score matched Study 019 and CINRG DNHS patients. Patients who received ataluren 40 mg/kg/day plus SoC in Study 019 took longer to reach predicted FVC <60% following LoA relative to patients receiving SoC alone in the CINRG DNHS (median duration of 4.9 years [n = 45] and 3.6 years [n = 35], respectively; p = 0.2190). Furthermore, a scatter plot of percentage predicted FVC over time since losing ambulation indicated a more gradual decline in respiratory function in patients treated with ataluren 40 mg/kg/day plus SoC compared with patients receiving SoC alone in the CINRG DNHS (Figure 3E).
In the propensity-score matched populations, 14 of 45 (31.1%) Study 019 patients and 24 of 45 (53.3%) patients in the CINRG DNHS experienced a decline to below the 50% predicted FVC threshold at a median age of 19.1 and 17.8 years, respectively (p = 0.0548) (Figure 3B), indicating that ataluren therapy may delay the decline in respiratory function to an FVC of <50% predicted by up to approximately 1 year in patients with nmDMD.
Among the matched populations, 3 of 45 (6.7%) Study 019 patients and 8 of 45 (17.8%) CINRG DNHS patients declined to below 30% predicted FVC. However, owing to the infrequency of events for this advanced-stage disease milestone, the median age for this milestone could not be estimated for patients in either Study 019 or CINRG DNHS (Figure 3C).
FVC below a threshold of 1 l is strongly predictive of mortality within 3 years and is associated with a fourfold increased risk of death [12,13]. One (2.2%) patient out of 45 in Study 019 and nine patients out of 45 (20.0%) in the CINRG DNHS experienced a decline in respiratory function below this critical threshold. Whereas the median age for this milestone could not be estimated for patients in Study 019, owing to the infrequency of events, the median age at decline to an FVC of <1 l for the matched CINRG DNHS patients was 21.9 years (Figure 3D).
As for the LoA analysis, a sensitivity analysis conducted using the age at diagnosis as the fourth covariate for both study populations generated similar data demonstrating the same trends to those from the respiratory function analyses presented here (Supplementary Figure 2).
Safety of ataluren
The safety profile of ataluren observed up to 336 weeks in Study 019 was consistent with other ataluren studies, and no new ataluren risks were identified. Overall, 91 of 94 patients (96.8%) experienced a total of 1282 TEAEs (Supplementary Table 3). The majority of TEAEs were mild or moderate in severity (54/94 [57.4%]) and TEAEs that were considered drug related were observed in 26 of 94 patients (27.7%). There were no life-threatening TEAEs. Thirty-one patients (33.0%) experienced serious adverse events (SAEs): the SAEs in all but one of these patients were considered by the investigator unrelated to ataluren. Two patients experienced two SAEs each that led to death; none of these events were considered by the investigator related to the study drug. Clinical laboratory assessments are reported in the Supplementary Materials.
Discussion
LoA and decline in respiratory function to critical thresholds are major disease milestones in DMD and prognostic of mortality risk [12,28]. Study 019 was a long-term safety study that also evaluated the age at LoA and decline in respiratory function in patients treated with ataluren 40 mg/kg/day. The findings presented here are consistent with ataluren delaying disease progression in both ambulatory and nonambulatory patients. Ataluren 40 mg/kg/day on top of SoC was associated with a delay in the age at LoA of approximately 2.2 years. Ataluren was also associated with an approximate 3.0-year delay in predicted FVC decreasing to <60% and an approximate 1.3-year delay in predicted FVC dropping below 50%. Ataluren may also delay predicted FVC from declining to below 30% and absolute FVC from dropping below 1 l; however, the data regarding patient age at these milestones are limited by the infrequency of these advanced respiratory events, particularly in ataluren-treated patients, who were not as progressed in their disease as CINRG DNHS patients. Longer follow-up of ataluren-treated patients is necessary to further investigate such advanced disease milestones.
These results support prior findings which indicate that ataluren can delay disease progression in patients with nmDMD. The findings are consistent with long-term data from the ataluren STRIDE Registry, an ongoing, multicenter, observational study in patients with nmDMD being treated with ataluren. The STRIDE Registry is designed to collect information on the safety and effectiveness of ataluren in routine clinical practice. In interim analyses, the delay in the median age at LoA in the ataluren STRIDE Registry, in which the mean ataluren exposure was 632.5 days, was found to be approximately 3.5 years compared with CINRG DNHS participants [23]. Data from both the Phase IIb and Phase III 48-week, randomized clinical studies are consistent with ataluren delaying disease progression in ambulatory patients, as evaluated by the 6-min walk test (6MWT) and timed-function tests [19,20]. Furthermore, meta-analyses of the data from the Phase IIb and Phase III clinical studies of ataluren also demonstrated statistically significant differences in the change in 6MWT data over 48 weeks of ataluren therapy relative to placebo, further supporting a role for ataluren in the slowing of disease progression [21]. Last, a separate totality of evidence analysis of the data from these two clinical trials of ataluren also provided supporting evidence for ataluren performing statistically significantly better than placebo [22].
The delay in LoA observed in the current study is highly clinically meaningful. Delaying disease milestones is of major importance, particularly since the age at which patients reach certain disease milestones such as the LoA predicts the onset of subsequent disease milestones and the loss of other functional abilities later in life, indicative of disease progression [9,28]. For example, LoA is associated with the beginning of the deterioration of the respiratory muscles and heart, and the development of scoliosis [6,9]. Age at LoA is also predictive of the age at which patients reach respiratory insufficiency [9] and age at progression to the absolute FVC <1 l threshold [28]. Therefore, the delay in LoA by 2.5 years in Study 019, compared with well-matched natural history controls, represents not only a highly meaningful prolongation of personal autonomy in daily life, but also a delay in the onset of subsequent disease milestones.
The delay in loss of respiratory function observed in Study 019 is also clinically important. The potential preservation of respiratory function in patients with DMD is important as respiratory failure is one of the common causes of death in this disease [25]. Recent long-term data from 270 patients with DMD showed that corticosteroids, irrespective of daily or intermittent dosing, significantly improved respiratory function by delaying the yearly decline in percentage predicted FVC, and resulted in a delayed need for noninvasive ventilation [35]. The data presented here thereby support the idea that the dystrophin-restoring mechanism of action of ataluren could be beneficial to patients with nmDMD throughout different stages of disease by providing a further benefit on top of that already conferred by corticosteroids in terms of preservation of respiratory function.
Of note, the ataluren-treated patient population studied here was comprised of a cohort of patients who received ataluren for different lengths of time. Of the 60 ataluren-treated patients, 27 received ataluren 40 mg/kg/day plus SoC in prior clinical trials and Study 019, whereas 33 patients received either placebo followed by ataluren 80 mg/kg/day (a non-efficacious dose), or ataluren 80 mg/kg/day only, plus SoC in prior clinical trials and thereby only began treatment with the approved ataluren dose of 40 mg/kg/day plus SoC upon entry to Study 019. Inclusion of the 33 patients in this overall group of 60 ataluren-treated patients is a conservative approach, because the duration of treatment with ataluren 40 mg/kg/day is much shorter for these patients than for the other 27 participants.
Furthermore, the age at first ataluren dose for the ataluren-treated cohort was variable; patients who received ataluren earlier in their lifetime may have had greater delays in age at loss of ambulation, given that they would have likely irreversibly lost less muscle in the natural history of the disease than older patients before they received ataluren. The age at first ataluren dose may therefore have influenced the effectiveness results presented in this analysis. No further analyses have been completed evaluating the effect of the age at first ataluren dose on the age at decline in function.
To enable analysis of the Study 019 efficacy data, propensity score matching was used to identify patients in Study 019 who were similar to those in the CINRG DNHS according to established predictors of disease progression [23,30]: age at first clinical symptoms, age at initiation of corticosteroids, duration of deflazacort use, and duration of other corticosteroid use [28,31,32]. Age at the onset of first symptoms is prognostic for severity of disease; in general, the younger the age at first symptoms, the more severe the disease. A 1-year increase in the age at first symptoms is associated with a 10% reduction in annual risk of LoA [31]. Age at first corticosteroid use, duration of corticosteroid use, and duration of deflazacort use represent key factors that are known to alter the course of the disease [28]. Corticosteroid therapy, if started at a young age, can delay disease progression: patients who started corticosteroid therapy under the age of 5 years performed better in terms of the North Star Ambulatory Assessment (NSAA) total score, relative to those who started treatment after the age of 5 years [32]. Another analysis has shown that patients with a cumulative corticosteroid treatment duration of at least 1 year demonstrate delayed occurrence of multiple disease progression milestones relative to those treated for less than 1 month or not treated at all, illustrating the consistent benefit of corticosteroid treatment across the lifespan of a patient [28].
As mentioned in the methodology section, the age at first symptoms was not recorded in Study 019, and so was unavailable for use as a covariate for propensity score matching with patients with DMD from the CINRG DNHS. Age at diagnosis was thus used as an alternative for this assessment. Accepting that these two outcomes are not the same, and that age at first symptoms is a more appropriate predictor of future disease progression [31], we are confident that selection of age at diagnosis is a conservative proxy for age at first symptoms. The risk assumed in this approach accepts the probability of selecting patients from the CINRG DNHS with a milder disease phenotype than matched Study 019 patients. This conservative approach is confirmed by the sensitivity analysis performed using age at diagnosis as the covariate for both groups: the median age at loss of ambulation for CINRG DNHS patients was 13.3 when using age at diagnosis as the covariate in Study 019 and age at first symptoms as the covariate in the CINRG DNHS; and 12.0 years when using age at diagnosis as the covariate for both groups. The consistent differences in age at LoA and respiratory function decline observed between Study 019 and CINRG DNHS cohorts, where CINRG DNHS patients reach all events at an earlier age than Study 019 patients, via both the main analyses and sensitivity analyses presented here support the overall conclusions drawn regarding a treatment benefit of ataluren.
A potential limitation of the present study is the difference in the definition of age at LoA, and thereby ‘nonambulatory’, between the Study 019 and CINRG DNHS cohorts. For patients in Study 019, the age at LoA was defined as the age at which disease progression to LoA was reported as an event or the age at the first of two consecutive visits at which the patient consistently took more than 30 s to run/walk 10 m, whichever occurred earlier; whereas for CINRG DNHS patients, age at LoA was defined as the age at self-reported full-time wheelchair use. However, this subtle difference in the way that age at LoA is defined is unlikely to influence the outcome of the analyses presented here, as both definitions ultimately reflect the age at which a patient is no longer able to walk and the duration of time that a patient may spend between the two stages described in the definitions would be very short and would not likely affect FVC values. Other limitations include the heterogeneity of the Study 019 and CINRG DNHS cohorts and lack of information on concomitant medications for the Study 019 cohort; additionally, the effect of genetic modifier genes was not explored in these analyses.
Conclusion
Under the current standard of care, DMD remains a disease with devastating consequences and a poor prognosis, with survival to the fourth decade of life for a small fraction of patients [5–7]. As shown in this study, ataluren treatment was associated with a delay in the age at LoA and decline in respiratory function in patients with nmDMD. The respiratory findings extend prior results that were primarily in ambulatory patients and indicate that ataluren also demonstrates treatment benefit in nonambulatory patients. The progressive and irreversible effects of DMD underscore the importance of early intervention with treatments that have the potential to slow physical deterioration and delay the natural course of this fatal disease, which is considered an important treatment benefit in the DMD patient community [36]. The delay in loss of motor function associated with ataluren is not only clinically important but is highly meaningful to patients and their families since the loss of a physical function in DMD is irreversible.
Summary points.
Ataluren is an orally bioavailable treatment for patients with nonsense mutation Duchenne muscular dystrophy (nmDMD) which promotes readthrough of an in-frame premature stop codon to enable the production of full-length dystrophin.
Study 019 was a long-term, 240-week safety study of ataluren use that also examined the efficacy of ataluren in patients with nmDMD.
Propensity score matching was used to identify patients from Study 019 and the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) who were similar in established predictors of disease progression.
Kaplan–Meier analyses of propensity-score matched patients demonstrated that there was a significant ∼2.2-year delay in the age at loss of ambulation (p = 0.0006) for patients receiving ataluren plus standard of care (SoC [palliative therapies and corticosteroid treatment]) in Study 019 relative to patients receiving SoC alone in the CINRG DNHS.
Ataluren plus SoC was also associated with a significant ∼3.0-year delay in predicted forced vital capacity (FVC) deteriorating to <60% relative to SoC alone.
The data support previous findings showing that ataluren plus SoC delays motor function and respiratory milestones of DMD progression in patients with nmDMD and extend prior results showing that ataluren demonstrates treatment benefit in nonambulatory as well as ambulatory patients.
Supplementary Material
Acknowledgments
The authors thank the patients and their families for their participation in the studies included in this analysis. The authors also thank individuals involved in the conduct of these studies and the collection of data, particularly principal investigators, clinical evaluators and study coordinators of Study 019 and CINRG DNHS.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/cer-2021-0196
Author contributions
F Muntoni and M Tulinius were members of the Study 019 Steering Committee and enrolled patients at their study sites. J Jiang and A Kristensen were responsible for data analysis. V Penematsa and F Bibbiani were responsible for monitoring the clinical study. CM McDonald has served as CINRG DNHS Study Chair and contributed to the overall Study 019 study design, data analysis and CINRG DNHS design. All authors reviewed data, drafted and critically reviewed the manuscript and approved the final version for submission.
Financial & competing interests disclosure
This work was supported by PTC Therapeutics. Study 019 was sponsored by PTC Therapeutics. The CINRG DNHS was funded by grants from the US Department of Education/NIDRR (#H133B031118, #H133B090001), the US Department of Defense (#W81XWH-09-1-0592), the National Institutes of Health (#UL1RR031988, #U54HD053177, #UL1RR024992, #U54RR026139, #2U54HD053177, #G12RR003051, #1R01AR061875, #RO1AR062380) and Parent Project Muscular Dystrophy. CM McDonald has acted as a consultant on clinical trials of DMD for Astellas, BioMarin, Capricor Therapeutics, Catabasis Pharmaceuticals, Edgewise Therapeutics, Eli Lilly and company, Epirium Bio (formerly Cardero Therapeutics), FibroGen, Gilead, Hoffmann-La Roche, Italfarmaco, Pfizer, PTC Therapeutics, Santhera Pharmaceuticals and Sarepta Therapeutics, and has received research support for clinical trials from BioMarin, Capricor Therapeutics, Eli Lilly and company, Italfarmaco, Pfizer, PTC Therapeutics, Hoffmann-La Roche, Santhera Pharmaceuticals and Sarepta Therapeutics. F Muntoni has received consultancy fees from AveXis, Biogen, Capricor Therapeutics, Catabasis Pharmaceuticals, Dyne Therapeutics, Novartis, Pfizer, PTC Therapeutics, Roche, Santhera Pharmaceuticals, Sarepta Therapeutics and Wave Therapeutics, and is supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children National Health Service Foundation Trust and University College London. V Penematsa, J Jiang, A Kristensen, E Goodwin, C Werner, James Li, R Able and P Trifillis are employees of PTC Therapeutics. F Bibbiani is a former employee of PTC Therapeutics. H Gordish-Dressman has served as a consultant for AGADA Biosciences, Audentes Therapeutics, ReveraGen BioPharma and Solid GT, and is a cofounder and part owner of TRiNDS. L Morgenroth reports no disclosures relevant to this manuscript. M Tulinius has received lecture fees from Biogen and PTC Therapeutics, has acted as a consultant on DMD clinical trials for BioMarin, Catabasis Pharmaceuticals, PTC Therapeutics, ReveraGen BioPharma and Sarepta Therapeutics, and as an advisory board member for AveXis, Biogen, PTC Therapeutics and Sarepta Therapeutics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing and editorial support were provided by E Colbeck, PhD, an employee of PharmaGenesis London, London, UK, and were funded by PTC Therapeutics.
Ethical conduct of research
Before any patient was enrolled and study-related data were collected, ethics committee approval of the protocol, informed consent form and all patient enrollment materials were obtained in each country and for each site, as applicable. The study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki, applicable privacy laws and local regulations for each participating site, as well as with the Guidelines for Good Pharmacoepidemiology Practices. The protocol was approved by the appropriate institutional review board/ethics committee and all parents/patients provided signed informed consent.
Data sharing statement
Individual patient data will not be made publicly available. The study protocol is available indefinitely to interested parties on request.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Contributor Information
Collaborators: M Ryan, K Jones, N Goemans, C Campbell, JK Mah, K Selby, B Chabrol, Y Pereon, T Voit, T Gidaro, U Schara, JB Kirschner, Y Nevo, GP Comi, E Bertini, E Mercuri, J Colomer, A Nascimento, JJ Vilchez, M Tulinius, T Sejersen, F Muntoni, K Bushby, and M Guglieri
References
Papers of special note have been highlighted as: •• of considerable interest
- 1.Bushby K, Finkel R, Birnkrant DJ et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9(1), 77–93 (2010a). [DOI] [PubMed] [Google Scholar]
- 2.Ellis JA, Vroom E, Muntoni F. 195th ENMC International Workshop: newborn screening for Duchenne muscular dystrophy 14–16th December, 2012, Naarden, The Netherlands. Neuromusc. Disord. 23(8), 682–689 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 7(1), 13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryder S, Leadley RM, Armstrong N et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J. Rare Dis. 12(1), 79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landfeldt E, Thompson R, Sejersen T, McMillan HJ, Kirschner J, Lochmuller H. Life expectancy at birth in Duchenne muscular dystrophy: a systematic review and meta-analysis. Eur. J. Epidemiol. 35(7), 643–653 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Passamano L, Taglia A, Palladino A et al. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myol. 31(2), 121–125 (2012). [PMC free article] [PubMed] [Google Scholar]
- 7.Rall S, Grimm T. Survival in Duchenne muscular dystrophy. Acta Myol. 31(2), 117–120 (2012). [PMC free article] [PubMed] [Google Scholar]
- 8.Kenneson A, Bobo JK. The effect of caregiving on women in families with Duchenne/Becker muscular dystrophy. Health Social Care Commun. 18(5), 520–528 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Humbertclaude V, Hamroun D, Bezzou K et al. Motor and respiratory heterogeneity in Duchenne patients: implication for clinical trials. Eur. J. Paediatr. Neurol. 16(2), 149–160 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Birnkrant DJ, Bushby K, Bann CM et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 17(4), 347–361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bushby K, Finkel R, Birnkrant DJ et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 9(2), 177–189 (2010). [DOI] [PubMed] [Google Scholar]
- 12.McDonald CM, Gordish-Dressman H, Henricson EK et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: long-term natural history with and without glucocorticoids. Neuromusc. Disord. 28(11), 897–909 (2018). [DOI] [PubMed] [Google Scholar]; •• Describes results from an analysis of changes in pulmonary function measures across time in patients with DMD treated with corticosteroids for more than 1 year compared with corticosteroid-naive patients in the CINRG DNHS.
- 13.Phillips MF, Quinlivan RC, Edwards RH, Calverley PM. Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am. J. Respir. Crit. Care Med. 164(12), 2191–2194 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Bladen CL, Salgado D, Monges S et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 36(4), 395–402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mah JK. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 12, 1795–1807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao QQ, McNally EM. The dystrophin complex: structure, function, and implications for therapy. Comp. Physiol. 5(3), 1223–1239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.PTC Therapeutics International Ltd. Summary of product characteristics, Translarna. European Medicines Agency, 2018. Available from: https://www.ema.europa.eu/en/documents/product-information/translarna-epar-product-information_en.pdf (2020).
- 18.Welch EM, Barton ER, Zhuo J et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 447(7140), 87–91 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Bushby K, Finkel R, Wong B et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 50(4), 477–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes the safety and efficacy results of ataluren in a Phase IIb randomized, double-blind, placebo-controlled trial.
- 20.McDonald CM, Campbell C, Torricelli RE et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390(10101), 1489–1498 (2017a). [DOI] [PubMed] [Google Scholar]; •• Describes the safety and efficacy results of ataluren in a Phase III randomized, double-blind, placebo-controlled trial.
- 21.Campbell C, Barohn RJ, Bertini E et al. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J. Comp. Eff. Res. 9(14), 973–984 (2020). [DOI] [PubMed] [Google Scholar]; •• A meta-analyses of the 6-minute walk test data from the Phase IIb and Phase III clinical studies of ataluren.
- 22.Li D, McDonald CM, Elfring GL et al. Assessment of treatment effect with multiple outcomes in 2 clinical trials of patients with duchenne muscular dystrophy. JAMA Netw. Open 3(2), e1921306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A totality of evidence analysis of data from the Phase IIb and Phase III clinical studies of ataluren.
- 23.Mercuri E, Muntoni F, Osorio AN et al. Safety and effectiveness of ataluren: comparison of results from the STRIDE Registry and CINRG DMD Natural History Study. J. Comp. Eff. Res. 9(5), 341–360 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes long-term safety and effectiveness data on ataluren from the ongoing, observational STRIDE Registry.
- 24.Muntoni F, Desguerre I, Guglieri M et al. Ataluren use in patients with nonsense mutation Duchenne muscular dystrophy: patient demographics and characteristics from the STRIDE Registry. J. Comp. Eff. Res. 8(14), 1187–1200 (2019). [DOI] [PubMed] [Google Scholar]; •• Describes demographics and disease characteristics of patients in the ongoing, observational ataluren STRIDE Registry.
- 25.Birnkrant DJ, Bushby K, Bann CM et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 17(3), 251–267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birnkrant DJ, Bushby K, Bann CM et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 17(5), 445–455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henricson EK, Abresch RT, Cnaan A et al. The cooperative international neuromuscular research group Duchenne natural history study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve 48(1), 55–67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald CM, Henricson EK, Abresch RT et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 391(10119), 451–461 (2018). [DOI] [PubMed] [Google Scholar]; •• Describes the long-term effects of corticosteroids on disease progression in patients with DMD in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS).
- 29.McDonald CM, Henricson EK, Abresch RT et al. The cooperative international neuromuscular research group Duchenne natural history study--a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods used. Muscle Nerve 48(1), 32–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inacio MC, Chen Y, Paxton EW, Namba RS, Kurtz SM, Cafri G. Statistics in brief: an introduction to the use of propensity scores. Clin. Orthop. Relat. Res. 473(8), 2722–2726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciafaloni E, Kumar A, Liu K et al. Age at onset of first signs or symptoms predicts age at loss of ambulation in Duchenne and Becker Muscular Dystrophy: Data from the MD STARnet. J. Pediatr. Rehab. Med. 9(1), 5–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ricotti V, Ridout DA, Scott E et al. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J. Neurol. Neurosurg. Psych. 84(6), 698–705 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Coca-Perraillon M. Local and global optimal propensity score matching. SAS Global Forum 185 (2007). https://support.sas.com/resources/papers/proceedings/proceedings/forum2007/185-2007.pdf [Google Scholar]
- 34.Finkel RS, Flanigan KM, Wong B et al. Phase IIa study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 8(12), e81302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trucco F, Domingos JP, Tay CG et al. Cardiorespiratory progression over 5 years and role of corticosteroids in Duchenne muscular dystrophy: a single-site retrospective longitudinal study. Chest 158(4), 1606–1616 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Peay HL, Hollin I, Fischer R, Bridges JF. A community-engaged approach to quantifying caregiver preferences for the benefits and risks of emerging therapies for Duchenne muscular dystrophy. Clin. Ttherapeut. 36(5), 624–637 (2014b). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.