

Abstract

Antibiotic resistance cassettes are indispensable tools in recombinant DNA technology, synthetic biology, and metabolic engineering. The genetic cassette encoding the TEM-1 β-lactamase (denoted Tn3.1) is one of the most commonly used and can be found in more than 120 commercially available bacterial expression plasmids (e.g., the pET, pUC, pGEM, pQE, pGEX, pBAD, and pSEVA series). A widely acknowledged problem with the cassette is that it produces excessively high titers of β-lactamase that rapidly degrade β-lactam antibiotics in the culture media, leading to loss of selective pressure, and eventually a large percentage of cells that do not have a plasmid. To address these shortcomings, we have engineered a next-generation version that expresses minimal levels of β-lactamase (denoted Tn3.1MIN). We have also engineered a version that is compatible with the Standard European Vector Architecture (SEVA) (denoted Ap (pSEVA#1MIN--)). Expression plasmids containing either Tn3.1MIN or Ap (pSEVA#1MIN--) can be selected using a 5-fold lower concentration of β-lactam antibiotics and benefit from the increased half-life of the β-lactam antibiotics in the culture medium (3- to 10-fold). Moreover, more cells in the culture retain the plasmid. In summary, we present two antibiotic-efficient genetic cassettes encoding the TEM-1 β-lactamase that reduce antibiotic consumption (an integral part of antibiotic stewardship), reduce production costs, and improve plasmid performance in bacterial cell factories.
Keywords: expression plasmid, genetic cassette, β-lactamase, directed evolution, translation initiation region, antibiotic stewardship
Introduction
β-Lactamases are a large family of proteins that inactivate β-lactam antibiotics, such as ampicillin and carbenicillin, by enzymatically cleaving the amide bond of the β-lactam ring.1,2 The TEM-1 β-lactamase was the first of the family to be discovered in the 1960s.3,4 It was encoded on the R1 plasmid in a wild-type isolate of Salmonella paratyphi B and cloned in the process of constructing the pBR322 plasmid (reviewed in ref (5)). Sequencing indicated that a 1216-nucleotide-long fragment from the Tn3 transposon of the R1 plasmid had been captured (herein referred to as Tn3.1). The Tn3.1 fragment contained the 861-nucleotide-long coding sequence for the TEM-1 β-lactamase (bla), as well as 208 nucleotides upstream and 147 nucleotides downstream.6 The upstream sequence contained the constitutive P3 promoter and a Shine–Dalgarno (SD) sequence that was positioned five nucleotides upstream of the AUG start codon (Figure 1A).
Figure 1.
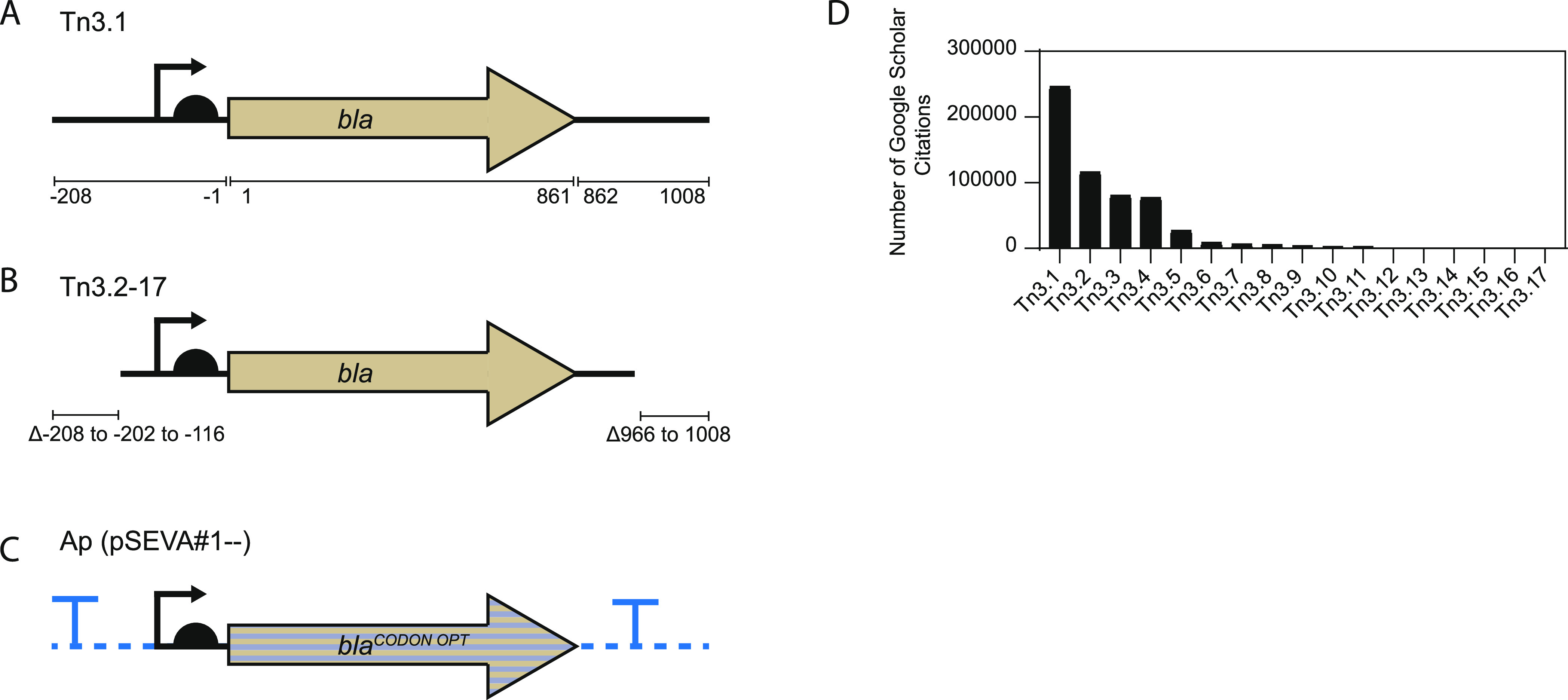
Commonly used genetic cassettes encoding the TEM-1 β-lactamase. (A) Tn3 fragment from the R1 plasmid of S. paratyphi B (herein called Tn3.1) is 1216 nucleotides long. It contains the bla coding sequence as well as 5′ and 3′UTRs. The 5′UTR contains a P3 promoter and Shine–Dalgarno sequence. Numbers correspond to the AUG start codon of bla. The full sequence of Tn3.1 is shown in Table 1. (B) Versions of the Tn3.1 fragment that are truncated in the 5′ and 3′ UTR are commonly used (herein called Tn3.2–Tn3.17). These versions may also contain nucleotide substitutions (summarized in Table 1). (C) The pSEVA collection contains a codon-varied genetic module encoding the TEM-1 β-lactamase and is flanked by transcriptional terminators and restriction enzyme recognition sites (D). Estimated frequency in the literature of the Tn3-based fragments. Expression plasmids containing the various fragments were searched on Google Scholar, and the number of citations was recorded.
The Tn3.1-based fragments are widely used as a selection marker in bacterial expression plasmids. For example, 49 commercially available expression plasmids contain the original Tn3.1 fragment (Table 1). Another 84 use versions that are slightly truncated and that contain nucleotide substitutions. One of these nucleotide substitutions deleted a PstI restriction enzyme recognition sequence, but the remainder have not, to our knowledge, been described in the literature. We refer to these altered fragments as Tn3.2–Tn3.17 (Figure 1B and Table 1). A variant of the Tn3.1 fragment has also recently been developed for the Standard European Vector Architecture (pSEVA).7,8 This version, which is referred to as Ap (pSEVA#1--), contains a codon-optimized version of bla that is insulated with transcriptional terminators (from the trpA gene of Escherichia coli and the gene VIII from phage fd) and is flanked by restriction enzyme recognition sites for multifragment assembly (SwaI and PshA1) in the SEVA system (Figure 1C). Google Scholar citations indicate that expression plasmids containing the Tn3.1–Tn3.17 fragments have been used in >581 000 studies. The most commonly used being Tn3.1, which has been used in >246 000 published studies (Figure 1D).
Table 1. Tn3 Fragments Used in Commercial Expression Plasmidsa.
| fragment | lengthQ | sequenceR | citationsS | expression plasmids using the fragment |
|---|---|---|---|---|
| Tn3.1 | 1–1216 | A | 246 266 | pBR332, pET1*, pET2*, pET3*, pET4*, pET5*, pET6*, pET7*, pET8*, pET11a-d, pET14b, pET15b, pET16b, pET17b, pET19b, pET-DEST42, pET100/D-TOPO, pET100/D-LacZ, pET101/D-LacZ, pET101/D-TOPO, pET102/D-LacZ, pET102/D-TOPO, pET104-DEST, pET104/GW/LacZ, pET104.1-DEST, pET104.1/D/GW-LacZ, pET151/D-TOPO, pET151/D/LacZ, pET160-DEST, pET160/GW/D-TOPO, pET161/GW-CAT, pET300/NT-GW/Ras Kinase, pET300/NT-DEST, pET301/CT-DEST, pET302/NT-his, pGEX-1 lambda T, pGEX-2T, pGEX-2TK, pGEX-3X, pGEX-4T-1, pGEX-4T-2, pGEX-4T-3, pGEX-5X-1, pGEX-6p-1, pGEX-6p-2, pGEX-6p-3 |
| Tn3.2 | 7–1216 | B, C | 116 221 | pGEM-1 pGEM-2, pGEM-4, pGEM-Luc, pUC12, pUC13, pUC18, pUC19, pUC21, pUC57, pUC118, pUC119, pUCX |
| Tn3.3 | 77–1216 | B | 80 658 | pET20b(+), pET21a-d(+), pET22(+), pET23a-d(+), pET25b(+), pET31b(+), pET32a-c(+), pET32 Ek/LIC, pET32 Xa/LIC, |
| Tn3.4 | 76–1216 | B, C | 77 342 | pGEM-5, pGEM-5Zf(+), pGEM-T, pGEM-T easy vector, pGEMT-3P2A, pGEMT-PTE2A |
| Tn3.5 | 6–1216 | B, C | 27 508 | pQE9, pQE16, pQE30, pQE31, pQE32, pQE40, pQE60, pQE70, pQE80-L, pQE81-L. pQE82-L |
| Tn3.6 | 7–1216 | B, C, D | 8930 | pGEM-3Z, pGEM-4Z |
| Tn3.7 | 111–1171 | B, C, O, P | 6688 | pETduet-1, pET43 Ek/LIC, pET43.1a(+), pET44a-c(+), pET45b(+), pET46 Ek/LIC, pET51b(+), pET51 Ek/LIC, pET52(+), pET52 Ek_LIC |
| Tn3.8 | 7–1216 | C to M | 5750 | pGEM-3Zf(+), pGEM-3Zf(−), pGEM-11zf(+), pGEM-11Zf(−), pGEMEX-1, pGEMEX-2 |
| Tn3.9 | 77–1216 | C to M | 3920 | pGEM-7Zf(+), pGEM-7Zf(−) |
| Tn3.10 | 116–1174 | C | 2930 | pBAD24 |
| Tn3.11 | 116–1174 | 2758 | pBAD18, pBAD30, pBAD-bHS, pBAD-EGFP | |
| Tn3.12 | 116–1216 | C | 1601 | pBAD7HisB-iRFP670, pBAD/HisD-TagRFP675, pBAD/Myc-HisA, pBAD/Myc-HisB, pBAD/Myc-HisC, pBAD/gii A, pBAD/gii-B, pBAD/gii-C, pBAD/gii/Calmod, pBAD/HisB |
| Tn3.13 | 77–1216 | B, C | 270 | pGEM-5Zf(−), pGEM-9Zf(−) |
| Tn3.14 | 87–1216 | B | 167 | pET303-CT-His-Rac Kinase, pET303-CT-His |
| Tn3.15 | 13–1216 | B, C, N | 104 | pUCsg-RNA, pUCC001 |
| Tn3.16 | 113–1216 | B | 64 | pBAD-DEST49, pBAD/Myc-His/LacZ, pBAD/D-TOPO |
| Tn3.17 | 113–1161 | O | 23 | pBAD18s |
A: 5′TTCTTGAAGACGAAAGGGCCTCGTGATACGCCTATTTTTATAGGTTAATGTCATGATAATAATGGTTTCTTAGACGTCAGGTGGCACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAATAACCCTGATAAATGCTTCAATAATATTGAAAAAGGAAGAGTATGAGTATTCAACATTTCCGTGTCGCCCTTATTCCCTTTTTTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAACGCTGGTGAAAGTAAAAGATGCTGAAGATCAGTTGGGTGCACGAGTGGGTTACATCGAACTGGATCTCAACAGCGGTAAGATCCTTGAGAGTTTTCGCCCCGAAGAACGTTTTCCAATGATGAGCACTTTTAAAGTTCTGCTATGTGGCGCGGTATTATCCCGTGTTGACGCCGGGCAAGAGCAACTCGGTCGCCGCATACACTATTCTCAGAATGACTTGGTTGAGTACTCACCAGTCACAGAAAAGCATCTTACGGATGGCATGACAGTAAGAGAATTATGCAGTGCTGCCATAACCATGAGTGATAACACTGCGGCCAACTTACTTCTGACAACGATCGGAGGACCGAAGGAGCTAACCGCTTTTTTGCACAACATGGGGGATCATGTAACTCGCCTTGATCGTTGGGAACCGGAGCTGAATGAAGCCATACCAAACGACGAGCGTGACACCACGATGCCTGCAGCAATGGCAACAACGTTGCGCAAACTATTAACTGGCGAACTACTTACTCTAGCTTCCCGGCAACAATTAATAGACTGGATGGAGGCGGATAAAGTTGCAGGACCACTTCTGCGCTCGGCCCTTCCGGCTGGCTGGTTTATTGCTGATAAATCTGGAGCCGGTGAGCGTGGGTCTCGCGGTATCATTGCAGCACTGGGGCCAGATGGTAAGCCCTCCCGTATCGTAGTTATCTACACGACGGGGAGTCAGGCAACTATGGATGAACGAAATAGACAGATCGCTGAGATAGGTGCCTCACTGATTAAGCATTGGTAACTGTCAGACCAAGTTTACTCATATATACTTTAGATTGATTTAAAACTTCATTTTTAATTTAAAAGGATCTAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTTCCACTGAGCGTCAGACCCC-3′.
B: G244 to A mutation in bla (V82 to I).
C: C545 to T mutation in bla (A182 to V); deletion of the PstI site.
D: G553 to C mutation in bla (A185 to P).
E: G226 to C, G227 to A, C228 to T mutations in bla (G76 to H).
F: G229 to A, G231 to A mutations in bla (A77 to T).
G: G232 to C mutation in bla (V78 to L).
H: G244 to A mutation in bla (G82 to H).
I: C275 to G mutation in bla.
J: A276 to G mutation in bla.
K: T277 to C mutation in bla (I93 to A).
L: A278 to G mutation in bla (I93 to A).
M: C281 to G mutation in bla (H94 to R).
N: G717 to T mutation in bla.
O: Nucleotide mutation in 5′UTR (−20 A to C).
P: Nucleotide mutation in 5′UTR (−93 A to C).
Q: Length relative to Tn3.1.
R: Nucleotide sequence of the Tn3.1 fragment is indicated by a B–P. Nucleotide changes from the Tn3.1 fragment are indicated by another letter. Numbering as depicted in Figure 1A, where the A of the AUG start codon for bla is denoted as +1.
S: Obtained from Google Scholar.
A widely acknowledged problem with Tn3.1-based fragments is that they produce excessively high levels of β-lactamase.10 As a result, cells are resistant to concentrations of β-lactam antibiotics that exceed the concentration required for selection. For example, most laboratory strains of E. coli are susceptible to <3 μg/mL of ampicillin. But when they harbor a medium-copy-number plasmid containing the Tn3.1 fragment, they are resistant to >5000 μg/mL.14 High-level production of β-lactamase contributes to the complete degradation of β-lactam antibiotics in culture media within 3 h, leading to loss of selective pressure.9−13 Cells that have lost the plasmid, as well as contaminating strains, can dominate the culture in the absence of a selection pressure.9,11,13,15,16 These problems could all be mitigated by reducing the production levels of β-lactamase from the Tn3-based cassettes.
High production levels of the TEM-1 β-lactamase from Tn3.1-based fragments are largely dependent on the efficiency of bla transcription and translation. Transcriptional initiation is mediated by the RNA polymerase of the host cell at the constitutive P3 promoter.6 Translational initiation is mediated by the 30S subunit of the ribosome at the translation initiation region (TIR),17 a stretch of approximately 30 nucleotides composed of the Shine–Dalgarno sequence, a linker region of five nucleotides, and the first five or six codons of the coding sequence.18 Production levels of the TEM-1 β-lactamase from Tn3.1-based fragments are also influenced by the copy number of the expression plasmid containing it.19,20 Herein, we have attempted to circumvent the problems associated with high-level production of β-lactamase by engineering a constitutively low-expressing version of the Tn3.1 fragment. We describe and characterize Tn3.1MIN, which differs from Tn3.1 by only four nucleotides in the TIR and which addresses all of the previously described problems with the Tn3.1-based fragments.
Results
The Tn3.1 Fragment Confers Resistance to High Concentrations of β-Lactam Antibiotics
Initially, the pET15b expression plasmid was used, which contains the Tn3.1 fragment and the coding sequence for a polyhistidine-tagged super-folder green fluorescent protein (pET15b-sfgfp) (Figure 2A). pET15b-sfgfp was transformed into the BL21(DE3) strain, grown in liquid culture to mid-exponential phase and plated on lysogeny broth (LB) agar containing different concentrations of ampicillin or carbenicillin. Colony counting indicated that the minimum inhibitory concentration required to kill 90% of cells (MIC90) was >700 μg/mL of ampicillin (Figure 2B, left panel) and >5000 μg/mL of carbenicillin (Figure 2B, right panel). These MIC90s are considerably higher than that needed for selection, as the MIC90 for the BL21(DE3) strain is <1 μg/mL for both ampicillin and carbenicillin (Figure 2C). A similar observation was made when using the BL21(DE3) pLysS strain (Figure S1, Supporting Information (SI)). These data support previous observations14 indicating that the Tn3.1 fragment confers resistance to excessively high concentrations of β-lactam antibiotics.
Figure 2.
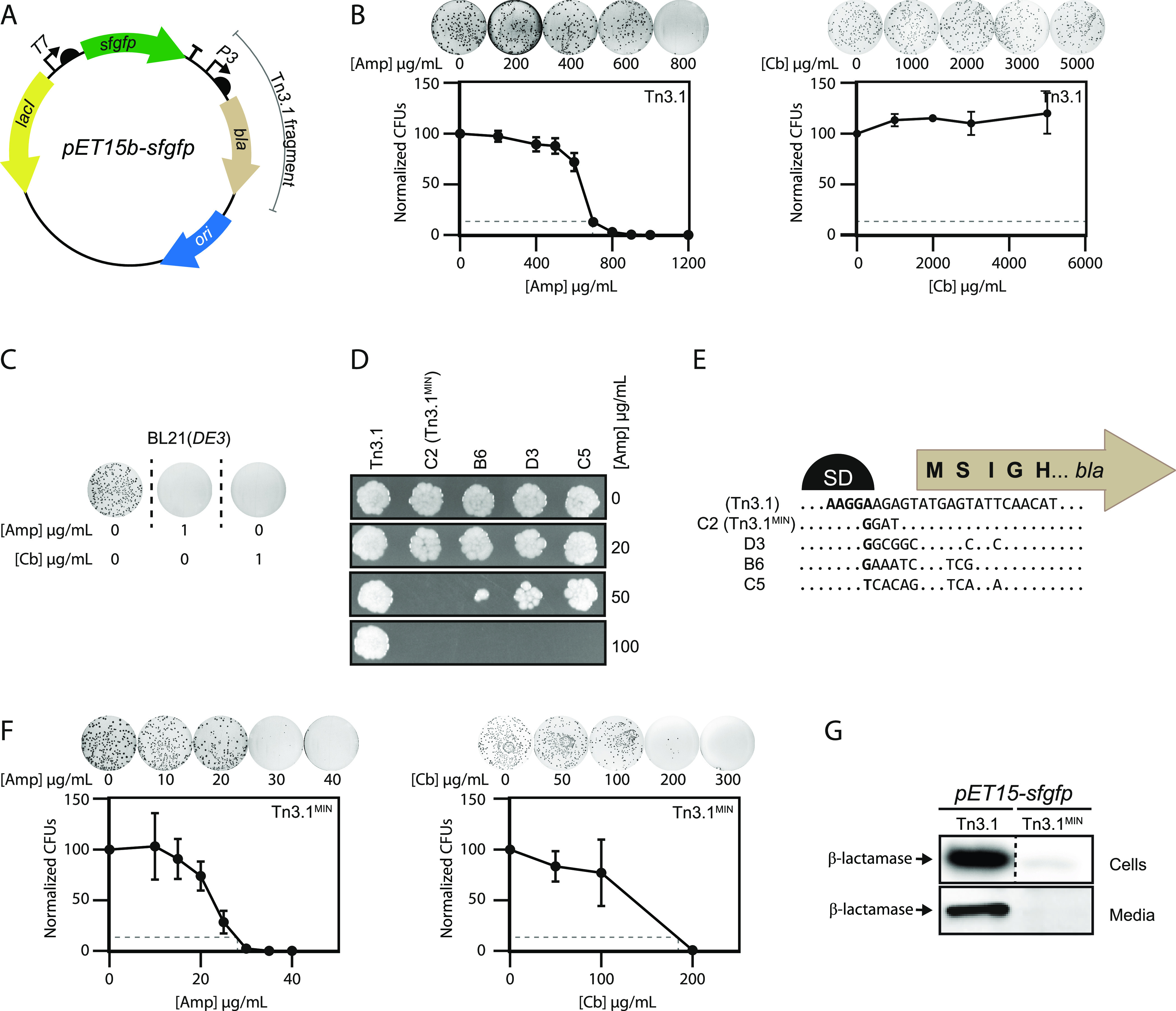
Tn3.1MIN reduces levels of the TEM-1 β-lactamase. (A) Illustration of the pET15b-sfgfp expression plasmid, which contains the Tn3.1 fragment. (B) BL21(DE3) harboring the pET15b-sfgfp (Tn3.1) expression plasmid was plated on LB agar containing different concentrations of ampicillin or carbenicillin. Colony numbers were normalized by the number of colonies that grew in the absence of antibiotics. The minimum inhibitory concentration (MIC90) required to kill 90% of cells was extrapolated from the curve (dotted line) and deemed to be approximately 700 μg/mL for ampicillin and >5000 μg/mL for carbenicillin. (C) BL21(DE3) cells (without an expression plasmid) were plated on LB agar containing no antibiotic or 1 μg/mL ampicillin or carbenicillin. As growth was not observed on 1 μg/mL ampicillin or carbenicillin, the MIC90 was deemed to be <1 μg/mL. (D) BL21(DE3) harboring the pET15b-sfgfp expression plasmids plated on LB agar containing different concentrations of ampicillin. The pET15b-sfgfp expression plasmids, denoted C2, B6, D3, and C5, were selected from a directed evolution process and contained Tn3.1 fragments with a different translation initiation region (TIR) for bla. C2 was chosen for further characterization and was named Tn3.1MIN. (E) Nucleotide sequence alignment of the TIR for bla in Tn3.1, Tn3.1MIN (C2), B6, D3, and C5. The TIR is defined as the nucleotide sequence from the Shine–Dalgarno (SD) region through to the fifth codon.18 (F) As in panel (B) except that BL21(DE3) harbored the pET15b-sfgfp (Tn3.1MIN) plasmid. The MIC90 was deemed to be <30 μg/mL for ampicillin and <200 μg/mL for carbenicillin. (G) Levels of TEM-1 β-lactamase in BL21(DE3) harboring the pET15b-sfgfp expression plasmid (Tn3.1 or Tn3.1MIN), or the culture media, were probed by Western blotting with antisera to the TEM-1 β-lactamase.
Reduced Production of the TEM-1 β-Lactamase from the Tn3.1 Fragment
We reasoned that the excessively high levels of resistance to β-lactam antibiotics are caused by high production titers of the TEM-1 β-lactamase from the Tn3.1 fragment. We utilized a directed evolution approach to identify a translation initiation region (TIR) that supported lower production yields, while retaining the constitutive P3 promoter. This approach was used as it gives a wide range of expression levels from a relatively small sequence library.21
Four new TIRs for the TEM-1 β-lactamase were selected from the library by plating on different concentrations of ampicillin. All reduced the level of resistance to below 100 μg/mL ampicillin (Figure 2D). The one conferring the lowest level of resistance (denoted C2) was chosen for further characterization. This TIR had four nucleotide changes upstream of the AUG start codon, which most likely changed the Shine–Dalgarno sequence (Figure 2E). Colony counting indicated that the MIC90s of BL21(DE3) harboring pET15b-sfgfp were reduced to <30 μg/mL of ampicillin (Figure 2F, left panel) and <200 μg/mL of carbenicillin (Figure 2F, right panel). Similar observations were made when using the BL21(DE3) pLysS strain (Figure S1, SI). Western blotting indicated that β-lactamase levels in both the cells and the media were reduced considerably (Figure 2G). Taken together, these data indicate that β-lactamase production levels from the Tn3.1 fragment can be reduced by the selection of a new TIR. We refer to the new version of the genetic cassette as Tn3.1MIN (MINimal production). Based on the MIC90s that we observed, we suggest that Tn3.1MIN, when integrated into medium-copy-number plasmids such as pET15b, are selected for at a concentration of 20 μg/mL of ampicillin and carbenicillin. These concentrations are 5-fold lower than that normally used for plasmid maintenance and >20-fold higher than that needed for selection against the BL21(DE3) strain lacking the plasmid.
Tn3.1MIN Increases the Half-Life of Antibiotics
A widely acknowledged problem with the Tn3.1-based cassettes is that the exceedingly high titers of β-lactamase cause rapid degradation of β-lactam antibiotics in culture media.9−11,13 This in turn leads to loss of selective pressure. As Tn3.1MIN reduced the production titers of β-lactamase, we were curious to know if the half-life of antibiotics increased. A single colony of BL21(DE3) harboring pET15b-sfgfp was inoculated into LB media containing ampicillin (20 μg/mL for Tn3.1MIN and 100 μg/mL for Tn3.1) and a semiquantitative mass spectrometry (MS) assay was used to monitor the concentration of ampicillin in the culture media during cultivation (Figure 3A). Rapid degradation of ampicillin was observed when the Tn3.1 cassette was integrated in pET15b-sfgfp. The t1/2 was calculated to be approximately 30 min, and the culture media was deemed to be above 1 μg/mL (the concentration required for selection) for 130 min (Figure 3B). When Tn3.1MIN was integrated in pET15b-sfgfp, the t1/2 was calculated to be approximately 3 h, and the culture media was deemed to be above 1 μg/mL for approximately 250 min (Figure 3B).
Figure 3.
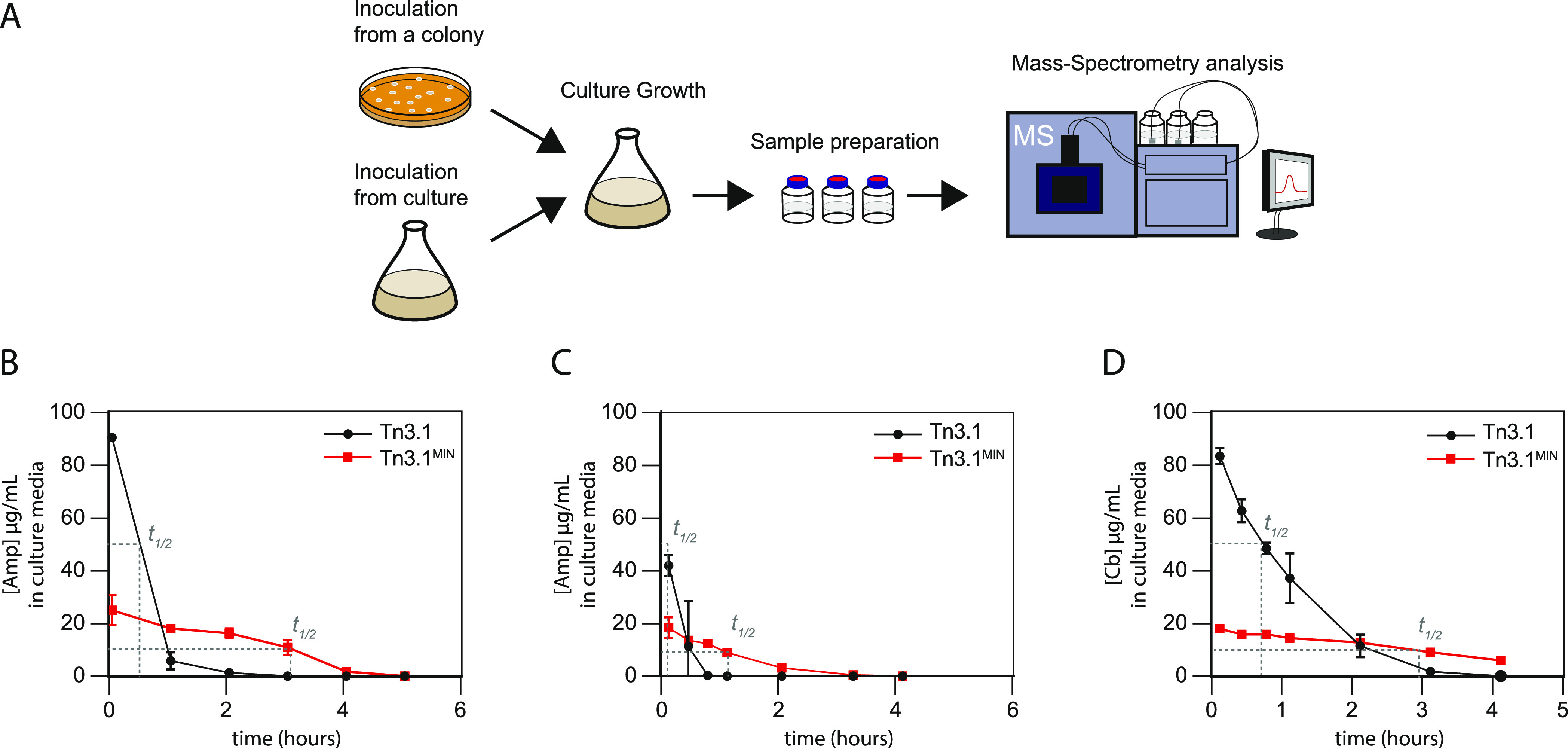
Tn3.1MIN increases the half-life of ampicillin and carbenicillin in the culture media. (A) Schematic of the experimental workflow used to assess the concentration of ampicillin and carbenicillin in the culture media. Either a single colony or an overnight culture was used to inoculate fresh LB media containing either 100 μg/mL (Tn3.1) or 20 μg/mL (Tn3.1MIN) ampicillin or carbenicillin. Aliquots were analyzed using a semiquantitative mass spectrometry approach. (B) Concentration of ampicillin in the culture media when a single colony of BL21(DE3) harboring pET15b-sfgfp was inoculated. Concentrations are plotted against culture time. (C) As for panel (B) except that an overnight culture was back-diluted 1:100. (D) As for panel (C) except that carbenicillin was used.
When overnight cultures of BL21(DE3) harboring pET15b-sfgfp were back-diluted 1:100 into LB media containing ampicillin or carbenicillin, as is typical for an experiment, we observed even shorter t1/2’s. When the Tn3.1 cassette was integrated in pET15b-sfgfp, the t1/2 of ampicillin was calculated to be 6 min, and the culture media was deemed to be above 1 μg/mL for approximately 70 min (Figure 3C). When Tn3.1MIN was integrated in pET15b-sfgfp, the t1/2 of ampicillin was calculated to be 63 min, and the culture media was deemed to be above 1 μg/mL for approximately 180 min (Figure 3C). When carbenicillin was used at the same concentrations, the t1/2 increased from 45 min (Tn3.1) to 175 min (Tn3.1MIN) (Figure 3D). These experiments indicate that the t1/2 of β-lactam antibiotics in the culture media was increased by 3–10-fold when using Tn3.1MIN, even though a 5-fold lower starting concentration was used.
Tn3.1MIN Helps Select for Cells That Harbor the Plasmid
In the absence of antibiotic selection, cells that have lost their plasmids during division can eventually dominate the culture.22,23 To investigate the rate of plasmid loss without antibiotic selection, cultures were plated on LB agar with or without ampicillin and CFU’s were compared (Figure 4A). When BL21(DE3) harboring the pET15b-sfgfp (Tn3.1) plasmid was cultured in the presence of 100 μg/mL ampicillin, we observed that almost all of the cells in the culture maintained the plasmid over a 22-h period (Figure 4B). The same observation was made when ampicillin was omitted from the culture (Figure 4B). When the same experiment was repeated, this time with the addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) to induce production of sfGFP (after 2 h of culturing), we observed that cells without the plasmid started to accumulate in the culture. And after 6 h of induction (8 h of culturing) approximately 50% of the cells in the culture did not have the plasmid. After 22 h of culturing, almost none of the cells in the culture had maintained the plasmid (Figure 4C). The same observation was made when ampicillin was omitted from the culture (Figure 4C). These data indicate that recombinant protein production results in an accumulation in plasmid-free cells within the time frame of a standard protein expression experiment.
Figure 4.
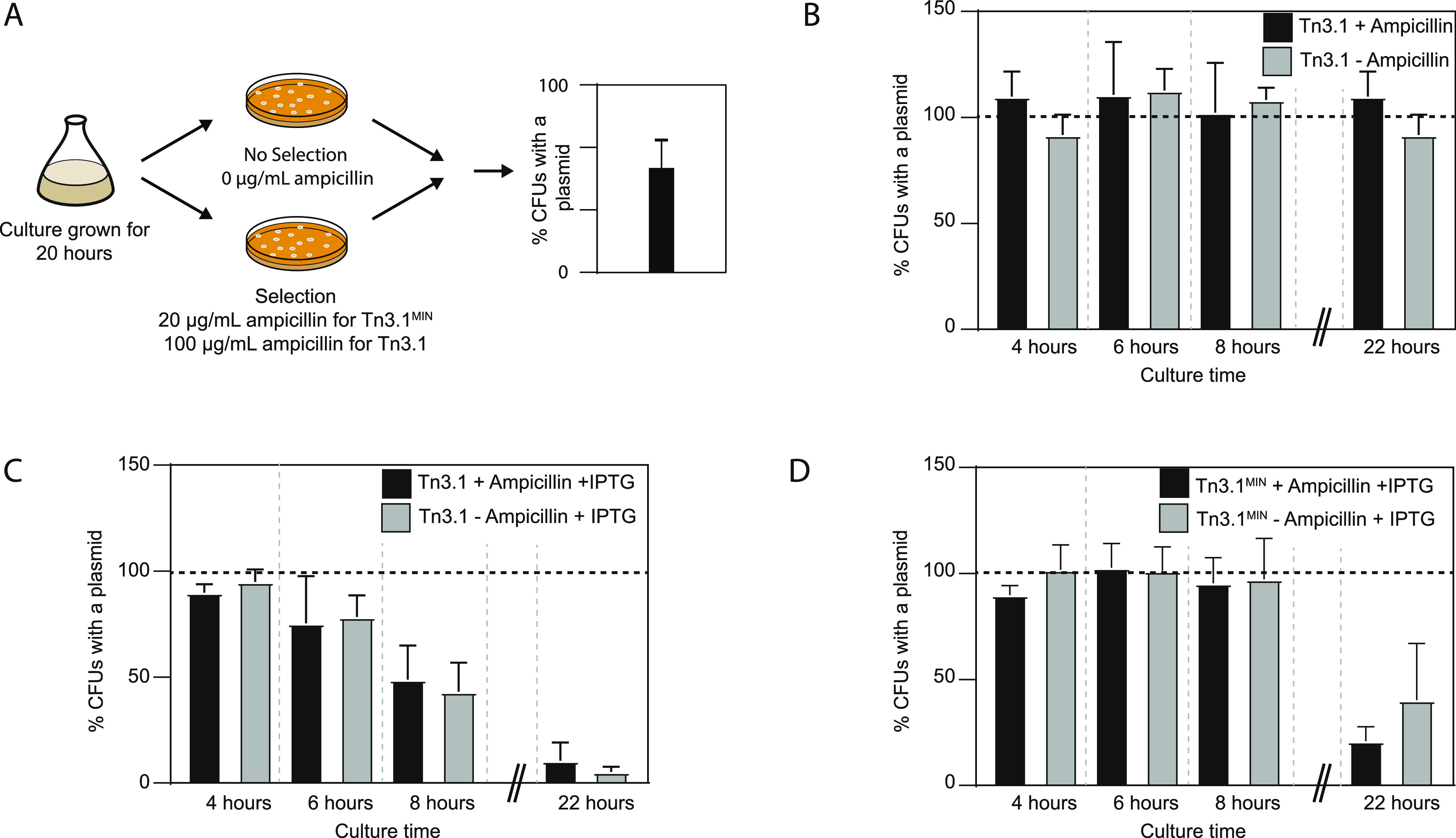
Tn3.1MIN helps cells to maintain the plasmid. (A) Schematic representation of the experimental workflow for determining the percentage of cells with a plasmid. This was determined by calculating the relative ratio of colonies on LB agar plates, with or without ampicillin selection. (B) Percentage of BL21(DE3) harboring pET15b-sfgfp (Tn3.1). In cultures with and without ampicillin, most cells maintained a plasmid after 20 h of cultivation. (C) After induction of sfGFP with IPTG, the majority of BL21(DE3) did not have the pET15b-sfgfp (or Tn3.1) plasmid. (D) As for panel (C) except that the pET15b-sfgfp (or Tn3.1MIN) plasmid was used. Here a larger proportion of cells in the culture harbored the plasmid. Data presented as mean ± standard deviation (s.d.) (n ≥ 3).
To determine if Tn3.1MIN could mitigate this problem, the same experiment was replicated with BL21(DE3) harboring the pET15b-sfgfp (Tn3.1MIN). It was observed that almost all of the cells in the culture maintained the plasmid after 6 h of induction (8 h of culturing). After 22 h of culturing, the majority of the cells did not contain a plasmid (Figure 4D). The same observations were made when ampicillin was omitted from the culture. These data indicate that the increased t1/2 of ampicillin was not the factor contributing to better plasmid maintenance in the culture. Nevertheless, Tn3.1MIN lowers the rate at which cells without a plasmid dominate the culture.
To determine whether the type of recombinant protein being produced had an impact on plasmid loss, a human protein that was soluble in the cytoplasm of E. coli (Mth1) and another that is known to form inclusion bodies (Neil3)24 were expressed. When the soluble Mth1 was produced, more cells with a plasmid were observed with Tn3.1MIN after 20 h of induction (8% for Tn3.1 vs 18% for Tn3.1MIN). When the inclusion body-prone Neil3 was achieved, cells without a plasmid completely dominated both cultures and differences between Tn3.1 and Tn3.1MIN could not be resolved (Figure S2, SI). Although plasmid loss was mitigated by Tn3.1MIN when sfGFP and Mth1 were produced, we did not observe any differences in production levels for these recombinant proteins (Figure S3, SI).
Tn3.1MIN Prevents Contamination
Back-diluted overnight cultures contain high titers of β-lactamase in the media that can degrade ampicillin directly as shown in Figure 3C. As a result, it is possible that cells without a plasmid, as well as contaminants, have a chance to grow and become dominant in the cell population.9,11,15,16 This possibility was explored by collecting the culture media from overnight cultures of BL21(DE3) harboring pET15b-sfgfp (Tn3.1 and Tn3.1MIN), and then back-diluting it 1:100 (without cells) into fresh LB media containing ampicillin (20 μg/mL for Tn3.1MIN and 100 μg/mL for Tn3.1). An ampicillin-sensitive “contaminant” was then spiked into the fresh LB media and its growth was monitored (Figure 5A). Although the contaminant should not be able to grow in the presence of ampicillin, growth was observed when media from a pET15b-sfgfp (Tn3.1) culture was back-diluted (Figure 5B). Growth of the contaminant was not observed when media from a pET15b-sfgfp (Tn3.1MIN) culture was back-diluted, indicating that concentrations of ampicillin remained above 1 μg/mL (needed for selection) long enough to eliminate the contaminant cells (Figure 5B). To ensure that the growth inhibition was not caused by metabolites in the spent media, ampicillin was omitted and subsequently growth was observed at an equivalent rate to when no supernatant was added (Figure 5C). Growth was not observed in the absence of supernatant when ampicillin was added at either 20 or 100 μg/mL (Figure 5D). These data indicate that the high levels of TEM-1 β-lactamase in the overnight growth media when using Tn3.1 result in degradation of the ampicillin so fast that contaminating cultures are not being selected against. This problem is mitigated by Tn3.1MIN.
Figure 5.
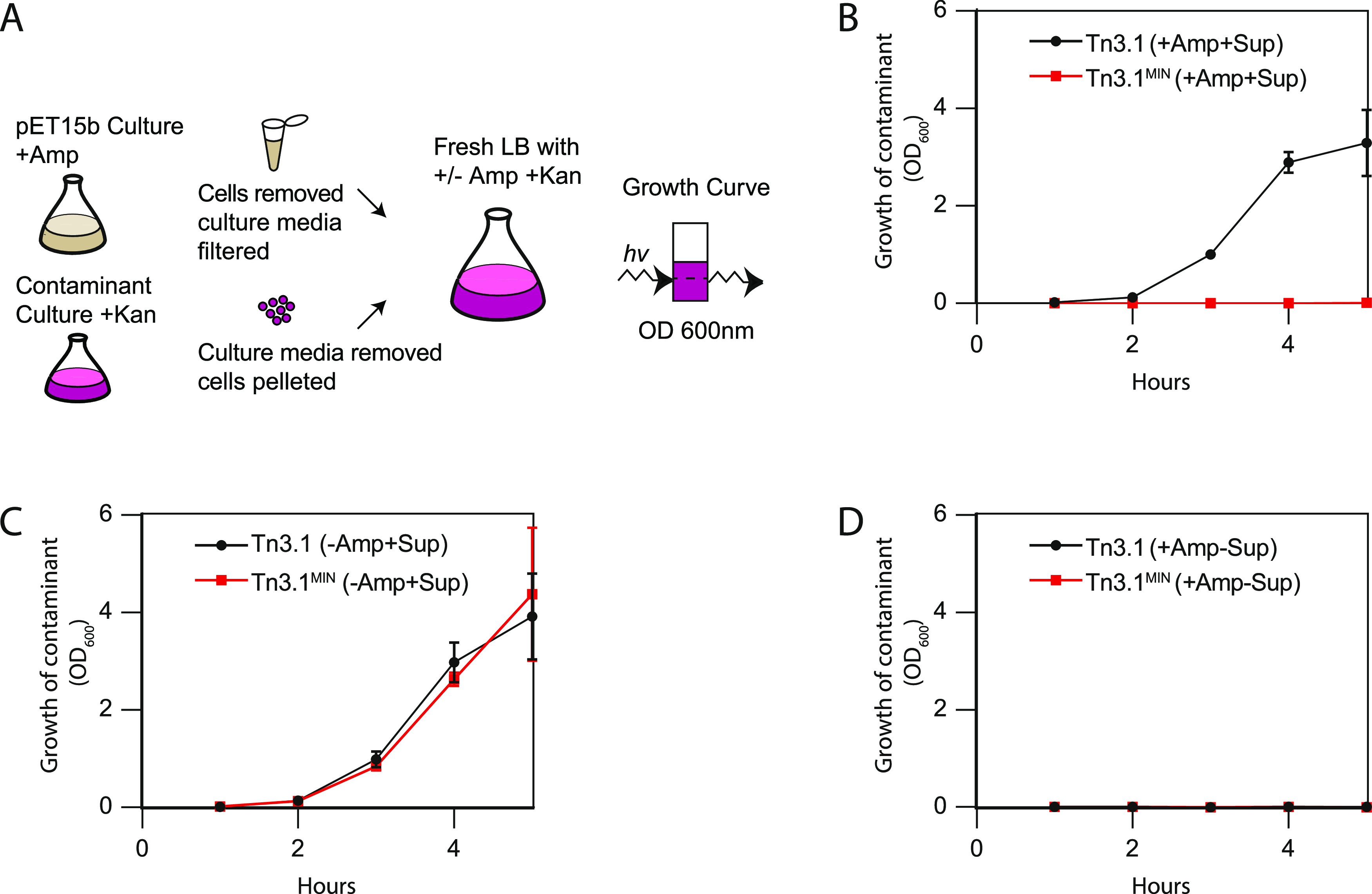
Tn3.1MIN prevents contamination. (A) Schematic representation of the experimental workflow for determining whether contaminants can survive after back-dilution. Supernatants from overnight cultures of BL21(DE3) harboring pET15b-sfgfp were back-diluted in the presence of a contaminant; BL21(DE3) harboring pET28a-mcherry. Growth of the contaminant was monitored by cell density. (B) Growth of the contaminant was monitored in LB media containing ampicillin (100 μg/mL for Tn3.1 or 20 μg/mL for Tn3.1MIN). The contaminant could grow when Tn3.1 was used but not Tn3.1MIN. (C) As for panel (B) except that ampicillin was omitted in the back-dilution. This control indicates that the contaminant can always grow in the absence of ampicillin, when the supernatants are present. (D) As for panel (B) except that the supernatants were omitted in the back-dilution. This control indicates that the contaminants cannot grow in the presence of ampicillin.
Next-Generation β-Lactamase Cassette for the pSEVA Series
The Standard European Vector Architecture (SEVA) contains a genetic cassette for β-lactamase, the Ap (pSEVA#1--) module, which differs from the Tn3.1 fragments in most other common plasmids (Figure 1). To determine if the Ap (pSEVA#1--) module also conferred high-level resistance to ampicillin, it was cloned with a pBR322 origin of replication to generate a pSEVA191 plasmid. pSEVA191 was then transformed into the MC1061 strain and spotted onto LB agar plates containing different concentrations of ampicillin (Figure 6A). Growth was observed on 1000 μg/mL ampicillin, which is more than 300 times higher than that needed to select against the MC1061 strain (Figure 6B). For comparison, cells containing the pET15b-sfgfp plasmid, which contained Tn3.1, also grew on a concentration of 1000 μg/mL ampicillin (albeit not as well). Western blotting of whole cells indicated that expression of β-lactamase from the Ap (pSEVA#1--) module was not as high as it was from the Tn3.1 fragment in the pET15b-sfgfp plasmid, but it was still easily detectable (Figure 6C). We therefore presume that the problems associated with high-level production of β-lactamase (as outlined above) are applicable to the Ap (pSEVA#1--) module.
Figure 6.
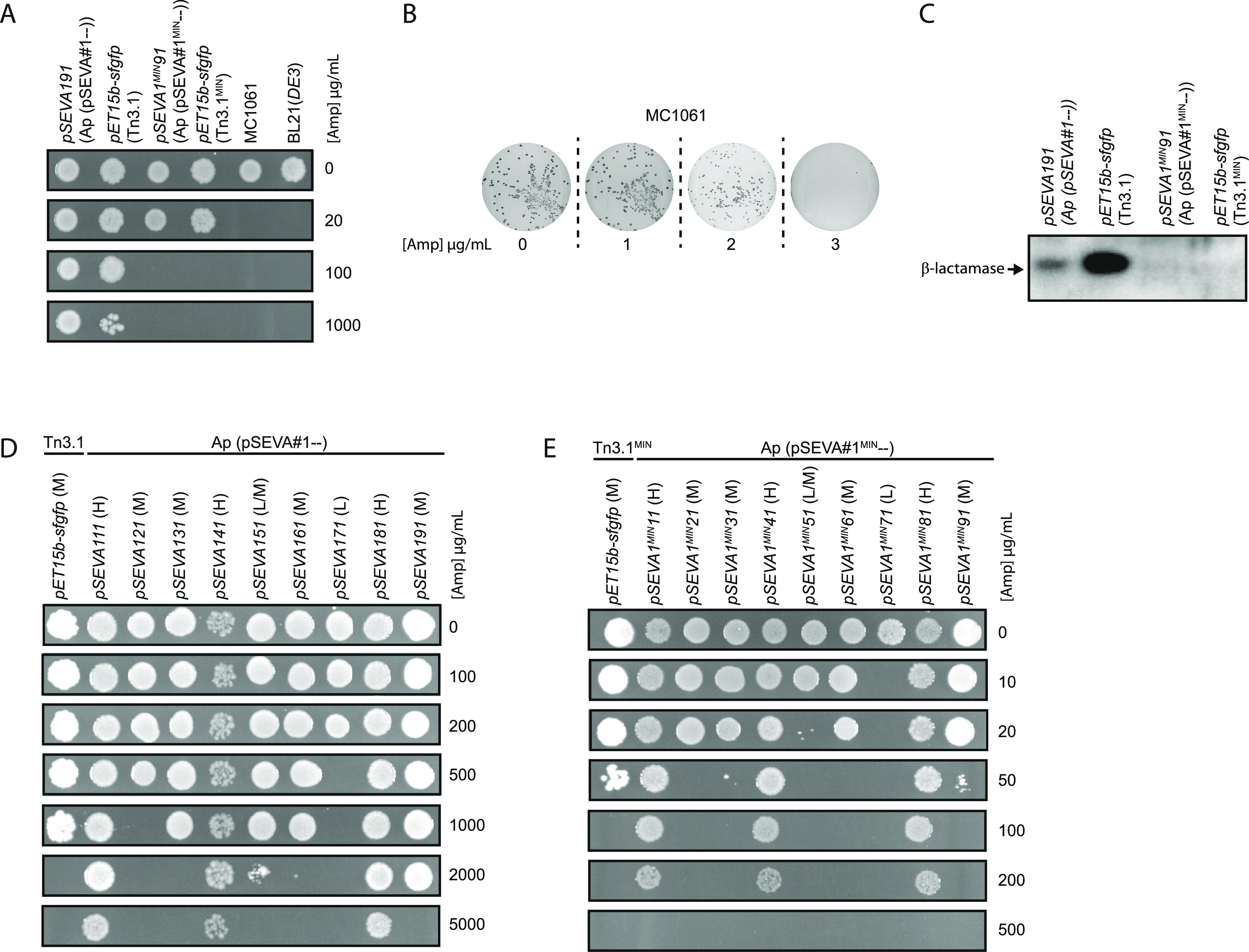
Characterization of Ap (pSEVA#1MIN--). (A) MC1061 cells harboring the pSEVA191 or pSEVA1MIN91 plasmid were spotted on LB agar plates with different concentrations of ampicillin. For comparison, BL21(DE3) harboring the pET15b-sfgfp (Tn3.1 and Tn3.1MIN) expression plasmid was also spotted. When Ap (pSEVA#1--) and Tn3.1 were used, cells could survive on 1000 μg/mL of ampicillin. When Ap (pSEVA#1MIN--) and Tn3.1MIN were used, cells could only survive on 20 μg/mL of ampicillin. (B) MC1061 cells (without an expression plasmid) were plated on LB agar containing 0, 1, 2, and 3 μg/mL ampicillin. As growth was not observed on 3 μg/mL ampicillin, the MIC90 was deemed to be <3 μg/mL. (C) Levels of TEM-1 β-lactamase were probed by Western blotting with antisera to the TEM-1 β-lactamase. β-Lactamase could only be observed when Ap (pSEVA#1--) and Tn3.1 were used. (D) Collection of pSEVA plasmids with Ap (pSEVA#1--) and different origins of replication were transformed into MC1061 cells and spotted on LB agar plates with different concentrations of ampicillin. For comparison, BL21(DE3) harboring the pET15b-sfgfp (Tn3.1) expression plasmid was also spotted. “L” denotes a low copy, “M” a medium copy, and “H” a high copy plasmid. (E) As in panel (D) except that all pSEVA vectors contained Ap (pSEVA#1MIN--) and pET15b-sfgfp contained Tn3.1MIN.
To circumvent these problems the Ap (pSEVA#1--) module was re-engineered so that it resembled the Tn3.1MIN fragment. Here the region from the 5′-end of the P3 promoter through to the stop codon of the codon-optimized bla was removed from the Ap (pSEVA#1--) module and replaced by the corresponding region from the Tn3.1MIN fragment. The transcriptional terminators and flanking restriction sites from Ap (pSEVA#1--) were retained. The new module is referred to as Ap (pSEVA#1MIN--). When cloned with a pBR322 origin of replication to generate the pSEVA1MIN91 plasmid, we observed growth on 20 μg/mL ampicillin but not 100 μg/mL (Figure 6A). This level of resistance is considerably reduced compared to the pSEVA191 plasmid. Western blotting indicated that the Ap (pSEVA#1MIN--) module reduced the expression of β-lactamase to an undetectable level (Figure 6C).
We exploited the pSEVA collection to determine how the Ap (pSEVA#1MIN--) worked with other origins of replication. A collection of pSEVA plasmids was constructed with nine different origins of replication (Table S1, SI). This included low (pSEVA171), medium (pSEVA121, pSEVA131, pSEVA151, pSEVA161, pSEVA191) and high copy (pSEVA111, pSEVA141, pSEVA181) variants. Initially, the Ap (pSEVA#1--) module was integrated and the cells were spotted on different concentrations of ampicillin. As noted previously,19,20 the level of resistance to ampicillin roughly correlated with copy number (Figure 6D), and aside from the low-copy-number pSEVA171, the level of resistance to ampicillin was unnecessarily high. When the Ap (pSEVA#1MIN--) module was integrated in the same plasmids, the level of resistance to ampicillin decreased by >25-fold in all cases (Figure 6E). For the low-copy-number pSEVA1MIN71 and the medium-copy-number pSEVA1MIN51, the level of resistance to ampicillin was <20 μg/mL, which is below the level we recommend. All other plasmids survived on more than 20 μg/mL of ampicillin. Moreover, the level of resistance to ampicillin conferred by the Ap (pSEVA#1MIN--) module roughly correlated with the copy number of the plasmids. Taken together, these data show that the Ap (pSEVA#1MIN--) module reduces β-lactamase expression levels, presumably mitigating the problems caused by excessive production. It can be used with medium and high copy origins of replication.
Discussion
Genetic cassettes encoding the TEM-1 β-lactamase (the Tn3.1-based fragments) have been used as selection markers in bacterial expression plasmids for more than 50 years.5 During this time, these fragments have been propagated, either unchanged (Tn3.1) or with minor changes (Tn3.2–Tn3.17), into more than 120 commercially available expression plasmids (see Table 1). And these expression plasmids have been used in >581 000 published studies. A widely acknowledged flaw with the cassettes is that they produce excessive amounts of β-lactamase, which rapidly degrade β-lactam antibiotics in the culture media, leading to loss of selective pressure.9−13 And in the absence of selection pressure, cells that have lost the plasmid can dominate the culture.22,23 In this study, we describe and characterize a next-generation version of the genetic cassette, which we refer to as Tn3.1MIN (MINimal expression). Tn3.1MIN contains only four nucleotide changes in the TIR, which reduces the amount of β-lactamase in both the cell and in the culture media. As a consequence, the t1/2 of β-lactam antibiotics in the culture media is increased and selection pressure is maintained for a longer period.
It is widely assumed that antibiotic selection pressure is essential for maintaining an expression plasmid. Our study challenges this dogma, as we observed that most cells harbored a plasmid even after 22 h of culture in the absence of ampicillin. However, when recombinant protein production was induced, we observed that most cells in the culture no longer harbored a plasmid after 6 h (8 h of culture). We reason that recombinant protein production was slowing the growth of cells harboring a plasmid and giving those that had lost the plasmid a significant growth advantage. This growth advantage was reduced when Tn3.1MIN was used and most cells harbored a plasmid after 6 h of induction (8 h of culture). Intriguingly similar results were observed in the absence of ampicillin; Thus, we reason that reduced expression of β-lactamase from the Tn3.1MIN fragment contributed to more cells harboring a plasmid by reducing the metabolic load. The increased half-life of ampicillin appeared to have little bearing.
Tn3.1MIN can be easily incorporated into expression plasmids that currently contain a Tn3.1 fragment by incorporating just four nucleotide changes in the translation initiation region. For those expression plasmids that currently contain a Tn3.2–Tn3.17 fragment, we reason that the same four nucleotide changes in the TIR would work similarly, but this has not been tested in this study. A more reliable approach would be to incorporate the entire 1216-nucleotide-long Tn3.1MIN fragment, as its performance has been characterized here. For the Standard European Vector Architecture,7,8 we designed a novel fragment that was based on Tn3.1MIN, which we have called Ap (pSEVA#1MIN--). Both Tn3.1MIN and Ap (pSEVA#1MIN--) confer a similar level of resistance to ampicillin (and carbenicillin).
Why should one include the Tn3.1MIN or Ap (pSEVA#1MIN--) fragment in an expression plasmid? Through the characterization presented in this paper three main advantages were identified: (1) The Tn3.1MIN fragment reduces antibiotic use by 5-fold. When the Tn3.1MIN fragment was integrated into medium-copy-number expression plasmids the working concentration of β-lactam antibiotics was 5-fold lower than the concentration typically recommended (i.e., 20 μg/mL instead of 100 μg/mL). This reduces antibiotic costs by 5-fold, which is particularly important if large culture volumes are used and/or if expression plasmids are used over long periods of time. Reducing antibiotic consumption is also an integral part of antibiotic stewardship and is being advocated by numerous healthcare and governmental bodies. (2) The Tn3.1MIN fragment improves plasmid performance. Primarily it reduces the amount of β-lactamase in the cell and in culture media. As a consequence, the t1/2 of β-lactam antibiotics in the culture media is increased by 3- to 10-fold and selection pressure is maintained for a longer period. Metabolic load is presumably decreased, and the point at which plasmid-less cells overtake the culture is delayed. This is particularly important when recombinant proteins are being produced, as cells without a plasmid can quickly outgrow those with a plasmid. (3) The Tn3.1MIN fragment improved selection against contaminants in the culture. When overnight cultures are back-diluted (1:100) they typically degrade β-lactam antibiotics so quickly that contaminants are able to grow.9,11 When a Tn3.1MIN fragment was used, contaminants were unable to grow as the β-lactam antibiotics were not degraded sufficiently quickly.
It has been demonstrated that excessive production of antibiotic resistance proteins limits the cell’s capacity to produce a recombinant protein.25 Panayotatos and co-workers tested this hypothesis in E. coli by lowering the production of the neomycin phosphotransferase (which confers resistance to kanamycin) using a promoter mutagenesis approach.14 They observed that the production of one recombinant protein was increased by 2-fold, but that there was no improvement for the other. Although the Tn3.1MIN fragment reduced the production of β-lactamase, and increased plasmid retention, we did not observe an increase in growth rate or in the production of the three recombinant proteins that were tested (i.e., sfGFP, Mth1, Neil3). This discrepancy has not been addressed in the current study, but we speculate that it could be explained by suppressing mutations that downregulate the T7 polymerase and which may mask improvements in plasmid retention.26
Bacterial expression plasmids are widely used in both academia and industry.27,28 It is a poorly acknowledged fact that the genetic modules used to construct them were cloned and developed in the 1960s, 1970s, and 1980s, when methods in molecular biology were in their infancy and knowledge about protein biogenesis was less advanced than it is today. Recent work has identified design flaws in some of these genetic modules, which hinder their performance. “Next-generation” versions have been developed, for example, in promoters,24,29 standardized TIRs,24,30 transcriptional terminators,31 and origins of replication.32 These “next-generation” genetic modules outperform the original modules. The Tn3.1MIN fragment developed here is an additional “next-generation” genetic module, which will contribute to making the expression plasmids and bacterial factories of the future more efficient.
Methods
Molecular Cloning
All polymerase chain reactions (PCR) were performed using the Q5 polymerase (New England Biolabs). All primers and DNA sequencing were carried out by Eurofins genomics (Germany). The pET15b-sfgfp expression plasmid was described in ref (24). The pET15b-neil3, pET15b-mth1 expression plasmids were generated by PCR amplification of the coding sequences and ligation by in vitro assembly33 in the MC1061 strain (K-12 F– λ– Δ(ara-leu)7697 [araD139]B/r Δ(codB-lacI)3 galK16 galE15 e14–mcrA0 relA1 rpsL150(StrR) spoT1 mcrB1 hsdR2(r–m+)). pSEVA111, pSEVA121, pSEVA131, pSEVA141, pSEVA151, pSEVA181, pSEVA191, pSEVA261, and pSEVA271 were generous gifts from Victor de Lorenzo and Esteban Martinez. pSEVA161 and pSEVA171 were generated by fragment shuffling from pSEVA261 and pSEVA271 into pSEVA111 using the restriction enzymes BoxI (PshAI) and SmiI (SwaI) (Thermo Fisher) as described in the SEVA system.7 The Ap (pSEVA#1--) fragment was generated by removing the region from the start of the P3 promoter to the stop codon of bla and replacing it with the analogous region from Tn3.1MIN. This process was also carried out by PCR amplification of the coding sequences and ligation by in vitro assembly.33 A list of plasmids is available in Table S1 and primers in Table S2.
Minimum Inhibitory Concentrations (MICs)
A single colony of MC1061, BL21(DE3) (B F–ompT gal dcm lon hsdSB(rB–mB–) λ(DE3 [lacI lacUV5-T7p07 ind1 sam7 nin5]) [malB+]K-12(λS)) or BL21(DE3) pLysS (B F–ompT gal dcm lon hsdSB(rB–mB–) λ(DE3 [lacI lacUV5-T7p07 ind1 sam7 nin5]) [malB+]K-12(λS) pLysS[T7p20 orip15A](CmR)) was inoculated into 5 mL of LB media with relevant antibiotics (Table S1) and incubated overnight with shaking at 37 °C. The cultures were then back-diluted 1:100 in fresh LB with relevant antibiotics in a 5 mL 24-well plate and grown to an OD600 between 0.3 and 0.7. The cultures were then serial diluted 1:10 000 (BL21(DE3) +/–pLysS) or 1:1 000 000 (MC1061) and 100 μL was plated onto LB agar plates with different concentrations of either ampicillin (Avantor) or carbenicillin (Formedium, U.K.). When BL21(DE3) pLysS cells were used, the plates also contained chloramphenicol (Alfa Aesar) at a concentration of 34 μg/mL. Images were taken using the upper white light in a GenoPlex (VWR International), and colonies were counted using OpenCFU.34 The MIC of antibiotic required to kill 90% of cells (MIC90) was determined from the colony numbers.
Spot Assays
A single colony of MC1061 was inoculated into 5 mL of LB media with relevant antibiotics (Table S1) and incubated overnight with shaking at 37 °C. The cultures were then back-diluted 1:100 in fresh LB with relevant antibiotics in a 5 mL 24-well plate and grown to an OD600 between 0.3 and 0.7. The cultures were then serially diluted 1:100 and 1 μL of each culture was spotted onto LB agar plates with different concentrations of ampicillin. Survival was deemed to be the highest concentration of ampicillin on which growth was observed.
Directed Evolution of the Translation Initiation Region (TIR)
The TIR for the gene encoding β-lactamase in pET15b-sfgfp was optimized using a directed evolution approach described previously.35,36 Briefly, forward and reverse degenerate primers were designed that allowed for all sequence possibilities in the six nucleotides preceding the AUG start codon, and restrained sequence possibilities (synonymous codon changes only) in the six nucleotides following the AUG start codon (Table S2, SI). The forward and reverse primers were overlapping by 18 nucleotides so that the subsequent PCR product could be re-ligated into a circular plasmid by in vivo assembly.33 Plasmid libraries with randomized TIRs were generated by PCR using the degenerate primers. The PCR cycle comprised 30 cycles of 95 °C for 30 s, 50 °C for 30 s, and 72 °C for 210 s. The resulting PCR product was treated with DpnI then transformed into MC1061 cells and grown overnight at 37 °C. The plasmid library was purified using an Omega Bio-Tek mini-prep kit.
The purified plasmid library was transformed into BL21(DE3) cells and susceptibility to ampicillin was determined on LB agar plates. Forty-eight random colonies were inoculated into 500 μL of LB media in a 96-well 2.2 mL growth plate containing 20 μg/mL of ampicillin. Cultures were grown until an OD600 of approximately 0.3, and 2 μL of the culture was then spotted on LB agar plates containing 20, 50, 100, 200, 300, and 400 μg/mL of ampicillin. Four colonies that survived only on <100 μg/mL of ampicillin were selected. The plasmids were purified and the TIR for the gene encoding β-lactamase was sequenced. One plasmid was selected for further characterization and was therefore sequenced to 97.4%. Aside from mutations in the TIR, no mutations were identified in the plasmid backbone.
Protein Expression
A single colony of BL21(DE3) harboring pET15b-sfgfp, pET15b-mth1, or pET15b-neil3 (Tn3.1 and Tn3.1MIN) were grown in LB media supplemented with appropriate antibiotics for 16–20 h at 37 °C with shaking. Thereafter, the overnight cultures were back-diluted 1: 100 in 5 mL 24-well plates and grown at 37 °C with shaking to an OD600 of 0.3–0.7. The cultures were then induced with 0.5 mM IPTG. The cultures were then incubated for 20 h at 37 °C with shaking.
Trichloroacetic Acid (TCA) Precipitation
TCA precipitations were carried out using a modified version of the method described in ref (37). In short, culture supernatants were mixed in a 1:4 ratio with 100% cold TCA and incubated on ice for 2 h, with occasional mixing. The precipitate was pelleted by centrifugation at 12 000g for 20 min, then washed in 200 μL of acetone—by resuspension, and then re-centrifugation at 17 000g for 20 min. The acetone was removed and the pellet was resuspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer.
SDS-PAGE and Western Blotting
SDS-PAGE was carried out on Tris-glycine 12% acrylamide and cast with a thickness of 1 mm. All were run using the Hoefer SE260 Mighty Small II Deluxe Mini Vertical Protein Electrophoresis Unit at 100 V for 3 h. For Western blotting, proteins were transferred onto a nitrocellulose membrane using a semidry Trans-Blot SD cell (Bio-Rad) for 1 h at 15 V. The nitrocellulose membranes were then incubated for either 1 h or overnight in 5% (w/v) nonfat milk (PanReac AppliChem) in Tris-buffered saline (TBS) (50 mM Tris, pH 7.4, 200 mM NaCl). His-tagged recombinant proteins were probed using the HisProbe-HRP conjugate (15165, Thermo Scientific) at a dilution of 1:10 000. β-Lactamase was probed using a mouse monoclonal primary antibody ((8A5.A10): sc-66062, Santa Cruz Biotechnology) at a 1:1000 dilution. For detection, a secondary anti-IgG Sheep Polyclonal Antibody HRP conjugate (NXA931, GE Healthcare) was used at a 1:3000 dilution. The SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific) was used as the substrate. Images were captured on an Azure c600 Imaging System (Azure Biosystems).
Semiquantitative Analysis of Ampicillin and Carbenicillin in LB Media by Electrospray Ionization Tandem Mass Spectrometry (MS/MS)
Growth Conditions
A single colony of BL21(DE3) harboring pET15b-sfgfp (TN3.1 or TN3.1MIN) was grown in 5 mL LB media supplemented with ampicillin or carbenicillin (100 μg/mL for TN3.1 and 20 μg/mL for TN3.1MIN) for 16–20 h at 37 °C with agitation at 185 rpm. Thereafter, the overnight cultures were back-diluted 1:100 in 5 mL of LB media, in 24-well plates and grown at 37 °C with agitation at 185 rpm. A 250 μL sample of each culture was taken for analysis at 0, 20, 40, 60, 120, 180, 240, and 300 min (see Sample Work-Up).38 Alternatively, a single colony of BL21(DE3) harboring pET15b-sfgfp (TN3.1 or TN3.1MIN) was grown in 5 mL of LB media supplemented with ampicillin (100 μg/mL for TN3.1 and 20 μg/mL for TN3.1MIN) and samples were taken (without back-dilution) for analysis at 0, 60, 120, 180, 240, 300, and 360 min. Proteins were removed in the same manner as above.
Sample Work-Up
Cells were pelleted from a 250 μL sample of each culture by centrifugation (14 000g, 1 min). A 200 μL aliquot of the supernatant was mixed with 20 μL of an internal standard (IS) dissolved in water (1 μg/mL carbenicillin for ampicillin analysis, or 0.1 μg/mL ampicillin for carbenicillin analysis). A 380 μL aliquot of acetonitrile was added immediately, the sample was shaken, and centrifuged at 17 000g for 10 min at 8 °C. A 30 μL aliquot of the resulting supernatant was diluted into 970 μL of 50% (v/v) acetonitrile. The sample was then filtered through a 0.45 or 0.20 μm poly(vinylidene difluoride) (PVDF) filter into a high-performance liquid chromatography (HPLC) vial. Prior to each analysis a calibration curve of 0.1–100 μg/mL for ampicillin and 1–100 μg/mL for carbenicillin (in fresh LB media) was prepared using the above-mentioned method.
Detection and Quantification of Ampicillin and Carbenicillin by Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS)
Analyses were performed using an ultraperformance liquid chromatography system (Waters) coupled to a Waters Xevo TQD Triple Quadrupole mass spectrometer with an electrospray ionization source. Quantitative analysis of the analytes was established by multiple reaction monitoring (MRM) in the positive mode. The tuning parameters, including collision energy, cone, and capillary voltages, were optimized by infusion of 1 mg/L solution of each analyte and IS in 50% (v/v) acetonitrile at a flow rate of 300 μL/min into the mass spectrometer.
LC-MS/MS analyses were performed by direct injection of 3 μL of sample (without a column) with 50% buffer A [2 mM ammonium acetate pH 4.7] and 50% buffer B [0.2% formic acid in acetonitrile] flowing at 0.3 mL/min. Nitrogen (650 L/h) and argon were used as the nebulizer and collision gases, respectively. Protonated molecular precursor [M + H]+ ions were identified at m/z 350.22 and 379.00 for ampicillin and carbenicillin, respectively. The most abundant ions corresponding to transitions ions 350 > 106 for ampicillin and 379 > 204 for carbenicillin were selected for quantification. An additional 4 for transitions for ampicillin and 3 for carbenicillin were monitored for verification when analyzing ampicillin. The transition of 350 > 114 was the only one used for verification of ampicillin (as the IS) when monitoring carbenicillin (the 3 transitions for carbenicillin remained unchanged). Optimum tuning parameters and the corresponding multiple reaction monitoring transitions are summarized in Table S3, SI. Mass Lynx software (version 4.1, Waters) was used for data processing and quantification was carried out manually.
LC-MS/MS Method Evaluation
Calibration curves were prepared prior to each analysis using fresh LB media spiked with different concentrations of the antibiotic of interest (see the Sample Work-Up section). Due to different sensitivities of each analyte, the linearities were obtained from 5 to 1000 ng/mL for ampicillin and 50–1000 ng/mL for carbenicillin with a coefficient of determination (r2) of 0.999. The limit of quantification (LOQ) for each analyte was considered as the lowest concentration of each calibration curve (S/N > 10).
Intraday quality controls (QC) were prepared as described in the Sample Work-Up section and measured at 5, 250, 400, and 500 ng/mL of ampicillin and 250, 400, and 500 ng/mL of carbenicillin in three replicates. The relative standard deviation (RSD) was calculated as 6–17% for ampicillin and 6–20% for carbenicillin (Table S4, SI).
Analyses of Potential Ion Suppression Effects of Internal Standards
Suppression of ampicillin signal by the IS was assessed by analyzing 1, 5, 10, 50, and 100 μg/mL ampicillin in LB in the presence and absence of 100 μg/mL carbenicillin. No significant suppression of the analyte was observed (Figure S4, SI). To assess the effects of spent media, curves were prepared in both fresh and spent LB media from BL21(DE3) using ampicillin and the IS. Briefly, cultures were grown for 4 h at 37 °C. Cells were removed by centrifugation at 3000g for 10 min, and calibration curves were prepared using 5, 10, 50, and 100 μg/mL of ampicillin. No significant difference between the curves was observed (Figure S4, SI).
Evaluation of Matrix Effect (ME) for the Detection and Quantification of Ampicillin and Carbenicillin in LB Media
To investigate the ME, the ratio of the peak area of the post extracted spiked LB media (Ae) to the peak area of standard solution (Ab) was calculated as the following equation
To measure the ME, three unspiked LB media samples were precipitated, diluted, and filtered as per the method described in the Sample Work-Up section. The filtrate was then evaporated under a stream of N2 gas and then reconstituted in aqueous solution containing 50, 100, and 150 ng/mL of the standards and the IS. ME was calculated by the equation above and was in the range of −15.2 to −31.5% for ampicillin and −33.3 to −41.5% for carbenicillin, indicative of suppression effects (Table S4, SI).
Assessment of Plasmid Maintenance
A single colony of BL21(DE3) harboring pET15b-sfgfp (Tn3.1 and Tn3.1MIN) was grown as described in the Protein Expression section. After induction with 0.5 mM IPTG, a serial dilution was carried out and 100 μL of cells was plated out on LB agar, with and without ampicillin (20 μg/mL for Tn3.1MIN and 100 μg/mL for Tn3.1). Colonies were counted using the OpenCFU program.34
Selection in Back-Diluted Cultures
A single colony of BL21(DE3) harboring pET28a-mcherry (aph) was grown in LB media supplemented with 50 μg/mL kanamycin for 16–20 h at 37 °C with shaking. The following morning, the culture was back-diluted 1:100 into 10 mL of fresh media with 50 μg/mL kanamycin. This 10 mL of culture was spiked with 100 μL of either (1) fresh LB media, or (2) spent cell-free media from overnight cultures of BL21(DE3) harboring either pET15b-sfgfp (Tn3.1) or pET15b-sfgfp (Tn3.1MIN). This volume corresponded to a 1:100 dilution and was obtained by centrifuging 2 mL of overnight culture from pET15b-sfgfp (Tn3.1) and pET15b-sfgfp (Tn3.1MIN) at 13 000g for 1 min, then filtering the supernatant through a 0.2 μm filter (Whatman, England). The cultures were supplemented with either (1) no additional antibiotics, or (2) 20 ug/mL of ampicillin for Tn3.1MIN and 100 ug/mL of ampicillin for Tn3.1. The growth of the BL21(DE3) harboring pET28a-mcherry was determined by measuring the OD600 in a 96-well SpectraMax m2e plate reader (Molecular Devices, U.K.).
Alignment of Tn3 Fragments
Nucleotide sequence alignments using the nucleotide BLAST (nBLAST) service from National Center for Biotechnology Information were conducted on the Tn3.1 fragment from ref (39) and sequence of vectors mentioned in Table 1 from Addgene or Thermo Fisher. The Tn3.1 fragment5 was aligned to whole plasmid sequences to estimate the length of the Tn3 fragments. When investigating the bla gene sequences, only the bla genes were aligned, not the whole fragments. For identification of differences in the P3 promoter region, the sequences from the 5′ end to the start codon of the bla gene were aligned.
Acknowledgments
The authors thank Patrick Shilling, Hwanmi Lim, and Pedro Marques de Sousa for technical advice, and Victor de Lorenzo for sharing the pSEVA collection. D.O.D. was supported by a grant from the Swedish Research Council (2017-00704) and the Carl Trygger stiftelse (CTS18:78). M.H.H.N. and C.N.B. were supported by a grant from the Novo Nordisk Foundation (NNF20CC0035580). S.J. was supported by a SciLifeLab Technology Development Grant from the Faculty of Science at Stockholm University to L.L.I.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.1c00393.
Plasmids and primers used in the study, as well as optimization parameters for the mass spectrometry (PDF)
Author Contributions
⊥ A.J.C. and D.K. contributed equally to this work. D.O.D. conceived the study. A.J.C., D.K., M.H.H.N., S.J., L.L.I., and D.O.D. designed the experiments. A.J.C., D.K., R.H., C.N.B., and S.J. performed the work. All authors analyzed the data. A.J.C. and D.O.D. wrote the paper with contributions from all other authors.
The authors declare the following competing financial interest(s): The directed evolution process and TIRs identified using it are patent-protected. The patents are the property of CloneOpt AB, of which D.O.D. and M.H.H.N. are shareholders.
Supplementary Material
References
- Fröhlich C.; Chen J. Z.; Gholipour S.; Erdogan A. N.; Tokuriki N. Evolution of β-lactamases and enzyme promiscuity. Protein Eng., Des. Sel. 2021, 34, gzab013 10.1093/protein/gzab013. [DOI] [PubMed] [Google Scholar]
- Salverda M. L. M.; De Visser J. A. G. M.; Barlow M. Natural evolution of TEM-1 β-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 2010, 34, 1015–1036. 10.1111/j.1574-6976.2010.00222.x. [DOI] [PubMed] [Google Scholar]
- Datta N.; Kontomichalou P. Penicillinase Synthesis Controlled By Infectious R Factors In Enterobacteriaceae. Nature 1965, 208, 239–241. 10.1038/208239a0. [DOI] [PubMed] [Google Scholar]
- Meynell E.; Datta N. Mutant Drug Resistant Factors of High Transmissibility. Nature 1967, 214, 885–887. 10.1038/214885a0. [DOI] [PubMed] [Google Scholar]
- Sutcliffe J. G. Complete Nucleotide Sequence of the Escherichia coli Plasmid pBR322. Cold Spring Harbor Symp. Quant. Biol. 1979, 43, 77–90. 10.1101/SQB.1979.043.01.013. [DOI] [PubMed] [Google Scholar]
- Sutcliffe J. G. Nucleotide sequence of the ampicillin resistance gene of Escherichia coli plasmid pBR322. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 3737–3741. 10.1073/pnas.75.8.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E.; Goñi-Moreno A.; Bartley B.; McLaughlin J.; Sánchez-Sampedro L.; Pascual Del Pozo H.; Prieto Hernández C.; Marletta A. S.; De Lucrezia D.; Sánchez-Fernández G.; Fraile S.; de Lorenzo V. SEVA 3.0: an update of the Standard European Vector Architecture for enabling portability of genetic constructs among diverse bacterial hosts. Nucleic Acids Res. 2020, 48, D1164–D1170. 10.1093/nar/gkz1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E.; Calles B.; Arévalo-Rodríguez M.; de Lorenzo V. pBAM1: an all-synthetic genetic tool for analysis and construction of complex bacterial phenotypes. BMC Microbiol. 2011, 11, 38 10.1186/1471-2180-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novagen . TB055 pET System Manual, 11th ed.; Novagen, 2011; pp 1–63. [Google Scholar]
- Korpimäki T.; Kurittu J.; Karp M. Surprisingly fast disappearance of β-lactam selection pressure in cultivation as detected with novel biosensing approaches. J. Microbiol. Methods 2003, 53, 37–42. 10.1016/S0167-7012(02)00213-0. [DOI] [PubMed] [Google Scholar]
- Studier F. W.; Moffatt B. A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- William Studier F.; Rosenberg A. H.; Dunn J. J.; Dubendorff J. W.. [6] Use of T7 RNA Polymerase to Direct Expression of Cloned Genes. Methods in Enzymology; Elsevier, 1990; pp 60–89. [DOI] [PubMed] [Google Scholar]
- Overton T. W. Recombinant protein production in bacterial hosts. Drug Discovery Today 2014, 19, 590–601. 10.1016/j.drudis.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Panayotatos N. Recombinant protein production with minimal-antibiotic-resistance vectors. Gene 1988, 74, 357–363. 10.1016/0378-1119(88)90169-2. [DOI] [PubMed] [Google Scholar]
- Yurtsev E. A.; Conwill A.; Gore J. Oscillatory dynamics in a bacterial cross-protection mutualism. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 6236–6241. 10.1073/pnas.1523317113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlin M. H.; Clark D. R.; McKenzie C.; Patel H.; Jackson N.; Kormanik C.; Powell C.; Bajorek A.; Myers D. A.; Dugatkin L. A.; Atlas R. M. Protection of Salmonella by ampicillin-resistant Escherichia coli in the presence of otherwise lethal drug concentrations. Proc. R. Soc. B Biol. Sci. 2009, 276, 3759–3768. 10.1098/rspb.2009.0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeing T. M.; Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature 2009, 461, 1234–1242. 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- Reeve B.; Hargest T.; Gilbert C.; Ellis T. Predicting Translation Initiation Rates for Designing Synthetic Biology. Front. Bioeng. Biotechnol. 2014, 2, 1–6. 10.3389/fbioe.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun H.-M.Enzymology Primer for Recombinant DNA Technology; Academic Press: San Diego, 1996; pp 595–613. [Google Scholar]
- Wong E. M.; Muesing M. A.; Polisky B. Temperature-sensitive copy number mutants of CoIE1 are located in an untranslated region of the plasmid genome. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 3570–3574. 10.1073/pnas.79.11.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh K.; Martínez V.; Toddo S.; Guntur S.; Herrgård M. J.; Elofsson A.; Nørholm M. H. H.; Daley D. O. Enhanced Protein Production in Escherichia coli by Optimization of Cloning Scars at the Vector-Coding Sequence Junction. ACS Synth. Biol. 2015, 4, 959–965. 10.1021/acssynbio.5b00033. [DOI] [PubMed] [Google Scholar]
- Chiang C.-S.; Bremer H. Stability of pBR322-derived plasmids. Plasmid 1988, 20, 207–220. 10.1016/0147-619X(88)90027-3. [DOI] [PubMed] [Google Scholar]
- Carroll A. C.; Wong A. Plasmid persistence: costs, benefits, and the plasmid paradox. Can. J. Microbiol. 2018, 64, 293–304. 10.1139/cjm-2017-0609. [DOI] [PubMed] [Google Scholar]
- Shilling P. J.; Mirzadeh K.; Cumming A. J.; Widesheim M.; Köck Z.; Daley D. O. Improved designs for pET expression plasmids increase protein production yield in Escherichia coli. Commun. Biol. 2020, 3, 214 10.1038/s42003-020-0939-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallehauge T. B.; Li S.; Pedersen L. E.; Ha T. K.; Ley D.; Andersen M. R.; Kildegaard H. F.; Lee G. M.; Lewis N. E. Ribosome profiling-guided depletion of an mRNA increases cell growth rate and protein secretion. Sci. Rep. 2017, 7, 40388 10.1038/srep40388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James J.; Yarnall B.; Koranteng A.; Gibson J.; Rahman T.; Doyle D. A. Protein over-expression in Escherichia coli triggers adaptation analogous to antimicrobial resistance. Microb. Cell Fact. 2021, 20, 13 10.1186/s12934-020-01462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosano G. L.; Morales E. S.; Ceccarelli E. A. New tools for recombinant protein production in Escherichia coli: A 5-year update. Protein Sci. 2019, 28, 1412–1422. 10.1002/pro.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosano G. L.; Ceccarelli E. A. Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 2014, 5, 172 10.3389/fmicb.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A. J.; Segall-Shapiro T. H.; Glassey E.; Zhang J.; Voigt C. A. Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 2019, 15, 196–204. 10.1038/s41589-018-0168-3. [DOI] [PubMed] [Google Scholar]
- Mirzadeh K.; Shilling P. J.; Elfageih R.; Cumming A. J.; Cui H. L.; Rennig M.; Nørholm M. H. H.; Daley D. O. Increased production of periplasmic proteins in Escherichia coli by directed evolution of the translation initiation region. Microb. Cell Fact. 2020, 19, 85 10.1186/s12934-020-01339-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairhofer J.; Wittwer A.; Cserjan-Puschmann M.; Striedner G. Preventing T7 RNA Polymerase Read-through Transcription—A Synthetic Termination Signal Capable of Improving Bioprocess Stability. ACS Synth. Biol. 2015, 4, 265–273. 10.1021/sb5000115. [DOI] [PubMed] [Google Scholar]
- Rouches M. V.; Xu Y.; Cortes L.; Lambert G. A Plasmid System with Tunable Copy Number. bioRxiv 2021, 2021.07.13.451660 10.1101/2021.07.13.451660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. F.; García-Nafría J. In vivo DNA assembly using common laboratory bacteria: A re-emerging tool to simplify molecular cloning. J. Biol. Chem. 2019, 294, 15271–15281. 10.1074/jbc.REV119.009109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann Q. OpenCFU, a New Free and Open-Source Software to Count Cell Colonies and Other Circular Objects. PLoS One 2013, 8, e54072 10.1371/journal.pone.0054072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh K.; Martínez V.; Toddo S.; Guntur S.; Herrgård M. J.; Elofsson A.; Nørholm M. H. H.; Daley D. O. Enhanced Protein Production in Escherichia coli by Optimization of Cloning Scars at the Vector–Coding Sequence Junction. ACS Synth. Biol. 2015, 4, 959–965. 10.1021/acssynbio.5b00033. [DOI] [PubMed] [Google Scholar]
- Daley D.; Mirzadeh K.; Toddo S.; Guntur S.. Selective Optimisation of a Ribosome Binding Site for Protein Production. EP3234146A1; US10,696,963B2, 2015.
- Koontz L.TCA Precipitation. Methods in Enzymology; Elsevier, 2014; pp 3–10. [DOI] [PubMed] [Google Scholar]
- Polson C.; Sarkar P.; Incledon B.; Raguvaran V.; Grant R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2003, 785, 263–275. 10.1016/S1570-0232(02)00914-5. [DOI] [PubMed] [Google Scholar]
- Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. 10.1093/nar/gkx1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.