

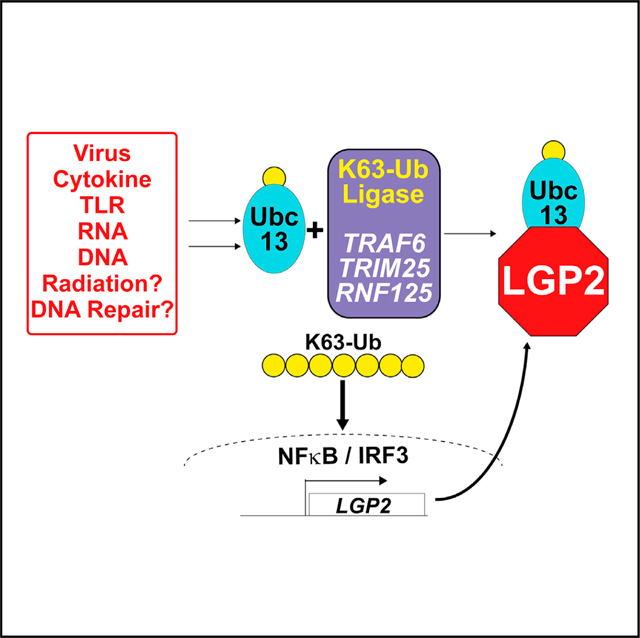
SUMMARY
Lysine 63-linked polyubiquitin (K63-Ub) chains activate a range of cellular immune and inflammatory signaling pathways, including the mammalian antiviral response. Interferon and antiviral genes are triggered by TRAF family ubiquitin ligases that form K63-Ub chains. LGP2 is a feedback inhibitor of TRAF-mediated K63-Ub that can interfere with diverse immune signaling pathways. Our results demonstrate that LGP2 inhibits K63-Ub by association with and sequestration of the K63-Ub-conjugating enzyme, Ubc13/UBE2N. The LGP2 helicase subdomain, Hel2i, mediates protein interaction that engages and inhibits Ubc13/UBE2N, affecting control over a range of K63-Ub ligase proteins, including TRAF6, TRIM25, and RNF125, all of which are inactivated by LGP2. These findings establish a unifying mechanism for LGP2-mediated negative regulation that can modulate a variety of K63-Ub signaling pathways.
In brief
Lenoir et al. identify the mechanism that the immune regulatory protein, LGP2, uses to inhibit IRF3 and NF-κB activation. LGP2 blocks K63 polyubiquitination required for immune signaling by binding and sequestering the K63-conjugating enzyme, Ubc13/UBE2N, affecting a wide range of antiviral, cytokine, and immune signaling pathways.
Graphical Abstract
INTRODUCTION
Polyubiquitin chains linked via lysine 63 (K63) mediate diverse cellular processes including DNA repair and numerous immune and inflammatory responses (Isaacson and Ploegh, 2009; Pontrelli et al., 2019). K63 ubiquitin (K63-Ub) chains are essential as a non-degradative signal activated by signal transduction pathways that culminate in the activation of master transcription regulators such as nuclear factor κB (NF-κB) and interferon regulatory factor 3 (IRF3). Like the proteasome-targeting K48-Ub system, E1 (Ub-activation), E2 (Ub-conjugation), and E3 (Ubligation) steps are needed to generate K63 polyubiquitin chains. The K63-specific E2 enzyme, Ubc13/UBE2N, is connected to fundamental cellular responses including pathogen recognition, inflammation, innate and adaptive immunity, cytokine and growth factor signaling, and DNA repair by distinct E3 complexes that polymerize K63-Ub in the context of specific regulatory networks.
In the innate antiviral response, E3 proteins such as tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6), tripartite motif containing 25 (TRIM25), and ring finger protein 125 (RNF125) are used to activate viral RNA sensors as well as inflammatory regulators such as NEMO and RIP1 (Chen et al., 2006; Ea et al., 2006; Gack et al., 2007; Heaton et al., 2016; Oshiumi et al., 2010; Wu and Arron, 2003; Zhao et al., 2005). K63-Ub stimulates immune and inflammatory gene expression from cytokine signaling pathways, including tumor necrosis factor α (TNFα), interleukin-17 (IL-17), CD40, IL-1β, and interferon (IFN) (Conze et al., 2008; Napetschnig and Wu, 2013). As a result, K63-Ub is an activator of gene regulatory networks essential for innate immunity and inflammation (Yamamoto et al., 2006).
In virus-infected cells, retinoic acid-inducible gene I (RIG-I)-like receptor (RLR) protein recognition of non-self RNAs in the cytosol triggers antiviral signal transduction through a mitochondria-localized receptor, mitochondrial antiviral signaling protein (MAVS). The RLR-family sensors RIG-I and melanoma differentiation-associated protein 5 (MDA5) form oligomeric complexes with the viral RNAs, and the RNA-activated sensors convert MAVS into an oligomeric signal transduction platform for antiviral responses. Activated MAVS can recruit TRAF family members TRAF2, TRAF3, TRAF5, and TRAF6 (Liu et al., 2013), K63-Ub ligases that enable recruitment of serine kinases IKKα, IKKβ, IKKε, and TBK1 that activate NF-κB and IRF3 (Liu et al., 2013). IRF3 and NF-κB are master regulators that can induce the expression of IFN-β and a network of antiviral and immune-stimulatory genes that restrict virus replication, initiate cell death, and induce IFN-JAK-STAT signaling and immune activity (Freaney et al., 2013; Perry et al., 2005; Rodriguez et al., 2014; Yoneyama et al., 2004).
While essential for restricting virus replication, inappropriate levels of antiviral gene expression are cytotoxic and contribute to autoimmune and inflammatory diseases that feature chronic IFN-stimulated gene expression signatures, including Aicardi-Goutières syndrome, type I diabetes, systemic lupus erythematosus, and rheumatoid arthritis (Crow, 2010). Accordingly, antiviral pathways include negative regulators and feedback mechanisms that attenuate signaling and re-establish basal homeostasis (Quicke et al., 2017).
Laboratory of genetics and physiology 2 (LGP2) is unique among the RLR proteins. It shares sequence conservation within the helicase domains and the C terminus with RIG-I and MDA5, but lacks the caspase activation and recruitment (CARD) region required for MAVS activation. Independent strains of LGP2-deficient mice were reported with disparate conclusions that indicated both positive and negative regulation of antiviral signaling and immune responses (Satoh et al., 2010; Suthar et al., 2012; Venkataraman et al., 2007). LGP2-deficient mice are more susceptible to viruses that are associated with MDA5 recognition, such as the picornaviruses encephalomyocarditis virus and poliovirus (Kato et al., 2006; Satoh et al., 2010; Venkataraman et al., 2007), suggesting that LGP2 may work together with MDA5 to promote efficient signal transduction. Mechanistic analysis demonstrated that the low basal level of LGP2 expression plays a role in stabilizing MDA5 RNA binding and ribonucleoprotein (RNP) filament cooperative assembly to enhance antiviral signaling (Bruns et al., 2014). The positive activities of LGP2 require RNA binding, ATP hydrolysis, and an intact C-terminal domain (Bruns et al., 2014; Parisien et al., 2018; Satoh et al., 2010).
LGP2 expression increases sharply in response to virus infection or treatment with antiviral mediators including poly(I:C) and IFN, and elevated LGP2 inhibits antiviral signaling dramatically (Komuro and Horvath, 2006; Rothenfusser et al., 2005; Saito et al., 2007). High LGP2 expression is associated with negative regulation of antiviral signaling, characteristic of a feedback inhibitor (Bamming and Horvath, 2009; Bruns et al., 2014; Rodriguez et al., 2014; Rothenfusser et al., 2005). LGP2 expression is strongly induced after cells are challenged with virus infection, IFN, or nucleic acid stimulation (Komuro and Horvath, 2006; Malur et al., 2012; Rothenfusser et al., 2005; Saito et al., 2007; Si-Tahar et al., 2014; Yoneyama and Fujita, 2007; Yoneyama et al., 2005), and suppresses signaling to prevent IRF3 and NF-κB activation (Parisien et al., 2018), interferes with IFN, pro-inflammatory, and antiviral gene synthesis (Parisien et al., 2018; Quicke et al., 2019; Satoh et al., 2010), and has the ability to regulate seemingly unrelated processes including Listeria monocytogenes-induced signaling (Pollpeter et al., 2011) and cancer cell radiotherapy sensitivity (Widau et al., 2014). Unlike positive regulation by LGP2, RNA binding, ATP hydrolysis, and the C-terminal domain are dispensable for LGP2 to function as a negative regulator (Parisien et al., 2018; Quicke et al., 2019). Consistent with this negative regulatory role, loss of LGP2, but not RIG-I and MDA5, in mouse bone marrow-derived macrophages was found to derepress NF-κB signaling and inflammatory responses following virus infection (Stone et al., 2019).
We recently reported that LGP2 has the ability to antagonize all the TRAF family members linked to antiviral signaling by inhibiting the ability of TRAF proteins to catalyze ubiquitination (Parisien et al., 2018). LGP2 disrupted antiviral signaling to IRF3 and NF-κB, and also inhibited NF-κB activity downstream of cGAMP/STING, TNFα, and IL-1β (Parisien et al., 2018). A well-correlated dataset demonstrated derepression of antiviral transcription in LGP2-deficient mouse dendritic cells, and identified another K63-Ub ligase, TRIM25, as an LGP2 inhibition target (Quicke et al., 2019). The ability of LGP2 to interfere with these wide-ranging targets invited further inquiry into the mechanism by which LGP2 could regulate the activity of a growing list of K63-Ub-mediated pathways.
New evidence indicates that LGP2 disrupts K63-Ub signaling by associating with the K63-Ub-conjugating enzyme Ubc13/UBE2N. Results show that LGP2 is able to disrupt K63-Ub by sequestering Ubc13/UBE2N to interfere with the essential interaction between Ubc13/UBE2N and TRAF6 as well as two other K63-Ub ligases, TRIM25 and RNF125. Conversely, in the absence of LGP2, interactions between TRAF6 and Ubc13/UBE2N are prolonged, resulting in extended transcription factor activation. LGP2 sequestration of Ubc13/UBE2N represents a unique means to broadly attenuate a wide number of cell signaling pathways.
RESULTS
Absence of LGP2 increases TNFα-stimulated gene transcription
LGP2 has been demonstrated to exert feedback control over the Ub ligase activity of TRAF proteins in the RLR-MAVS pathway, and mouse or human cells lacking LGP2 give rise to greater gene expression through IRF3 and NF-κB (Parisien et al., 2018; Quicke et al., 2019). LGP2-deficient human 2fTGH fibrosarcoma cells were generated by CRISPR/Cas9 editing in the first coding exon of human LGP2 (exon 3), using the non-homologous endjoining mechanism. In these cells, we find that the LGP2 open reading frame is disrupted after 8 amino acids by a 56-nt deletion that resolves in a +2 frameshift that contains a stop codon after 14 residues. In addition, we observe a dramatically decreased LGP2 mRNA level in these cells (Figures 1and S1). qRT-PCR verifies derepression of IFN-β and ISG15 mRNA expression in LGP2 knockout (KO) cells infected with Sendai virus (SeV) (Figures 1A and 1B). To examine the ability of LGP2 to regulate a more distantly related TRAF-dependent signaling pathway, we evaluated TNFα signaling. TNFα binds to the TNFR and activates TRAF2- and TRAF5-dependent phosphorylation and degradation of IκBα, allowing NF-κB to translocate to the nucleus and drive inflammatory gene expression. Both intact and LGP2-deficient lines respond to TNFα, resulting in Ser32 phosphorylation and degradation of IκBα (Figure 1C). IκBα Ser32 is phosphorylated within 30 min of TNFα stimulation, but by 1 h, LGP2-deficient cells exhibit enhanced phosphorylation level. IκBα abundance dramatically decreases at 30 min and returns to baseline after 1 h in wild-type (WT) cells. LGP2-deficient cells exhibit more efficient IκBα degradation that persists until 4 h. Analysis of TNFα response genes by qRT-PCR revealed increased levels of C-C motif ligand 2 (CCL2), CCL5, and IL-1β mRNAs induced by TNFα in the absence of LGP2, consistent with derepression of NF-κB in the absence of LGP2 (Figures 1D–1F). LGP2 mRNA is not induced by TNFα stimulation (Figure 1G). As observed with virus or double-stranded RNA (dsRNA)-induced antiviral genes, TNFα-induced NF-κB activity is subject to LGP2 negative regulation.
Figure 1. Loss of LGP2 increases antiviral and TNFα responses.

(A and B) 2fTGH cell lines with (WT) and without (KO) LGP2 expression were infected with 5 PFU/cell SeV for 10 h prior to RNA isolation and qRT-PCR to measure mRNA levels of: (A) IFN-β and (B) ISG15. Dot plots represent technical replicates from one of three independent experiments ± standard deviation. *p < 0.05, **p < 0.005, ***p < 0.0005 by two-tailed Student’s t test.
(C) 2fTGH cell lines with (WT) and without (KO) LGP2 expression were stimulated with TNFα and lysed at the time of stimulation (0), and after 30 min, 1 h, 2 h, 4 h, and 6 h. Lysates were subjected to immunoblot with antiserum for IκBα, p-IκBα (Ser32), and GAPDH.
(D–G) 2fTGH cell lines with (WT) and without (KO) LGP2 expression were stimulated for up to 6 h with TNFα prior to RNA isolation and qRT-PCR to measure mRNA levels of (D) CCL2, (E) CCL5, (F) IL-1β, and (G) LGP2 (exon 12/13). Dot plots represent technical replicates from one of three independent experiments ± standard deviation. *p < 0.05, **p < 0.005, ***p < 0.0005 by two-tailed Student’s t test. Additional qPCR experiments and bar graphs are shown in Figures S1 and S2.
LGP2 disrupts TRIM25 and RNF125 K63 ubiquitination
Several E3 proteins with dual K63-Ub and K48-Ub ligase activities have been identified as regulators of antiviral signaling (Rehwinkel andGack,2020).Oneofthese,TRIM25,isthoughttomediateK63Ub chains that regulate RIG-I and MAVS (Gack et al., 2007), but can also catalyze K48-Ub (Castanier et al., 2012). TRIM25 was identified as an LGP2 interaction partner and inhibition target (Quicke et al., 2019). Another, RNF125, is implicated in regulation of RIG-I via K48-Ub, but also catalyzes K63-Ub (Tang et al., 2020; Zhao et al., 2005). The ability of LGP2 to interfere with these E3 proteins was compared with TRAF6 in cell-based Ub transfer assays. In the total lysate ( Figures S3A–3C), TRAF6- mediated WT-Ub and K63-only Ub conjugation was inhibited by LGP2 (17% and 45% of control, respectively), but K48-Ub was unaffected by TRAF6. Immunoprecipitation analysis indicates that TRAF6 is modified by both WT-Ub and K63-Ub conjugates, but is not a K48-Ub ligase (Figures 2A–2C). For TRIM25, a dual-specificity Ub ligase, the total lysates reveal effects of LGP2 inhibition of WT-Ub and K63-Ub (86% and 73%, respectively), but not K48-Ub (110%; Figures S3D–S3F). Immunoprecipitation indicates that WT-Ub and K63-Ub conjugation are inhibited by LGP2 (15% and 33% of control, respectively), but K48-Ub is relatively unaffected (84%; Figures 2D–2F). We also note an LGP2-dependent interference with TRIM25 modification with WT-Ub. RNF125 is similarly affected by LGP2, with a decrease in WT-Ub and K63-Ub conjugation in the total lysates (29% and 70%), but not K48-Ub (Figures S3G–S3I). Immunoprecipitation indicates that LGP2 interferes with conjugation of WT-Ub and K63-Ub (78%and 42% of control), but K48-Ub levels remained at 92% of control (Figures 2G–2I). Additionally, LGP2 was found to co-precipitate with both TRIM25 and RNF125. In a complementary experiment, endogenous K63-Ub and K48-Ub were analysed by immunoblot with specific antisera in WT and LGP2-deficient cells. Results indicate higher levels of K63-Ub species in the absence of LGP2 after either SeV infection or TNFα stimulation (152% and 126% of WT controls, respectively; Figures 2J and 2K). These findings extend the potential for LGP2 regulation to diverse K63-Ub ligases outside the TRAF family and support the conclusion that LGP2 broadly yet specifically targets K63-Ub. Identification of these additional LGP2 inhibition targets only underscores the mechanistic conundrum of how a single protein can interfere with K63-Ub in multiple cellular contexts mediated by unrelated Ub ligases.
Figure 3. LGP2 interferes with TRAF6 Ub ligase independent of TRAF6 interaction.

(A) Diagrammatic representation of full-length (FL) TRAF6 and truncated variants, illustrating positions of RING domain, Zn finger, TRAF-N, and TRAF-C domains and highlighting the 8-amino-acid (8aa) difference critical for LGP2 co-precipitation.
(B) HEK293T cells were transfected with expression vectors for HA-tagged LGP2 protein along with the FLAG-tagged TRAF6 proteins, and lysates were subjected to FLAG M2 immunoaffinity purification and immunoblotting to detect LGP2 or TRAF6 proteins in both the lysate and the immunoprecipitate (IP).
(C) HEK293T cells were transfected with (+) FLAG-tagged TRAF6, 351 (amino acids 1–351) and 359 (amino acids 1–359), HA-tagged ubiquitin, and 6His-tagged LGP2 as indicated. Lysates were subjected to FLAG immunoaffinity purification and blotting with anti-HA for ubiquitin, anti-LGP2 for LGP2, anti-FLAG for TRAF6, 351, and 359, and anti-GAPDH as a loading control. For all panels, representative experiments are shown of n ≥ 3 replicates.
(D) HEK293T cells were transfected with (+) FLAG-tagged TRAF6, 351 (amino acids 1–351) and 359 (amino acids 1–359), HA-tagged K63-only Ub, and 6His-tagged LGP2 as indicated. Lysates were subjected to FLAG immunoaffinity purification and blotting with anti-HA for K63-only ubiquitin, antiLGP2 for LGP2, anti-FLAG for TRAF6, 351, and 359, and anti-GAPDH as a loading control. For all panels, representative experiments are shown of n ≥ 3 replicates.
(E–G) NF-κB luciferase reporter gene assays with (+) or without (−) expression of FLAG-tagged TRAF6 (E), 351 (F), or 359 (G) with or without 4 ng, 20 ng, 80 ng, or 400 ng of HA-tagged LGP2 vector. Cells were harvested at 24 h post transfection for luciferase assays. Bars represent average values (n = 3) ± standard deviation. Corresponding immunoblots are shown in Figure S5. ***p < 0.0005 by two-tailed Student’s t test.
Figure 2. LGP2 interferes with TRAF6-, TRIM25-, and RNF125-induced K63 ubiquitination.

(A–I) HEK293T cells were transfected with expression vectors for FLAG-tagged TRAF6 (A–C), TRIM25 (D–F), or RNF125 (G–I) along with HA-tagged ubiquitin with all lysines (WTUb; A, D, and G), K63-only ubiquitin (K63Ub; B, E, and H), or K48-only ubiquitin (K48Ub; C, F, and I), and 6His-tagged LGP2. Lysates were subjected to FLAG M2 immunoaffinity purification and immunoblotting with specific antisera to detect HA-ubiquitins, LGP2, and FLAG-tagged TRAF6, TRIM25, or RNF125. Quantification of ubiquitin levels is indicated below in square brackets. Representative experiment of n ≥ 3 replicates. Additional control immunoblots are shown in Figure S3, and additional experiments in Figure S4.
(J and K) 2fTGH cell lines with (WT) and without (KO) LGP2 expression were infected with SeV (J, 5 PFU/cell) for 10 h or stimulated with TNFα (K, 30 min). Samples were lysed with 1% SDS and sonicated for 2 min. Lysates were diluted and subjected to immunoblot with antiserum for K63 ubiquitin (K63Ub), K48 ubiquitin (K48Ub), or GAPDH. Quantification of ubiquitin levels is indicated below in square brackets.
LGP2 inhibition of K63-Ub is independent of TRAF6 interaction
TRAF proteins have a characteristic domain structure that includes an N-terminal RING domain linked to Zn finger and N-and C-terminal TRAF homology domains (TRAF-N and TRAF-C; see Figure 3A). Interference with TRAF-mediated Ub transfer was previously correlated with LGP2 co-precipitation with the TRAF-C domain. Analysis of additional TRAF6 truncations revealed that proteins including residues 1–359 supported LGP2 co-precipitation, but further truncation by 8 amino acids to residues 1–351 did not (Figure 3B; compare 359 with 351). TRAF6 and the truncated TRAF6 proteins all contain an intact RING domain and are thereby active in auto-poly ubiquitination to generate Ub-conjugated forms (Fu et al., 2018) (Figure 3C). LGP2 expression completely suppresses the Ub ligase activity of all the TRAF6 proteins, and remarkably does so irrespective of its ability to co-precipitate with them, as demonstrated with both WT ubiquitin and K63-only ubiquitin (Figures 3Cand 3D, top panels).All of these TRAF6 proteinsare able to activate NF-κB-mediated transcription, but LGP2 expression suppresses them (Figures 3E–3G). These findings define the regions proximal to TRAF6 amino acids 351–359 as fundamental for LGP2 interference that is dissociable and independent of LGP2-TRAF6 co-precipitation.
LGP2 inhibits TRAF6, TRIM25, and RNF125 association with Ubc13/UBE2N
The best characterized K63-Ub-conjugating enzyme, Ubc13/UBE2N, is responsible for the majority of reported K63-Ub modifications in immune signaling, and its regulation is a crucial control point for controlling NF-κB activation (Wertz and Dixit, 2010). Ubc13/UBE2N interacts with the RING domain to catalyze Ub transfer and isopeptide bond formation, but RING interactions alone can only generate short K63-Ub oligomers (Yin et al., 2009). Prior investigation of TRAF6 K63-Ub ligase processivity identified amino acids in the vicinity of 351–359 as an essential component of an oligomerization-primed high-affinity binding site for Ubc13/UBE2N interaction. This region stabilizes the active conformation of the TRAF6-Ubc13 complex and enables more efficient and processive synthesis of long K63-Ub chains (Hu et al., 2017; Yang et al., 2004). The WT and truncated TRAF6 proteins are able to use endogenous Ubc13/UBE2N to form K63-Ub species, albeit with TRAF6 351 exhibiting lower processivity (Figure 3C). Likewise, these TRAF proteins all coprecipitate with endogenous Ubc13/UBE2N (Figure 4A), consistent with RING domain-mediated interactions (Fu et al., 2018). LGP2 itself was also able to precipitate the endogenous Ubc13/UBE2N, but the closely related RIG-I protein did not (Figure 4A). In agreement, immunoprecipitation of endogenous Ubc13 revealed association with endogenous LGP2 after SeV infection (Figure 4B). Similar results were obtained in vitro using purified proteins. TRAF6, TRAF6 1–351, LGP2, and RIG-I were FLAG-purified from transfected cell lysates with M2 affinity gel and eluted with FLAG peptide, as shown in the Coomassie blue stain (Figure 4C). These purified proteins were mixed with purified 6His-Ubc13 (obtained from R&D Systems), as shown in Figure 4D, prior to further purification. The mixture was subjected to either nickel-nitrilotriacetate (Ni-NTA) purification and imidazole elution (Figure 4E) or FLAG M2 affinity gel purification and SDS elution (Figure 4F) to identify Ubc13 binding partners. When 6His-Ubc13 is pulled down with Ni-NTA, we recover TRAF6, TRAF6 1–351, and LGP2 as Ubc13/UBE2N interactors, but not RIG-I (Figure 4E). In the complementary experiment, affinity purification of the FLAG-tagged proteins reveals that 6His-Ubc13 associates with TRAF6, TRAF6 1–351, and LGP2, but not RIG-I (Figure 4F). These complementary experiments support a direct interaction between LGP2 and Ubc13/UBE2N.
Figure 4. LGP2 association with Ubc13/UBE2N disrupts the TRAF6, TRIM25, and RNF125 signaling complexes.

(A) HEK293T cells were transfected with expression vectors for FLAG-tagged TRAF6 (6), 351, 359, RIG-I, or LGP2. Lysates were subjected to FLAG immunoaffinity purification, and detection of co-precipitation was carried out with anti-Ubc13/UBE2N immunoblot, anti-FLAG immunoblot for TRAF6, 351, 359, RIG-I, and LGP2, and anti-GAPDH control.
(B) 2fTGH cells were subjected to SeV infection (5 PFU/cell) and lysates prepared at the indicated time points. Lysates were immunoprecipitated with Ubc13/UBE2N antiserum or rabbit immunoglobulin G (IgG) control, and interaction partners detected with immunoblot with antiserum for endogenous Ubc13/UBE2N, LGP2, and GAPDH.
(C) FLAG-tagged TRAF6 (6), TRAF6 1–351 (351), LGP2, or RIG-I were expressed in HEK293T cells, immunoaffinity purified on M2 agarose, and eluted with 33 FLAG peptide. Twenty-four micrograms of these purified FLAG proteins was visualized with Coomassie blue stain.
(D) Purified FLAG-tagged TRAF6 (6), TRAF6 1–351 (351), LGP2, or RIG-I (2 mg) was mixed with purified 6His-Ubc13 (2 mg; Boston Biochem) in a 1:1 ratio and subjected to anti-FLAG or anti-Ubc13 immunoblot.
(E) Ni-NTA purification of a 1:1 mixture of FLAG proteins and 6His-Ubc13. Co-purification of TRAF6 (6), TRAF6 1–351 (351), and LGP2 was detected with anti-FLAG antiserum.
(F) FLAG purification of a 1:1 mixture of FLAG proteins and 6His-Ubc13. Co-purification of 6His-Ubc13 was detected with anti-Ubc13 antiserum.
(G) HEK293T cells were transfected with expression vectors for FLAG-tagged TRAF6 (6), 351, 359, or TRAF-C, HA-tagged Ubc13/UBE2N, and 6His-tagged LGP2. Lysates were subjected to FLAG immunoaffinity purification (IP) and co-precipitation analyzed by immunoblot with anti-LGP2, anti-FLAG to detect TRAFs, anti-HA for Ubc13/UBE2N, and anti-GAPDH loading control. For all panels, representative experiments are shown of n ≥ 3 replicates.
(H–J) HEK293T cells were transfected with expression vectors for FLAG-tagged TRAF6 (H), TRIM25 (I), or RNF125 (J), along with increasing amounts of expression vector for HA-tagged Ubc13/UBE2N. Lysates were subjected to FLAG immunoaffinity purification, and detection of co-precipitation was carried out with antiserum (for LGP2), FLAG (for TRAF6, TRIM25, or RNF125), and anti-HA (for Ubc13/UBE2N), and anti-GAPDH loading control. For all panels, representative experiments are shown of n ≥ 3 replicates.
The ability of the TRAF6 variants to co-precipitate with expressed Ubc13/UBE2N was verified (Figure 4G, center lanes). Dramatically, co-expression of LGP2 completely eliminated the TRAF6-Ubc13/UBE2N co-precipitation (Figure 4G, right lanes). Irrespective of LGP2-TRAF6 co-precipitation, LGP2 is able to disrupt the TRAF6-Ubc13/UBE2N signaling complex.
To determine whether LGP2 was more generally able to disrupt the interaction between Ubc13/UBE2N and K63-Ub ligases, we analyzed TRAF6, TRIM25, and RNF125 in Ubc13/UBE2N co-immunoprecipitation assays (Figures 4H–4J). Increased expression of LGP2 results in coordinate dissociation of Ubc13/UBE2N co-precipitation with all the E3 proteins tested. As we have previously demonstrated LGP2 interference with RNA/RLR/MAVS, DNA/cGAS/STING, TNFα, and IL-1β pathways (Parisien et al., 2018), we conclude that LGP2’s ability to disrupt E2-E3 interactions affects a broad range of K63 ubiquitin-mediated cellular signaling pathways.
Ubc13/UBE2N association with LGP2 requires helicase subdomain Hel2i
LGP2 negative regulation has been characterized as independent of RNA binding and ATP hydrolysis (Bruns et al., 2014), and a C-terminally truncated form of LGP2 lacking these activities (LGP2-H) was previously shown to inhibit TRAF-dependent ubiquitination and antiviral signaling (Parisien et al., 2018; Quicke et al., 2019). LGP2 and LGP2-H were both able to co-precipitate with Ubc13/UBE2N, but RIG-I and MDA5 were not (Figure 5A), confirming that Ubc13/UBE2N interaction satisfies the established criteria for an LGP2 negative regulation target. Incubation of lysates with RNase A had no effect on these associations (Figure S8). To more clearly define the Ubc13/UBE2N-binding region of LGP2, we evaluated a series of truncations that sequentially remove the C-terminal domain and the helicase (Hel) subdomains for their ability to co-precipitate endogenous Ubc13/UBE2N (Figure 5B). Results indicate that removal of the LGP2 C-terminal domain (LGP2H) and Hel2 domain (ΔH2) does not affect Ubc13 co-precipitation, but removal of the Hel2i domain eliminates Ubc13/UBE2N association (Figure 5C). In support of this finding, all of the Ubc13/UBE2N-interacting LGP2 fragments are able to negatively regulate antiviral signaling to the IFN-β promoter (Figure 5D).
Figure 5. Helicase subdomain Hel2i of LGP2 mediates Ubc13/UBE2N association.

(A) HEK293T cells were transfected with expression vectors for FLAG-tagged LGP2 (L), LGP2-H (H), RIG-I (R), or MDA5 (M) with (+) or without (−) HA-tagged Ubc13/UBE2N. Lysates were subjected to HA immunoaffinity purification, and detection of co-precipitation was carried out with anti-FLAG for LGP2, LGP2-H, RIG-I, or MDA5, anti-HA for Ubc13/UBE2N, or anti-GAPDH loading control.
(B) Diagrammatic representation of full-length (FL) LGP2 and truncated variants, illustrating positions of conserved motifs and helicase 1, helicase 2i, and helicase 2 regions.
(C) HEK293T cells were transfected with expression vectors for FLAG-tagged LGP2 (FL), LGP2-H (H), ΔH2, ΔH2i, ΔII/III, and QiΔ. Lysates were subjected to FLAG immunoaffinity purification, and detection of co-precipitation was carried out with anti-Ubc13/UBE2N immunoblot, anti-FLAG immunoblot for LGP2, LGP2-H, ΔH2, ΔH2i, ΔII/III, and QiΔ, and anti-GAPDH control.
(D) −110 IFN-β luciferase reporter gene assay with LGP2, LGP2-H, ΔH2, ΔH2i, ΔII/III, and QiΔ infected with SeV (5 PFU/cell) for 14 h. Cells were harvested after 14 h SeV infection for luciferase assays. Corresponding immunoblots and bar graph are shown in Figure S6.
LGP2 sequesters Ubc13/UBE2N in an independent complex
An immunoprecipitation experiment was designed to help elucidate the mechanism of LGP2 interference. Expression of epitope-tagged TRAF6, Ubc13/UBE2N, and LGP2 alone or in combination with each other was interrogated with tag-specific affinity purification and immunoblotting. Purification of 6Histagged LGP2 revealed precipitation of co-expressed FLAG-tagged TRAF6 or HA-tagged Ubc13/UBE2N (Figure 6A). When all three proteins were expressed together, LGP2 co-precipitated both TRAF6 and Ubc13/UBE2N. This may represent independent Ubc13/UBE2N and TRAF complexes with LGP2 or a tripartite complex formed by LGP2 nucleating both targets. Under the same conditions, precipitation of FLAGTRAF6 (Figure 6B) revealed that TRAF6 is able to co-precipitate Ubc13/UBE2N or LGP2, but in the presence of LGP2, Ubc13/UBE2N was no longer detected in association with TRAF6. Complementarily, precipitation of Ubc13/UBE2N (Figure 6C) revealed co-precipitation of TRAF6 or LGP2, but in the presence of LGP2, TRAF6 was no longer detected in association with Ubc13/UBE2N. These results support the concept that LGP2 has the ability to compete with TRAF6 for Ubc13/UBE2N.
Figure 6. LGP2 sequesters Ubc13/UBE2N in a TRAF6-independent complex.

(A–C) HEK293T cells were transfected with (+) or without () expression vectors for FLAG-tagged TRAF6, HA-tagged Ubc13/UBE2N, and 6His-tagged LGP2 as indicated, and lysates were subjected to immunoaffinity purification with Ni-NTA beads for LGP2 (A), FLAG M2 affinity matrix for TRAF6 (B), or HA affinity beads for Ubc13/UBE2N (C). Analysis of co-precipitation was carried out with antiserum for LGP2, FLAG (for TRAF6), HA (for Ubc13/UBE2N), and GAPDH loading control. For all panels, representative experiments are shown of n ≥ 3 replicates.
(D) Workflow diagram depicting the experimental scheme for immunoprecipitation (IP)/re-immunoprecipitation (Re-IP) experiments.
(E) Re-immunoprecipitation of eluates (top) and supernatants (bottom) from FLAG-TRAF6 immunoaffinity purification with anti-HA affinity matrix to captureUbc13/UBE2N and its partners. Analysis of co-precipitation was carried out with antiserum for LGP2, FLAG (for TRAF6), and HA (for Ubc13/UBE2N). For all panels, representative experiments are shown of n ≥ 3 replicates. Corresponding control immunoblots are shown in Figure S7.
(F) Re-immunoprecipitation of eluates (top) and supernatants (bottom) from FLAG-TRAF6 immunoaffinity purification with Ni-NTA matrix to capture 6His-LGP2 and its partners. Analysis of co-precipitation was carried out with antiserum for LGP2, FLAG (for TRAF6), and HA (for Ubc13/UBE2N). For all panels, representative experiments are shown of n ≥ 3 replicates. Corresponding control immunoblots are shown in Figure S7.
To determine the composition of the LGP2 interference complex(es) and the fate of Ubc13/UBE2N that is dissociated from TRAF6 by LGP2, we carried out elution and re-immunoprecipitation experiments to examine interactions in both the TRAF6 precipitates and the unbound supernatants (Figure 6D). TRAF6 was subjected to FLAG purification with M2 affinity matrix to collect TRAF6 and its partners (as in Figure 6B) and unbound supernatants (TRAF6-free) collected separately. FLAG peptide eluates and supernatants were subjected to a second immunoprecipitation for HA-tagged Ubc13/UBE2N (Figure 6E) or His-tagged LGP2 (Figure 6F). The re-precipitated eluates and supernatants were then analyzed by immunoblot to detect LGP2, Ubc13/UBE2N, and TRAF6.
Re-immunoprecipitation of the FLAG-TRAF6 eluate for Ubc13/UBE2N (Figure 6E) verified the TRAF6:Ubc13/UBE2N complex in the absence of LGP2, but this complex was strikingly missing when the proteins were expressed together with LGP2. Instead, most of the Ubc13/UBE2N was found in the supernatants, free of TRAF6, and associated with LGP2.
Re-immunoprecipitation of the FLAG-TRAF6 eluates for LGP2 (Figure 6F) verified trace amounts of TRAF6:LGP2 co-precipitation observed in prior overexpression assays (Parisien et al., 2018), but Ubc13/UBE2N was not detected in these samples. Instead, analysis of the TRAF6-free supernatant revealed the majority of LGP2 in complex with Ubc13/UBE2N independent of TRAF6. Together, these data indicate that LGP2 is able to form stable inhibitory complexes with Ubc13/UBE2N that sequester it from TRAF6.
Loss of LGP2 prolongs TRAF6-Ubc13/UBE2N interactions
The principal conclusion from these experiments is that LGP2 negative regulation involves disrupting the TRAF6-Ubc13/UBE2N K63-Ub transfer complex to terminate downstream signaling. To test the ability of LGP2 to regulate the TRAF6-Ubc13/UBE2N complex in a native context, we evaluated the consequences of LGP2 deficiency by examining the endogenous proteins using natural activation conditions following virus infection. Human fibrosarcoma cells and LGP2-deficient daughter cells were infected with SeV and lysates prepared over a 14-h time course. LGP2 was not detected in uninfected cells, but accumulated in responseto infection inthe WTbut not KO cells (Figure 7A). Immunoprecipitation of TRAF6 over this time course revealed a small amount of Ubc13/UBE2N detected at the time of infection (t = 0) that increased dramatically by 8 h post infection (hpi) (Figure 7B). In the intact WT cells, the activated TRAF6-Ubc13/UBE2N complex resolved toward basal levels by 12 hpi and was undetectable at 14 hpi. In contrast, LGP2 deficiency resulted in a prolonged TRAF6-Ubc13/UBE2N complex that remained intact beyond 14 hpi (Figure 7C). In agreement, virus-induced activating IRF3 Ser396 phosphorylation was sustained in the absence of LGP2 (Figure 7C), resulting in higher IRF3 nuclear accumulation (Figure S8). Together, these data fully support the conclusion that LGP2 dissociates Ubc13/UBE2N from TRAF6, acting as a constraint on antiviral signaling that controls transcription factor activation.
Figure 7. Loss of LGP2 prolongs TRAF6-Ubc13/UBE2N association.

(A) 2fTGH cell lines with (WT) and without (KO) LGP2 expression were infected with Sendai virus (SeV, 5 PFU/cell) and lysates prepared at the time of inoculation (0), and 12 and 14 h post infection prior to immunoblot with antiserum for LGP2 and GAPDH. Relative LGP2 protein abundance is indicated, normalized to GAPDH.
(B) Intact 2fTGH cells (WT) were subjected to SeV infection (5 PFU/cell) and lysates prepared at the indicated time points. Lysates were immunoprecipitated with TRAF6 antiserum or rabbit IgG control, and interaction partners detected with immunoblot with antiserum for Ubc13/UBE2N, TRAF6, phosphorylated IRF3 (pIRF3) (Ser396), total IRF3, and GAPDH.
(C) LGP2-deficient 2fTGH cells (KO) were subjected to SeV infection (5 PFU/cell) and lysates prepared at the indicated time points. Lysates were immunoprecipitated with TRAF6 antiserum or rabbit IgG control, and interaction partners detected with immunoblot with antiserum for Ubc13/UBE2N, TRAF6, pIRF3 (Ser396), total IRF3, and GAPDH.
(D) Working model for LGP2 feedback interference with Ubc13/UBE2N. At steady state, K63-Ub ligases such as TRAF6 (depicted) TRIM25, or RNF125 can engage Ubc13 via their RING domain. Stimulation with virus, inflammatory cytokines, or other activators leads to TRAF6 oligomerization and creates a site for Ubc13/UBE2N engagement that leads to processive ubiquitination, which activates transcription factors NF-κB and IRF3. Feedback inhibition by increased LGP2 abundance engages Ubc13/UBE2N through helicase subdomain Hel2i, preventing further TRAF6-mediated K63-ubiquitination, attenuating signaling, and disrupting transcription.
DISCUSSION
Antiviral responses are a powerful and effective means to curb virus replication and trigger innate and adaptive immune systems. To prevent adverse consequences of hyperactive signaling or prolonged IFN activation, feedback mechanisms have evolved to re-establish homeostatic gene expression. We have deduced that feedback inhibition mediated by the LGP2 protein is achieved by sequestering the Ub-conjugating enzyme, Ubc13/UBE2N, to reduce K63 polyubiquitination and attenuate IRF3 and NF-κB activation. Identification of Ubc13/UBE2N as a target of LGP2 inhibition importantly helps explain how a single protein has the ability to interfere with an extensive range of seemingly unrelated proteins including TRAF6, TRIM25, and RNF125, downstream of signaling pathways induced by TNFα, IL-1β, cGAS/STING, RLR/MAVS/IFN (Parisien et al., 2018), RANTES/MAPK (Si-Tahar et al., 2014), T cell activation (Suthar et al., 2012), and cancer radiotherapy (Lhuillier et al., 2021; Widau et al., 2014). All of these pathways require K63-Ub to activate downstream signaling to transcription factors, and Ubc13/UBE2N provides a unifying target for the pervasive LGP2 negative regulation observed in this broad range of biological contexts (Figure 7D).
Ubc13/UBE2N is well known as an important regulator of many immune and inflammatory signals as well as DNA repair responses and is linked to Toll-like receptor (TLR), IFN, CD40L/BAFF, transforming growth factor β, and mitogen-activated protein kinase (MAPK) pathways (Hodge et al., 2016). Phenotypes of Ubc13/UBE2N-deficient mice indicate that it has functions in nearly all immune cell types (Wu and Karin, 2015). As an essential mediator of inflammation, Ubc13/UBE2N is an attractive therapeutic target and has applications in cancer treatment or alleviation of anticancer therapy resistance, treatment of chronic inflammation, or controlling responses to virus infections. Other cellular regulators of Ubc13/UBE2N have been described, notably the OTUB family proteins, A20 (TNFΑIP3) and OTUB1, which can inactivate Ubc13/UBE2N signaling to NF-κB from TRAF, TLR, IL-1β, TNFα, and DNA repair pathways (Nakada et al., 2010; Shembade and Harhaj, 2010; Shembade et al., 2010). A20 and OTUB1 disrupt Ubc13/UBE2N signaling by assembling complexes that can deubiquitinate and induce proteolysis of both K48 and K63 linked polymers. LGP2 does not share homology with the ubiquitin-like or ovarian tumor (OTU) functional domains of these proteins, and although there is currently no evidence that LGP2 can catalyze proteolytic cleavage of ubiquitin conjugates, it is likely that further investigation will uncover additional components and/or post-translational modifications required for LGP2-mediated K63-Ub regulation.
As an innate immune regulator, LGP2 is unusual in its ability to perform as both an inhibitor and activator of antiviral signaling (Rodriguez et al., 2014). In uninfected cells, the low basal concentration of LGP2 acts to nucleate MDA5-dsRNA interactions that lead to RNP filaments that engage MAVS to initiate TRAF-mediated activation of NF-κB and IRF3 (Bruns et al., 2014). In addition to IFN and antiviral gene expression, LGP2 is strongly induced by virus infection or IFN stimulation, and at higher concentration acts as a potent feedback inhibitor (Parisien et al., 2018). LGP2 feedback inhibition has been characterized at both molecular and cellular levels (Esser-Nobis et al., 2020; Rodriguez et al., 2014). LGP2 is reported to form inhibitory interactions proximal to MAVS, but also dynamically relocates between microsomes and mitochondria to engage multiple targets. The connection between LGP2 subcellular movement and K63-Ub regulation is yet to be fully appreciated.
In the case of TRAF6 (Figure 7D), signal-induced activation is known to result in oligomerization that produces a high-affinity binding site for Ubc13/UBE2N in a coiled-coil region near the TRAFN-TRAFC domain interface that enables more processive K63-Ub chain formation (Hu et al., 2017; Yang et al., 2004). Results indicate that LGP2 is able to engage Ubc13/UBE2N through the LGP2 Hel2i domain and sequester it from TRAF6 interaction. In RIG-I, the Hel2i domain is thought to be part of the autoinhibitory mechanism that prevents constitutive antiviral signaling. However, the key contact sites for RIG-I Hel2i regulation are not well conserved in MDA5 and LGP2 (Rawling and Pyle, 2014), and LGP2 has no CARD region. Without these constraints, LGP2 has apparently evolved a distinct Hel2i function in negative regulation of K63 Ub by engagement of Ubc13/UBE2N. This inhibitory complex interferes with signaling and prevents perpetuation of downstream transcription. In contrast, loss of LGP2 prevents repression of TRAF6 signaling, resulting in longer association with Ubc13/UBE2N and greater transcriptional activity. The implication of Ubc13/UBE2N in mediating diverse immune and stress response pathways suggests that LGP2 regulation might provide insight into therapeutic targeting approaches for acute and chronic diseases.
Limitations of the study
This paper describes the mechanism by which LGP2 interferes with Ubc13/UBE2N to disrupt K63 ubiquitination. While this mechanism is independent of RNA binding and the C-terminal domain, previous reports have implicated RNA binding and C-terminal properties as potential inhibitory mechanisms (Rothenfusser et al., 2005; Saito et al., 2007). The present studies do not entirely rule out the ability of these alternative pathways to contribute to the overall function of LGP2 as a feedback regulator but do underscore the importance of K63 regulation as a primary and independent target.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Curt M. Horvath (horvath@northwestern.edu).
Materials availability
Plasmids generated in this study are available upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
BOSC cells (ATCC# CRL-11270), HEK293T cells (ATCC# CRL-3216), 2fTGH cells (George Stark), and 2fTGH LGP2 Cas9 knockouts (KO) were grown in DMEM supplemented in 10% cosmic calf serum and 1% penicillin/streptomycin at 37°C and 5% CO2. KO cells were described previously (Parisien et al., 2018) For generation and analysis of CRISPR KO cells, gRNA target sequences were identified in exon 3 of LGP2 genomic DNA using Target Finder (http://crispr.mit.edu/). The gRNAs with the top three scores and least offtarget binding were selected from exon3.
gRNA#1 (GAGCTTCGGTCCTACCAAT), gRNA#2 (CTTCGGTCCTACCAATGGG), gRNA#3 (GGGTCTTCCCGGCACCCGT). Each gRNA was then incorporated into a 455-bp DNA fragment synthesized as a gBlock (IDT) and cloned into pCR-Blunt II-TOPO vector (Invitrogen, cat# 450245). All three gRNAs and hCas9 (Addgene #41815) were transfected into cells and selected with 500 μg/mL G418. Genetic edit was validated by sequencing and by mismatch-cleavage assay (IndelCheck, GeneCopoeia, cat# ICPE-050). Positive clones were ultimately screened by immunoblot with a LGP2 antibody after virus induction (Proteintech, cat# 11355–1-AP).Cells are routinely tested for mycoplasma contamination and regularly restored from early-passage frozen stocks.
METHOD DETAILS
Virus and cytokines
Infection of cells with Sendai virus (Sendai Virus, Cantell strain, MOI = 5PFU/cell) was performed in serum-free media. After 1 h, cells were washed, placed in growth medium supplemented with 2% CCS, and harvested at the indicated time points. Treatment of cells with recombinant human tumor necrosis factor alpha (TNFα, R&D systems, cat# 210-TA-005) was performed using 10 ng per 1 mL in 10% CCS media.
RT-qPCR
Total RNA was extracted using the TRIzol Reagent (Invitrogen, cat# 15596018). Samples were treated with DNAse I (Invitrogen, cat# AM2224), and 1–5 μg of total RNA was primed with random primers and reverse transcribed using SuperScript III (Invitrogen, cat# 18080085). Gene expression was measured by quantitative real-time PCR (qPCR) using the Mx3005P SYBR Green real-time PCR system (Agilent) and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data are representative of multiple experiments and plotted as mean values of technical replicates with error bars representing standard deviation in technical replicates.
Plasmid generation
FLAG-tagged TRAF6 and 351, HA-tagged ubiquitin and HA-tagged K63 ubiquitin were described previously (Parisien et al., 2018). FLAG-tagged TRAF6 mutants 1–393 (393), 1–387 (387), 1–371 (371), and 1–359 (359) were created through site-directed mutagenesis with stop codons inserted at amino acids 394, 388, 372, and 360 respectively. FLAG-tagged LGP2 (FL), LGP2-H (H) were described previously (Parisien et al., 2018). FLAG-tagged ΔH2, ΔH2i, ΔII/III, and QiΔ were created through site-directed mutagenesis with stop codons inserted at amino acids 352, 178, 138, and 129 respectively. pcDNA3.0-HA-UbcH13 and pFlagCMV2-EFP (TRIM25) was obtained from Dong-Er Zhang (Addgene plasmid #12461 and plasmid #12449). pRK5-HA-Ubiquitin-K48 was obtained from Ted Dawson (Addgene plasmid #17605). FLAG-tagged LGP2, FLAG-tagged RNF125, and 6His-tagged LGP2 were described previously (Bamming and Horvath, 2009; Komuro and Horvath, 2006).
Immunoprecipitation
For co-immunoprecipitation experiments, FLAG-tagged, HA-tagged, and 6His-tagged plasmids were transfected into HEK293T cells by the calcium phosphate method. Twenty-four hours later, cells were harvested by first washing with cold phosphate-buffered saline and then lysed with whole cell extract buffer (WCEB) consisting of 50 mM Tris, 280 mM NaCl, 0.5% NP-40, 0.2 mM EDTA, 2 mM EGTA, 10% glycerol, 1 mM DTT, 2.5 mM sodium vanadate, and protease inhibitors. For FLAG-tagged proteins, lysates were incubated with FLAG M2 affinity beads (Sigma, cat# A2220) overnight. For 6His-tagged proteins, lysates were washed and eluted with SDS sample buffer or 50% WCEB and 50% 2M imidazole. Then, they were separated by SDS–PAGE and processed for immunoblotting. For immunoprecipitating endogenous Ubc13 from FLAG M2 beads, samples were incubated with M2 beads for twenty-four hours and washed twice with WCEB buffer. For immunoblotting, the separated proteins were transferred to nitrocellulose and probed with commercial primary antibodies recognizing FLAG (Sigma, cat# F3165), HA (Sigma, cat# H3663), DHX58/LGP2 (Abcam, cat# ab67270), Ubc13 (ThermoFisher, Cat# 37–1100), TRAF6 (Cell Signaling Cat#8028, ProteinTech, Cat#66498), IκBα (Santa Cruz Biotechnology, cat# sc-371), p- IκBα (Ser32) (Cell Signaling, cat# 2859S), p-IRF3 (Cell Signaling, Cat# 29047S), IRF3 (Santa Cruz Biotechnologies, 492Cat#sc-376455), or GAPDH (Santa Cruz Biotechnology, cat# sc-47724).
For immunoprecipitation:re-immunoprecipitation experiments, lysates described previously were incubated with FLAG M2 affinity beads (Sigma, cat# A2220) overnight. The supernatant was collected in a separate tube and the beads were washed and eluted with 3X FLAG peptide (ApeBio cat# A6001) for 8 h. Supernatants and eluates were incubated with either HA affinity beads (Sigma, cat# E6779) or Ni-NTA beads (Qiagen) overnight. Ni-NTA beads were charged according to manufacturer’s instructions. Supernatants and eluates were washed and eluted with either SDS sample buffer (HA affinity beads) or 50% WCEB and 50% 2M imidazole. Then, they were separated by SDS–PAGE and processed for immunoblotting as described above.
For endogenous immunoprecipitation experiments, 2fTGH and KO cells were subjected to SeV infection for 0 h, 8 h, 10 h, 12 h, and 14 h. After infection, cells were harvested and lysed with WCEB for 30 min. Then 50 μg of sample was placed in one tube and set aside as the total lysate. Another 50 μg of sample was combined with 5 μg of rabbit polyclonal TRAF6 antibody (Cell Signaling Technologies cat# 8028S) and incubated while rotating at 4°C for 2 h. After rotating, 40 μL of Protein A/G PLUS agarose beads (Santa Cruz cat# sc2003) were added to each sample and incubated while rotating at 4°C overnight. The next day, samples were spun down at 8200 g for 4 min and washed with WCEB. This was repeated three times. After the second wash, 40 μL of 5X PLB was added to each sample and boiled for 5 min. Samples were then used for immunoblot. For endogenous K63-Ub and K48-Ub experiments, 2fTGH and KO cells were lysed with 100 μL of Tris pH 8.0 + 1% SDS. Then samples were sonicated at 4°C for 10 s on and 10 s off, until samples were fully lysed. Samples were then spun down at 10,000 rpm for 10 min and diluted in 500 μL of WCEB. For cell fractionation, cells were washed 3 times in cold PBS, resuspended in 1x Hypotonic Buffer (20 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2), and incubated on ice for 15 min 10% NP40 was added to samples and vortexed. Samples were centrifuged at 3000 rpm for 10 min and the supernatant was saved (cytoplasmic fraction). The pellet was resuspended in Cell Extraction buffer (10 mM Tris, pH 7.5, 2 mM Na3VO4, 100 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% glycerol, 1 mM EGTA, 0.1% SDS, 1 mM NaF, 0.5% deoxycholate, 20 mM Na4P2O7) for 30 min on ice, vortexing every 10 min, and then centrifuged at 14,000 rpm for 20 min (nuclear fraction).
Proteins were visualized by enhanced chemiluminescence (PerkinElmer, cat# NEL105001EA) and imaged using a UVP BioSpectrum MultiSpectral Imaging System. Digital files are printed on thermal paper for laboratory notebook records and exported to Adobe Photoshop and Illustrator for cropping and figure assembly. Intact digital files are only subjected to minor contrast and quality modifications and applied to entire image and shown intact without lane splicing. Figures indicate 3 biological replicates, and the figure presented is a representative of those replicates.
Quantification of immunoblots
Protein band intensities were quantified using the Area Density Analysis tool through the UVP BioSpectrum MultiSpectral Imaging System software. Calculations for protein band intensities were [ ] = (total density of band of interest)/(total density of E3 + WTUb/K63Ub/K48Ub).
Reporter gene assays
HEK293T cells were transfected with the NFκB (43 PRDII) or 110 IFNβ luciferase reporter gene along with Renilla luciferase vector and other expression vectors as indicated. In samples where <600 ng of DNA was transfected, DNA amounts were equalized using salmon sperm DNA (Invitrogen, cat# 15632–011) in order to bring the total amount of DNA to 600 ng. At 24 h post-transfection, cells were harvested and assayed for firefly and Renilla luciferase activities. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, cat# E1960). Relative luciferase activity was calculated by dividing the firefly luciferase values by those of the Renilla luciferase. Data are plotted as mean values, with error bars representing standard deviation. Figures indicate two to three biological replicates, the figure presented is a representative of those replicates, and statistical analysis was done using a two-tailed Student’s t-test.
QUANTIFICATION AND STATISTICAL ANALYSIS
For luciferase reporter gene assays, figures are representative of ≥3 biological replicates and each of these experiments contains three technical replicates. Data are plotted as mean values of these replicates, with error bars representing standard deviation calculated using a two-tailed Student’s t-test. The two-tailed Student’s t-test is a standard statistical test for measuring the significance of the results from these assays. p values calculated from the two-tailed Student’s t-test conform to the standards set by the field.
For RT–qPCR, data presented are representative of ≥3 independent experiments and plotted as mean values of technical replicates, averaged before any statistical inference test is performed, with error bars representing standard deviation in technical replicates. Statistical analysis was done using a two-tailed Student’s t-test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-FLAG antibody | Sigma Aldrich | Cat#A2220 AB_439685 |
| Anti-HA antibody | Sigma Aldrich | Cat#H3663 |
| Anti-LGP2 antibody | Abcam | Cat#ab67270 |
| Anti-Ubc13 antibody | Thermo Fisher | Cat# 37–1100 AB_2533298 |
| Anti-TRAF6 (D21G3) antibody | Cell Signaling | Cat#8028 |
| Anti-TRAF6 antibody | ProteinTech | Cat#66498–1-Ig AB_2881862 |
| Anti-pIRF3 (Ser396) antibody | Cell Signaling | Cat#4947 |
| Anti-IRF3 antibody | Santa Cruz Biotechnologies | Cat# sc-3764SS |
| Anti-IκBα antibody | Santa Cruz Biotechnology | Cat# sc-371 |
| Anti-plκBα (Ser32) antibody | Cell Signaling | Cat# 28S9S |
| Anti-GAPDH antibody | Santa Cruz Biotechnologies | Cat# sc-47724 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Sendai Virus, Cantell Strain | Dr. Peter Palese | |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Human His6 UBE2N (Ubc13)/Uev1a Complex Recombinant Protein | R&D Systems | Cat#E2664100 |
| 3X (DYKDDDDK) Peptide | Apex Bio | Cat#A6001 |
| Human Tumor Necrosis Factor Alpha (TNFα) cytokine | PeproTech | Cat#300–01A |
| FLAG M2 affinity beads | Sigma Aldrich | Cat# A2220 |
| Ezview™ Red Anti-HA Affinity Gel | Sigma Aldrich | Cat# E6779 |
| Protein A/G PLUS agarose beads | Santa Cruz Biotechnologies | Cat# sc-2003 |
| Ni-NTA His Bind Resin | Qiagen | Cat# 70666 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Enhanced Chemiluminescence | Perkin Elmer | Cat# NEL 105001EA |
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1960 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: HEK293T | ATCC | CRL-3216 |
| Human: 2fTGH | Dr. George Stark | NA |
| Human: BOSC23 | ATCC | CRL-11270 |
| Human: 2fTGH LGP2 CRISPR Cas-9 | Parisien et al. (2018) | NA |
| knockout | ||
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer: GAPDH Forward:ACAGTCAGCCGCATCTTCTT | This paper | NA |
| Primer GAPDH Reverse:ACGACCAAATCCGTTGACTC | This paper | NA |
| Primer CCL5 Forward:CTGCTTTGCCTACATTGCCC | This paper | NA |
| Primer CCL5 Reverse:TCGGGTGACAAAGACGACTG | This paper | NA |
| Primer CCL2 Forward: CCCCAGTCACCTGCTGTTAT | This paper | NA |
| Primer CCL2 Reverse: GCTTCTTTGGGACACTTGCT | This paper | NA |
| Primer IL-1 β Forward: CCTACTCACTTAAAGCCCGC | This paper | NA |
| Primer IL-1 β Reverse: GAAGCGGTTGCTCATCAGAATG | This paper | NA |
| Primer LGP2 Exon 7/8 Forward: ATCACAGGGAGCACGTCACT | This paper | NA |
| Primer LGP2 Exon 7/8 Reverse: CTCTGGGCCATGAGTTGC | This paper | NA |
| Primer LGP2 Exon 12/13 Forward: GGCACCCACCATGTCAAT | This paper | NA |
| Primer LGP2 Exon 12/13 Reverse: CCCAGGCTTCCAGTCCTT | This paper | NA |
|
| ||
| Recombinant DNA | ||
|
| ||
| FLAG TRAF6 | Parisien et al. (2018) | NA |
| FLAG TRAF6 1–351 (351) | This paper | NA |
| FLAG TRAF6 1–359 (359) | This paper | NA |
| FLAG TRAF6 1–371 (371) | This paper | NA |
| FLAG TRAF6 1–387 (387) | This paper | NA |
| FLAG TRAF6 1–393 (393) | This paper | NA |
| pFLAGCMV2-EFP (TRIM25) | Zou and Zhang, 2006 | Addgene Plasmid#12449 |
| FLAG RNF125 | ||
| FLAG RIG-I | Bamming and Horvath (2009) | NA |
| FLAG LGP2 | Bamming and Horvath (2009) | NA |
| FLAG LGP2-H | Parisien et al. (2018) | NA |
| FLAG LGP2 ΔH2 | This paper | NA |
| FLAG LGP2 ΔH2i | This paper | NA |
| FLAG LGP2 ΔII/III | This paper | NA |
| FLAG LGP2 QiΔ | This paper | NA |
| 6His LGP2 | Bamming and Horvath (2009) | NA |
| pRK-HA-Ubiquitin-K48 | Lim et al. (2005) | Addgene Plasmid,#17605 |
| HA-Ubiquitin | Parisien et al. (2018) | NA |
| HA-Ubiquitin-K63 | Lim et al. (2005) | Addgene Plasmid,#17606 |
| HA-Ubc13 | Zou et al. (2005) | Addgene Plasmid,#12461 |
Highlights.
Immune sensor LGP2 is able to inhibit a wide variety of K63 ubiquitin ligases
The target of LGP2 inhibition is the K63 conjugating enzyme, Ubc13/UBE2N
The helicase 2i domain of LGP2 mediates the interaction with Ubc13/UBE2N
Loss of LGP2 prolongs TRAF6-Ubc13/UBE2N interaction and IRF3/NF-κB activity
ACKNOWLEDGMENTS
We are grateful to members of the Horvath lab for their guidance and helpful comments on this work and the manuscript, and Mehul Suthar (Emory) for helpful discussions. Supported by NIH grants R21AI148949 and R01GM111652 to C.M.H. J.J.L. was supported by Cellular and Molecular Basis of Disease Training grant (NIH T32 GM008061).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110175.
REFERENCES
- Bamming D, and Horvath CM (2009). Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J. Biol. Chem. 284, 9700–9712. 10.1074/jbc.M807365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns AM, Leser GP, Lamb RA, and Horvath CM (2014). The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 55, 771–781. 10.1016/j.molcel.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanier C, Zemirli N, Portier A, Garcin D, Bidere N, Vazquez A, and Arnoult D. (2012). MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 10, 44. 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Bhatia D, Chang Q, and Castranova V. (2006). Finding NEMO by K63-linked polyubiquitin chain. Cell Death Differ 13, 1835–1838. 10.1038/sj.cdd.4402014. [DOI] [PubMed] [Google Scholar]
- Conze DB, Wu CJ, Thomas JA, Landstrom A, and Ashwell JD (2008). Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-kappaB activation. Mol. Cell Biol 28, 3538–3547. 10.1128/MCB.02098-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MK (2010). Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res. Ther. 12, S5. 10.1186/ar2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, and Chen ZJ (2006). Activation of IKK by TNFαlpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22, 245–257. 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Esser-Nobis K, Hatfield LD, and Gale M Jr. (2020). Spatiotemporal dynamics of innate immune signaling via RIG-I-like receptors. Proc. Natl. Acad. Sci. U S A. 117, 15778–15788. 10.1073/pnas.1921861117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freaney JE, Kim R, Mandhana R, and Horvath CM (2013). Extensive cooperation of immune master regulators IRF3 and NFkappaB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep. 4, 959–973. 10.1016/j.celrep.2013.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu TM, Shen C, Li Q, Zhang P, and Wu H. (2018). Mechanism of ubiquitin transfer promoted by TRAF6. Proc. Natl. Acad. Sci. U S A. 115, 1783–1788. 10.1073/pnas.1721788115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, and Jung JU (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Heaton SM, Borg NA, and Dixit VM (2016). Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 213, 1–13. 10.1084/jem.20151531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CD, Spyracopoulos L, and Glover JN (2016). Ubc13: the Lys63 ubiquitin chain building machine. Oncotarget 7, 64471–64504. 10.18632/oncotarget.10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Xu J, Xie X, Zhou Y, Tao P, Li H, Han X, Wang C, Liu J, Xu P, et al. (2017). Oligomerization-primed coiled-coil domain interaction with Ubc13 confers processivity to TRAF6 ubiquitin ligase activity. Nat. Commun. 8, 814. 10.1038/s41467-017-01290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson MK, and Ploegh HL (2009). Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 5, 559–570. 10.1016/j.chom.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. (2006). Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105. [DOI] [PubMed] [Google Scholar]
- Komuro A, and Horvath CM (2006). RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol. 80, 12332–12342. 10.1128/JVI.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhuillier C, Rudqvist NP, Yamazaki T, Zhang T, Charpentier M, Galluzzi L, Dephoure N, Clement CC, Santambrogio L, Zhou XK, et al. (2021). Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J. Clin. Invest 131. 10.1172/JCI138740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, et al. (2005). Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25, 2002–2009. 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chen J, Cai X, Wu J, Chen X, Wu Y-T, Sun L, and Chen ZJ (2013). MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2. 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malur M, Gale M Jr., and Krug RM (2012). LGP2 downregulates interferon production during infection with seasonal human influenza A viruses that activate interferon regulatory factor 3. J. Virol. 86, 10733–10738. 10.1128/JVI.00510-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada S, Tai I, Panier S, Al-Hakim A, Iemura S, Juang YC, O’Donnell L, Kumakubo A, Munro M, Sicheri F, et al. (2010). Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 466, 941–946. 10.1038/nature09297. [DOI] [PubMed] [Google Scholar]
- Napetschnig J, and Wu H. (2013). Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 42, 443–468. 10.1146/annurev-biophys083012-130338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiumi H, Miyashita M, Inoue N, Okabe M, Matsumoto M, and Seya T. (2010). The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 8, 496–509. 10.1016/j.chom.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Parisien JP, Lenoir JJ, Mandhana R, Rodriguez KR, Qian K, Bruns AM, and Horvath CM (2018). RNA sensor LGP2 inhibits TRAF ubiquitin ligase to negatively regulate innate immune signaling. EMBO Rep. 19. 10.15252/embr.201745176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry AK, Chen G, Zheng D, Tang H, and Cheng G. (2005). The host type I interferon response to viral and bacterial infections. Cell Res. 15, 407–422. 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- Pollpeter D, Komuro A, Barber GN, and Horvath CM (2011). Impaired cellular responses to cytosolic DNA or infection with Listeria monocytogenes and vaccinia virus in the absence of the murine LGP2 protein. PLoS One 6, e18842. 10.1371/journal.pone.0018842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontrelli P, Conserva F, and Gesualdo L. (2019). The role of lysine 63-linked ubiquitylation in health and disease. In Ubiquitin Proteasome System—Current Insights into Mechanism of Cellular Regulation and Disease, Summers M, ed.. 10.5772/intechopen.83659. [DOI] [Google Scholar]
- Quicke KM, Diamond MS, and Suthar MS (2017). Negative regulators of the RIG-I-like receptor signaling pathway. Eur. J. Immunol. 47, 615–628. 10.1002/eji.201646484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quicke KM, Kim KY, Horvath CM, and Suthar MS (2019). RNA Helicase LGP2 negatively regulates RIG-I signaling by preventing TRIM25-mediated caspase activation and recruitment domain ubiquitination. J. Interferon Cytokine Res. 10.1089/jir.2019.0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawling DC, and Pyle AM (2014). Parts, assembly and operation of the RIG-I family of motors. Curr. Opin. Struct. Biol. 25, 25–33. 10.1016/j.sbi.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, and Gack MU (2020). RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551. 10.1038/s41577-020-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez KR, Bruns AM, and Horvath CM (2014). MDA5 and LGP2: Accomplices and antagonists of antiviral signal transduction. J. Virol. 88, 8194–8200. 10.1128/jvi.00640-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, and Fitzgerald KA (2005). The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 175, 5260–5268. 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, and Gale M Jr. (2007). Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. U S A. 104, 582–587. 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, and Takeuchi O. (2010). LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. U S A 107, 1512–1517. 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, and Harhaj E. (2010). A20 inhibition of NFkappaB and inflammation: targeting E2:E3 ubiquitin enzyme complexes. Cell Cycle 9, 2481–2482. 10.4161/cc.9.13.12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Ma A, and Harhaj EW (2010). Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327, 1135–1139. 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Tahar M, Blanc F, Furio L, Chopy D, Balloy V, Lafon M, Chignard M, Fiette L, Langa F, Charneau P, and Pothlichet J. (2014). Protective role of LGP2 in influenza virus pathogenesis. J. Infect Dis. 210, 214–223. 10.1093/infdis/jiu076. [DOI] [PubMed] [Google Scholar]
- Stone AEL, Green R, Wilkins C, Hemann EA, and Gale M Jr. (2019). RIG-I-like receptors direct inflammatory macrophage polarization against West Nile virus infection. Nat. Commun. 10, 3649. 10.1038/s41467-019-11250-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Ramos HJ, Brassil MM, Netland J, Chappell CP, Blahnik G, McMillan A, Diamond MS, Clark EA, Bevan MJ, and Gale M Jr. (2012). The RIG-I-like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity 37, 235–248. 10.1016/j.immuni.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Tu S, Lin G, Guo H, Yan C, Liu Q, Huang L, Tang N, Xiao Y, Pope RM, et al. (2020). Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J. Exp. Med. 217. 10.1084/jem.20182091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, and Barber GN (2007). Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 178, 6444–6455. [DOI] [PubMed] [Google Scholar]
- Wertz IE, and Dixit VM (2010). Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect. Biol. 2, a003350. 10.1101/cshperspect.a003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widau RC, Parekh AD, Ranck MC, Golden DW, Kumar KA, Sood RF, Pitroda SP, Liao Z, Huang X, Darga TE, et al. (2014). RIG-I-like receptor LGP2 protects tumor cells from ionizing radiation. Proc. Natl. Acad. Sci. U S A. 111, E484–E491. 10.1073/pnas.1323253111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, and Arron JR (2003). TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. Bioessays 25, 1096–1105. 10.1002/bies.10352. [DOI] [PubMed] [Google Scholar]
- Wu X, and Karin M. (2015). Emerging roles of Lys63-linked polyubiquitylation in immune responses. Immunol. Rev. 266, 161–174. 10.1111/imr.12310. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Okamoto T, Takeda K, Sato S, Sanjo H, Uematsu S, Saitoh T, Yamamoto N, Sakurai H, Ishii KJ, et al. (2006). Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat. Immunol. 7, 962–970. 10.1038/ni1367. [DOI] [PubMed] [Google Scholar]
- Yang K, Zhu J, Sun S, Tang Y, Zhang B, Diao L, and Wang C. (2004). The coiled-coil domain of TRAF6 is essential for its auto-ubiquitination. Biochem. Biophys. Res. Commun. 324, 432–439. 10.1016/j.bbrc.2004.09.070. [DOI] [PubMed] [Google Scholar]
- Yin Q, Lin SC, Lamothe B, Lu M, Lo YC, Hura G, Zheng L, Rich RL, Campos AD, Myszka DG, et al. (2009). E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16, 658–666. 10.1038/nsmb.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, and Fujita T. (2007). Function of RIG-I-like receptors in antiviral innate immunity. J. Biol. Chem. 282, 15315–15318. 10.1074/jbc.R700007200. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr., Akira S, et al. (2005). Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, and Fujita T. (2004). The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737. [DOI] [PubMed] [Google Scholar]
- Zhao H, Li CC, Pardo J, Chu PC, Liao CX, Huang J, Dong JG, Zhou X, Huang Q, Huang B, et al. (2005). A novel E3 ubiquitin ligase TRAC-1 positively regulates T cell activation. J. Immunol. 174, 5288–5297. 10.4049/jimmunol.174.9.5288. [DOI] [PubMed] [Google Scholar]
- Zou W, Papov V, Malakhova O, Kim KI, Dao C, Li J, and Zhang DE (2005). ISG15 modification of ubiquitin E2 Ubc13 disrupts its ability to form thioester bond with ubiquitin. Biochem. Biophys. Res. Commun. 336, 61–68. 10.1016/j.bbrc.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Zou W, and Zhang DE (2006). The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 281, 3989–3994. 10.1074/jbc.M510787200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.