

Abstract
Cancer cells almost universally harbor constitutively active Phosphatidylinositol-3 Kinase (PI3K) Pathway activity via mutation of key signaling components and/or epigenetic mechanisms. Scores of PI3K Pathway inhibitors are currently under investigation as putative chemotherapeutics. However, feedback and stem cell mechanisms induced by PI3K Pathway inhibition can lead to reduced treatment efficacy. To address therapeutic barriers, we examined whether JAKi would reduce stem gene expression in a setting of PI3K Pathway inhibition in order to improve treatment efficacy. We targeted the PI3K Pathway with NVP-BEZ235 (dual PI3K and mTOR inhibitor) in combination with the Janus Kinase inhibitor JAKi in glioblastoma (GBM) and basal-like breast cancer (BBC) cell lines. We examined growth, gene expression, and apoptosis in cells treated with NVP-BEZ235 and/or JAKi. Growth and recovery assays showed no significant impact of dual treatment with NVP-BEZ235/JAKi compared to NVP-BEZ235 treatment alone. Gene expression and flow cytometry revealed that single and dual treatments induced apoptosis. Stem gene expression was retained in dual NVP-BEZ235/JAKi treatment samples. Future in vivo studies may give further insight into the impact of combined NVP-BEZ235/JAKi treatment in GBM and BBC.
Keywords: GBM, PI3K, stem phenotypes, apoptosis
Introduction
In 2008 Weinberg and colleagues discovered that certain poorly-differentiated aggressive cancers such as glioblastoma multiforme (GBM) and basal-like breast cancer (BBC) harbor an embryonic stem cell-like gene expression signature1. These cancers express stem genes such as OCT4 (Octamer-binding Transcription Factor 4), SOX2 (SRY-Box Transcription Factor 2) and NANOG (Nanog Homeobox)1–4. Stem genes contribute to poor prognosis in GBM and BBC5, 6. Of note, we and others found that like embryonic stem cells (ESC), the PI3K (Phosphatidylinositol 3-kinase) Pathway is fundamentally rewired in GBM and BBC7–11. Transcription factor FOXO1 (Forkhead box O 1) was functional despite constitutively active PI3K (against the paradigm) in GBM and BBC7. Furthermore, FOXO1 promoted stem gene expression in GBM and BBC cancer cells via a similar mechanism described in ESC7, 10. PI3K inhibitor treatment induced stem gene expression in GBM and BBC cells7, 8.
GBM is a prevalent type of tumor of the central nervous system12, 13. GBM has high metastatic abilities and an aggressive phenotype which poses a challenge on surgical removal, giving patients an average 15 months of survival14, 15. Breast cancer can be classified based on gene expression signatures into distinct subtypes such as luminal A, luminal B, BBC, HER2 positive and normal-like16–19. BBC comprises approximately 10-17% of breast cancers in the United States and has characteristics in common with myoepithelial cells of the breast20–22. Frequently, BBC lacks expression of estrogen, progesterone and HER2 receptors, making these cancers unresponsive to hormonal-based therapies23, 24. BBC is associated with poor prognosis25–27. In addition to harboring stem-like signatures, GBM and BBC are associated with self-renewal, metastasis, and chemotherapeutic resistance1. Despite therapeutic advances, targeting the bulk of tumorigenic cells does not exclude patients from future metastasis and relapse28–30. Targeting pathways that drive the stem signatures in GBM and BBC may ultimately decrease patient relapse31–33.
The PI3K Pathway is almost universally activated in cancer by mutations to promote growth and survival34, 35. Endogenously, the PI3K Pathway is activated by growth factors binding to receptor tyrosine kinases (RTKs). Active PI3K converts phosphatidylinositol-4,5, bisphosphate (PIP2) to phosphatidylinositol-3,4,5 trisphosphate (PIP3)36. Tumor suppressor PTEN encodes a phosphatase that converts PIP3 to PIP2 to down-regulate this pathway37–39. Second messenger PIP3 membrane-recruits and activates AKT which phosphorylates FOXO transcription factors (FOXO −1, −3, and −4) among other substrates40. FOXO transcription factors regulate metabolism, apoptosis, tumor suppression and cellular differentiation41–43. FOXO transcription factors also promote stem characteristics in embryonic stem (ES), BBC, and GBM cells in part by activating OCT4 gene expression7, 10. In GBM and BBC, PI3K Pathway genes are characteristically mutated to activate signaling output. PTEN is commonly mutated to an inactive form, whereas the RTK gene EGFR (Epidermal Growth Factor) is mutated to encode an active form leading to increased growth and survival44–46.
Given the central role of the PI3K pathway in driving tumor formation and progression, it is a common chemotherapeutic target34, 47–49. PI3K pathway targeted therapies in GBM and BBC have improved in selectivity and stability while also decreasing in toxicity50, 51. EGFR inhibitor erlotinib slowed GBM progression in a fraction of patients52–54. An enhanced PI3K inhibitor that targets both PI3K and mTOR (mammalian target of rapamycin), NVP-BEZ235 improved survival outcomes and elicited advanced antitumor effects in GBM55–58. However, we and others have observed that targeting the PI3K pathway leads to the induction of stem genes in GBM and BBC cells, potentially promoting cancer recurrence7, 8
Stem gene expression in GBM and BBC is activated by numerous mechanisms including the Janus Kinase 1/Janus Kinase 2 (JAK1/2) and STAT3 (Signal Transducer and Activator of Transcription 3) Pathway59, 60. STAT3 is endogenously activated by JAK1/2 upon cytokine binding to cognate receptors. JAK1/2 phosphorylates STAT3 on tyrosine 705, leading to homodimerization and regulation of target genes in the nucleus61. The JAK/STAT3 Pathway becomes constitutively active in cancer cells to accelerate tumor progression61. In GBM and BBC, activating mutations in JAK1, JAK2 and EGFR lead to constitutively active STAT346, 62–65. Increased cytokine signaling also contributes to STAT3 activation in these cancers66–69. Active STAT3 during tumorigenesis contributes to proliferation and hinders apoptosis70–72. STAT3 sustains cancer stem cells and promotes metastasis73–78. In pluripotent stem cells JAK1 activates STAT3, which in turn directly binds to the OCT4 promoter to induce transcription59. Reduced STAT3 expression in pluripotent stem cells led to a loss in stem cell marker alkaline phosphatase59. Based on this, we reasoned that JAK inhibition may reduce stem characteristics in GBM and BBC cells.
JAK inhibitors have proved effective at decreasing invasiveness and tumorigenesis in GBM and BBC in preclinical studies79–81. Pacritinib increased survival in GBM orthotopic xenograft studies82. Ruxolitinib decreased migration and colony formation in GBM cells79. AZD1480 reduced GBM xenograft growth83. Similarly, JAK inhibition with ruxolitinib was examined as a therapeutic for BBC with limited success84. Although ruxolitinib was able to diminish STAT3 activation in BBC, it was not efficacious as a single agent therapy84.
One barrier to effectively employing PI3K targeted therapies in GBM and BBC is the induction of stem genes, which may ultimately promote resistance and/or recurrence7, 8. We hypothesized that targeting the PI3K Pathway (to suppress growth and survival) in combination with JAKi (to suppress growth and stem gene expression) may have a combinatorial impact on GBM and BBC.
Materials and Methods
Cell culture
Cell lines were obtained from ATCC (American Type Culture Collection, Manassas, VA) and grown under standard conditions (5% CO2, 10% FBS (fetal bovine serum), with 5% antibiotic/antimycotic (Thermo Fisher, Waltham, MA)). Cell lines were tested for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza, Basel Switzerland, cat: LT07-218); all experiments were done with mycoplasma negative cells. U87MG cells were propagated in MEM (Minimal Essential Medium). DBTRG and BT549 cells were propagated in RPMI (Roswell Park Memorial Institute 1640 Medium). LN18, U118MG, A172, MDA-MB-468 and LN229 cells were propagated in DMEM (Dulbecco’s Modified Eagle Medium).
Drug Treatments
Cells were plated at a density of 15,000 cells per mL and were treated for indicated number of days with indicated drugs for growth assays. Recovery assays included indicated drug treatments added to cells plated at 2,700 cells per mL. Recovery assay samples had fresh media added after six days of drug treatment; these samples were allowed to recover for six days in fresh media before staining to assess cellular growth after treatment. Treatments were investigated in triplicate (in numerous independent experiments) and stained with crystal violet. Plates were aspirated of media then washed with 0.5 ml of 1x phosphate buffered saline (PBS) before being stained with 0.5 ml of crystal violet stain (0.5% crystal violet in buffered formalin) and incubated for 15 minutes. The stain was aspirated, and wells were washed 3 times with 0.5 ml of 1x PBS. After collections were completed, crystal violet-stained plates were solubilized using 0.5 ml on each well of 10% acetic acid and placed on shaker for 1 hour. Solubilized samples were transferred to 96 well plates and quantified on a spectrophotometer at 590 nm. Quantified plates were analyzed with a Tukey Test on vassarstats.net for statistical significance. Error bars were added using the standard error of the collection readouts. NVP-BEZ235 was purchased from Sigma (Saint Louis, MO), and utilized at a final concentration of 50 nM in growth and recovery experiments. JAKi was purchased from Sigma and used at a final concentration of 2 μM. NVP-BEZ235 and JAKi were dissolved in DMSO.
Western Blot
Cell lines were treated with 50 nM NVP-BEZ235, 2 μM JAKi and/or DMSO (solvent) for 24 hours. Total protein was obtained by rinsing cells with 1XPBS (phosphate buffered saline) followed by direct lysis in 2x sample buffer (125 mM Tris-HCL at pH 6.8, 2% sodium dodecyl sulfate (SDS), 10% 2-mercaptoethanol, 20% glycerol, 0.05% bromophenol blue, 8 M urea); 2x sample buffer was added to each well and cells scraped with a cell scraper. The lysate was collected from each well, placed into 1.5 mL microcentrifuge tubes and heated for 10 minutes at 95°C in a dry-bath heat block. Protein lysates were separated by sodium dodecyl sulfate – polyacrylamide gel electrophoresis (SDS-PAGE) at 100V for 1 hour. The protein was then transferred onto a polyvinylidene fluoride (PVDF) membrane for an hour and 30 minutes then blocked in a 5% milk solution (Carnation powdered milk, 1X Tris-buffered saline with Tween 20 (TBST) for an hour. The membrane was incubated with indicated primary antibody overnight at 4°C then, washed for 20 minutes with TBST in 5-minute intervals. The blot was then incubated with secondary antibodies for 1.5 hours. The membrane was washed again for 20 minutes in 5-minute intervals and allowed to develop using SuperSignal West Dura Extended Duration Substrate luminol solution (Pierce Biotechnology, Waltham, MA) for 5 minutes. A Bio Rad ChemDoc XRS+ Molecular Imager was utilized for protein detection (Bio Rad Hercules, CA). Densitometry was performed using Image Lab Software (Bio Rad Hercules, CA). Relative expression was calculated using total band volume of a protein of interest divided by the GAPDH total band volume for the protein sample. Band densities were depicted relative to the DMSO solvent control. Antibodies were obtained from Cell Signaling Technologies (Danvers, MA): total STAT3 (9139T), phospho STAT3 tyrosine 705 (9145T), and phospho AKT serine 473 (9271S) and total AKT (4691S). GAPDH (G-9) was obtained from Santa Cruz Biotechnology Inc., Dallas, TX.
Quantitative Real Time PCR
Total RNA was prepared using the Qiagen RNeasy kit (Hilden, Germany), which was then used to generate cDNA with Superscript Reverse Transcriptase II (Invitrogen, Carlsbad, CA). Samples (cDNAs) were analyzed using (Power SYBR Green Master Mix, Applied Biosystems, Foster City, CA) and the Illumina Eco Real-time system (San Diego, CA). Expression levels were normalized to GAPDH in gene expression experiments and calculated using 2−ΔΔCT method 85. Primer sequences are detailed in supplemental Table S1. All drug treatments utilized 50 nm NVP-BEZ235 and 2 μM JAKi. Time points for RNA collection were optimized for each cell line. U87MG and LN229 RNA lysates were collected after five days of drug treatment. BT549 RNA lysates for stem genes were collected after five days of drug treatment. BT549 RNA lysates for apoptosis were collected after 24 hours. MDA-MB-468 RNA lysates were collected after three days of drug treatment.
Flow Cytometry
All cell lines were treated with 50 nM NVP-BEZ235, 2 μM JAKi and/or DMSO. U87MG cells were treated with drug for five days before assessing apoptosis, whereas LN229, BT549 and MDA-MB-468 cells were treated with drug for three days before assessing apoptosis. Growth medium was aspirated after indicated treatment. Cells were incubated with 2 mL of 0.25% trypsin each for 5 minutes at room temperature. 1.5 mL of each plate was transferred into 1.7 mL microcentrifuge tubes and centrifuged at Gx0.2 for 10 minutes. Trypsin supernatant was aspirated, cells were resuspended with 1xPBS and then centrifuged at Gx0.2 for 10 minutes. 1xPBS supernatant was aspirated and cells in microcentrifuge tubes were then resuspended with dilute binding buffer (Molecular Probes FITC Annexin V/Dead Cell Apoptosis Kit from Thermo Fisher, Waltham, MA). 100 μl of each sample diluted with binding buffer were added to a fresh microcentrifuge tube. Each tube had 2 μl of Annexin V FITC and 1 μl of PI added and incubated for 15 minutes in the dark. 400 μl of binding buffer was added after the 15-minute incubation and cells were sorted by BD FACS Celesta flow cytometer (BD Biosciences, San Jose, CA).
RESULTS
Treatment with NVP-BEZ235 or JAKi led to a reduction in GBM and BBC cells
To assess the impact of combined PI3K pathway and JAK inhibition on cancer cell growth and survival, we treated U87MG GBM, LN229 GBM, BT549 BBC, and MDA-MB-468 BBC cancer cell lines with vehicle (DMSO), 50 nM NVP-BEZ235, and/or 2 μM JAKi (Figure 1A–D) for five days. Plates were stained with crystal violet at indicated time points and data were analyzed by ANOVA with Tukey HSD Test. In the U87MG cell line on day 5, each treatment (JAKi, NVP-BEZ235 and combination of JAKi with NVP-BEZ235) was found to have reduced cell numbers compared to the DMSO vehicle (P < 0.01). JAKi single treatment had a significantly lower growth inhibition (P <0.01) when compared to single NVP-BEZ235 and double treatment of JAKi/NVP-BEZ235. However, there was no significant difference between the single NVP-BEZ235 treatment and the JAKi/NVP-BEZ235 double treatment for cell growth. In the BT549, LN229 and MDA-MB-468 cell lines on day five, each treatment (JAKi, NVP-BEZ235 and combination of JAKi with NVP-BEZ235) was found to negatively impact growth compared to the DMSO vehicle (P < 0.01). However, there was no significant difference between JAKi, NVP-BEZ235, and JAKi/NVP-BEZ235 treatments in growth assays (Figure 1A–D).
Figure 1. NVP-BEZ235 or JAKi reduced Growth in Examined GBM and BBC Cell Lines.
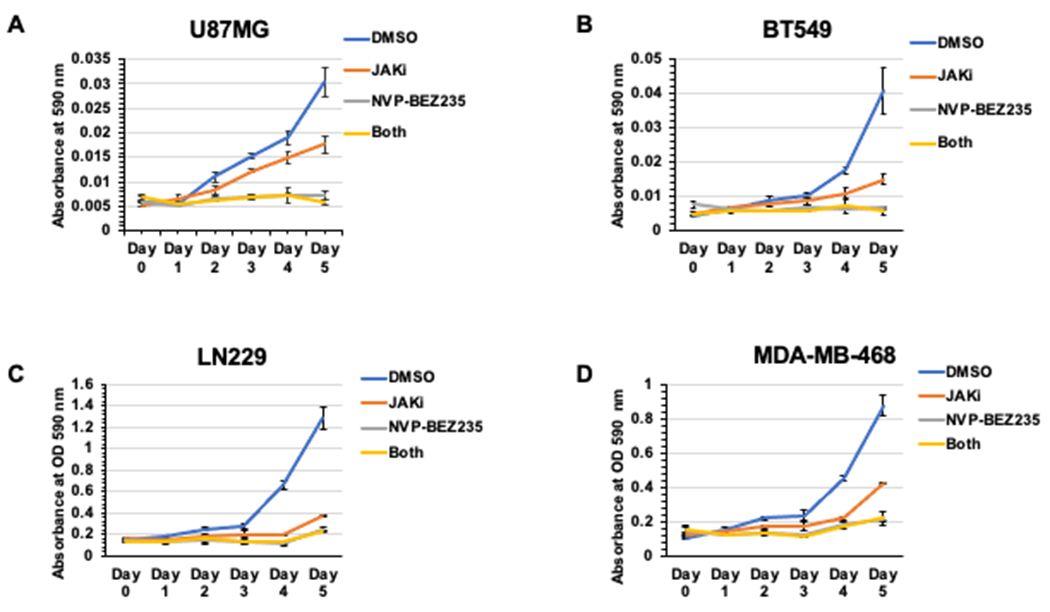
A. Growth assays with U87MG cells treated with 50 nM NVP-BEZ235 and/or 2 μm JAKi. All treatments significantly impacted growth compared to DMSO control with no increased impact from dual NVP-BEZ235/JAKi treatment (compared to single NVP-BEZ235 alone) based on ANOVA with Tukey HSD Test. JAKi treatment impacted growth significantly less than NVP-BEZ235. B-D. Growth assays with BT549, LN229 and MDA-MB-468 cells treated with 50 nM NVP-BEZ235 and/or 2μm JAKi. All treatments significantly impacted growth compared to DMSO control. JAKi treatment was not significantly different compared to NVP-BEZ235 or NVP-BEZ235/JAKi based on ANOVA with Tukey HSD Test.
Protein lysates were utilized to assess inhibition of STAT3 (via 2 μM JAKi treatment) and AKT activation (via PI3K/mTOR inhibition, 50 nM NVP-BEZ235 treatment) with GAPDH as a loading control using western blot and densitometry analyses (Figure 2A–F). Lysates collected from NVP-BEZ235 drug-treated samples had reduced P-AKT (S473), whereas extracts collected from the JAKi drug-treated cells showed reduced P-STAT3 (Y705).
Figure 2. Western Blot Analysis for NVP-BEZ235 or JAKi Treatments.

A. Total protein lysates were collected 24 hours post NVP-BEZ235 (50 nM) and/or JAKi (2μM) treatment in U87MG and BT549 cells and examined by western blot analysis with indicated antibodies. B-C. Densitometry was performed using Image Lab Software (Bio-Rad) for U87MG and BT549 western blot data as described in the Materials and Methods. P-AKT (S473) was reduced in NVP-BEZ235 treatment samples, whereas P-STAT3 (Y705) was reduced in JAKi treatment samples. D. Total protein lysates were collected 24 hours post NVP-BEZ235 (50 nM) and/or JAKi (2 μM) treatment in LN229 and MDA-MB-468 cells and examined by western blot analysis with indicated antibodies. E-F. Densitometry was performed using Image Lab Software (Bio-Rad) for LN229 and MDA-MB-468 western blot data. P-AKT (S473) was reduced in NVP-BEZ235 treatment samples, whereas P-STAT3 (Y705) was reduced in JAKi treatment samples.
GBM cells showed reduced cell number in recovery assays with PI3K or JAK Inhibition
Given the similarities between the single NVP-BEZ235 combined NVP-BEZ235/JAKi treatments in growth assays, we sought to further investigate the phenotype by performing recovery assays. We treated indicated cell lines with vehicle (DMSO), NVP-BEZ235 alone (50 nM), JAKi alone (2 μM) or combined NVP-BEZ235/JAKi for six days. After drug treatment, we added fresh media and allowed the cells to recover for six days to determine differences in cell survival. The recovery assays would allow cytostatic cells to grow back, highlighting potential differences between cytotoxic and cytostatic impacts. Cells were stained with crystal violet as described in the Materials and Methods to assess cell numbers following treatment and recovery. We observed significantly decreased cell numbers with single and double treatment samples (NVP-BEZ235 and/or JAKi) based on ANOVA with Tukey HSD Test with reference to vehicle (DMSO) controls in all cell lines examined: GBM cell lines U87MG, DBTRG, LN18, LN229 and U118MG, as well as BBC cell lines BT549 and MDA-MB-468 (Figure 3A–G). However, there was no significant change between NVP-BEZ235 and NVP-BEZ235/JAKi treatments in recovery assays for any of the cell lines examined by Tukey HSD Test (Figure 3A–G).
Figure 3. NVP-BEZ235 or JAKi treatment reduced Cell Numbers in Recovery Assays.

Recovery assays were performed with GBM and BBC cell lines in which cells were treated with indicated drugs for six days and were then allowed to recover in complete media without drug for six days. A-G. All drug treatments in each examined cell line significantly impacted cell numbers compared to DMSO control (denoted *), with no increased impact from dual NVP-BEZ235/JAKi treatment compared to NVP-BEZ235 alone based on ANOVA with Tukey HSD Test. NVP-BEZ235 was abbreviated NVP. MDA-MB-468 was abbreviated 468.
Treatment with NVP-BEZ235 or JAKi led to increased apoptosis in U87MG cells
Gene expression analyses were performed to investigate mechanisms driving cell loss in U87MG, LN229, BT549 and MDA-MB-468 samples treated with the NVP-BEZ235 and/or JAKi. We initially hypothesized that the dual treatment would potentially cause a shift to reroute FOXO factors to induce apoptotic genes at the expense of stem genes. Stem factor OCT4 is activated in part by STAT386, 87. FOXO1 was also previously shown to activate OCT4 gene expression in stem cells as well as at least some GBM and BBC cancer cell lines7, 10. We tracked apoptotic genes that FOXO factors were known to induce: TRAIL, BIM, and FAS88. We found evidence of apoptotic gene induction by each drug treatment in each of the cell lines examined compared to DMSO controls, except for JAKi and combined NVP-BEZ235/JAKi treatments in MDA-MB-468 (Figures 4A–B and S1 A–B). In contrast to our hypothesis, all examined cell lines retained OCT4 expression as a stem marker in combination NVP-BEZ235/JAKi treated samples (Figures 4 C–D and S1 C–D). Some of the cell lines retained expression of additional markers such as SOX2, NANOG and ALPL (Alkaline Phosphatase Biomineralization Associated), (Figures 4 C–D and S1 C–D). Therefore, the combined NVP-BEZ235/JAKi treatment succeeded at inducing apoptotic genes but failed to suppress stem gene expression.
Figure. 4. Treatment of examined GBM or BBC cells with Dual PI3K Inhibitor NVP-BEZ235 and/or JAKi Induced Apoptotic Gene Expression.

A-B. Gene expression of FOXO-induced apoptotic targets (determined by qRT-PCR relative to GAPDH control) in U87MG and BT549 cells. NVP-BEZ235 treatment led to induction of BIM and TRAIL. C. Expression of stem genes from drug treatment samples by qRT-PCR (relative to GAPDH control). Stem genes OCT4, SOX2, NANOG and ALPL were induced in U87MG NVP-BEZ235 treatment samples with or without JAKi. Single agent treatment with JAKi led to reduced SOX2 and NANOG. D. Expression of OCT4 and NANOG in BT549 cells with indicated drug treatments. OCT4 and NANOG remained induced with the combined NVP-BEZ235/JAKi treatment based on ANOVA with Tukey HSD Test (denoted *).
To further examine apoptotic induction by NVP-BEZ235 and/or JAKi treatment, we performed flow cytometry. The marker for early apoptosis annexin-V conjugated with green fluorescent dye FITC was used to bind to externally exposed phospholipid phosphatidylserine thereby detecting membrane depolarization89. A DNA intercalating agent and late marker of apoptosis propidium iodide (PI) was used to detect cell permeability90. Cells stained with annexin-V FITC and/or PI were counted as apoptotic. Treatment with NVP-BEZ235 alone or the combination of NVP-BEZ235/JAKi led to similar levels of apoptosis in BT549 cells compared to DMSO controls (Figure 5A–E). JAKi alone, NVP-BEZ235 alone, and combined NVP-BEZ235/JAKi treatments (compared to DMSO control) led to apoptotic induction in U87MG, LN229 and MDA-MD-468 cell lines based on ANOVA with Tukey HSD Test (Figure 5E).
Figure 5. Flow Cytometric Analyses Revealed Apoptosis Induction by NVP-BEZ235 and/or JAKi in examined GBM and BBC cell lines.

Cells treated with indicated drugs were stained with AnnexinV-FITC and PI and were examined by flow cytometric analyses. A-D. Treatment of BT549 cells with NVP-BEZ235 alone or combined NVP-BEZ235/JAKi led to increased AnnexinV-FITC and PI staining. E. The percentage of apoptotic cells was determined by flow cytometric analyses for indicated drug-treated cell lines. AnnexinV-FITC and/or PI staining was counted as positive for apoptosis. Treatment with JAKi alone, NVP-BEZ235 alone, or combined NVP-BEZ235/JAKi led to significantly increased apoptosis compared to DMSO controls in U87MG, LN229 and MDA-MB-468 cells, whereas NVP-BEZ235 alone or in combination with JAKi led to apoptosis in BT549 cells based on ANOVA with Tukey HSD Test (denoted *).
CONCLUSION
The PI3K Pathway is almost universally constitutively active in GBM and BBC due to pathway mutations37, 44, 46. Activation of the PI3K Pathway drives tumor formation, progression, and metastasis, making it an ideal target for therapeutic intervention34, 36, 47. The use of PI3K inhibitors to treat GBM and BBC was previously shown to significantly induce cell death91–93. Cell cycle arrested remnants could develop resistance to treatments to become metastatic94. The 50 nM dose of NVP-BEZ235 employed in this work was utilized in clinical settings and pre-clinical studies55, 56. Similar doses of NVP-BEZ235 examined in GBM cell lines (U87MG, U251, T98G and SGH44) induced G1 cell cycle arrest and apoptosis55. NVP-BEZ235 treatment also reduced chemoresistance to temozolomide in GBM models and enhanced radiosensitivity of GBM stem cells55, 56. Low doses of NVP-BEZ235 (10-100 nM) led to either growth arrest or cell death in BBC cell lines95, 96. STAT3 inhibition led to a decrease in temozolomide resistance in GBM97. Likewise, inhibition of JAK/STAT led to decreased invasiveness in GBM and BBC83, 84. Our study employed 2μM JAKi based on work by Marotta et al in a BBC setting78. Clinical trials are underway to examine the impact of JAK inhibition as a therapeutic for GBM and BBC79, 84.
Although the combined treatment of NVP-BEZ235/JAKi had a limited impact in our study, others have found potent efficacies in acute lymphoblastic leukemia and B cell malignancies98–100. Signaling redundancies that induce stem characteristics may underpin the lack of efficacy in GBM and BBC cells.
Examined GBM and BBC cells retained expression of stem genes in dual treatment (NVP-BEZ235 and JAKi) samples. Therefore, inhibition of JAKi was not sufficient to block stem gene expression in the NVP-BEZ235 treated samples (Figure 4C–D and S1 C–D). The impact of STAT3 on stem gene expression is context dependent59, 101, 102. It remains to be determined if the increase in stem gene expression with NVP-BEZ235 treatment resulted from the survival of cells that express stem genes (such as cancer stem cells surviving and non-cancer stem cells undergoing apoptosis), leading to an enrichment in cancer stem cells. Alternatively, JAK-independent mechanisms may drive stem gene expression in this setting or the cells that express these stem genes may be resistant to drug treatment (perhaps via drug efflux). The timing of drug treatment may also contribute to therapeutic efficacy. Treatment of GBM with NVP-BEZ235 for one hour prior to with ionizing radiation strongly enhanced radiosensitivity103. In contrast, a 24-hour NVP-BEZ235 treatment prior to IR treatment led to G1 arrest and exhibited less DNA damage; there was no enhancement of IR sensitivity with the 24-hour treatment103. Delineating molecular mechanisms that drive stem characteristics and therapeutic resistance is key to improving chemotherapeutic efficacy for GBM and BBC.
Supplementary Material
Figure S1. Treatment of LN229 and MDA-MB-468 cells with Dual PI3K Inhibitor NVP-BEZ235 Induced Apoptotic Gene Expression. A. Gene expression of FOXO-induced apoptotic targets (determined by qRT-PCR relative to GAPDH control) in LN229 cells. BIM and FAS were induced by NVP-BEZ235. B. NVP-BEZ235 treatment led to induction of TRAIL gene expression by q-RT-PCR in MDA-MB-468 cells. C-D. Expression of stem genes from drug treatment samples by qRT-PCR (relative to GAPDH control). Stem genes OCT4 and SOX2 were induced in LN229 NVP-BEZ235 treatment samples with or without JAKi. D. Expression of OCT4 and ALPL in MDA-MB-468 cells with indicted drug treatments. OCT4 and ALPL remained induced with the combined NVP-BEZ235/JAKi treatment based on ANOVA with Tukey HSD Test (denoted *).
Highlights.
Treatment of a set of glioblastoma multiforme (GBM) and basal-like breast cancer (BBC) cell lines with PI3K inhibitor NVP-BEZ235, JAKi or NVP-BEZ235/JAKi combination led to reduced cell numbers with no increased impact for the combination treatment.
Flow cytometric and gene expression analyses revealed that NVP-BEZ235, JAKi or NVP-BEZ235/JAKi treatments led to induction of apoptosis in examined GBM and BBC cell lines.
Sustained stem gene expression was observed with NVP-BEZ235/JAKi combination treatment in examined GBM and BBC cell lines.
Acknowledgments
The authors would like to thank the UTRGV Department of Biology and COS for their support, reagents and expertise.
Funding
This work was supported by NIH 1SC3GM132053-02 (M.K.), HHMI 52007568 (N.V.), USDA Step 2 2015-38422-24061(A.L.), USDA H.S.I. 2016-38422-25760 (M.K. and N.V), UTRGV College of Sciences (COS) Seed Grant (M.K.), NSF Advance 1209210 (MX.), and NSF 1463991 (M.K.).
Abbreviations
- PI3K
Phosphatidylinositol 3 Kinase
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- AKT
protein kinase B
- FOXO
Forkhead box subfamily O
- ES
embryonic stem
- GBM
glioblastoma multiforme
- BBC
Basal breast cancer
- HER2
Human Epidermal Growth Factor Receptor 2
- OCT4
Octamer-binding Transcription factor 4
- SOX2
SRY-Box Transcription Factor 2
- MEM
minimal essential media
- DMEM
Dulbecco’s Modified Eagle Medium
- RPMI
Roswell Park Memorial Institute (RPMI) 1640 Medium
- FBS
fetal bovine serum
- PBS
Phosphate buffered saline
- NANOG
Nanog Homeobox
- ALPL
Alkaline Phosphatase Liver
- TRAIL
TNF-related apoptosis-inducing ligand
- BIM
Bcl-2-like protein 11
- FAS
TNF Receptor Superfamily, Member 6
- STAT3
Signal transducer and activator of transcription 3
- JAK2
Janus Kinase 2
- mTOR
mammalian target of rapamysin
- EGFR
Epidermal Growth Factor Receptor
- RTK
Receptor tyrosine kinase
- IBC
Institutional Biosafety Committee
Footnotes
Ethics approval
Work was performed with Institutional Biosafety Committee approval from the University of Texas Rio Grande Valley: Registration number: 2016-003-IBC.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All cell lines and additional data prepared from this work are available upon request.
References
- 1.Ben-Porath I; Thomson MW; Carey VJ; Ge R; Bell GW; Regev A; Weinberg RA, An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008, 40 (5), 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerra-Rebollo M; Garrido C; Sanchez-Cid L; Soler-Botija C; Meca-Cortes O; Rubio N; Blanco J, Targeting of replicating CD133 and OCT4/SOX2 expressing glioma stem cells selects a cell population that reinitiates tumors upon release of therapeutic pressure. Sci Rep 2019, 9 (1), 9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zvelebil M; Oliemuller E; Gao Q; Wansbury O; Mackay A; Kendrick H; Smalley MJ; Reis-Filho JS; Howard BA, Embryonic mammary signature subsets are activated in Brca1−/− and basal-like breast cancers. Breast Cancer Res 2013, 15 (2), R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Dhfyan A, Embryonic signature in breast cancers; Pluripotency roots of cancer stem cells. Saudi Pharm J 2013, 21 (2), 229–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng N; Zhang D; Liu X, Prognostic significance of expression of Myogenin and OCT4 in glioma. Minerva Pediatr 2018. [DOI] [PubMed] [Google Scholar]
- 6.Ikushima H; Todo T; Ino Y; Takahashi M; Saito N; Miyazawa K; Miyazono K, Glioma-initiating cells retain their tumorigenicity through integration of the Sox axis and Oct4 protein. J Biol Chem 2011, 286 (48), 41434–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez E; Vazquez N; Lopez A; Fanniel V; Sanchez L; Marks R; Hinojosa L; Cuello V; Cuevas M; Rodriguez A; Tomson C; Salinas A; Abad M; Holguin M; Garza N; Arenas A; Abraham K; Maldonado L; Rojas V; Basdeo A; Schuenzel E; Persans M; Innis-Whitehouse W; Keniry M, The PI3K pathway impacts stem gene expression in a set of glioblastoma cell lines. J Cancer Res Clin Oncol 2020, 146 (3), 593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones NM; Rowe MR; Shepherd PR; McConnell MJ, Targeted inhibition of dominant PI3-kinase catalytic isoforms increase expression of stem cell genes in glioblastoma cancer stem cell models. Int J Oncol 2016, 49 (1), 207–16. [DOI] [PubMed] [Google Scholar]
- 9.Liang R; Rimmele P; Bigarella CL; Yalcin S; Ghaffari S, Evidence for AKT-independent regulation of FOXO1 and FOXO3 in haematopoietic stem and progenitor cells. Cell Cycle 2016, 15 (6), 861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X; Yalcin S; Lee DF; Yeh TY; Lee SM; Su J; Mungamuri SK; Rimmele P; Kennedy M; Sellers R; Landthaler M; Tuschl T; Chi NW; Lemischka I; Keller G; Ghaffari S, FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat Cell Biol 2011, 13 (9), 1092–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keniry M; Pires MM; Mense S; Lefebvre C; Gan B; Justiano K; Lau YK; Hopkins B; Hodakoski C; Koujak S; Toole J; Fenton F; Calahan A; Califano A; DePinho RA; Maurer M; Parsons R, Survival factor NFIL3 restricts FOXO-induced gene expression in cancer. Genes Dev 2013, 27 (8), 916–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olson JJ; Nayak L; Ormond DR; Wen PY; Kalkanis SN; Ryken TC; Committee ACJG, The role of targeted therapies in the management of progressive glioblastoma : a systematic review and evidence-based clinical practice guideline. J Neurooncol 2014, 118 (3), 557–99. [DOI] [PubMed] [Google Scholar]
- 13.Olson JJ; Nayak L; Ormond DR; Wen PY; Kalkanis SN; Committee ACJG, The role of cytotoxic chemotherapy in the management of progressive glioblastoma : a systematic review and evidence-based clinical practice guideline. J Neurooncol 2014, 118 (3), 501–55. [DOI] [PubMed] [Google Scholar]
- 14.Cheray M; Begaud G; Deluche E; Nivet A; Battu S; Lalloue F; Verdier M; Bessette B, Cancer Stem-Like Cells in Glioblastoma. In Glioblastoma, De Vleeschouwer S, Ed. Brisbane (AU), 2017. [PubMed] [Google Scholar]
- 15.Coleman N; Ameratunga M; Lopez J, Development of Molecularly Targeted Agents and Immunotherapies in Glioblastoma: A Personalized Approach. Clin Med Insights Oncol 2018, 12, 1179554918759079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorlie T; Wang Y; Xiao C; Johnsen H; Naume B; Samaha RR; Borresen-Dale AL, Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms. BMC Genomics 2006, 7, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahr A; Holtrich U; Solbach C; Scharl A; Strebhardt K; Karn T; Kaufmann M, Molecular classification of breast cancer patients by gene expression profiling. J Pathol 2001, 195 (3), 312–20. [DOI] [PubMed] [Google Scholar]
- 18.Sotiriou C; Neo SY; McShane LM; Korn EL; Long PM; Jazaeri A; Martiat P; Fox SB; Harris AL; Liu ET, Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A 2003, 100 (18), 10393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reis-Filho JS; Pusztai L, Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet 2011, 378 (9805), 1812–23. [DOI] [PubMed] [Google Scholar]
- 20.Yehiely F; Moyano JV; Evans JR; Nielsen TO; Cryns VL, Deconstructing the molecular portrait of basal-like breast cancer. Trends Mol Med 2006, 12 (11), 537–44. [DOI] [PubMed] [Google Scholar]
- 21.Turner NC; Reis-Filho JS, Basal-like breast cancer and the BRCA1 phenotype. Oncogene 2006, 25 (43), 5846–53. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y; Liu J; Raj-Kumar PK; Sturtz LA; Praveen-Kumar A; Yang HH; Lee MP; Fantacone-Campbell JL; Hooke JA; Kovatich AJ; Shriver CD; Hu H, Development and validation of prognostic gene signature for basal-like breast cancer and high-grade serous ovarian cancer. Breast Cancer Res Treat 2020, 184 (3), 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.She QB; Gruvberger-Saal SK; Maurer M; Chen Y; Jumppanen M; Su T; Dendy M; Lau YK; Memeo L; Horlings HM; van de Vijver MJ; Isola J; Hibshoosh H; Rosen N; Parsons R; Saal LH, Integrated molecular pathway analysis informs a synergistic combination therapy targeting PTEN/PI3K and EGFR pathways for basal-like breast cancer. BMC Cancer 2016, 16, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Domagala P; Huzarski T; Lubinski J; Gugala K; Domagala W, PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast Cancer Res Treat 2011, 127 (3), 861–9. [DOI] [PubMed] [Google Scholar]
- 25.Pazaiti A; Fentiman IS, Basal phenotype breast cancer: implications for treatment and prognosis. Womens Health (Lond) 2011, 7 (2), 181–202. [DOI] [PubMed] [Google Scholar]
- 26.Cassol L; Silveira Graudenz M; Zelmanowicz A; Cancela A; Werutsky G; Rovere RK; Garicochea B, Basal-like immunophenotype markers and prognosis in early breast cancer. Tumori 2010, 96 (6), 966–70. [PubMed] [Google Scholar]
- 27.Arnes JB; Begin LR; Stefansson I; Brunet JS; Nielsen TO; Foulkes WD; Akslen LA, Expression of epidermal growth factor receptor in relation to BRCA1 status, basal-like markers and prognosis in breast cancer. J Clin Pathol 2009, 62 (2), 139–46. [DOI] [PubMed] [Google Scholar]
- 28.Murat A; Migliavacca E; Gorlia T; Lambiv WL; Shay T; Hamou MF; de Tribolet N; Regli L; Wick W; Kouwenhoven MC; Hainfellner JA; Heppner FL; Dietrich PY; Zimmer Y; Cairncross JG; Janzer RC; Domany E; Delorenzi M; Stupp R; Hegi ME, Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 2008, 26 (18), 3015–24. [DOI] [PubMed] [Google Scholar]
- 29.Sin WC; Lim CL, Breast cancer stem cells-from origins to targeted therapy. Stem Cell Investig 2017, 4, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu Z; Pestell TG; Lisanti MP; Pestell RG, Cancer stem cells. Int J Biochem Cell Biol 2012, 44 (12), 2144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Budillon A; Curley S; Fusco R; Mancini R, Identification and Targeting of Stem Cell-Activated Pathways in Cancer Therapy. Stem Cells Int 2019, 2019, 8549020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cruz-Lozano M; Gonzalez-Gonzalez A; Marchal JA; Munoz-Muela E; Molina MP; Cara FE; Brown AM; Garcia-Rivas G; Hernandez-Brenes C; Lorente JA; Sanchez-Rovira P; Chang JC; Granados-Principal S, Hydroxytyrosol inhibits cancer stem cells and the metastatic capacity of triple-negative breast cancer cell lines by the simultaneous targeting of epithelial-to-mesenchymal transition, Wnt/beta-catenin and TGFbeta signaling pathways. Eur J Nutr 2019, 58 (8), 3207–3219. [DOI] [PubMed] [Google Scholar]
- 33.Yang L; Shi P; Zhao G; Xu J; Peng W; Zhang J; Zhang G; Wang X; Dong Z; Chen F; Cui H, Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther 2020, 5 (1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong KK; Engelman JA; Cantley LC, Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev 2010, 20 (1), 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan TL; Cantley LC, PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008, 27 (41), 5497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fruman DA; Chiu H; Hopkins BD; Bagrodia S; Cantley LC; Abraham RT, The PI3K Pathway in Human Disease. Cell 2017, 170 (4), 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J; Yen C; Liaw D; Podsypanina K; Bose S; Wang SI; Puc J; Miliaresis C; Rodgers L; McCombie R; Bigner SH; Giovanella BC; Ittmann M; Tycko B; Hibshoosh H; Wigler MH; Parsons R, PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275 (5308), 1943–7. [DOI] [PubMed] [Google Scholar]
- 38.Maehama T; Dixon JE, PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol 1999, 9 (4), 125–8. [DOI] [PubMed] [Google Scholar]
- 39.Maehama T; Dixon JE, The tumor suppressor PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 1998, 273 (22), 13375–8. [DOI] [PubMed] [Google Scholar]
- 40.Brunet A; Bonni A; Zigmond MJ; Lin MZ; Juo P; Hu LS; Anderson MJ; Arden KC; Blenis J; Greenberg ME, Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96 (6), 857–68. [DOI] [PubMed] [Google Scholar]
- 41.Martins R; Lithgow GJ; Link W, Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15 (2), 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carter ME; Brunet A, FOXO transcription factors. Curr Biol 2007, 17 (4), R113–4. [DOI] [PubMed] [Google Scholar]
- 43.Greer EL; Brunet A, FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24 (50), 7410–25. [DOI] [PubMed] [Google Scholar]
- 44.Saal LH; Gruvberger-Saal SK; Persson C; Lovgren K; Jumppanen M; Staaf J; Jonsson G; Pires MM; Maurer M; Holm K; Koujak S; Subramaniyam S; Vallon-Christersson J; Olsson H; Su T; Memeo L; Ludwig T; Ethier SP; Krogh M; Szabolcs M; Murty VV; Isola J; Hibshoosh H; Parsons R; Borg A, Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet 2008, 40 (1), 102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keniry M; Parsons R, The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 2008, 27 (41), 5477–85. [DOI] [PubMed] [Google Scholar]
- 46.Pires MM; Hopkins BD; Saal LH; Parsons RE, Alterations of EGFR, p53 and PTEN that mimic changes found in basal-like breast cancer promote transformation of human mammary epithelial cells. Cancer Biol Ther 2013, 14 (3), 246–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo J; Manning BD; Cantley LC, Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 2003, 4 (4), 257–62. [DOI] [PubMed] [Google Scholar]
- 48.Lang F; Wunderle L; Badura S; Schleyer E; Bruggemann M; Serve H; Schnittger S; Gokbuget N; Pfeifer H; Wagner S; Ashelford K; Bug G; Ottmann OG, A phase I study of a dual PI3-kinase/mTOR inhibitor BEZ235 in adult patients with relapsed or refractory acute leukemia. BMC Pharmacol Toxicol 2020, 21 (1), 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu H; Shi Y; Jiao X; Yang G; Wang R; Yuan Y, Synergistic antitumor effect of dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with cisplatin on drug-resistant non-small cell lung cancer cell. Oncol Lett 2020, 20 (6), 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X; Wu C; Chen N; Gu H; Yen A; Cao L; Wang E; Wang L, PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7 (22), 33440–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lau YK; Du X; Rayannavar V; Hopkins B; Shaw J; Bessler E; Thomas T; Pires MM; Keniry M; Parsons RE; Cremers S; Szabolcs M; Maurer MA, Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget 2014, 5 (21), 10503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raizer JJ; Giglio P; Hu J; Groves M; Merrell R; Conrad C; Phuphanich S; Puduvalli VK; Loghin M; Paleologos N; Yuan Y; Liu D; Rademaker A; Yung WK; Vaillant B; Rudnick J; Chamberlain M; Vick N; Grimm S; Tremont-Lukats IW; De Groot J; Aldape K; Gilbert MR; Brain Tumor Trials C, A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J Neurooncol 2016, 126 (1), 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Claver A; Lorente M; Mur P; Campos-Martin Y; Mollejo M; Velasco G; Melendez B, Gene expression changes associated with erlotinib response in glioma cell lines. Eur J Cancer 2013, 49 (7), 1641–53. [DOI] [PubMed] [Google Scholar]
- 54.Yalon M; Rood B; MacDonald TJ; McCowage G; Kane R; Constantini S; Packer RJ, A feasibility and efficacy study of rapamycin and erlotinib for recurrent pediatric low-grade glioma (LGG). Pediatr Blood Cancer 2013, 60 (1), 71–6. [DOI] [PubMed] [Google Scholar]
- 55.Yu Z; Xie G; Zhou G; Cheng Y; Zhang G; Yao G; Chen Y; Li Y; Zhao G, NVP-BEZ235, a novel dual PI3K-mTOR inhibitor displays anti-glioma activity and reduces chemoresistance to temozolomide in human glioma cells. Cancer Lett 2015, 367 (1), 58–68. [DOI] [PubMed] [Google Scholar]
- 56.Wang WJ; Long LM; Yang N; Zhang QQ; Ji WJ; Zhao JH; Qin ZH; Wang Z; Chen G; Liang ZQ, NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol Sin 2013, 34 (5), 681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Halatsch ME; Schmidt U; Behnke-Mursch J; Unterberg A; Wirtz CR, Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat Rev 2006, 32 (2), 74–89. [DOI] [PubMed] [Google Scholar]
- 58.Maira SM; Stauffer F; Brueggen J; Furet P; Schnell C; Fritsch C; Brachmann S; Chene P; De Pover A; Schoemaker K; Fabbro D; Gabriel D; Simonen M; Murphy L; Finan P; Sellers W; Garcia-Echeverria C, Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 2008, 7 (7), 1851–63. [DOI] [PubMed] [Google Scholar]
- 59.Do DV; Ueda J; Messerschmidt DM; Lorthongpanich C; Zhou Y; Feng B; Guo G; Lin PJ; Hossain MZ; Zhang W; Moh A; Wu Q; Robson P; Ng HH; Poellinger L; Knowles BB; Solter D; Fu XY, A genetic and developmental pathway from STAT3 to the OCT4-NANOG circuit is essential for maintenance of ICM lineages in vivo. Genes Dev 2013, 27 (12), 1378–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hall J; Guo G; Wray J; Eyres I; Nichols J; Grotewold L; Morfopoulou S; Humphreys P; Mansfield W; Walker R; Tomlinson S; Smith A, Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 2009, 5 (6), 597–609. [DOI] [PubMed] [Google Scholar]
- 61.Sansone P; Bromberg J, Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol 2012, 30 (9), 1005–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tate JG; Bamford S; Jubb HC; Sondka Z; Beare DM; Bindal N; Boutselakis H; Cole CG; Creatore C; Dawson E; Fish P; Harsha B; Hathaway C; Jupe SC; Kok CY; Noble K; Ponting L; Ramshaw CC; Rye CE; Speedy HE; Stefancsik R; Thompson SL; Wang S; Ward S; Campbell PJ; Forbes SA, COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019, 47 (D1), D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quezado M; Ronchetti R; Rapkiewicz A; Santi M; Blumenthal DT; Rushing EJ, Chromogenic in situ hybridization accurately identifies EGFR amplification in small cell glioblastoma multiforme, a common subtype of primary GBM. Clin Neuropathol 2005, 24 (4), 163–9. [PubMed] [Google Scholar]
- 64.Hao Z; Guo D, EGFR mutation: novel prognostic factor associated with immune infiltration in lower-grade glioma; an exploratory study. BMC Cancer 2019, 19 (1), 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xin Y; Han FG; Chen D; Chen J; Fang CX; Lou L; Liu H, A metaanalysis study of the association between EGFR rs2252586 mutation and the risk of glioma. Cell Mol Biol (Noisy-le-grand) 2017, 63 (11), 116–118. [DOI] [PubMed] [Google Scholar]
- 66.Loeffler S; Fayard B; Weis J; Weissenberger J, Interleukin-6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp1. Int J Cancer 2005, 115 (2), 202–13. [DOI] [PubMed] [Google Scholar]
- 67.Peng D; Tanikawa T; Li W; Zhao L; Vatan L; Szeliga W; Wan S; Wei S; Wang Y; Liu Y; Staroslawska E; Szubstarski F; Rolinski J; Grywalska E; Stanislawek A; Polkowski W; Kurylcio A; Kleer C; Chang AE; Wicha M; Sabel M; Zou W; Kryczek I, Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res 2016, 76 (11), 3156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim MS; Lee WS; Jeong J; Kim SJ; Jin W, Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 2015, 6 (37), 40158–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang L; Han S; Sun Y, An IL6-STAT3 loop mediates resistance to PI3K inhibitors by inducing epithelial-mesenchymal transition and cancer stem cell expansion in human breast cancer cells. Biochem Biophys Res Commun 2014, 453 (3), 582–7. [DOI] [PubMed] [Google Scholar]
- 70.Judd LM; Bredin K; Kalantzis A; Jenkins BJ; Ernst M; Giraud AS, STAT3 activation regulates growth, inflammation, and vascularization in a mouse model of gastric tumorigenesis. Gastroenterology 2006, 131 (4), 1073–85. [DOI] [PubMed] [Google Scholar]
- 71.Sumita N; Bito T; Nakajima K; Nishigori C, Stat3 activation is required for cell proliferation and tumorigenesis but not for cell viability in cutaneous squamous cell carcinoma cell lines. Exp Dermatol 2006, 15 (4), 291–9. [DOI] [PubMed] [Google Scholar]
- 72.Zhang YW; Wang LM; Jove R; Vande Woude GF, Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21 (2), 217–26. [DOI] [PubMed] [Google Scholar]
- 73.Doheny D; Sirkisoon S; Carpenter RL; Aguayo NR; Regua AT; Anguelov M; Manore SG; Arrigo A; Jalboush SA; Wong GL; Yu Y; Wagner CJ; Chan M; Ruiz J; Thomas A; Strowd R; Lin J; Lo HW, Combined inhibition of JAK2-STAT3 and SMO-GLI1/tGLI1 pathways suppresses breast cancer stem cells, tumor growth, and metastasis. Oncogene 2020, 39 (42), 6589–6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pindiprolu SH; Pindiprolu S, CD133 receptor mediated delivery of STAT3 inhibitor for simultaneous elimination of cancer cells and cancer stem cells in oral squamous cell carcinoma. Med Hypotheses 2019, 129, 109241. [DOI] [PubMed] [Google Scholar]
- 75.Galoczova M; Coates P; Vojtesek B, STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett 2018, 23, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim SY; Kang JW; Song X; Kim BK; Yoo YD; Kwon YT; Lee YJ, Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal 2013, 25 (4), 961–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chung SS; Aroh C; Vadgama JV, Constitutive activation of STAT3 signaling regulates hTERT and promotes stem cell-like traits in human breast cancer cells. PLoS One 2013, 8 (12), e83971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marotta LL; Almendro V; Marusyk A; Shipitsin M; Schemme J; Walker SR; Bloushtain-Qimron N; Kim JJ; Choudhury SA; Maruyama R; Wu Z; Gonen M; Mulvey LA; Bessarabova MO; Huh SJ; Silver SJ; Kim SY; Park SY; Lee HE; Anderson KS; Richardson AL; Nikolskaya T; Nikolsky Y; Liu XS; Root DE; Hahn WC; Frank DA; Polyak K, The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest 2011, 121 (7), 2723–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Delen E; Doganlar O, The Dose Dependent Effects of Ruxolitinib on the Invasion and Tumorigenesis in Gliomas Cells via Inhibition of Interferon Gamma-Depended JAK/STAT Signaling Pathway. J Korean Neurosurg Soc 2020, 63 (4), 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mukthavaram R; Ouyang X; Saklecha R; Jiang P; Nomura N; Pingle SC; Guo F; Makale M; Kesari S, Effect of the JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres. J Transl Med 2015, 13, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stechishin OD; Luchman HA; Ruan Y; Blough MD; Nguyen SA; Kelly JJ; Cairncross JG; Weiss S, On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro Oncol 2013, 15 (2), 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jensen KV; Cseh O; Aman A; Weiss S; Luchman HA, The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS One 2017, 12 (12), e0189670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McFarland BC; Ma JY; Langford CP; Gillespie GY; Yu H; Zheng Y; Nozell SE; Huszar D; Benveniste EN, Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther 2011, 10 (12), 2384–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stover DG; Gil Del Alcazar CR; Brock J; Guo H; Overmoyer B; Balko J; Xu Q; Bardia A; Tolaney SM; Gelman R; Lloyd M; Wang Y; Xu Y; Michor F; Wang V; Winer EP; Polyak K; Lin NU, Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Livak KJ; Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25 (4), 402–8. [DOI] [PubMed] [Google Scholar]
- 86.Wang L; Jiang Z; Huang D; Duan J; Huang C; Sullivan S; Vali K; Yin Y; Zhang M; Wegrzyn J; Tian XC; Tang Y, JAK/STAT3 regulated global gene expression dynamics during late-stage reprogramming process. BMC Genomics 2018, 19 (1), 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Misra SK; De A; Pan D, Targeted Delivery of STAT-3 Modulator to Breast Cancer Stem-Like Cells Downregulates a Series of Stemness Genes. Mol Cancer Ther 2018, 17 (1), 119–129. [DOI] [PubMed] [Google Scholar]
- 88.Calnan DR; Brunet A, The FoxO code. Oncogene 2008, 27 (16), 2276–88. [DOI] [PubMed] [Google Scholar]
- 89.Shounan Y; Feng X; O’Connell PJ, Apoptosis detection by annexin V binding: a novel method for the quantitation of cell-mediated cytotoxicity. J Immunol Methods 1998, 217 (1-2), 61–70. [DOI] [PubMed] [Google Scholar]
- 90.Liao TT; Jia RW; Shi YL; Jia JW; Wang L; Chua H, Propidium iodide staining method for testing the cytotoxicity of 2,4,6-trichlorophenol and perfluorooctane sulfonate at low concentrations with Vero cells. J Environ Sci Health A Tox Hazard Subst Environ Eng 2011, 46 (14), 1769–75. [DOI] [PubMed] [Google Scholar]
- 91.Glienke W; Maute L; Wicht J; Bergmann L, The dual PI3K/mTOR inhibitor NVP-BGT226 induces cell cycle arrest and regulates Survivin gene expression in human pancreatic cancer cell lines. Tumour Biol 2012, 33 (3), 757–65. [DOI] [PubMed] [Google Scholar]
- 92.Hill R; Kalathur RK; Callejas S; Colaco L; Brandao R; Serelde B; Cebria A; Blanco-Aparicio C; Pastor J; Futschik M; Dopazo A; Link W, A novel phosphatidylinositol 3-kinase (PI3K) inhibitor directs a potent FOXO-dependent, p53-independent cell cycle arrest phenotype characterized by the differential induction of a subset of FOXO-regulated genes. Breast Cancer Res 2014, 16 (6), 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kampa-Schittenhelm KM; Heinrich MC; Akmut F; Rasp KH; Illing B; Dohner H; Dohner K; Schittenhelm MM, Cell cycle-dependent activity of the novel dual PI3K-MTORC1/2 inhibitor NVP-BGT226 in acute leukemia. Mol Cancer 2013, 12, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zwang Y; Jonas O; Chen C; Rinne ML; Doench JG; Piccioni F; Tan L ; Huang HT; Wang J; Ham YJ; O’Connell J; Bhola P; Doshi M; Whitman M ; Cima M; Letai A; Root DE; Langer RS; Gray N; Hahn WC, Synergistic interactions with PI3K inhibition that induce apoptosis. Elite 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brachmann SM; Hofmann I; Schnell C; Fritsch C; Wee S; Lane H; Wang S; Garcia-Echeverria C; Maira SM, Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci U S A 2009, 106 (52), 22299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cai J; Xia J; Zou J; Wang Q; Ma Q; Sun R; Liao H; Xu L; Wang D; Guo X, The PI3K/mTOR dual inhibitor NVP-BEZ235 stimulates mutant p53 degradation to exert anti-tumor effects on triple-negative breast cancer cells. FEBS Open Bio 2020, 10 (4), 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kohsaka S; Wang L; Yachi K; Mahabir R; Narita T; Itoh T; Tanino M; Kimura T; Nishihara H; Tanaka S, STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther 2012, 11 (6), 1289–99. [DOI] [PubMed] [Google Scholar]
- 98.Tasian SK; Teachey DT; Li Y; Shen F; Harvey RC; Chen IM; Ryan T; Vincent TL; Willman CL; Perl AE; Hunger SP; Loh ML; Carroll M; Grupp SA, Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2017, 129 (2), 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim JH; Kim WS; Park C, Interleukin-6 mediates resistance to PI3K-pathway-targeted therapy in lymphoma. BMC Cancer 2019, 19 (1), 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fiskus W; Verstovsek S; Manshouri T; Smith JE; Peth K; Abhyankar S; McGuirk J; Bhalla KN, Dual PI3K/AKT/mTOR inhibitor BEZ235 synergistically enhances the activity of JAK2 inhibitor against cultured and primary human myeloproliferative neoplasm cells. Mol Cancer Ther 2013, 12 (5), 577–88. [DOI] [PubMed] [Google Scholar]
- 101.Kim JH; Choi HS; Kim SL; Lee DS, The PAK1-Stat3 Signaling Pathway Activates IL-6 Gene Transcription and Human Breast Cancer Stem Cell Formation. Cancers (Basel) 2019, 11 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yin X; Zhang BH; Zheng SS; Gao DM; Qiu SJ; Wu WZ; Ren ZG, Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of Stat3/Snail signaling. J Hematol Oncol 2015, 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kuger S; Graus D; Brendtke R; Gunther N; Katzer A; Lutyj P; Polat B; Chatterjee M; Sukhorukov VL; Flentje M; Djuzenova CS, Radiosensitization of Glioblastoma Cell Lines by the Dual PI3K and mTOR Inhibitor NVP-BEZ235 Depends on Drug-Irradiation Schedule. Transl Oncol 2013, 6 (2), 169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Treatment of LN229 and MDA-MB-468 cells with Dual PI3K Inhibitor NVP-BEZ235 Induced Apoptotic Gene Expression. A. Gene expression of FOXO-induced apoptotic targets (determined by qRT-PCR relative to GAPDH control) in LN229 cells. BIM and FAS were induced by NVP-BEZ235. B. NVP-BEZ235 treatment led to induction of TRAIL gene expression by q-RT-PCR in MDA-MB-468 cells. C-D. Expression of stem genes from drug treatment samples by qRT-PCR (relative to GAPDH control). Stem genes OCT4 and SOX2 were induced in LN229 NVP-BEZ235 treatment samples with or without JAKi. D. Expression of OCT4 and ALPL in MDA-MB-468 cells with indicted drug treatments. OCT4 and ALPL remained induced with the combined NVP-BEZ235/JAKi treatment based on ANOVA with Tukey HSD Test (denoted *).
Data Availability Statement
All cell lines and additional data prepared from this work are available upon request.