

Summary
We report a droplet microfluidic method to target and sort individual cells directly from complex microbiome samples and to prepare these cells for bulk whole-genome sequencing without cultivation. We characterize this approach by recovering bacteria spiked into human stool samples at a ratio as low as 1:250 and by successfully enriching endogenous Bacteroides vulgatus to the level required for de novo assembly of high-quality genomes. Although microbiome strains are increasingly demanded for biomedical applications, a vast majority of species and strains are uncultivated and without reference genomes. We address this shortcoming by encapsulating complex microbiome samples directly into microfluidic droplets and amplifying a target-specific genomic fragment using a custom molecular TaqMan probe. We separate those positive droplets by droplet sorting, selectively enriching single target strain cells. Finally, we present a protocol to purify the genomic DNA while specifically removing amplicons and cell debris for high-quality genome sequencing.
Keywords: droplet microfluidics, single-cell analysis, genomics, genome, gut microbiome, functional analysis, metagenomics, bacteria
Graphical abstract
Highlights
-
•
Rare microbial high-quality genomes can be obtained by culture-free enrichment
-
•
Microfluidic workflow benchmarked to work directly with complex microbiome samples
-
•
Genomic DNA molecules can be cleaned from abundant amplicons for sequencing
-
•
Metagenomic target selection demonstrated based on the meta mOTUs tool
Motivation
The microbiome is an increasingly important research target. In particular, the gut microbiome is linked to >100 disease states and contains several thousand species. In addition to computational innovations for sequencing data, complementary experimental high-throughput methods to perform precision genomics are urgently needed. Microfluidic droplet screens offer increased resolution; ultra-high throughput; reductions in sample and reagent usage, cost, and contamination; and the possibility to add functional enrichment. An increasing diversity of microfluidic tools for bacterial single-cell analysis exists in the context of controlled laboratory cultures. Here we describe a complete microfluidic workflow for targeted genome sequencing directly from frozen complex microbiome samples.
Pryszlak et al. present an ultra-high-throughput droplet microfluidic workflow to enrich target bacteria from the complex background of millions of gut microbiota, including steps to clean their genomic DNA for whole-genome sequencing. Their cultivation-free method enables the recovery of high-quality genomes from rare target cells (<1%) in microbiome samples.
Introduction
The microbiome is a complex and increasingly important research target. In particular, the gut microbiome is linked to over 100 disease states or syndromes (Gupta et al., 2016; Louis et al., 2014; Nobu et al., 2015; Vos and Vos, 2012) and contains at least several thousand species (Almeida et al., 2019). A person's microbiome influences how the body metabolizes drugs (Zimmermann et al., 2019a; 2019b) and therefore has an impact on success or failure of any pharmacological intervention. Current microbiome studies are mainly driven by innovations in the analysis of metagenomic sequencing data. It has, for instance, recently become possible to reconstruct metagenomically assembled genomes (MAGs) of abundant taxa in metagenomic datasets (Almeida et al., 2019; Nayfach et al., 2019; Parks et al., 2017). However, beyond abundance limitations, MAGs have been found to be prone to chimeric assemblies (Chen et al., 2020). Metagenomic approaches are also limited in resolving mobile elements, e.g., showing which species share the same genes due to horizontal gene transfer, information of increasing relevance in medical research on antibiotic resistance and toxin encoding genes.
More complementary experimental high-throughput approaches are needed to perform precision genomics. Beyond the benefits of increased resolution through targeted experiments (Cross et al., 2019; Grieb et al., 2020), microfluidic droplet screens are able to provide the required throughput (Prakadan et al., 2017) to study complex ecosystems such as microbiomes (Tauzin et al., 2020; Terekhov et al., 2018). Experiments in droplets can also substantially lower the amount of material and reagents used, cost, and contamination (Hosic et al., 2016; Nishikawa et al., 2015) and provide additional information through functional enrichment (Kintses et al., 2010; Tauzin et al., 2020; Terekhov et al., 2018; Yaginuma et al., 2019). An increasing diversity of microfluidic tools for bacterial single-cell analysis exists in the context of laboratory cultures. However, few natural isolates from complex microbiomes are available as laboratory cultures (Almeida et al., 2019).
In terms of cultivation-free techniques for the genomic analysis of microbiota cells, generating single amplified genomes (SAGs) in microfluidic gel microdroplets (Chijiiwa et al., 2020) or fused emulsions (Lan et al., 2017; Zheng et al., 2020) has been achieved, with barcoding either inside droplets or after isolation in wells. This approach has yielded novel information on uncultivated and low-abundance strains at a single-cell level with the trade-off of low genome coverage and amplification biases. SAG generation is not targeted and would not allow specifically looking for microbes carrying particular genes of interest. Genetic targeting has been demonstrated by earlier studies of PCR in droplets to generate a sequence-dependent fluorescence signal, used for PCR-activated droplet sorting (PADS) (Lim et al., 2015, 2017). This was shown for plasmids and phage sequences in E. coli laboratory cultures and more recently for genetic libraries cloned into E. coli (Xu et al., 2020). However, it was never applied to complex microbiomes of unknown, highly diverse biochemical composition, additionally hindered by sample contaminants such as fibers and particles found in stool samples.
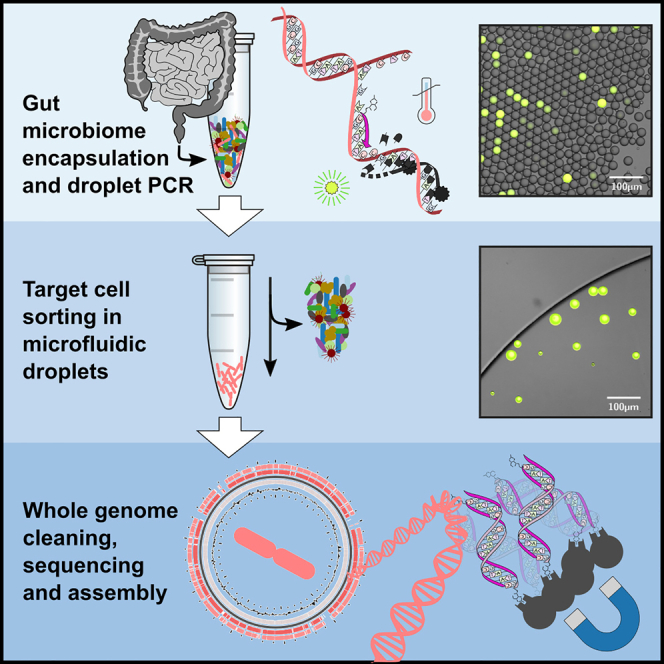
To combine strengths of culture-free approaches and hypothesis-driven targeted approaches, we have established a genomics resource where a desired strain is enriched without cultivation and where genome fragments are sequenced in bulk to recover high-quality genomes. We demonstrate a workflow that directly processes complex, potentially medically relevant microbiome samples, which are far less homogeneous and defined than laboratory cultures. By testing directly on complex samples, we aim to make our development a useful resource for the microbiome research community. While building on PADS methods, we show here how to apply them directly to microbiome samples, how to target bacteria without reference genome, how the method can be used to estimate the absolute abundance of a particular species, and how the abundant amplicon sequences used for droplet identification can be removed prior to genome sequencing. Our resource’s workflow is summarized in Figure 1.
Figure 1.

Microfluidic workflow. Microfluidic chip designs, microscope images of their use, and schematics of molecular mechanisms
(Top left) DDDroplet generation chip and photo of 39 μm-droplet generation with cells and reagents in oil. The arrow indicates the flow direction. Top center: illustration of emulsion PCR in a thermocycler. If the target DNA sequence is present, it allows the binding of biotinylated primers and probes, thus the synthesis of corresponding amplicons. During strand extension, the TaqMan probes are cleaved, releasing fluorescent molecules.
(Top right) Droplet sorting chip design and microscopy images. The microscopy pictures on the right show an emulsion before sorting and during sorting and the positive droplet enrichment post sorting. Here, fluorescent droplets indicate the presence of the gut bacterium B. vulgatus inside the droplet. Multiple microscopy images of the droplet sorting junction at different time points were overlaid and colored to demonstrate droplet flow-traces. In the right image, a small black arrow indicates the location of the nearby sorting electrode, and a bright green spot indicates the upstream location of fluorescence detection. The scale bars correspond to 100 μm.
(Bottom) Illustration of the removal of abundant amplicons with biotin-binding Streptavidin beads (after pooling the sorted positive droplets) to purify genomic DNA for sequencing library preparation. Library preparation further involves whole-genome amplification (WGA) after binding adapter primers to the randomly broken genomic DNA fragments, followed by DNA fragment size selection.
Results
Absolute cell counts of microbiome samples give measure of cell density for encapsulation
The density of cells in the stool samples processed here were quantified with flow cytometry, in order to ensure that the samples can be diluted to a reproducible density (here, ca. 133,000 cells per microliter) suitable for single-cell encapsulation into monodisperse droplets. To achieve volumetric microbial density measures, the stained and diluted samples were mixed with reference beads (polystyrene colloids) of a known concentration. The reference bead count in the flow cytometric analysis then indicated the analyzed volume of sample suspension. Results are shown in Figure 2A, generated according to the method detailed under “stool sample preparation and cell counting.” Flow cytometric analysis with unspecific DNA stains has previously been shown to be an appropriate analysis tool for such complex samples (Props et al., 2017; Vandeputte et al., 2017). The cell density between stool samples varied roughly within the same order of magnitude (see Figure 2A). In particular, the sample, #e, the only infant stool sample used, had a low cell number per volume compared with adult samples, which agreed with our expectations. The cell density in stool samples was much higher than that of typical liquid cultures.
Figure 2.

Cell density and encapsulation. Bacterial density based on flow cytometry measurements and microscopy images of emulsions
(A) Number of Syto BC-stained cells counted per volume of stool sample before dilution, and two overnight culture aliquots of bacterial cultures Bacillus subtilis (B.s.) and Bacteroides vulgatus (B.v.), here shown increased by a factor of 50 to visualize them despite the lower cell densities compared with stool. Measurements were performed in triplicates and the mean and standard deviation are shown for each sample.
(B and C) Microscopy images of droplet emulsion containing diluted B. subtilis culture post droplet PCR. The images are overlays of the microscope's fluorescence and brightfield channel as well as the contours of positive fluorescent areas, as returned from the droplet analysis tool described in the STAR Methods section. The scale bars correspond to 100 μm.
Primer-pairs and molecular probes can be made specific to target species
In order to target any microbial strain from a sample of interest, with or without a reference genome, we introduced the use of marker gene variants of the meta mOTUs V2.0 (Milanese et al., 2019) tool for primer and probe design. Single-copy phylogenetic marker gene based operational taxonomic units (mOTUs) quantitatively analyze the relative abundance of taxa in the sample by mapping shotgun metagenomic data to single-copy, non-16s rRNA marker genes. Marker gene sequences were obtained directly from shotgun metagenomic sequencing data, without requiring reference databases. Specific to these sequences, we designed primer pairs and, as a second layer of specificity, also TaqMan probe sequences. After designing several alternative primers and probes, they were shortlisted experimentally through testing in qPCR assays, gel electrophoresis, and Sanger sequencing of PCR products (see STAR Methods, primer and probe design and testing). Finally, in microfluidic droplets, the TaqMan probe fluorescence lit up as expected for the corresponding bacterial culture (see Figures 2B and 2C), specifically for the few spike-in culture cells in the presence of many other alternative bacterial sequences in stool (see Figures 3D–3F), as expected from earlier qPCR benchmarking. The best performing sequences for our target organisms are given in the key resources table as part of STAR Methods.
Figure 3.

Recovery of spike-in cells by droplet sorting. Fluorescence activated single-cell resolution droplet sorting of B. subtilis cells, which where spiked into stool samples at different ratios, of an emulsion with 1 in 4 droplets occupied by a cell. (A and D) About 1 B. subtilis cell in 50 microbiota cells, at 25% droplets occupied, means 1 positive droplet in ca. 200, ratio 0.005; (B and E) 1:100 cells, droplet ratio 0.0025; (C and F) 1:250 cells, droplet ratio 0.001
(A and B) Density plots of droplet sorting data with the applied sorting gates highlighted in red and with percentage of events within the gate. Sections of data are shown as these experiments were run over long time frames (ca. 10–20 h of sorting per sample), and gate boundaries had to be adjusted every few hours due to temperature changes and slightly varying droplet sizes. Repeated measurements yielded similar results.
(D–F) Microscopy overlay images of fluorescence and brightfield channel, showing positive droplets (containing B. subtilis culture) and negative droplets (empty or stool microbiota) post droplet PCR. The scale bars correspond to 100 μm.
(G) DNA sequencing data results of B. subtilis culture spiked into stool samples. The target read abundance is shown by sample. The unknown fraction (gray) could not be assigned to any bacterial genomes in our database, as further confirmed with Kraken assignments.
(H) Mapping of target DNA sequencing reads to the B. subtilis reference genome, showing the genome coverage of different samples.
Stoichiometry of genetic single-cell bacteria assay in microfluidic droplets
The number of cells per droplet follows a Poisson distribution and cannot be controlled tightly with passive encapsulation. To conduct single-cell experiments in droplets, the cell suspension of known density was therefore generally diluted to a ratio of 1 cell in 4 volumes of a monodisperse droplet (here, chosen at ca. 30 pL) to optimiseoptimize the fraction of droplets hosting single cells. This volume dilution ratio (0.25) corresponds to the Poisson parameter (λ) of the cell distribution in droplets. At this droplet occupancy ratio, the double-encapsulation of suspended cells into the same droplet is rare (2.4%) (Collins et al., 2015).
To estimate the efficiency of the PCR assay, in other words to which degree positive droplet ratios corresponded to theoretical occupancy values, we here encapsulated Bacillus subtilis culture stock at a lower and a higher dilution (λ = 1 and 0.0625) compared with standard experiments. To reach desired dilution ratios, an adjusted volume of Bacillus subtilis cell suspension was added to the droplet encapsulation PCR mix as detailed in the respective STAR Methods Table, given the culture stock cell density as determined by flow cytometry (ca. two million cells per microliter; see Figure 2A). We then assessed the positive droplet ratio of resulting emulsions post PCR, on the basis of a custom ImageJ macro (see STAR Methods, microscopy and evaluating positive droplet ratios). The observed positive ratios were 48% ± 6% (1915 out of 3,970 completely imaged droplets in total across several fields of view; see representative image section in Figure 2B) and 5% ± 1% (243 out of 5,105 droplets; see Figure 2C), respectively. The Poisson distribution returns theoretical percentages of 63% and 6% of occupied droplets for these cell encapsulation parameters, matching the observed values well and indicating an assay efficiency of 80%.
Single microbiota cells can be identified and sorted in droplets
Cells of the laboratory isolate of soil bacterium Bacillus subtilis, which are not found naturally in the gut, were spiked into stool samples at 3 different ratios of 1 in 50, 1 in 100, and 1 in 250 cells and encapsulated into microfluidic droplets. Given the low mixing ratio and the droplet occupancy of about 1 cell per 4 droplets, the positive ratio of the PCR assay could not be quantified by microscopy. However, we did observe rare positive droplets as expected (see Figures 3A–3F).
We then sorted a total of 7 B. subtilis spike-in emulsions, each belonging to 1 of 3 dilution ratios, to enrich target droplets. For this purpose, droplets were reinjected into the sorting chip (Figure 1, top right) and deflected by dielectrophoresis into the collection channel of the chip (Figure 1, top right center) whenever their signal fell within our fluorescence thresholds in terms of fluorescence intensity and signal length (Figures 3A–3C). When inspected by microscopy, the sorted positive emulsion mostly showed exclusively positive droplets (Figure 1, top right bottom) but the exact ratio was difficult to assess as many droplets merged or split downstream of the sorting junction, destabilized by the electric field of the sorting electrodes. The relative number of positive droplets inside the sorting gates decreased as expected from higher spike-in ratios (Figure 3A) to lower ratios (Figure 3C). It should be noted that droplet emulsions became less monodisperse during thermocycling, and that the droplet sizes separated out partially in the syringe given the long sorting time frames, so that the average droplet size decreased over time and sorting boundaries needed to be adjust dynamically every few hours due to this size drift as well as temperature changes affecting the laser and detectors.
Amplicons can be removed from low-abundance genomic DNA
As a byproduct of the PCR-based species detection, amplicons enrich and become by far the most abundant DNA fragments inside droplets, whereas a droplet only contains a single copy of genomic DNA. Sorted positive droplets are thus dominated by the already known amplicon sequence. We therefore also developed a workflow for amplicon clean-up from sorted and pooled positive droplets while maintaining the low-abundance target genomes.
We used biotinylated primers for PCR, to incorporate a chemical handle into amplicons with which they could be selectively bound to magnetic Streptavidin beads, to remove them from un-labelled genomic DNA, as illustrated in Figure 1, bottom, and detailed in the STAR Methods section amplicon removal and sequencing library preparation. After three rounds of Streptavidin bead purification and consecutive genomic DNA re-concentration on unselectively binding SPRI beads, amplicons were reduced to an undetectable amount (see electropherograms in Figure S1). Even after DNA amplification during Illumina sequencing library preparation, when genomic DNA become abundant, the amplicon concentration was very low or absent from recorded electropherograms. The electropherograms also revealed that the genomic DNA, molecules of several mega base pairs (bp), already fragmented into pieces of ca. 300 to 3,000 bp during thermocycling in droplets. There was no need for additional DNA fragmentation steps before preparing the sequencing library.
Whole-genome sequencing output provides a rough estimation of bacteria species abundance
In a simple mixing experiment, we tested the quantitative accuracy of sequencing read counts as a prediction of input cell number ratios, as a proxy for later purity assessments of DNA libraries prepared from sorted droplets. For this purpose, we mixed bacteria cultures Bacteroides vulgatus PC510 strain and Bacillus subtilis strain 168 BS168_ctg based on their cell density as determined by flow cytometry. Cells were mixed at a ratio of (1) 10:1 and (2) 1:1, thermocycled in droplets and sequencing libraries prepared in accordance to our overall workflow (see STAR Methods, amplicon removal and sequencing library preparation) but without droplet sorting to enrich a target. Sequencing resulted in 376,322 and 441,707 high-quality paired reads for samples (1) and (2), respectively. Reads of the two mixed B. vulgatus and B. subtilis culture samples were mapped to their reference genomes (see STAR Methods) to calculate their relative abundance in each sample. B. vulgatus had higher relative abundance in both samples, with the ratios of B. vulgatus to B. subtilis 286:33 in sample 1) and 253:201 in (2), revealing an average relative divergence to the original mixing ratio of 21%.
Alongside Bacillus subtilis, Bacteroides vulgatus culture was used in this particular benchmark because its wild strains also frequently occur in the gut and such endogenous strains were targeted during droplet sorting of later experiments.
Sequencing of enriched samples shows high target abundance
With the above error rate of sequencing-based cell-ratio quantification in mind, we sequenced the 7 sorted spike-in samples (discussed in the section, single microbiota cells can be identified and sorted in droplets) and binned recovered sequences to estimate the target enrichment in experiments. This typically showed a high enrichment level, with >98% of recovered reads being assigned to B. subtilis (4/7 samples; see Figure 3G). However, some samples showed DNA reads of other origin, e.g., the sample 1:50-A had extremely few reads overall (78,000 read pairs, the average of all 7 samples being 1,640,780 read pairs), indicating insufficient levels of input DNA, and reads from this sample were not assigned to any taxa (including no B. subtilis) by the Kraken pipeline.
The spike-in experiments also demonstrated which minimum number of sorted droplets of the target species (corresponding roughly to cells at the beginning of library preparation) is necessary to recover a high-quality genome, which ultimately limits our overall assay sensitivity in terms of target strain abundance, given the ability to sort a maximal number of droplets per experiment. Indeed, all samples except 1:50-A resulted into a relatively even genomic coverage of B. subtilis (Figure 3H), indicating that an assembly of this genome is possible based on our recovered reads. It is of note that a few regions seemed to be recovered at a higher rate in all experiments independently. These could represent duplicated genome regions and they did not overly imbalance the experiment. The good target genome coverage, even by our most diluted sample (1:250 spike-in), indicates that only about 4,000 sorted target cells (here, from ca. 1 million droplets) were needed to recover sufficient reads of a target genome.
High-quality de novo genome of endogenous microbiota bacterium was assembled
After the above extensive spike-in benchmarking of the method, we applied our precision genomics approach to the endogenous stool bacterium Bacteroides vulgatus to enrich and sequence its genome de novo. This bacterium, typical for western gut microbial communities, was chosen as a target because several high-quality genomes (Moss et al., 2020) and safety class 1 laboratory cultures are available and allowed us to compare our findings. The targeted experiment was performed twice using aliquots from the same stool sample, with PADS data and images shown in Figure 4B. No bacterial cultures where involved. Sequencing of the two final genomic DNA libraries, together occupying half an Illumina MiSeq run, resulted in (1) 460,513 and (2) 1,045,142 high-quality paired reads, respectively. Mapped read abundance for sorted stool samples showed high relative abundance of two endogenous Bacteroides vulgatus strains, PC510 and ATCC 8482, in the total pool of sequencing reads. The read abundance for PC510 was (1) 262 and (2) 603 and for ATCC 8482 (1) 116 and (2) 332. Assembly of reads resulted in a total assembly length of 11,200,633 bp and 19,045 scaffolds, with 526 contigs >2,500 bp for sample (1). Sample (2) assembled 36,865 scaffolds with a total length of 21,451,804 bp and 708 contigs >2,500 bp.
Figure 4.

Enrichment and sequencing of endogenous target genome. Workflow summary of the precision genomics approach to enrich target endogenous species from its microbiome community with microfluidic droplets and summary of the two Bacteroides vulgatus genomes assembled in our experiments
(A) Workflow schematic and genomes statistics.
(B) Two biological replicate experiments of fluorescence activated single-cell droplet sorting of endogenous Bacteroides vulgatus cells in stool samples, of an emulsion, with about 1 in 4 droplets occupied by a cell. (B, left) Density plots of droplet sorting data with the applied sorting gates highlighted in red and with percentage of events within the gate. Sections of data are shown. (B, right) Microscopy overlay images of fluorescence and brightfield channel, showing positive droplets (containing endogenous B. vulgatus) and negative droplets (empty or other stool microbiota) post droplet PCR. The scale bars correspond to 100 μm.
(C) The two assembled genomes were mapped to a reference genome using the blastx algorithm, illustrating (https://server.gview.ca/) the substantial genome coverage achieved here.
Binning of these scaffolds >2,500 bp resulted in 1 medium- and 1 high-quality genome, 1 from each sample, designated as B_vulg_1 and B_vulg_2. We performed genome quality benchmarks on our sequencing results using available reference genomes of the strains to which our reads mapped. Taxonomy completeness and contamination were determined by CheckM (Parks et al., 2015). CheckM's bacteroidales marker set calculated B_vulg_1 as 88.78% complete (90% needed for high-quality score), with 0.56% contamination, and B_vulg_2 as 99.25% complete with no contamination. Another medium-quality MAG (70% complete) was also binned from sample (2) belonging to the family of Enterobacteriaceae. The statistics and coverage of the two assembled target genomes in comparison to the reference strain ATCC 8482 of the same species are summarized in Figures 4A and 4C. Larger mapping gaps (Figure 4C) can most likely be attributed to local sequence differences between the strain encountered in the donor versus the reference strain.
Compared with the reference sequence Bacteroides vulgatus ATCC 8482, genome B_vulg_1 had an average nucleotide identity similarity of 95.2% and B_vulg_2 had 1 of 95.5% (this difference likely reflects their difference of genome completeness); 16s rRNA was found in both genomes and showed a similarity to B. vulgatus ATCC 8482 of 97.4%, supporting the fact that our genomes are the same species as ATCC8482 but are likely a different strain. The amino acid sequences of both genomes and B. vulgatus ATCC 8482 were analyzed using the KEGG Automatic Annotation Server to identify metabolic similarity. Our analysis showed consistency in the enzyme presence across their central carbon metabolism, energy pathways, and transporters, suggesting that despite being different strains, their genomes can perform similar metabolic functions to B. vulgatus ATCC 8482.
Discussion
We have demonstrated how the same frozen gut microbiome (stool) samples of previous metagenomic analyses can be prepared for single-cell genetic assays in microfluidic droplets. A bacteria cultivation-free workflow using complex gut microbiome samples was demonstrated and benchmarked with in-droplet digital PCR, droplet sorting, genomic library preparation, and sequencing data analysis. Evidence has been provided that single cells can be targeted, endogenous target species can be enriched through droplet sorting, amplicon sequences from the genomic DNA can be removed, and whole genomes can be sequenced to obtain high-quality and low-contamination drafts of the strains of the target bacterial species. The method was applied to the endogenous gut species Bacteroides vulgatus, and a high-quality, low-contamination genome was recovered (without using or requiring reference genomes). We believe this method to be an important resource for the scientific community in their mission to advance from cell cultures to apply precision genomics methods directly to complex samples of medical or biotechnological relevance. In the following paragraphs, performance and limitations in comparison to other methods are discussed.
Typically, molecular DNA probes are designed to target a variable region of the 16S RNA marker gene, e.g, (Baccari et al., 2020; Zwielehner et al., 2011), based on sequences in reference databases or amplicon sequence variants (ASVs) in samples (Callahan et al., 2017). However, the variable regions of the 16S RNA gene are short and therefore provide little space for highly specific probe design. Accordingly, 16S variable region sequences as routinely processed with Illumina sequencing do not allow distinguishing between species or strains of interest. In contrast, the single-copy marker gene approach, meta mOTUs V2 (Milanese et al., 2019) enables a fast species level assessment of the microbial diversity and relative abundances of a microbiome sample even without reference databases. It uses ten marker genes, which areare longer than the 16S RNA gene. In addition to advantages in resolution, relative abundances based on these single-copy genes are also more accurate than those based on the 16S RNA gene, because the latter can be present in multiple copies per cell. In this work, we successfully demonstrate the reference-free use of meta mOTUs to design specific primers and molecular probes directly on shotgun metagenomic data from a sample, specifically on the comparably large sequence space of the ten marker genes within the metagenomic data. The meta mOTUs accuracy of relative abundances furthermore contributes valuable information when assessing if target species have a sufficient abundance in the sample to yield a high-quality result given the experimental power available. This method can be directly applied to new targets also in other habitats.
The best known culture-free method to obtain individual genomes from a microbiome sample is the generation of SAGs (Chijiiwa et al., 2020; Lan et al., 2017; Zheng et al., 2020) from single cells. A few hundred genomes can be recovered in parallel with low contamination when using high-throughput droplet microfluidic methods for SAGs, and the most abundant species may be represented with several SAGs that can be pooled to increase genome quality (Chijiiwa et al., 2020). Because this method is not targeted, however, it is not certain that any specific genome is among the results, and the genomes have a low average quality (Gawad et al., 2016). The presented targeted approach here is, in contrast, more likely to recover a high-quality genome of the target species, thereby complementing existing SAG methods. In this precision genomics resource, we show with sequencing data of spike-in bacterial culture that bacteria can be enriched sufficiently even at spike-in ratios as low as 1:250, with as few as 4,000 enriched target cells, to achieve a ca. 40× coverage of the target genome, which is suitable for a reliable genome assembly.
Absolute quantification of bacterial load and the abundance of target species is important complementary information for metagenomic data (Props et al., 2017; Vandeputte et al., 2017). The droplet digital PCR (ddPCR) approach has the advantage that it can be used for the absolute enumeration of targeted bacterial cells within a volume of complex environmental sample, in addition to recovering the target genomes later on as shown here. ddPCR has previously been used for the quantitative detection of various bacterial pathogens (Li et al., 2018), DNA copy number (Hindson et al., 2011), and viral sequence abundances (Long and Berkemeier, 2021; Martinez-Hernandez et al., 2019), even outperforming qPCR assays (Hindson et al., 2013; Yang et al., 2014), and with the possibility to multiplex targets (Ackerman et al., 2020). For absolute cell quantification by ddPCR, an estimate of the assay efficiency is needed to conclude the cell number from an observed positive droplet ratio. We conclude from our data that the assay provides a visual readout of approximately 80% of the theoretical number of droplets occupied with target cells (see results, stoichiometry of genetic single-cell bacteria assay in microfluidic droplets), this efficiency was stable between different samples and bacteria tested. There are a number of experimental factors that limit the assay efficiency to below 100% and cause some level of variation between experiments: (1) temperature-only-based cell lysis efficiency during thermocycling, (2) PCR efficiency <100% (Lim et al., 2015), (3) manual fluorescence intensity thresholding setting against background, and (4) error in bacterial density measurement and dilution of input culture.
High throughput is important for experiments when analyzing diverse microbiome samples at the single-cell resolution. Although very high microfluidic droplet sorting rates up to 30 kHz (Sciambi and Abate, 2014) have been reported for ideal microfluidic droplet sorters, real screens in biology often require much more conservative sorting settings, reducing the overall throughput to values, such as 300 Hz (Josephides et al., 2019), for instance, because of droplet pre-incubation off-chip followed by reinjection into a sorting chip causing much less regular spacing. Another important technical parameter for sorting efficiency and binning is the polydispersity of droplets, which increases during PCR thermocycling through droplet merger and shrinking of some droplets. The use of robust surfactants is key to stabilizing droplets during thermocycling, and additional measures such as mineral oil topping can further reduce evaporation. However, if the mineral oil is not removed carefully afterward, residues of other oils can obstruct channels of the sorting chip later on, which is why we did not use them in this protocol. Preventive measures may be necessary when the recorded fluorescence signal of the emulsion blurs too much to bin a positive droplet population. In our sorting algorithm, we furthermore exclude merged droplets based on their doubled signal length. Optimizing the sorting process for each sample and increasing the sorting rate from our study (ca. 250 Hz) to higher rates such as 1 kHz could probably decrease the sensitivity limit about 4-fold when sorting for the same duration (here, ca. 8 h, sorting of ca. 7 m droplets, and carrying ca. 1.5 m cells, or allow setting more stringent sorting conditions to reduce the number of false positive droplets close to zero.
It has been shown that microfluidic droplets are particularly well suited to cultivate microorganisms (Dichosa et al., 2014; Jiang et al., 2016; Mahler et al., 2021; Zengler et al., 2002), including slow growing species. While broadly effective growth media are not available for most habitats, many gut microbiota specifically have been shown to in principle grow well in a small set of media (Tramontano et al., 2018; Watterson et al., 2020; Yousi et al., 2019). Nevertheless, a majority of bacteria and even more so archaea (Sun et al., 2019) have not been cultivated in the laboratory (Bodor et al., 2020; Epstein, 2013; Hug et al., 2016), cultivation biases are not well characterized, and special anaerobic equipment as well as fresh samples are needed for many species. We therefore developed a cultivation-free resource, which can be directly applied to other habitats. The method does not even rely on purifying cells or keeping them alive, which allowed us, e.g., to analyze samples stored for months in −80°C freezers and to work with anaerobic gut bacteria under standard aerobic laboratory conditions. Available cultures and reference genomes do help, however, to benchmark primer and probe designs for their specificity, ensure the lysis efficiency of cells, and compare results.
While we developed this method, significant progress has been made in purely computational tools to assemble genomes from metagenomic datasets (Alneberg et al., 2018; Bowers et al., 2017), including long read data (Bertrand et al., 2019; Moss et al., 2020). Such MAGs could serve as a helpful complementary resource to SAGs and genomes recovered with this resource. On the one hand, the vast number of available MAGs can help to identify species of interest as well as aid to assess probe sequence specificity. Sorted genomes, on the other hand, may be used to improve the quality of special MAGs, such as contamination and chimeric sequences. Sorted genomes come with less foreign DNA, which decreases the amount of contamination in the generated bins, while also simplifying and speeding up binning and assembly. Furthermore, the cell sorting physically co-enriches mobile genetic elements that can often not be clearly assigned to genomes.
Last but not least, as the field aims to move from functional predictions to ecotypes of species, we require single-cell approaches (Hatzenpichler et al., 2020) to combine genetic analysis with functional screens (Berg et al., 2020). Droplet microfluidic methods allow the use of fluorescent probes indicating functional properties of cells as trigger for droplet sorting (Xi et al., 2017) to enrich not only for the presence of genetic target sequences as shown here but also industrially relevant enzymatic activities (Hosokawa et al., 2015; Tauzin et al., 2020; Zeng et al., 2020), cell-surface proteins via antibodies (Cross et al., 2019), functionalized quantum dots (Feng and Qian, 2018), or other probes targeting cell surfaces and bioactive content (Terekhov et al., 2018). Such methods can also be extended to microfluidic chambers (Leung et al., 2012). These approaches can be combined with the presented resource to achieve powerful multi-omic readouts. But even without combination, this method allows a direct target enrichment of functional sequences, e.g. all taxa with a genetic region indicating esterase activity, particular secondary metabolites, or other desirable functions, alleviating resolution problems associated with sequencing depth of untargeted libraries. The droplet sorting approach is particularly attractive to target mobile genetic elements, such as viral sequences (Lim et al., 2017; Martinez-Hernandez et al., 2019) or transposons for antibiotic resistance (Zlitni et al., 2020), which can often not be clearly associated in metagenomic data (Bertrand et al., 2018).
Limitations of the study
This experimental approach to enrich and sequence genomes represents a sophisticated workflow that requires complex instrumentation and reagents. We have also observed batch variability in the Bio-Rad droplet generation oil, which in rare cases may lead to droplet PCR failure. Handling individual cells and DNA also provides very little intermediate visual feedback on the status of the experiment after the droplet sorting step, and therefore success or failure often only becomes apparent after DNA library amplification or when analyzing sequenced genomic data. Furthermore, applying the method to unknown microbiota targets experimentally means little knowledge of the success rate of cell lysis during thermocycling, probe suitability, and potential adhesion to other cells that may be co-enriched as genetic contaminants.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| B. vulgatus cell culture | Lab stock | Bacteroides vulgatus ATCC 8482 |
| B. subtilis cell culture | Lab stock | Bacillus subtilis str. 168 BS168 |
| Biological samples | ||
| Human stool sample | Korpela et al., 2018 | Stool sample from baby collected in Luxembourg. https://www.ebi.ac.uk/ena/browser/view/SAMEA104445596 |
| Deposited data | ||
| Data and scripts (CheckM, Binning, Contig KEGG-KO table, Chip designs, PADS data plotting via GitHub) | This paper, deposited in the Open Science Framework | https://doi.org/10.17605/OSF.IO/6TMZK |
| FIJI plugin code and imaging data | This paper, deposited in Zenodo | https://doi.org/10.5281/zenodo.5748533 |
| Genomic data | This paper, deposited in ENA | Project accession number PRJEB48713; sample 1 https://www.ebi.ac.uk/ena/browser/view/ERS8538067 and sample 2 https://www.ebi.ac.uk/ena/browser/view/ERS8538068 |
| Oligonucleotides | ||
| Forward primer with 5’ Mod: BtnTg for Bacteroides vulgatus and dorei | Sigma-Aldrich | CAAGCTGAGAAAAGCAGCCAAA |
| Probe sequence with Iowa Black FQ and intermediate ZEN quencher and 5’ 6-FAM fluorophore for Bacteroides vulgatus and dorei | IDT | AGTGGCAGTAGCCGGAGGGGTA TCAGCCA |
| Reverse primer with 5’ Mod: BtnTg for Bacteroides vulgatus and dorei | Sigma-Aldrich | GAATGAGTTACGAAGCCCGTTG |
| Forward primer with 5’ Mod: BtnTg for Bacillus subtilis | Sigma-Aldrich | TCGTGCTGAGACAGTTGCTT |
| Probe sequence with Iowa Black FQ and intermediate ZEN quencher and 5’ 6-FAM fluorophore for Bacillus subtilis | IDT | TTGCGGGCGGCGGTATGGCAGGAGCT |
| Reverse primer with 5’ Mod: BtnTg for Bacillus subtilis | Sigma-Aldrich | TCTTTCCCTTCAAGGCGGAC |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christoph Merten (christoph.merten@epfl.ch).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Microbe strains
Bacteroides vulgatus ATCC 8482, from −80°C frozen glycerol stock, was grown on LB-agarose streak plates at 37°C overnight, followed by liquid LB media cultivation in a shaking incubator at 37°C overnight before use. Bacillus subtilis str. 168 BS168 was directly inoculated into liquid culture without plate cultivation. Aliquots of the microbial cultures were frozen without the addition of glycerol at −80°C for experimental use and cell counting at different time points. Immediately before use, cells were washed in filtered (0.22 μm) 0.9% saline solution to replace LB media and remove free floating DNA.
Method details
Stool sample preparation and cell counting
0.5 ml stool sample (measured using a volume scaled syringe cylinder) was suspendedsuspended in 4.5 ml of filtered (0.22 μm) 0.9% saline solution. The sample was then further diluted to obtain a final dilution of 1:50. Sterilised glass rattler beads (generic) were added and samples were homogenized on a Vortex mixer for 1 min in order to suspend the sample and detach microbiota from dietary fibres. The homogenization step was repeated until faecal pellets were completely dissolved. Suspension was filtrated through a 40 μm Cell Strainer (Falcon) to remove all remaining large organic particles and clumps, using gravity flow only. Aliquots were prepared from this suspension and frozen at −80°C.
Flow cytometric cell counting of stool and cell cultures was performed with a modified protocol of van der Waaij et al. (1994), using bacteria staining with SYTO® BC and reference beads to assess the measured liquid volume, following the suppliers protocol (Invitrogen B7277 Bacteria Counting Kit for flow cytometry). Flow cytometry was performed on a LSR Fortessa bench top analyser (BD), using the 488 nm laser line and the 530/30-A detector.
Primer and probe design and testing
Primers and probes were designed with the Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/) and the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/tools/primer-blast/) primer designing tools based on the mOTUs V2 (Milanese et al., 2019) marker gene sequences of the species of interest. Probes were designed using the same tools and PrimerQuest (https://www.idtdna.com/pages/tools/primerquest). To work well in this protocol, the annealing temperature should ideally be 60°C for the primer and 70°C for the probe (the length of amplicon should ideally be 80–150 bp long), which was further checked with various online in-silico PCR simulation software tools. Candidate sequences were also extensively specificity-checked by blasting against large sequence databases of Microbiota (NCBI and internal). Several primer candidates were ordered (Sigma-Aldrich) and efficiency and specificity tested in a qPCR assay in triplicates against a number of purified DNA pools (human DNA, mix of E.coli strains, mix of 31 gut microbiota strains, DNA from stool samples), all diluted and aliquoted. We added 0.5 ng of DNA to each reaction well, which corresponded to optimal signals in our experiments. qPCR tests were performed with Syber green master mix including ROX reference dye. The most efficient primer pairs free of PCR side products and false-negatives where product-confirmed by gel-electrophoresis and Sanger sequencing of amplicons (Eurofins) and ordered again (from Sigma-Aldrich), this time with biotinylation for the actual experiments. TaqMan probes were purchased at IDT (sequence with 3’ Iowa Black FQ, intermediate ZEN quencher and 5’ 6-FAM fluorophore). Final primers and probes were tested in wells as well as droplets, using the PCR mix given in the “Table of cell encapsulation mix for PCR”. The best performing sequences for target organisms in this study are shown in the key resources table.
Preparation of microfluidic chips
Two microfluidic devices, as shown in Figure 1, were drawn in AutoCAD (Autodesk) and printed black on UV-transparent polymer film (by SelbaTech) at a resolution of 25400 dpi. Master moulds of the microfluidic chips were then fabricated using soft-lithography on 100 mm diameter silicon wafers (Silicon Materials) with the negative photoresist SU-8 2025 (MicroChem) and a mask aligner (Suess MicroTec MJB3). The droplet generation and sorting chips were produced with a channel height of 35 um, while the sorting chip is a two-layer design with a taller droplet inlet chamber of 100 um (using photoresist SU-8 2075, MicroChem). After standard baking and developing steps, a degassed PDMS and curing agent mix (Sylgard 184, Dow Corning Corp.) was poured onto the master moulds and cured overnight at 65°C. The silicone elastomer chips were then peeled off the master, cut and punched with biopsy punches (Harris Unicore) for tubing ports, and then bonded to microscopy glass slides (Thermo Fisher Scientific Inc.; droplet generation chips) or onto ITO glass (Delta Technologies LTD; ground electrode for sorting chips), after surface activation in an oxygen plasma oven (Femto, Diener electronic GmbH). The electrode channels of the sorting ships were filled with Indium-solder (0.5 mm Solder Wire Indalloy19, Indium corporation of America) on a hotplate at ca. 85°C. Finally, before use, microfluidic channels were rinsed with Aquapel (Autoserv, Germany) and dried to increase hydrophobicity. Polytetrafluoroethylene (PTFE) tubing (Adtech Polymer Engineering Ltd & APT Advanced Polymer Tubing GmbH) was used to connect syringes containing liquids to inlet ports on the chips.
Single-cell encapsulation into droplets and digital PCR
Stool sample aliquots, TATAA Probe GrandMaster Mix (TA02-625), primer-mix, probe and dNTP-mix aliquots were defrosted and mixed according to the “Table of cell encapsulation mix for PCR”. Prior to mixing the 800 μl volume, the stool sample aliquot was washed to remove potentially free-floating DNA contaminants. To do so, the same volume of PBS buffer was added to the cell suspension, vortexed, and pelleted in a centrifuge for 12 min at 3000 g, after which the supernatant was discarded. The pellet was finally resuspended in PBS buffer corresponding to the original suspension volume by pipetting up and down 20 times. The PCR mix was optimized for best amplification efficiency in the droplets taking into account surfactant presence and the small volume of the reaction.
Table of cell encapsulation mix for PCR
| Aqueous reagent | Volume | Concentration/comment |
|---|---|---|
| Cell suspension aliquot | e.g. 50 μL | Volume adjusted according to cell suspension aliquot density to encapsulate on average one cell in every fourth or fifth droplet (ca. 30 pl/droplet) |
| 10 μM primer mix | 55 μL | 0.69 μM final concentration of each primer (forward and reverse, aliquots mixed in advance at 10 μM of each primer) |
| 100 μM TaqMan probe | 20 μL | 2.5 μM final concentration |
| Taq hotstart polymerase | 2 uL | To improve efficiency of late PCR cycles |
| 100 μM mix of each dNTP | 6 μL | 0.75 μM final concentration |
| Master mix | 440 μL | |
| Nuclease-free Water | Until 800 μL |
The aqueous phase containing cells and PCR reagents and the oil phase (Bio-RAD droplet generation oil “for probes” or “for EvaGreen”, depending on batch quality) were injected into the droplet generation chip using syringe pumps (Harvard apparatus). Droplets were produced within 60 min by flow focusing at flow rates of ca.1000 μL/h for the aqueous phase and two-fold higher oil flow rates (with a droplet generation rate of ca. 8 kHz). Droplets were collected and thermocycled off Chip in a 96 well plate (low profile skirted, Bio-RAD) according to the settings in the “Table of droplet PCR thermocycle settings”, followed by storage at 37°C in the dark overnight (up to five days) for further downstream sorting procedures. Droplets without cells and with B. subtilis culture instead of stool were processed in parallel as quality control.
Table of droplet PCR thermocycle settings
| Time | Temperature | Conditions |
|---|---|---|
| 95°C | 15 min | Hold for cell lysis (Rosa et al., 2010), from here on sample biosafety level is reduced to L1. |
| 95°C | 3 sec | 45× |
| 60°C∗ | 16 sec | |
| 72°C | 2 min | Hold |
| 37°C | infinite |
∗Each program needs to be adjusted to probes/primers annealing temperature.
Microfluidic fluorescence-activated droplet sorting
In order to specifically enrich fluorescent droplets, sorting was performed with a set-up as previously described (Chaipan et al., 2017; Debs et al., 2012). Droplets were reinjected into the sorting device at a frequency of ca. 100 Hz–250 Hz (flow rates 15–30 μL/h) using syringe pumps (Harvard apparatus) along with two spacer sheath oil inlets (ddPCR™ Droplet Reader Oil, Bio-RAD) at twenty times higher flow rates. To detect the fluorescent 6-FAM signal coming from cleaved TaqMan probes during the PCR reaction, droplets were excited using a blue diode laser (488 nm, 20 mW; Melles Griot) and the fluorescence intensity was measured at and upwards of the emission wavelength of 514 nm using a PMT (Hamamatsu). A custom LabVIEW software was used to enable dynamic adjustments of PMT gain (0–1 V), droplet fluorescence width and intensity thresholds for sorting, electrode voltage (1–1.5 kV), AC pulse frequency (30–40 kHz), pulse duration and delay.
Microscopy and evaluating positive droplet ratios
Fluorescence microscopy was performed to analyse droplet fluorescence. Microscopy slides (Thermo Fisher Scientific) were wetted with Droplet Generation Oil (Bio-RAD) and droplets were imaged directly on slides or inside broad microfluidic imaging channels on slides using a 10-fold objective with a Nikon Ti-E widefield microscope. Images in the brightfield and green fluorescence channels were taken sequentially.
For the analysis, an ImageJ macro was developed (GitLab: https://gitlab.com/marco.r.cosenza/simple_droplet_tools) to support the measurement of droplet fluorescence intensity. A permanent copy of the software has been deposited in Zenodo and its DOI is listed in the key resources table.
Amplicon removal and sequencing library preparation
Droplets from the sorting experiment were collected in one tube and frozen at −20. Excess oil was removed with a syringe and thin needle on dry ice (to keep the sorted sample, now a small ice crystal, from entering the syringe). At ambient temperature, 10–30 μl DNAse free water and 4–10 μl PFO (1H,1H,2H,2H-Perfluoro-1-octanol, Sigma-Aldrich), was added to collect the sample, dilute the salt concentration and break the emulsion (remove most excess oil). Next, the biotinated PCR amplicons were removed from the sample by three washes on magnetic Streptavidin C1 Dynabeads (Invitrogen). The binding protocol was used according to the manufacturer's instructions, with an DNA immobilisation incubation time of 15 min each run to ensure binding of long amplicons at low DNA concentrations. To collect genomic DNA and not the amplicons, we collected the supernatant of the binding assay and disposed the beads.
The amplicon-cleaned genomic DNA was desalted and cleaned by binding to magnetic SPRI beads (Beckman Coulter) (0.95×), and then the NebNext ULTRA II kit (New England Biolabs) protocol was followed for Illumina sequencing library preparation, increasing incubation times three-fold to account for low DNA concentrations. This protocol involves the following steps: DNA fragmentation end repair, adaptor ligation (adapters need to be diluted 25-fold due to very low DNA concentration), SPRI bead clean-up without size selection, PCR enrichment of fragments (equivalent to whole genome amplification, 13 PCR cycles used here), and SPRI bead clean-up without size selection. Before and after amplification, the DNA was quantified with qBit (Invitrogen) and Bioanalyzer (Agilent), initially proving the amplicon removal (no fragments at all will most likely be visible at this step due to low concentrations) and later showing the DNA fragment size distribution as displayed in Figure S1. The genomic DNA fragments during droplet PCR and was usually in a good size range for sequencing, however, further fragment size selection with SPRI beads might be necessary in particular when several barcoded samples have to be pooled. At last, the sample or sample-pool was sequenced with a 150 or 250 paired end kit on a MiSeq instrument (Illumina).
Sequencing based assessment of target bacterial enrichment and genome coverage
Reads obtained from the shotgun metagenomic sequencing of 8 metagenomic samples were quality-filtered by removing reads shorter than 70% of the maximum expected read length (100 bp), with an observed accumulated error >2 or an estimated accumulated error >2.5 with a probability of ≥0.01 (Puente-Sánchez et al., 2016), or >1 ambiguous position. Reads were trimmed if base quality dropped below 20 in a window of 15 bases at the 3′ end, or if the accumulated error exceeded 2 using the sdm read filtering software (Hildebrand et al., 2014). To obtain the coverage profile over the target B. subtilis genome, filtered reads were mapped using Bowtie2 v 2.3.4.1 (Langmead and Salzberg, 2012), with the parameters“--no-unal --end-to-end --score-min L,-0.6,-0.6”. Resulting bam files were sorted, duplicates removed and indexed using Samtools 1.3.1 (Li et al., 2009). Reads mapping with a mapping quality <20, <95% nucleotide identity or <75% overall alignment length were filtered using custom Perl scripts. From these depth profiles were created using bedtools v2.21.0 which were translated with a custom C++ program “rdCov” (GitHub: https://github.com/hildebra/rdCover) into average coverage in a 50 bp window. To estimate the abundance of different genera, Kraken (Wood and Salzberg, 2014) profiles were created for these samples, using default parameters and the default databases “minikraken” available from https://ccb.jhu.edu/software/kraken/. Abundance and genome coverage profiles were visualized in R 3.6.2.
Genome sequencing based relative abundance assessment
Sequencing results were first checked using FastQC v0.11.8 (Andrews, 2010). Relative abundance of mixed bacterial culture samples was calculated by mapping reference genomes for Bacteroides vulgatus PC510 (ADKO01000001.1- ADKO01000117.1), Bacteroides vulgatus ATCC 8482 (UYXB01000001.1- UYXB01000005.1) and Bacillus subtilis subsp. subtilis str. 168 BS168_ctg (ABQK01000001.1- ABQK01000005.1) against all for samples. Mapping was done using the BWA v0.7.15 (Li and Durbin, 2009) commands ‘bwa mem’ and Samtools v1.7 (Li et al., 2009) command ‘samtools view -hb’ and ‘samtools sort’. Generated bam files were compiled using the script ‘jgi_summarize_bam_contig_depths –outputDepth depth.txt ∗.bam’. Contig length calculated in Depth.txt were summed to get genome size. Read abundance for each contig was averaged to calculated average read abundance. Average read abundance was divided by genome size to normalized and multiplied 1×107 by for comparison between samples.
Genome assembly and analysis
Sequencing results were first checked using FastQC v0.11.8 (Andrews, 2010). Both sorted endogenous B. vulgatus reads enriched from a stool sample were trimmed using the commands ‘interleave-fastq’ (Garrison and Boisvert, 2014) and ‘sickle pe -c interleaved.fastq -t sanger -m combo.fastq -s singles.fastq’ (Joshi and Fass, 2014). Samples were then individually assembled using the Spades v3.14.1 program (Nurk et al., 2013) with the command ‘spades.py --12 combo.fastq -s singles.fastq -o output’. Assembled contigs for both samples were mapped to both sorted stool samples using BWA and Samtools commands previously described under ‘Genome sequencing based relative abundance assessment’. Generated bam files were compiled using the script ‘jgi_summarize_bam_contig_depths –outputDepth depth.txt ∗.bam’ and then binned using the metabat v2:2.15 command ‘metabat -i scaffolds.fasta -a depth.txt --seed 1987 -o output –unbinned’ (Kang et al., 2015). Completeness and Contamination of resulting metagenomic assembled genomes (MAGs) was determined by CheckM v1.0.13 (Parks et al., 2015) using the ‘lineage_wf’ flag. The amino acid sequences of both genomes, B_vulg_1, B_vulg_2, and B. vulgatus ATCC 8482 were analyzed using the KEGG Automatic Annotation Server to identify metabolic similarity. A search was run using the blastx and prokaryotic option, resulting in a table of the contig name for each genome and the KEGG orthology identifier of the best matched gene.
Quantification and statistical analysis
This study contains a large number of direct quantifications (cells, droplets, DNA fragments, volumes, speed, and distances) throughout the results section, but only basic statistical concepts (percentage, mean, and ratios) were used to describe these results.
Acknowledgments
The authors thank the EMBL Genomics and Flow Cytometry Core Facility teams for training and advice. We are grateful to the support by Nassos Typas and his group, in particular Sarela Garcia Santamarina and Matylda Zietek, for the provision of cultures, space, and advice. We also thank Alessio Milanese for discussions on the use of the meta mOTUs V2 tool. All authors are grateful for funding and support by the EMBL. A.P., T.W., and M.R.C .have been funded by EIPOD, the European Commission Marie Curie Program with EMBL (T.W.: EI3POD, grant no. 664726). T.W. also acknowledges funding by ANID-FONDECYT 11200666. F.H.'s salary is funded by the BBSRC institute strategic program Gut Microbes and Health BB/r012490/1 and its constituent project BBS/e/F/000Pr10355. This project has also received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 948219). E.K. was funded by the European Research Council (ERC-AdG-669830 MicrobioS).
Author contributions
C.M., P.B., and A.P.: conceptualization; C.M., P.B., A.P., V.B., T.W., and E.K.: methodology and validation; A.P. and T.W.: investigation; C.M. and P.B.: supervision; C.M., P.B., A.P., and T.W.: funding acquisition; M.R.C., K.W.S., and F.H.: software; K.W.S., F.H., A.P., and T.W.: data analysis; T.W.: validation, review, writing – original draft and revision, and visualization; all: reviewing and editing.
Declaration of interests
The authors declare no competing interests.
Published: December 28, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100137.
Contributor Information
Peer Bork, Email: peer.bork@embl.org.
Christoph A. Merten, Email: christoph.merten@epfl.ch.
Supplemental information
Data and code availability
-
•
Data, microfluidic chip designs, lists and, figure scripts (CheckM, Binning, Contig KEGG-KO table, PADS data plotting via GitHub) have been deposited in the Open Science Framework. Metagenomic sequences and assembled genomes have been deposited in the ENA database. The corresponding DOIs and permanent identifiers are listed in the key resources table.
-
•
The FIJI plugin to analyze droplets along with the images used in this publication have been made available on Zenodo. The DOI is given in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ackerman C.M., Myhrvold C., Thakku S., Freije C.A., Metsky H.C., Yang D.K., Ye S.H., Boehm C.K., Kosoko-Thoroddsen T.-S.F., Kehe J., et al. Massively multiplexed nucleic acid detection with Cas13. Nature. 2020:1–6. doi: 10.1038/s41586-020-2279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A., Mitchell A.L., Boland M., Forster S.C., Gloor G.B., Tarkowska A., Lawley T.D., Finn R.D. A new genomic blueprint of the human gut microbiota. Nature. 2019;568:499–504. doi: 10.1038/s41586-019-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alneberg J., Karlsson C.M.G., Divne A.-M., Bergin C., Homa F., Lindh M.V., Hugerth L.W., Ettema T.J.G., Bertilsson S., Andersson A.F., Pinhassi J. Genomes from uncultivated prokaryotes: a comparison of metagenome-assembled and single-amplified genomes. Microbiome. 2018;6:173. doi: 10.1186/s40168-018-0550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. Babraham Institute; 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data. [Google Scholar]
- Baccari O., Elleuch J., Barkallah M., Boukedi H., Ayed N.B., Hammami A., Fendri I., Abdelkafi S. Development of a new TaqMan-based PCR assay for the specific detection and quantification of Simkania negevensis. Mol. Cell Probe. 2020;53:101645. doi: 10.1016/j.mcp.2020.101645. [DOI] [PubMed] [Google Scholar]
- Berg G., Rybakova D., Fischer D., Cernava T., Vergès M.-C.C., Charles T., Chen X., Cocolin L., Eversole K., Corral G.H., et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. 2020;8:103. doi: 10.1186/s40168-020-00875-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D., Shaw J., Kalathiappan M., Ng A.H.Q., Muthiah S., Li C., Dvornicic M., Soldo J.P., Koh J.Y., Tek N.O., et al. Nanopore sequencing enables high-resolution analysis of resistance determinants and mobile elements in the human gut microbiome. Biorxiv. 2018;456905 doi: 10.1101/456905. [DOI] [Google Scholar]
- Bertrand D., Shaw J., Kalathiyappan M., Ng A.H.Q., Kumar M.S., Li C., Dvornicic M., Soldo J.P., Koh J.Y., Tong C., et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 2019;37:937–944. doi: 10.1038/s41587-019-0191-2. [DOI] [PubMed] [Google Scholar]
- Bodor A., Bounedjoum N., Vincze G., Kis Á., Laczi K., Bende G., Szilágyi Á., Kovács T., Perei K., Rákhely G. Challenges of unculturable bacteria: environmental perspectives. Rev. Environ. Sci. Bio Technology. 2020;19:1–22. doi: 10.1007/s11157-020-09522-4. [DOI] [Google Scholar]
- Bowers R.M., Kyrpides N.C., Stepanauskas R., Harmon-Smith M., Doud D., Reddy T.B.K., Schulz F., Jarett J., Rivers A.R., Eloe-Fadrosh E.A., et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017;35:725–731. doi: 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B.J., McMurdie P.J., Holmes S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. Isme J. 2017;11:2639–2643. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaipan C., Pryszlak A., Dean H., Poignard P., Benes V., Griffiths A.D., Merten C.A. Single-virus droplet microfluidics for high-throughput screening of neutralizing epitopes on HIV particles. Cell. Chem. Biol. 2017;24:751–757. doi: 10.1016/j.chembiol.2017.05.009. [DOI] [PubMed] [Google Scholar]
- Chen L.-X., Anantharaman K., Shaiber A., Eren M.A., Banfield J.F. Accurate and complete genomes from metagenomes. Genome Res. 2020;30:315–333. doi: 10.1101/gr.258640.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chijiiwa R., Hosokawa M., Kogawa M., Nishikawa Y., Ide K., Sakanashi C., Takahashi K., Takeyama H. Single-cell genomics of uncultured bacteria reveals dietary fiber responders in the mouse gut microbiota. Microbiome. 2020;8:5. doi: 10.1186/s40168-019-0779-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins D.J., Neild A., deMello A., Liu A.-Q., Ai Y. The Poisson distribution and beyond: methods for microfluidic droplet production and single cell encapsulation. Lab Chip. 2015;15:3439–3459. doi: 10.1039/c5lc00614g. [DOI] [PubMed] [Google Scholar]
- Cross K.L., Campbell J.H., Balachandran M., Campbell A.G., Cooper S.J., Griffen A., Heaton M., Joshi S., Klingeman D., Leys E., et al. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat. Biotechnol. 2019;37:1314–1321. doi: 10.1038/s41587-019-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debs B.E., Utharala R., Balyasnikova I.V., Griffiths A.D., Merten C.A. Functional single-cell hybridoma screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. USA. 2012;109:11570–11575. doi: 10.1073/pnas.1204514109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichosa A.E., Daughton A.R., Reitenga K.G., Fitzsimons M.S., Han C.S. Capturing and cultivating single bacterial cells in gel microdroplets to obtain near-complete genomes. Nat. Protoc. 2014;9:608–621. doi: 10.1038/nprot.2014.034. [DOI] [PubMed] [Google Scholar]
- Epstein S. The phenomenon of microbial uncultivability. Curr. Opin. Microbiol. 2013;16:636–642. doi: 10.1016/j.mib.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Feng H., Qian Z. Functional carbon quantum dots: a versatile platform for chemosensing and biosensing. Chem. Rec. 2018;18:491–505. doi: 10.1002/tcr.201700055. [DOI] [PubMed] [Google Scholar]
- Garrison E., Boisvert S. 2014. Interleave FASTQ.https://github.com/ekg/interleave-fastq/blob/master/LICENSE [Google Scholar]
- Gawad C., Koh W., Quake S.R. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 2016;17:175–188. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- Grieb A., Bowers R.M., Oggerin M., Goudeau D., Lee J., Malmstrom R.R., Woyke T., Fuchs B.M. A pipeline for targeted metagenomics of environmental bacteria. Microbiome. 2020;8:21. doi: 10.1186/s40168-020-0790-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Allen-Vercoe E., Petrof E.O. Fecal microbiota transplantation: in perspective. Ther. Adv. Gastroenter. 2016;9:229–239. doi: 10.1177/1756283x15607414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzenpichler R., Krukenberg V., Spietz R.L., Jay Z.J. Next-generation physiology approaches to study microbiome function at single cell level. Nat. Rev. Microbiol. 2020;18:241–256. doi: 10.1038/s41579-020-0323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand F., Tadeo R., Voigt A.Y., Bork P., Raes J. LotuS: an efficient and user-friendly OTU processing pipeline. Microbiome. 2014;2:30. doi: 10.1186/2049-2618-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson B.J., Ness K.D., Masquelier D.A., Belgrader P., Heredia N.J., Makarewicz A.J., Bright I.J., Lucero M.Y., Hiddessen A.L., Legler T.C., et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson C.M., Chevillet J.R., Briggs H.A., Gallichotte E.N., Ruf I.K., Hindson B.J., Vessella R.L., Tewari M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods. 2013;10:1003–1005. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosic S., Murthy S.K., Koppes A.N. Microfluidic sample preparation for single cell analysis. Anal. Chem. 2016;88:354–380. doi: 10.1021/acs.analchem.5b04077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa M., Hoshino Y., Nishikawa Y., Hirose T., Yoon D.H., Mori T., Sekiguchi T., Shoji S., Takeyama H. Droplet-based microfluidics for high-throughput screening of a metagenomic library for isolation of microbial enzymes. Biosens. Bioelectron. 2015;67:379–385. doi: 10.1016/j.bios.2014.08.059. [DOI] [PubMed] [Google Scholar]
- Hug L.A., Baker B.J., Anantharaman K., Brown C.T., Probst A.J., Castelle C.J., Butterfield C.N., Hernsdorf A.W., Amano Y., Ise K., et al. A new view of the tree of life. Nat. Microbiol. 2016;1:16048. doi: 10.1038/nmicrobiol.2016.48. [DOI] [PubMed] [Google Scholar]
- Jiang C.-Y., Dong L., Zhao J.-K., Hu X., Shen C., Qiao Y., Zhang X., Wang Y., Ismagilov R.F., Liu S.-J., Du W. High-throughput single-cell cultivation on microfluidic streak plates. Appl. Environ. Microb. 2016;82:2210–2218. doi: 10.1128/aem.03588-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephides D., Davoli S., Whitley W., Ruis R., Salter R., Gokkaya S., Vallet M., Matthews D., Benazzi G., Shvets E., et al. Cyto-mine: an integrated, picodroplet system for high-throughput single-cell analysis, sorting, dispensing, and monoclonality assurance. Slas Technology. 2019;25:177–189. doi: 10.1177/2472630319892571. [DOI] [PubMed] [Google Scholar]
- Joshi N., Fass J. 2014. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files. [Google Scholar]
- Kang D.D., Froula J., Egan R., Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. Peerj. 2015;3:e1165. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintses B., Vliet L.D., Devenish S.R., Hollfelder F. Microfluidic droplets: new integrated workflows for biological experiments. Curr. Opin. Chem. Biol. 2010;14:548–555. doi: 10.1016/j.cbpa.2010.08.013. [DOI] [PubMed] [Google Scholar]
- Korpela K., Costea P., Coelho L.P., Kandels-Lewis S., Willemsen G., Boomsma D.I., Segata N., Bork P. Selective maternal seeding and environment shape the human gut microbiome. Genome Res. 2018;28:561–568. doi: 10.1101/gr.233940.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F., Demaree B., Ahmed N., Abate A.R. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat. Biotechnol. 2017;35:640–646. doi: 10.1038/nbt.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K., Zahn H., Leaver T., Konwar K.M., Hanson N.W., Pagé A.P., Lo C.-C., Chain P.S., Hallam S.J., Hansen C.L. A programmable droplet-based microfluidic device applied to multiparameter analysis of single microbes and microbial communities. Proc. Natl. Acad. Sci. USA. 2012;109:7665–7670. doi: 10.1073/pnas.1106752109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Bai R., Zhao Z., Tao L., Ma M., Ji Z., Jian M., Ding Z., Dai X., Bao F., Liu A. Application of droplet digital PCR to detect the pathogens of infectious diseases. Biosci. Rep. 2018;38 doi: 10.1042/bsr20181170. BSR20181170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Subgroup. 1000 Genome Project Data Processing The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.W., Lance S.T., Stedman K.M., Abate A.R. PCR-activated cell sorting as a general, cultivation-free method for high-throughput identification and enrichment of virus hosts. J. Virol. Methods. 2017;242:14–21. doi: 10.1016/j.jviromet.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.W., Tran T.M., Abate A.R. PCR-activated cell sorting for cultivation-free enrichment and sequencing of rare microbes. PLoS ONE. 2015;10:e0113549. doi: 10.1371/journal.pone.0113549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S., Berkemeier B. Ultrasensitive detection and quantification of viral nucleic acids with raindance droplet digital PCR (ddPCR) Methods. 2021 doi: 10.1016/j.ymeth.2021.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P., Hold G.L., Flint H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014;12:661–672. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- Mahler L., Niehs S.P., Martin K., Weber T., Scherlach K., Hertweck C., Roth M., Rosenbaum M.A. Highly parallelized droplet cultivation and prioritization on antibiotic producers from natural microbial communities. Elife. 2021;10:e64774. doi: 10.7554/elife.64774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Hernandez F., Garcia-Heredia I., Gomez M.L., Maestre-Carballa L., Martínez J.M., Martinez-Garcia M. Droplet digital PCR for estimating absolute abundances of widespread pelagibacter viruses. Front Microbiol. 2019;10:1226. doi: 10.3389/fmicb.2019.01226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanese A., Mende D.R., Paoli L., Salazar G., Ruscheweyh H.-J., Cuenca M., Hingamp P., Alves R., Costea P.I., Coelho L., et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019;10:1014. doi: 10.1038/s41467-019-08844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss E.L., Maghini D.G., Bhatt A.S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 2020;38:701–707. doi: 10.1038/s41587-020-0422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S., Shi Z., Seshadri R., Pollard K.S., Kyrpides N.C. New insights from uncultivated genomes of the global human gut microbiome. Nature. 2019;568:505–510. doi: 10.1038/s41586-019-1058-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa Y., Hosokawa M., Maruyama T., Yamagishi K., Mori T., Takeyama H. Monodisperse picoliter droplets for low-bias and contamination-free reactions in single-cell whole genome amplification. PLoS ONE. 2015;10:e0138733. doi: 10.1371/journal.pone.0138733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobu M.K., Narihiro T., Rinke C., Kamagata Y., Tringe S.G., Woyke T., Liu W.-T. Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor. Isme J. 2015;9:1710–1722. doi: 10.1038/ismej.2014.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S., Bankevich A., Antipov D., Gurevich A., Korobeynikov A., Lapidus A., Prjibelsky A., Pyshkin A., Sirotkin A., Sirotkin Y., et al. In: Research in Computational Molecular Biology. RECOMB 2013. Lecture Notes in Computer Science, Lecture Notes in Computer Science. Deng M., Jiang R., Sun F., Zhang X., editors. 2013. Assembling genomes and mini-metagenomes from highly chimeric reads; pp. 158–170. [DOI] [Google Scholar]
- Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D.H., Rinke C., Chuvochina M., Chaumeil P.-A., Woodcroft B.J., Evans P.N., Hugenholtz P., Tyson G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017;2:1533–1542. doi: 10.1038/s41564-017-0012-7. [DOI] [PubMed] [Google Scholar]
- Prakadan S.M., Shalek A.K., Weitz D.A. Scaling by shrinking: empowering single-cell “omics” with microfluidic devices. Nat. Rev. Genet. 2017;18:345–361. doi: 10.1038/nrg.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Props R., Kerckhof F.-M., Rubbens P., Vrieze J., Sanabria E., Waegeman W., Monsieurs P., Hammes F., Boon N. Absolute quantification of microbial taxon abundances. Isme J. 2017;11:584–587. doi: 10.1038/ismej.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente-Sánchez F., Aguirre J., Parro V. A novel conceptual approach to read-filtering in high-throughput amplicon sequencing studies. Nucleic Acids Res. 2016;44:e40. doi: 10.1093/nar/gkv1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa G., Clasen T., Miller L. Microbiological effectiveness of disinfecting water by boiling in rural Guatemala. Am. J. Trop. Med. Hyg. 2010;82:473–477. doi: 10.4269/ajtmh.2010.09-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciambi A., Abate A.R. Accurate microfluidic sorting of droplets at 30 kHz. Lab Chip. 2014;15:47–51. doi: 10.1039/c4lc01194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Liu Y., Pan J., Wang F., Li M. Perspectives on cultivation strategies of archaea. Microb. Ecol. 2019:1–15. doi: 10.1007/s00248-019-01422-7. [DOI] [PubMed] [Google Scholar]
- Tauzin A.S., Pereira M.R., Vliet L.D.V., Colin P.-Y., Laville E., Esque J., Laguerre S., Henrissat B., Terrapon N., Lombard V., et al. Investigating host-microbiome interactions by droplet based microfluidics. Microbiome. 2020;8:141. doi: 10.1186/s40168-020-00911-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terekhov S.S., Smirnov I.V., Malakhova M.V., Samoilov A.E., Manolov A.I., Nazarov A.S., Danilov D.V., Dubiley S.A., Osterman I.A., Rubtsova M.P., et al. Ultrahigh-throughput functional profiling of microbiota communities. Proc. Natl. Acad. Sci. USA. 2018;115:201811250. doi: 10.1073/pnas.1811250115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano M., Andrejev S., Pruteanu M., Klünemann M., Kuhn M., Galardini M., Jouhten P., Zelezniak A., Zeller G., Bork P., et al. Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat. Microbiol. 2018;3:514–522. doi: 10.1038/s41564-018-0123-9. [DOI] [PubMed] [Google Scholar]
- Vandeputte D., Kathagen G., D’hoe K., Vieira-Silva S., Valles-Colomer M., Sabino J., Wang J., Tito R.Y., Commer L., Darzi Y., et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551:507–511. doi: 10.1038/nature24460. [DOI] [PubMed] [Google Scholar]
- Vos W.M., Vos E.A. Role of the intestinal microbiome in health and disease: from correlation to causation. Nutr. Rev. 2012;70:S45–S56. doi: 10.1111/j.1753-4887.2012.00505.x. [DOI] [PubMed] [Google Scholar]
- Waaij L.A., Mesander G., Limburg P.C., Waaij D. Direct flow cytometry of anaerobic bacteria in human feces. Cytometry. 1994;16:270–279. doi: 10.1002/cyto.990160312. [DOI] [PubMed] [Google Scholar]
- Watterson W.J., Tanyeri M., Watson A.R., Cham C.M., Shan Y., Chang E.B., Eren A.M., Tay S. Droplet-based high-throughput cultivation for accurate screening of antibiotic resistant gut microbes. Elife. 2020;9:e56998. doi: 10.7554/elife.56998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D.E., Salzberg S.L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H.-D., Zheng H., Guo W., Gañán-Calvo A.M., Ai Y., Tsao C.-W., Zhou J., Li W., Huang Y., Nguyen N.-T., Tan S. Active droplet sorting in microfluidics: a review. Lab Chip. 2017;17:751–771. doi: 10.1039/c6lc01435f. [DOI] [PubMed] [Google Scholar]
- Xu P., Modavi C., Demaree B., Twigg F., Liang B., Sun C., Zhang W., Abate A.R. Microfluidic automated plasmid library enrichment for biosynthetic gene cluster discovery. Nucleic Acids Res. 2020;48:e48. doi: 10.1093/nar/gkaa131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaginuma K., Aoki W., Miura N., Ohtani Y., Aburaya S., Kogawa M., Nishikawa Y., Hosokawa M., Takeyama H., Ueda M. High-throughput identification of peptide agonists against GPCRs by co-culture of mammalian reporter cells and peptide-secreting yeast cells using droplet microfluidics. Sci. Rep. UK. 2019;9:10920. doi: 10.1038/s41598-019-47388-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R., Paparini A., Monis P., Ryan U. Comparison of next-generation droplet digital PCR (ddPCR) with quantitative PCR (qPCR) for enumeration of Cryptosporidium oocysts in faecal samples. Int. J. Parasitol. 2014;44:1105–1113. doi: 10.1016/j.ijpara.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Yousi F., Kainan C., Junnan Z., Chuanxing X., Lina F., Bangzhou Z., Jianlin R., Baishan F. Evaluation of the effects of four media on human intestinal microbiota culture in vitro. Amb Express. 2019;9:69. doi: 10.1186/s13568-019-0790-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W., Guo L., Xu S., Chen J., Zhou J. High-throughput screening technology in industrial biotechnology. Trends Biotechnol. 2020;38:888–906. doi: 10.1016/j.tibtech.2020.01.001. [DOI] [PubMed] [Google Scholar]
- Zengler K., Toledo G., Rappe M., Elkins J., Mathur E., Short J., Keller M. Cultivating the uncultured. Proc. Natl. Acad. Sci. USA. 2002;99:15681–15686. doi: 10.1073/pnas.252630999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W., Zhao S., Yin Y., Zhang H., Needham D.M., Evans E.D., Dai C.L., Lu P.J., Alm E.J., Weitz D.A. Microbe-seq: high-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome. Biorxiv. 2020 doi: 10.1101/2020.12.14.422699. [DOI] [PubMed] [Google Scholar]
- Zimmermann M., Zimmermann-Kogadeeva M., Wegmann R., Goodman A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570:462–467. doi: 10.1038/s41586-019-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M., Zimmermann-Kogadeeva M., Wegmann R., Goodman A.L. Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science. 2019;363:eaat9931. doi: 10.1126/science.aat9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlitni S., Bishara A., Moss E.L., Tkachenko E., Kang J.B., Culver R.N., Andermann T.M., Weng Z., Wood C., Handy C., et al. Strain-resolved microbiome sequencing reveals mobile elements that drive bacterial competition on a clinical timescale. Genome Med. 2020;12:50. doi: 10.1186/s13073-020-00747-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwielehner J., Lassl C., Hippe B., Pointner A., Switzeny O.J., Remely M., Kitzweger E., Ruckser R., Haslberger A.G. Changes in human fecal microbiota due to chemotherapy analyzed by taqman-pcr, 454 sequencing and pcr-dgge fingerprinting. PLoS ONE. 2011;6:e28654. doi: 10.1371/journal.pone.0028654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data, microfluidic chip designs, lists and, figure scripts (CheckM, Binning, Contig KEGG-KO table, PADS data plotting via GitHub) have been deposited in the Open Science Framework. Metagenomic sequences and assembled genomes have been deposited in the ENA database. The corresponding DOIs and permanent identifiers are listed in the key resources table.
-
•
The FIJI plugin to analyze droplets along with the images used in this publication have been made available on Zenodo. The DOI is given in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.