

Abstract

The ability to sample multiple reactions on the same single enzyme is important to link rare intermediates with catalysis and to unravel the role of conformational changes. Despite decades of efforts, however, the single-molecule characterization of nonfluorogenic enzymes during multiple catalytic turnovers has been elusive. Here, we show that nanopore currents allow sampling the dynamic exchange between five structural intermediates during E. coli dihydrofolate reductase (DHFR) catalysis. We found that an endosteric effect promotes the binding of the substrate to the enzyme with a specific hierarchy. The chemical step then switched the enzyme from the closed to the occluded conformation, which in turn promotes the release of the reduced cofactor NADP+. Unexpectedly, only a few reactive complexes lead to catalysis. Furthermore, second-long catalytic pauses were observed, possibly reflecting an off-path conformation generated during the reaction. Finally, the free energy from multiple cofactor binding events were required to release the product and switch DHFR back to the reactive conformer. This catalytic fueled concerted mechanism is likely to have evolved to improve the catalytic efficiency of DHFR under the high concentrations of NADP+ in E. coli and might be a general feature for complex enzymatic reactions where the binding and release of the products must be tightly controlled.
Keywords: ClyA nanopore, DHFR, single-molecule, enzymology, conformational dynamics
Introduction
The mesmerizing power of enzymes to catalyze chemical reactions has fascinated scientists for over a century. The first crystal structure of an enzyme, lysozyme,1,2 confirmed that enzymes fold into a three-dimensional structure that stabilizes the transition state of the catalyzed reaction. Fifty years on, however, scientists are not yet able to rationally design enzymes.3−5 The failure of catalytic antibodies to catalyze reactions with enzyme-like efficiency6 suggested that transition-state stabilization is only part of the picture.7 In the meanwhile, other characteristics of enzymes emerged. The first is that enzymes are soft structures, and their flexibility and dynamics are important factors to efficiently catalyze reactions.8−12 Another is that enzymes might adopt more than one thermally stable conformation,13−16 which might be linked to catalysis.17−19 As last, another intriguing characteristic of enzymes is that they appear to have large structures stabilized by a network of hundreds of weak interactions, the slight disruption of which (sometimes one) can have profound effects on the catalytic efficiency of the enzyme.20−22 Indeed, the link between long range dynamics and catalysis has been proposed but also fiercely debated.23−27
Dihydrofolate reductase (DHFR) has been used extensively as a model enzyme for investigating the link between structure, dynamics, and catalysis.28−34 The enzyme catalyzes the reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF) using nicotinamide adenine dinucleotide phosphate (NADPH) as the cofactor (Figure 1A). Five intermediates have been identified in the steady-state catalytic cycle35 and characterized by X-ray crystallography36−40 and NMR:29,31,32,41,42 E:NADPH, E:NADPH:DHF, E:NADP+:THF, E:THF, and E:NADPH:THF (Figure 1B). Conformational changes are observed in the Met20 loop (residues 9–24) between the Michaelis complex E:NADPH:DHF [modeled by E:NADP+:folate in structural studies] and the product complex [modeled by E:NADP+:THF].36 The Met20 loop adopts two main conformations: the “closed” conformation in which the loop packs tightly against the nicotinamide ring of the cofactor, and the “occluded” conformation in which the loop sterically blocks the nicotinamide-binding pocket. The holoenzyme (E:NADPH) and the model Michaelis complex (sampled by E:NADP+:folate) are in the closed conformation, whereas the product complexes adopt the occluded conformation (Figure 1B).29,32,36
Figure 1.
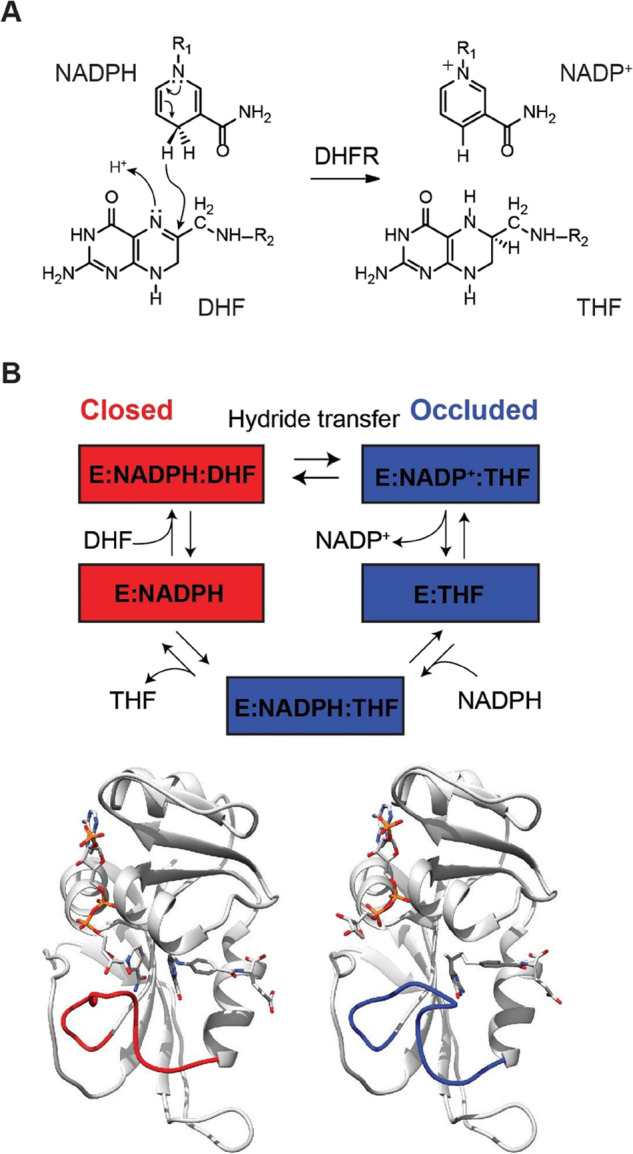
DHFR reaction. (A) DHFR-catalyzed reaction. (B) DHFR catalytic cycle as identified by X-ray crystallography and NMR spectroscopy for the Met20 loop conformation. The cycle starts with DHFR in the complex with NADPH (E:NADPH, PDB 1RX1), then dihydrofolate binds (E:NADPH:DHF, PDB 1RX2). In both of these structures, DHFR adopts the closed conformation, where the Met20 loop (red loop) is packed against the nicotinamide ring of the cofactor. After the reaction, the enzyme switches to the occluded conformation whereby the Met20 loop sterically hinders the nicotinamide-ribose binding pocket. Release of NADP+ (PDB 1RC4) and rebinding of NADPH (PDB 1RX6) precede the release of THF (PDB 1RX5). Below are structures of the closed ternary complex E:NADP+:FOL: (left, PDB 1RX2) and occluded ternary complex E:NADPH:5,10-dideazaTHF (ddTHF) (right, PDB 1RX6) illustrating the conformational changes of the M20 loop (shown in red and blue, respectively) that occur upon hydride transfer.
We have recently shown that DHFR can be studied by single-molecule current recordings. Cysteine-free DHFR molecules were extended with a positivity charged C-terminal polypeptide tag, and two negatively charged residues were introduced to the surface (named here as DHFRtag) to induce nanopore capture.43,44 Importantly, the confinement inside the nanopore, and the effect of the electric field does not affect the thermodynamic stability of the protein within the nanopore.43,45−50 We found that DHFRtag exists in ground-state conformations or conformers that have different affinities for various ligands.44 Intriguingly, the exchange between conformers was promoted by ligands that stabilized the transition-state configurations, suggesting that the conformers might have a role in the catalytic cycle of the enzyme. Here, we sample the catalytic reaction of DHFR at the single-molecule level during multiple turnovers to address the link between conformer exchange and catalysis.
Results
Binding of Substrate Ligands to DHFRtag
In nanopore experiments, the lipid bilayer defines a cis and a trans environment (Figure 2A) that can contain different solutions. Under a negative external applied potential (with the sign referring to the trans electrode/solution, Figure 2A), DHFRtag molecules added to the cis solution enter inside a single protein nanopore by the effect of the electroosmotic flow. The entry of the protein is observed by the step-wise reduction of the negative current, from the open pore current (Io) to a blocked pore current value (IB) (Figure 2A). Since IB values showed ∼5% variation from pore to pore, we used instead the fractional residual current percent (Ires% = IB/Io × 100), which showed less variation (∼2.5%). In this work, the DHFR reaction was probed by adding ligands to the cis or trans solutions. The cofactor NADPH or its deuterated (NADPD) or reduced (NADP+) forms were added to the cis solution. This allowed preincubation of DHFR with the cofactor, which is shown to reduce hysteresis.51 The slow-reacting substrate folate (FOL), the substrate dihydrofolate (DHF), and the product tetrahydrofolate (THF) were added to the trans side of the nanopore and encounter DHFR only inside the nanopore. NADPH and NADPD bind to the closed configuration of the enzyme,36 while FOL, DHF, and THF bind to the occluded configuration of DHFR36,52 (Figures 1B and 2H). The binding of ligands induced discrete current enhancement from the basal blocked current of DHFRtag (Ires% = 75.7 ± 0.3) (Figure 2A,B) and are expressed throughout as ΔIres% (Figure 2, Table 1, and Figures S1–S5). The binding of substrates induced lower current enhancement compared to the binding of the cofactor (Figure 2H). From the duration of the blockades, the association and dissociation rate constants of all ligands can be measured (Table 1). The values measured inside the nanopore corresponded well to bulk values,44 indicating that the environment of the nanopore did not affect the structure and function of the enzyme. Figure 2E shows that tetrahydrofolate has two types of blockades, both having the same residual current but different durations (dissociation rates: E1:THF = 1.39 ± 0.2 s–1 and E2:THF = 27.5 ± 2.0 s–1, Table 1 and Figure S3). We previously showed that DHFR exists in at least two conformers with different affinities for ligands.44 Therefore, it is likely that the two dwell times correspond to the binding of THF to two conformers, which, most likely, correspond to the closed and occluded conformation of DHFR (see later). The binding of NADP+ to DHFRtag was not observed, because of its fast dissociation constant (koff = 300 s–1).35
Figure 2.

Binding of ligands to DHFRtag inside the ClyA nanopore. (A) Typical ionic current blockades provoked by the capture of a single DHFRtag molecule (50 nM, cis) by ClyA-AS at −80 mV. On the left of the trace is the surface representation of a ClyA-AS nanopore (gray) embedded in a planar lipid bilayer (yellow), DHFRtag, which is colored according to the vacuum electrostatics (PyMOL). (B–G) Expansions of typical blockades of DHFRtag in the absence of ligands (B) and in the presence of 33.6 μM folate [trans, (C)] 5 μM dihydrofolate [trans, (D)] 12.6 μM tetrahydrofolate [trans, (E)] 26.6 μM NADPH [cis, (F)] and 22.5 μM NADPD [cis, (G)]. (H) Table showing the connection between the observed current levels and the different structures of DHFR as obtained by X-ray crystallography. All current traces were collected in 250 mM KCl, 15 mM Tris–HCl pH 7.8 at 25 °C, by applying a Bessel low-pass filter with a 2 kHz cutoff and sampled at 10 kHz. All traces except the recordings in A were filtered digitally with a Gaussian low-pass filter with a 100 Hz cutoff.
Table 1. Current Blockades and Kinetic Constants of DHFRtag upon Addition of NADPH, NADPD (cis), Folate, Dihydrofolate, and/or Tetrahydrofolate (trans)a.
| ligand | Ires-bound (%) | Ires-apo (%) | ΔIres (%) | ΔIres (pA) | kon (μM–1 s–1) | koff (s–1) | KDapp(μM) |
|---|---|---|---|---|---|---|---|
| folate | 75.6 ± 0.5 | 75.8 ± 0.5 | 0.79 ± 0.02 | 2.1 ± 0.1 | 1.2 ± 0.3 | 63.4 ± 3.2 | 53 ± 14 |
| DHF | 76.0 ± 0.2 | 76.9 ± 0.2 | 0.80 ± 0.01 | 2.1 ± 0.1 | 4.5 ± 0.6 | 18.7 ± 4.1 | 4.2 ± 1.1 |
| THF | 76.5 ± 0.2 | 77.2 ± 0.2 | 0.75 ± 0.02 | 1.9 ± 0.1 | 1.9 ± 0.6 | 27.5 ± 2.0 (closed) | 14 ± 4.7 |
| 1.39 ± 0.2 (occluded) | 0.73 ± 0.3 | ||||||
| NADPH | 76.9 ± 0.4 | 75.3 ± 0.2 | 1.35 ± 0.04 | 3.6 ± 0.1 | 0.23 ± 0.01 | 8.91 ± 0.8 | 39 ± 3.9 |
| NADPD | 76.9 ± 0.1 | 75.9 ± 0.1 | 1.38 ± 0.03 | 3.6 ± 0.2 | 0.20 ± 0.06 | 7.48 ± 0.1 | 37 ± 11 |
| NADP+ + folate | 77.5 ± 0.1 | 75.8 ± 0.1 | 1.76 ± 0.04 | 4.7 ± 0.3 | 34.1 ± 1.5 | ||
| NADPH + folate | 77.6 ± 0.3 | 75.7 ± 0.3 | 1.89 ± 0.05 | 5.0 ± 0.1 | 136 ± 18 | ||
| NADPD + folate | 77.9 ± 0.1 | 76.0 ± 0.1 | 1.81 ± 0.06 | 4.7 ± 0.1 | 30.7 ± 4.1 | ||
| NADP+ + DHF | 77.2 ± 0.5 | 75.6 ± 0.4 | 1.60 ± 0.08 | 4.5 ± 0.3 | 129 ± 25 | ||
| NADPH + DHF | 78.2 ± 0.5 | 76.4 ± 0.6 | 1.79 ± 0.07 | 4.9 ± 0.1 | 56.1 ± 12 | ||
| NADPD + DHF | 77.4 ± 0.2 | 75.5 ± 0.2 | 1.74 ± 0.15 | 4.8 ± 0.2 | 54.1 ± 25 | ||
| NADP+ + THF (closed) | 76.8 ± 0.2 | 75.6 ± 0.2 | 1.62 ± 0.02 | 4.4 ± 0.1 | 105 ± 20 | ||
| NADP+ + THF (occluded) | 77.2 ± 0.2 | 75.6 ± 0.2 | 1.25 ± 0.01 | 3.4 ± 0.1 | 175 ± 30 | ||
| NADPH + THF (closed) | 78.0 ± 0.1 | 76.2 ± 0.1 | 1.80 ± 0.03 | 4.6 ± 0.1 | 120 ± 8.7 | ||
| NADPH + THF (occluded) | 77.4 ± 0.1 | 76.2 ± 0.1 | 1.28 ± 0.09 | 3.0 ± 0.1 | 158 ± 17 | ||
| NADPD + THF (closed) | 77.5 ± 0.2 | 75.8 ± 0.2 | 1.80 ± 0.02 | 4.8 ± 0.1 | 103 ± 5.0 | ||
| NADPD + THF (occluded) | 76.9 ± 0.2 | 75.8 ± 0.2 | 1.23 ± 0.05 | 3.3 ± 0.1 | 177 ± 19 |
Errors are given as standard deviation between independent pores (N = 3). All current traces were collected in 250 mM KCl, 15 mM Tris–HCl pH 7.8 at room temperature (25 °C) at −80 mV. (L) and (H) indicate the low- and high-affinity off rates of THF.
Ternary Complex Formation from the Closed Conformation
The reactive configuration, where the enzyme is in closed configuration, was probed using the slow-reactive substrate folate (33.6 μM, trans) in combination with NADP+ (1.5 μM, cis, Figures 3A top and S6), NADPH (26.6 μM, cis, Figures 3A middle and S7), or NADPD (24.2 μM, cis, Figures 3A bottom and S8). The formation of the ternary complexes was observed as transient current enhancements from the enzyme:FOL or enzyme:NADPH level (Figures 3C and S6–S8). The single-molecule nature of the nanopore experiment allowed measuring the sequential order of binding and releasing of the ligands to and from the enzyme (Figure 3B). When NADPH and folate were used, the majority (86.2 ± 4.2%) of the collected events leading to the ternary complex appeared from the closed (E:NADPH) configuration. Since at the ligand concentration tested here both binding sites have the same probability of being occupied, these results indicate that the binding of a substrate affects the affinity of the enzyme for the cofactor, or vice versa. This endosteric effect allows the ternary complex to be formed hierarchically from the closed to the occluded conformation. Once the Michaelis complex was formed, in 86.0 ± 4.1% of measured events, folate was released before NADPH (koff = 135.8 ± 18.3 s–1), indicating that folate has a lower affinity for the reactive configuration than NADPH. By contrast, when NADP+ and folate were sampled, in 90.3 ± 1.7% of the observed events, NADP+ was released before folate (koff = 34.1 ± 1.5 s–1, Figure 3A,B), indicating that the reactive configuration of DHFR has a higher affinity for folate than for NADP+. Hence, moving from E:NADPH:FOL to E:NADP+:FOL, which mimics the progression of the chemical step, the affinity of DHFR for the cofactor decreases, while the affinity for folate increases. When NADPD was sampled, in 99.2 ± 0.1% of the events, the ternary complex was formed from the NADPD level, and in 99.6 ± 0.1% of the events, the dissociation produced a NADPD-bound configuration (koff = 30.7 ± 4.1 s–1) (Figure 3A,B). Since deuterium makes stronger hydrogen bonds than hydrogen,53 the reactive configuration is stabilized by a hydrogen bond between the pro-R hydrogen of the product and the substrate. No product formation was observed when using slow-reactive folate.
Figure 3.

Formation of the ternary complex in the closed configuration. (A) Typical current blockades induced by DHFRtag (50 nM, cis) in the presence of folate (33.6 μM, trans) and NADP+ (1.5 μM, cis) [(A) top], folate (33.6 μM, trans) and NADPH (26.6 μM, cis) [(A) middle], and folate (30.9 μM, trans) and NADPD (24.2 μM, cis) [(A) bottom]. The current level of the apoenzyme, folate-bound level, NADPH/NADPD-bound level, and ternary complex level are shown as green, blue, red, and orange lines, respectively. (B) Scheme indicating the hierarchy of ligand binding to DHFRtag under the different conditions. (C) Table showing the connection between the observed current levels and the different structures of DHFR as obtained by X-ray crystallography. All current traces were collected in 250 mM KCl, 15 mM Tris–HCl pH 7.8 at room temperature (25 °C), by applying a Bessel low-pass filter with a 2 kHz cutoff and sampled at 10 kHz. The traces were filtered digitally with a Gaussian low-pass filter with a 100 Hz cutoff.
Ternary Complex Formation from the Occluded Conformation: Product Release and Reverse Reaction
We showed in Figure 2E that THF can bind with high affinity (E2:THF) or low affinity (E1:THF) to DHFR (Figure 2E, Figures 4A top and S3). In the presence of THF (10 μM, trans) and NADPH (0.3 mM, cis), we observed two classes of ternary complex blockades (Figure 4A). When THF was bound to the high-affinity conformation, additional reversible current events were observed (E2:THF:NADPH, ΔIres% = 0.47 ± 0.01%, Figures 4 bottom, S9, and S10). Increasing the concentration of NADPH (to 0.6 or 0.9 mM) increased the frequency of the transient events but not their duration (Figures 4A,C and S9–S11), indicating that the additional current enhancements reflected transient formation of the occluded ternary product E2:NADPH:THF. As observed in ensemble experiments,54,55 the dwell time of the E2:NADPH:THF complex, which reflects the release of the product of the reaction, decreased with increasing NADPH concentration (Figures 4D and S9–S11), confirming that the release of THF is facilitated by the binding of NADPH. The amplitude of the ionic current level of the E2:THF:NADPH ternary complex (ΔIres% = 1.28 ± 0.09%) was similar to that of the E:NADPH binary complex (ΔIres% = 1.35 ± 0.04%, Figures 3C and 4E). This observation is consistent with the reported relaxation dispersion experiments,54,55 which showed that the E:NADPH:THF complex transiently samples a closed excited state whereby the backbone conformation closely resembles the ground state of the closed E:NADPH complex.
Figure 4.

Formation of the ternary complex in the occluded configuration. (A) Typical tetrahydrofolate-induced blockades (12.6 μM, trans) [(A) top], in the presence of NADP+ (8.8 μM, cis) [(A) middle], and in the presence of NADPH (973 μM, cis) [(A) bottom]. The current level of the apoenzyme, tetrahydrofolate-bound level, NADPH-tetrahydrofolate-bound level, NADPH-bound level, and ternary complex level are shown as green, blue, purple, red, and orange lines, respectively. (B) Full point histogram of a E:NADPH:THF complex. (C) Relative occupancy of NADPH in the ternary complex (E:NADPH:THF) at different NADPH concentrations. (D) Dependency of the dwell time of tetrahydrofolate (trans) versus the concentration of NADPH (cis). The black diamonds are obtained when tetrahydrofolate and NADPH were used in the experiment; the squares are obtained when the reaction was followed (see later). (E) Table showing the connection between the observed current levels and the different structure of DHFR as obtained by X-ray crystallography. All current traces were collected in 250 mM KCl, 15 mM Tris–HCl pH 7.8 at room temperature (25 °C), by applying a Bessel low-pass filter with a 2 kHz cutoff and sampled at 10 kHz. The traces were filtered digitally with a Gaussian low-pass filter with a 100 Hz cutoff.
When THF was bound to the low-affinity conformation E1:THF, additional reversible NADP+ or NADPH current events were also observed (ΔIres% = 1.62 ± 0.02 and 1.80 ± 0.03%, respectively, Figures 4A middle and bottom, 4E, S9, S10, S12, and S13). These values were similar to the blockades recorded for the NADPH and DHF/FOL ternary complexes, which are in the closed conformation (Figure 3C), suggesting they represent a ternary complex with DHFR in the closed conformation. It follows that E1:THF and E2:THF most likely represent the binding of THF to the closed and occluded conformation of DHFR, respectively. Interestingly, NADP+ bound more often and strongly (Table 1, Figures 4A and S12) to the low-affinity conformer (E1, closed configuration) than the high-affinity conformer (E2, occluded configuration), indicating that switching from the closed to the occluded conformation reduced the affinity of the enzyme for NADP+ and consequentially reduced the probability for the reverse reaction (THF + NADP+ → DHF + NADPH) to occur.
Forward Catalyzed Reaction
The catalytic cycle was sampled using excess of NADPH (∼1.0 mM, cis) compared to DHF (low μM range, trans, Figures 5 and S14), which reflects the physiological concentrations of reactants. Under these conditions, multiple reactions from individual enzymes were observed, as shown by the appearance of the typical THF blockades (pink asterisks, Figure 5). Figure 6 shows a long recording of the catalyzed reaction providing an overview of the catalytic proficiency of the enzyme during nearly four minutes. The reaction rate was measured by counting the number of products formed over the time of the protein blockade (Table S1). Interestingly, When the reaction was sampled using a 10-fold excess of DHF compared to NADPH, the majority (58.2 ± 7.9%) of reactions still occurred from the NADPH-bound level but only after large current rearrangements (Figures 5D, S15, and S16). This observation is compatible with the enzyme rearranging its structure to switch from the occluded configuration to the closed configuration and confirms that the reaction is hierarchical, as it preferentially occurs from the closed state despite the initial order of binding of the substrates.
Figure 5.

DHFR-catalyzed reaction measured by nanopore recordings. (A–C) Selected traces showing the details of the current levels of the catalytic reaction. NADPH (0.7 mM) and dihydrofolate (DHF) (4.3 μM) were added to the cis and trans chambers, respectively. The pink squares indicate the rearrangement in the transition-state conformation leading to the reaction. The current levels of the apoenzyme, tetrahydrofolate-bound, NADPH-bound, and ternary complex conformations are highlighted with a green, blue, red and yellow line, respectively. The blue underlining (C) shows representative long ternary complex. (D) Current trace of a catalyzed reaction in which DHF (50.5 μM, trans) is in excess compared to NADPH (5.5 μM, cis). The pink asterisks are indicating the occurrence of the product, tetrahydrofolate. Current traces were collected in 250 mM KCl, 15 mM Tris–HCl pH 7.15 (A–C) or 7.8 (D) at room temperature (25 °C), by applying a Bessel low-pass filter with a 2 kHz cutoff and sampled at 10 kHz. The trace was filtered digitally with a Gaussian low-pass filter with a 100 Hz cutoff.
Figure 6.

Continuous recording of DHFRtag conformational changes during the catalyzed reactions. NADPH (1.0 mM) and dihydrofolate (DHF) (1.9 μM) were added to the cis and trans chambers, respectively. The pink asterisks are indicating the occurrence of the product, tetrahydrofolate. Current traces were collected in 250 mM KCl, 15 mM Tris–HCl pH 7.8 at room temperature (25 °C), by applying a Bessel low-pass filter with a 2 kHz cutoff and sampled at 10 kHz. The trace was filtered digitally with a Gaussian low-pass filter with a 100 Hz cutoff.
The single-molecule observation revealed that not all the ternary complexes lead to a reaction (Figures 5A and 6B). The percentage of nonreactive configurations increased with the pH (from 62.6 ± 1.9% at pH 7.15 to 89.0 ± 2.8% at pH 9.1, Table S1 and Figures S14, S17, and S18), while the duration of the nonreactive configurations remained constant over the pH range tested (∼20 ms, Table S1). In DHFR, the protonation of DHF is the rate-limiting step.56 Since the duration of the reactive configuration is constant over the pH, these data suggest that the protonation of the substrate happens before the M20 loop closes over the reactants, most likely as soon as DHF binds to the enzyme.
The use of NADPD instead of NADPH did not have a significant effect on the product release (Figures S11 and S20–S22) nor the dwell time of the ternary complex (Table S1). However, the presence of NADPD reduced by ∼two-fold the number of reactions per second compared to NADPH (Table S1), showing the existence of a kinetic isotope effect, comparable to the kinetic isotope effect measured in bulk.57,58 We also found that in about 10% of the recorded events, long ternary complexes were observed (1.64 ± 1.47 s, Table S1, Figures 5C, 6B, S14, S17, and S18). These long ternary complex events were not observed under nonreactive conditions (e.g., when THF or FOL were sampled with NADPH or NADP+, Figures 3A, 4A, S6–S10, S12, and S19), and their number was reduced by about threefold when NADPD was used with NADPH (Table S1, and Figures S20–S22), indicating they are likely the consequence of the chemical step.
Discussion and Conclusions
Recently, we found that DHFR exists in slow-converting conformers with different affinities for ligands.44 In this work we follow the DHFR reaction at the single-molecule level during multiple turnover conditions. The long observation times of the nanopore experiment revealed that, occasionally, DHFR populates an additional long-lasting and inactive conformation, which most likely corresponds to an alternative fold of DHFR. The latter is not observed under nonreactive conditions (e.g., using the slow-reacting folate, Figures 3A, S6–S10, S12, S13, and S19), indicating that this is likely to be the result of the catalytic step. This observation suggests an intriguing hypothesis in which the enthalpy generated by the chemical step might induce local unfolding in the enzyme. Thus, the soft interactions in folded enzymes might play a role in dissipating the excess energy generated during the catalytic step.
The single-molecule experiments allowed measuring the kinetic properties of individual conformers, unraveling the details of the DHFR catalytic cycle. Crystallographic work established that NADPH binds to the closed conformation and DHF to the occluded conformation of DHFR.36 We found that the binding of ligands is hierarchical, from the closed to the occluded conformation of DHFR (Figure 3B) and not vice versa (Figures S15 and S16). This observation is compatible with a mechanism in which the binding of NADPH increases the affinity of the enzyme for the substrate, or the binding of the substrate decreases the affinity for NADPH (Figure 3A,B). This endosteric effect allows the hierarchical formation of the reactive ternary complex from the closed state (Figures 3A and 6). The reactive complex is formed for about 20 ms (Figures 5A,B, 6, S14, and S17–S22 and Table 1). If the substrate does not protonate, most likely during the initial binding to the enzyme, DHF is released back to solution and the enzyme remains in the closed conformation (Figure 5A). If the substrate protonates and the reaction occurs, the enzyme switches to the THF-bound occluded conformation, which promotes the release of NADP+. Since conformer exchange only happens during catalysis, these results indicate that the free energy of the chemical step is used to transit from the closed to the occluded conformation. Most notably, we also found that NADP+ has a higher affinity for the substrate-bound closed conformation (E1:THF) than for the product-bound occluded conformation (E2:THF)(Figures 4A and S12). Therefore, conformer exchange reduces the affinity of the enzyme for NADP+ as well as the probability for the backward reaction (THF → DHF). In the final step, NADPH binds to E2:THF and promotes the switching back to the reactive closed configuration. THF has lower affinity for the closed conformation than for the occluded conformation (Figures 4A and S3), indicating that the free energy of cofactor binding is expended to help release the product.
This work shows that the soft interactions in DHFR have a more sophisticated role in catalysis rather than just stabilizing the transition state of the catalyzed reaction, as generally assumed.59,60 Endosteric interactions in DHFR control the hierarchy of ligand binding and conformer exchange. Within this framework, the free energy of the reaction and cofactor is expended to favor the forward reaction at the expenses of the backward reaction. The cost is a slowed steady-state catalysis, where several ternary complexes are required to release the product or to induce the catalytic switch from the closed to the occlude conformation. This mechanism is likely to have evolved to control the affinity of the enzyme for NADP+. In E. coli cells, the concentrations of NADP+ and NADPH are equal. This is in contrast with human cells, where the concentration of NADPH is about 100-fold higher than NADP+. Although the structure of human DHFR is almost identical to that of E. coli DHFR, its sequence is highly divergent. Both DHFR enzymes have similar kinetics involving the same catalytic cycle with its intermediates. However, 35% of the human enzyme also populates a second catalytic cycle, with E:NADP+:THF being the diverging point, where NADP+ remains bound to the enzyme and THF is released first.61,62 Interestingly, the Met20 loop of human DHFR always remains in the closed configuration. Hence, the catalytically accessed occluded configuration of E. coli DHFR, most likely evolved to promote the release of the NADP+ and to prevent the backward catalyzed reaction in order to allow efficient albeit slower catalysis at high intracellular concentration of NADP+.
Acknowledgments
We thank the Allemann Group (Cardiff University) for providing the plasmid containing the alcohol dehydrogenase from Thermoanaerobacter brockii. We thank the European Research Council for financial support to N.S.G.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c04388.
Materials and methods; pH dependency of kinetic properties of DHFR (Table S1); Figures with continuous recordings of induced currents levels to DHFRtag, dependency of the dwell time of THF with the concentration of the cofactor under different conditions (Figures S1–S22) (PDF)
Author Contributions
N.S.G. performed the experiments and analyzed the data presented in the manuscript. N.S.G. and G.M. wrote the manuscript. G.M. supervised the project.
The authors declare no competing financial interest.
Supplementary Material
References
- Blake C. C. F.; Koenig D. F.; Mair G. A.; North A. C. T.; Phillips D. C.; Sarma V. R. Structure of Hen Egg-White Lysozyme: A Three-Dimensional Fourier Synthesis at 2 Å Resolution. Nature 1965, 206, 757–761. 10.1038/206757a0. [DOI] [PubMed] [Google Scholar]
- Blake C. C.; Johnson L. N.; Mair G. A.; North A. C.; Phillips D. C.; Sarma V. R. Crystallographic Studies of the Activity of Hen Egg-White Lysozyme. Proc. R. Soc. London, Ser. B 1967, 167, 378–388. 10.1098/rspb.1967.0035. [DOI] [PubMed] [Google Scholar]
- Chen K.; Arnold F. H. Engineering New Catalytic Activities in Enzymes. Nat. Catal. 2020, 3, 203–213. 10.1038/s41929-019-0385-5. [DOI] [Google Scholar]
- Arnold F. H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem., Int. Ed. 2018, 57, 4143–4148. 10.1002/anie.201708408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chica R. A.; Doucet N.; Pelletier J. N. Semi-Rational Approaches to Engineering Enzyme Activity: Combining the Benefits of Directed Evolution and Rational Design. Curr. Opin. Biotechnol. 2005, 16, 378–384. 10.1016/j.copbio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Hilvert D. Critical Analysis of Antibody Catalysis. Annu. Rev. Biochem. 2000, 69, 751–793. 10.1146/annurev.biochem.69.1.751. [DOI] [PubMed] [Google Scholar]
- Mader M. M.; Bartlett P. A. Binding Energy and Catalysis: The Implications for Transition-State Analogs and Catalytic Antibodies. Chem. Rev. 1997, 97, 1281–1302. 10.1021/cr960435y. [DOI] [PubMed] [Google Scholar]
- Bigot S.; Saleh O. A.; Cornet F.; Allemand J.-F.; Barre F. Oriented Loading of FtsK on KOPS. Nat. Struct. Mol. Biol. 2006, 13, 1026–1028. 10.1038/nsmb1159. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; Dyson H. J.; Wright P. E. An NMR Perspective on Enzyme Dynamics. Chem. Rev. 2006, 106, 3055–3079. 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- Cannon W. R.; Singleton S. F.; Benkovic S. J. A Perspective on Biological Catalysis. Nat. Struct. Mol. Biol. 1996, 3, 821–833. 10.1038/nsb1096-821. [DOI] [PubMed] [Google Scholar]
- Eisenmesser E. Z. Enzyme Dynamics During Catalysis. Science 2002, 295, 1520–1523. 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- Richard J. P. Protein Flexibility and Stiffness Enable Efficient Enzymatic Catalysis. J. Am. Chem. Soc. 2019, 141, 3320–3331. 10.1021/jacs.8b10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. C.; Tawfik D. S. Conformational Diversity and Protein Evolution–a 60-Year-Old Hypothesis Revisited. Trends Biochem. Sci. 2003, 28, 361–368. 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- Hammes G. G. Multiple Conformational Changes in Enzyme Catalysis †. Biochemistry 2002, 41, 8221–8228. 10.1021/bi0260839. [DOI] [PubMed] [Google Scholar]
- Petrović D.; Risso V. A.; Kamerlin S. C. L.; Sanchez-Ruiz J. M. Conformational Dynamics and Enzyme Evolution. J. R. Soc., Interface 2018, 15, 20180330 10.1098/rsif.2018.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Tawfik D. S. Protein Dynamism and Evolvability. Science 2009, 324, 203–207. 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. Evolutionary Aspects of Enzyme Dynamics. J. Biol. Chem. 2014, 289, 30205–30212. 10.1074/jbc.R114.565515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell E.; Kaltenbach M.; Correy G. J.; Carr P. D.; Porebski B. T.; Livingstone E. K.; Afriat-Jurnou L.; Buckle A. M.; Weik M.; Hollfelder F.; Tokuriki N.; Jackson C. J. The Role of Protein Dynamics in the Evolution of New Enzyme Function. Nat. Chem. Biol. 2016, 12, 944–950. 10.1038/nchembio.2175. [DOI] [PubMed] [Google Scholar]
- Bar-Even A.; Milo R.; Noor E.; Tawfik D. S. The Moderately Efficient Enzyme: Futile Encounters and Enzyme Floppiness. Biochemistry 2015, 54, 4969–4977. 10.1021/acs.biochem.5b00621. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Catalytic Efficiency of Enzymes: A Theoretical Analysis. Biochemistry 2013, 52, 2012–2020. 10.1021/bi301515j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rod T. H.; Radkiewicz J. L.; Brooks C. L. Correlated Motion and the Effect of Distal Mutations in Dihydrofolate Reductase. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 6980–6985. 10.1073/pnas.1230801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan P. T. R.; Lutz S.; Benkovic S. J. Coupling Interactions of Distal Residues Enhance Dihydrofolate Reductase Catalysis: Mutational Effects on Hydride Transfer Rates †. Biochemistry 2002, 41, 12618–12628. 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- Agarwal P. K.; Billeter S. R.; Rajagopalan P. T.; Benkovic S. J.; Hammes-Schiffer S. Network of Coupled Promoting Motions in Enzyme Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 2794–2799. 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabha G.; Lee J.; Ekiert D. C.; Gam J.; Wilson I. A.; Dyson H. J.; Benkovic S. J.; Wright P. E. A Dynamic Knockout Reveals That Conformational Fluctuations Influence the Chemical Step of Enzyme Catalysis. Science 2011, 332, 234–238. 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveridge E. J.; Behiry E. M.; Guo J.; Allemann R. K. Evidence That a “dynamic Knockout” in Escherichia coli Dihydrofolate Reductase Does Not Affect the Chemical Step of Catalysis. Nat. Chem. 2012, 4, 292–297. 10.1038/nchem.1296. [DOI] [PubMed] [Google Scholar]
- Luk L. Y. P.; Javier Ruiz-Pernía J.; Dawson W. M.; Roca M.; Loveridge E. J.; Glowacki D. R.; Harvey J. N.; Mulholland A. J.; Tuñón I.; Moliner V.; Allemann R. K. Unraveling the Role of Protein Dynamics in Dihydrofolate Reductase Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16344–16349. 10.1073/pnas.1312437110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S. M. C.; Clouthier C. M.; Park J.; Gagné D.; Berghuis A. M.; Doucet N.; Pelletier J. N. Maintenance of Native-like Protein Dynamics May Not Be Required for Engineering Functional Proteins. Chem. Biol. 2014, 21, 1330–1340. 10.1016/j.chembiol.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Schnell J. R.; Dyson H. J.; Wright P. E. Structure, Dynamics, and Catalytic Function of Dihydrofolate Reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. The Dynamic Energy Landscape of Dihydrofolate Reductase Catalysis. Science 2006, 313, 1638–1642. 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Radkiewicz J. L.; Brooks C. L. Protein Dynamics in Enzymatic Catalysis: Exploration of Dihydrofolate Reductase. J. Am. Chem. Soc. 2000, 122, 225–231. 10.1021/ja9913838. [DOI] [Google Scholar]
- Falzone C. J.; Wright P. E.; Benkovic S. J. Dynamics of a Flexible Loop in Dihydrofolate Reductase from Escherichia coli and Its Implication for Catalysis. Biochemistry 1994, 33, 439–442. 10.1021/bi00168a007. [DOI] [PubMed] [Google Scholar]
- Osborne M. J.; Schnell J.; Benkovic S. J.; Dyson H. J.; Wright P. E. Backbone Dynamics in Dihydrofolate Reductase Complexes: Role of Loop Flexibility in the Catalytic Mechanism †. Biochemistry 2001, 40, 9846–9859. 10.1021/bi010621k. [DOI] [PubMed] [Google Scholar]
- Mhashal A. R.; Pshetitsky Y.; Eitan R.; Cheatum C. M.; Kohen A.; Major D. T. Effect of Asp122 Mutation on the Hydride Transfer in E. coli DHFR Demonstrates the Goldilocks of Enzyme Flexibility. J. Phys. Chem. B 2018, 122, 8006–8017. 10.1021/acs.jpcb.8b05556. [DOI] [PubMed] [Google Scholar]
- Kozlowski R.; Zhao J.; Dyer R. B. Acceleration of Catalysis in Dihydrofolate Reductase by Transient, Site-Specific Photothermal Excitation. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2014592118 10.1073/pnas.2014592118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierke C. A.; Johnson K. A.; Benkovic S. J. Construction and Evaluation of the Kinetic Scheme Associated with Dihydrofolate Reductase from Escherichia coli. Biochemistry 1987, 26, 4085–4092. 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Kraut J. Loop and Subdomain Movements in the Mechanism of Escherichia coli Dihydrofolate Reductase: Crystallographic Evidence †, ‡. Biochemistry 1997, 36, 586–603. 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- Nagae T.; Yamada H.; Watanabe N. High-Pressure Protein Crystal Structure Analysis of Escherichia coli Dihydrofolate Reductase Complexed with Folate and NADP +. Acta Crystallogr., Sect. D: Struct. Biol. 2018, 74, 895–905. 10.1107/S2059798318009397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystroff C.; Kraut J. Crystal Structure of Unliganded Escherichia coli Dihydrofolate Reductase. Ligand-Induced Conformational Changes and Cooperativity in Binding. Biochemistry 1991, 30, 2227–2239. 10.1021/bi00222a028. [DOI] [PubMed] [Google Scholar]
- Bystroff C.; Oatley S. J.; Kraut J. Crystal Structures of Escherichia coli Dihydrofolate Reductase: The NADP+ Holoenzyme and the Folate.Cntdot. NADP+ Ternary Complex. Substrate Binding and a Model for the Transition State. Biochemistry 1990, 29, 3263–3277. 10.1021/bi00465a018. [DOI] [PubMed] [Google Scholar]
- Cao H.; Gao M.; Zhou H.; Skolnick J. The Crystal Structure of a Tetrahydrofolate-Bound Dihydrofolate Reductase Reveals the Origin of Slow Product Release. Commun. Biol. 2018, 1, 226. 10.1038/s42003-018-0236-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne M. J.; Venkitakrishnan R. P.; Dyson H. J.; Wright P. E. Diagnostic Chemical Shift Markers for Loop Conformation and Substrate and Cofactor Binding in Dihydrofolate Reductase Complexes. Protein Sci. 2009, 12, 2230–2238. 10.1110/ps.03219603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. Millisecond Timescale Fluctuations in Dihydrofolate Reductase Are Exquisitely Sensitive to the Bound Ligands. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1373–1378. 10.1073/pnas.0914163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems K.; Ruić D.; Biesemans A.; Galenkamp N. S.; Van Dorpe P.; Maglia G. Engineering and Modeling the Electrophoretic Trapping of a Single Protein Inside a Nanopore. ACS Nano 2019, 13, 9980. 10.1021/acsnano.8b09137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galenkamp N. S.; Biesemans A.; Maglia G. Directional Conformer Exchange in Dihydrofolate Reductase Revealed by Single-Molecule Nanopore Recordings. Nat. Chem. 2020, 12, 481. 10.1038/s41557-020-0437-0. [DOI] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; Moeyaert B.; Cheley S.; Bayley H.; Maglia G. An Engineered ClyA Nanopore Detects Folded Target Proteins by Selective External Association and Pore Entry. Nano Lett. 2012, 12, 4895–4900. 10.1021/nl3024438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; De Maeyer M.; Maglia G. Tuning the Size and Properties of ClyA Nanopores Assisted by Directed Evolution. J. Am. Chem. Soc. 2013, 135, 13456–13463. 10.1021/ja4053398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesemans A.; Soskine M.; Maglia G. A Protein Rotaxane Controls the Trans Location of Proteins Across a ClyA Nanopore. Nano Lett. 2015, 15, 6076–6081. 10.1021/acs.nanolett.5b02309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meervelt V.; Soskine M.; Singh S.; Schuurman-Wolters G. K.; Wijma H. J.; Poolman B.; Maglia G. Real-Time Conformational Changes and Controlled Orientation of Native Proteins Inside a Protein Nanoreactor. J. Am. Chem. Soc. 2017, 139, 18640–18646. 10.1021/jacs.7b10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; Maglia G. Single-Molecule Analyte Recognition with ClyA Nanopores Equipped with Internal Protein Adaptors. J. Am. Chem. Soc. 2015, 137, 5793–5797. 10.1021/jacs.5b01520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galenkamp N. S.; Soskine M.; Hermans J.; Wloka C.; Maglia G. Direct Electrical Quantification of Glucose and Asparagine from Bodily Fluids Using Nanopores. Nat. Commun. 2018, 9, 4085. 10.1038/s41467-018-06534-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccanari D. P.; Joyner S. S. Dihydrofolate Reductase Hysteresis and Its Effect on Inhibitor Binding Analyses. Biochemistry 1981, 20, 1710–1716. 10.1021/bi00510a002. [DOI] [PubMed] [Google Scholar]
- Venkitakrishnan R. P.; Zaborowski E.; McElheny D.; Benkovic S. J.; Dyson H. J.; Wright P. E. Conformational Changes in the Active Site Loops of Dihydrofolate Reductase during the Catalytic Cycle. Biochemistry 2004, 43, 16046–16055. 10.1021/bi048119y. [DOI] [PubMed] [Google Scholar]
- Scheiner S.; Čuma M. Relative Stability of Hydrogen and Deuterium Bonds. J. Am. Chem. Soc. 1996, 118, 1511–1521. 10.1021/ja9530376. [DOI] [Google Scholar]
- Oyen D.; Fenwick R. B.; Stanfield R. L.; Dyson H. J.; Wright P. E. Cofactor-Mediated Conformational Dynamics Promote Product Release From Escherichia coli Dihydrofolate Reductase via an Allosteric Pathway. J. Am. Chem. Soc. 2015, 137, 9459–9468. 10.1021/jacs.5b05707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyen D.; Fenwick R. B.; Aoto P. C.; Stanfield R. L.; Wilson I. A.; Dyson H. J.; Wright P. E. Defining the Structural Basis for Allosteric Product Release from E. coli Dihydrofolate Reductase Using NMR Relaxation Dispersion. J. Am. Chem. Soc. 2017, 139, 11233–11240. 10.1021/jacs.7b05958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villafranca J. E.; Howell E. E.; Oatley S. J.; Warren M. S.; Kraut J. A Structure–Function Study of Dihydrofolate Reductase by Protein Engineering. Philos. Trans. R. Soc., A 1986, 317, 405–413. 10.1098/rsta.1986.0050. [DOI] [Google Scholar]
- Morrison J. F.; Stone S. R. Mechanism of the Reaction Catalyzed by Dihydrofolate Reductase from Escherichia coli: PH and Deuterium Isotope Effects with NADPH as the Variable Substrate. Biochemistry 1988, 27, 5499–5506. 10.1021/bi00415a017. [DOI] [PubMed] [Google Scholar]
- Stojković V.; Perissinotti L. L.; Willmer D.; Benkovic S. J.; Kohen A. Effects of the Donor–Acceptor Distance and Dynamics on Hydride Tunneling in the Dihydrofolate Reductase Catalyzed Reaction. J. Am. Chem. Soc. 2012, 134, 1738–1745. 10.1021/ja209425w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. A 21st Century Revisionist’s View at a Turning Point in Enzymology. Nat. Chem. Biol. 2009, 5, 543–550. 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- Ma B.; Kumar S.; Tsai C. J.; Hu Z.; Nussinov R. Transition-State Ensemble in Enzyme Catalysis: Possibility, Reality, or Necessity?. J. Theor. Biol. 2000, 203, 383–397. 10.1006/jtbi.2000.1097. [DOI] [PubMed] [Google Scholar]
- Bhabha G.; Ekiert D. C.; Jennewein M.; Zmasek C. M.; Tuttle L. M.; Kroon G.; Dyson H. J.; Godzik A.; Wilson I. A.; Wright P. E. Divergent Evolution of Protein Conformational Dynamics in Dihydrofolate Reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. 10.1038/nsmb.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleman J. R.; Beard W. A.; Delcamp T. J.; Prendergast N. J.; Freisheim J. H.; Blakley R. L. Atypical Transient State Kinetics of Recombinant Human Dihydrofolate Reductase Produced by Hysteretic Behavior. Comparison with Dihydrofolate Reductases from Other Sources. J. Biol. Chem. 1989, 264, 2625–2633. 10.1016/S0021-9258(19)81659-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.