

Abstract
Measurements of neuronal activity across brain areas are important for understanding the neural correlates of cognitive and motor processes such as attention, decision-making and action selection. However, techniques that allow cellular resolution measurements are expensive and require a high degree of technical expertise, which limits their broad use. Wide-field imaging of genetically encoded indicators is a high-throughput, cost-effective and flexible approach to measure activity of specific cell populations with high temporal resolution and a cortex-wide field of view. Here we outline our protocol for assembling a wide-field macroscope setup, performing surgery to prepare the intact skull and imaging neural activity chronically in behaving, transgenic mice. Further, we highlight a processing pipeline that leverages novel, cloud-based methods to analyze large-scale imaging datasets. The protocol targets laboratories that are seeking to build macroscopes, optimize surgical procedures for long-term chronic imaging and/or analyze cortex-wide neuronal recordings. The entire protocol, including steps for assembly and calibration of the macroscope, surgical preparation, imaging and data analysis, requires a total of 8 h. It is designed to be accessible to laboratories with limited expertise in imaging methods or interest in high-throughput imaging during behavior.
Introduction
Simultaneous recordings of activity across brain areas have recently enabled unprecedented insights into the coordination of neuronal activity during both spontaneous1–3 and cognitive behaviors4–8. One of the first methods used to unravel the spatial organization of large-scale cortical activity was wide-field imaging9,10. Originally, hemodynamic signals were used as a proxy for neuronal activity, and the large field of view and noninvasive nature of wide-field imaging made it ideal for mapping cortical responses to sensory stimulation. However, hemodynamic signals have limited spatiotemporal resolution and specificity11–14. An alternative is using fluorescent activity indicators, such as calcium- or voltage-indicators, which can be genetically encoded, have high signal-to-noise ratio and provide direct insights into the activity of specific cell types5,15–17. The widespread availability of transgenic mouse lines combined with advancements in recording techniques allows imaging the entire dorsal cortex with high spatial and temporal resolution.
The ability to record neural activity from a large number of cortical areas simultaneously with genetically encoded indicators enabled exciting possibilities beyond sensory mapping that have just recently come to the forefront. In particular, recordings from the entire dorsal cortex profoundly affected how we consider trial-to-trial variability and neural engagement during cognitive behaviors3–7,18. Alongside developments of neural activity indicators, new surgical preparations enable the acquisition of neural signals through the intact skull19,20, thereby reducing the potential for brain trauma, inflammation and tissue regrowth. This makes wide-field imaging ideally suited for longitudinal studies and high-throughput assays. However, a major bottleneck is that wide-field imaging produces massive datasets that are not trivial to preprocess, analyze, store and share with the community. Recent advances in computational methods, such as techniques for dimensionality reduction and statistical modeling, provide new opportunities for compressing, denoising and exploring wide-field data21–23.
Here we provide a detailed protocol for our high-throughput imaging pipeline, which includes assembling a wide-field macroscope along with a step-by-step guide for the surgical preparation, data acquisition and analysis, and allows repeated imaging of most of the dorsal cortex of transgenic animals expressing calcium indicators4. Taken together, this protocol aims to circumvent the main barriers to implementing longitudinal imaging of cortex-wide neuronal activity by introducing a novel, overarching platform that allows any investigator to build a wide-field setup, measure neural activity, and analyze signals on the cloud, thus readily generating new observations about widespread cortical activity.
Overview of the procedure
Our protocol consists of four main stages (Fig. 1a). In the first stage, we describe how to assemble and calibrate the wide-field macroscope (Steps 1–24). In the second stage, we highlight our procedures for skull clearing (Steps 25–40). In the third stage, we describe procedures for imaging neural activity in behaving mice expressing genetically encoded calcium indicators (Steps 41–46). In the last stage, we describe how to deploy analyses on the cloud-based NeuroCAAS platform (http://neurocaas.org), using the website or the graphical user interface (Steps 47–49).
Fig. 1 |. Overview of the procedure described in this protocol.
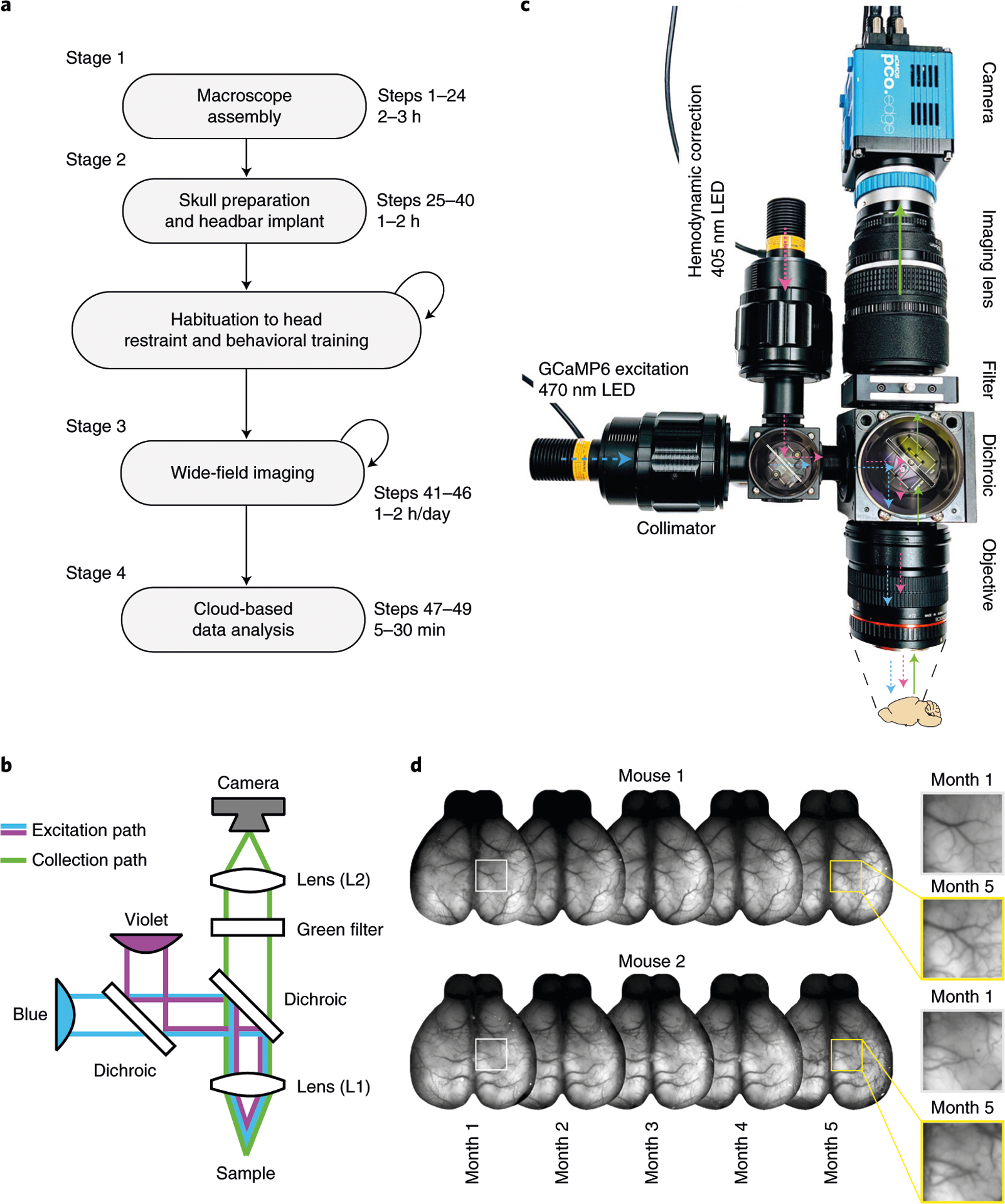
a, Main stages of a wide-field experiment. Description of the animal preparation in Steps 25–40, imaging in Steps 41–46 and data preprocessing in Steps 47–49. Animal habituation depends on the behavioral assay and is not discussed here. Data analysis beyond preprocessing depends on the scientific question; see the anticipated results section for examples. b, Diagram of the macroscope in tandem-lens configuration. c, Overview of the macroscope described in Steps 1–24 with light path diagram. Colors indicate the wavelength of the light in the corresponding path (violet: 405 nm; blue: 470 nm). d, Implant stability over 5 months. Cyanoacrylate maintains the skull semitransparent chronically, enabling long-term monitoring of neuronal activity. Left insets show the blood vessels magnified at month 1 and month 5. Animal experiments followed NIH guidelines and were approved by the Institutional Animal Care and Use Committee of Cold Spring Harbor Laboratory.
Applications
Our protocol can be used to probe the impact of behavior on neural activity across cortical areas, to establish the flow of activity in distant areas (e.g., in cortex-wide functional mapping experiments14), to perform longitudinal studies of cortical function or to measure activity of distinct cortical cell types by taking advantage of specific transgenic lines or injection strategies24,25. Wide-field imaging can also be used in high-throughput automated experiments26,27 and to assess the impact of drugs on cortical activity or in disease models28. Finally, the protocol can be easily extended for high-speed imaging of voltage indicators29, for multicolor imaging of multiple fluorescent indicators5,30 and for optogenetic manipulation5,19.
Comparison with other methods
Wide-field imaging provides several advantages over conventional two-photon microscopy: large field of view, high temporal resolution (kilohertz range possible), low cost and reduced complexity. However, these come at the expense of limited imaging depth and lack of single-cell resolution. Another advantage over two-photon microscopy is the long working distance that makes wide-field imaging easier to combine with electrophysiology3,31. Further, when compared with functional ultrasound32 and magnetic resonance imaging33, wide-field imaging provides higher temporal resolution and the possibility to target specific cell subpopulations.
The skull clearing technique has major advantages over cranial window preparations for longitudinal studies. Surgeries for implanting cranial windows require a high level of surgical proficiency, and optical clearance might degrade over time because of bone regrowth or infection. This limits the experimental throughput and increases the number of animals needed for a given study.
NeuroCAAS enables cloud-based deployment of recently developed algorithms for analyzing wide-field data that complement and extend traditional methods. Further, NeuroCAAS aims to remove the burden of installing software packages that often have elaborate dependencies and/or acquiring and maintaining expensive computing hardware. In addition, it allows analyses to be executed in parallel, which is a scalable alternative to running analyses sequentially on a single machine where independent datasets cannot be processed simultaneously. An alternative to the cloud that is similarly scalable is a high-speed computing cluster. Due to the high cost of operating a high-speed computer cluster, some institutions operate and maintain clusters that make computing resources available to local personnel. However, these facilities can be costly to access and are available only to a subset of institutions with neuroscience laboratories. Finally, deploying analyses on a computer cluster often requires knowledge of how to operate the scheduling software that organizes computing resources, which can impose a barrier for some users and is greatly simplified in the cloud platform.
Limitations
The depth of imaging through the intact cleared skull is limited by light scattering and therefore biased towards activity of superficial cortical layers. Placing the focal plane deeper into the cortex can blur the image but does allow more fluorescence from deeper cortical layers (e.g., Fig. 2d). In addition, novel molecular tools promise to overcome this limitation by expressing indicators exclusively in the somatic compartment34. This eliminates the contribution of dendritic or axonal processes to the emitted fluorescence.
Fig. 2 |. Acquisition in rolling shutter, dual color excitation mode and synchronization.
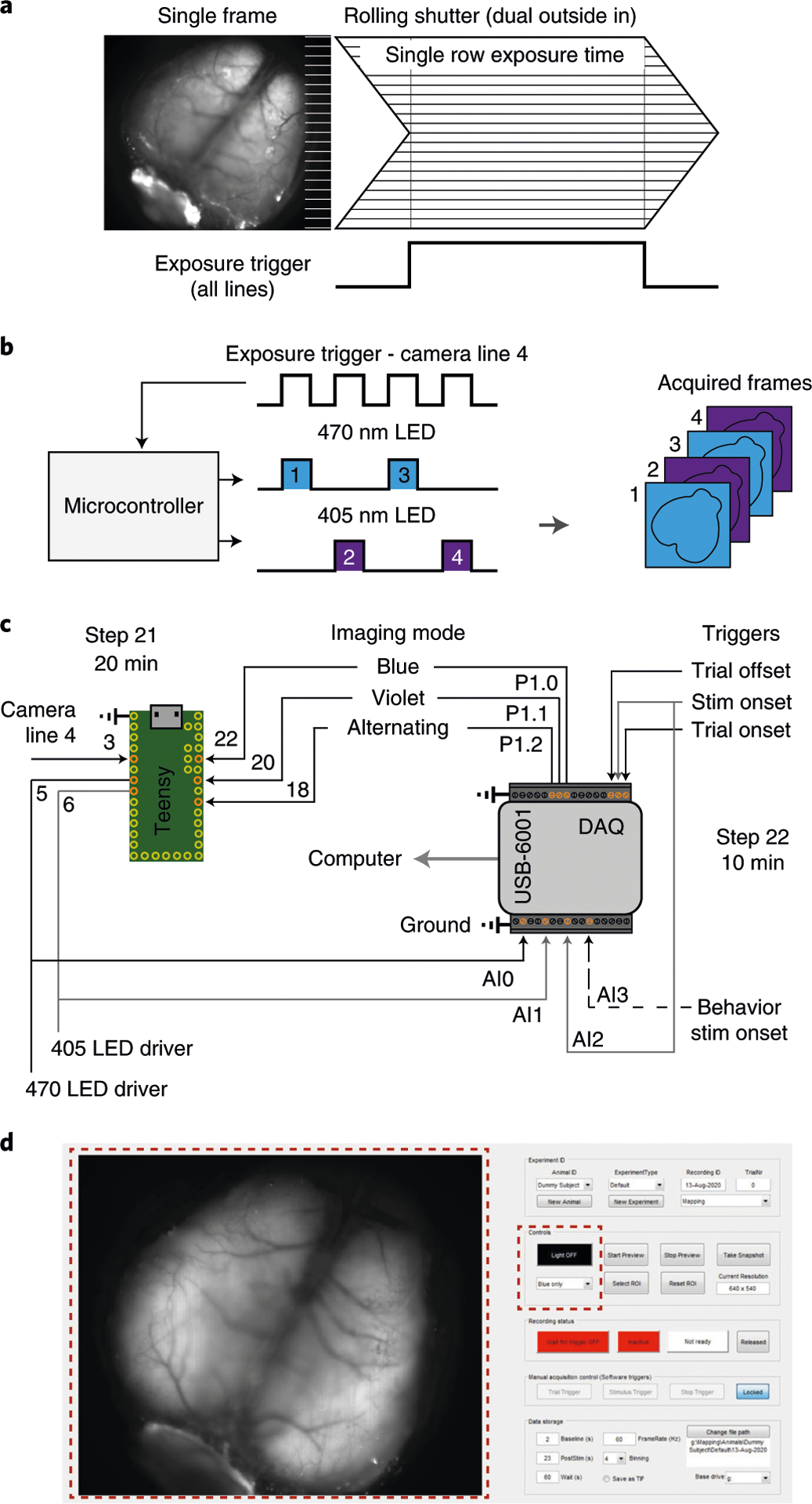
a, Illustration of single-frame acquisition with a rolling shutter camera in ‘dual outside-in’ mode. Common exposure time of all lines is restricted to part of the frame duration. The dorsal cortex is in focus as in Step 44 for imaging. b, Excitation light is restricted to the period of common exposure of all lines and alternated between 470 nm and 405 nm. The microcontroller alternates light according to the camera exposure input. c, Connection diagram for the microcontroller circuit and integration with the data acquisition system for synchronizing with a behavioral setup. Dashed line is optional. Gray lines are to disambiguate overlap. d, Graphical user interface of the acquisition software. Red, dashed squares highlight the preview (left) and LED control (right) panels. A detailed description of the software is at https://github.com/churchlandlab/WidefieldImager.
A key limitation is that wide-field signals reflect the activity pooled over many neurons. Therefore, features encoded by the coordinated activity of different neurons in a local population and not reflected in the bulk activity of an area are elusive. Nonetheless, pooling activity over many neurons might be beneficial when measuring weak signals correlated across many neurons (e.g., inconspicuous sensory stimuli or arousal-related activity).
The NeuroCAAS platform allows the deployment of a set of complex analyses on optimized hardware without requiring expertise in programing or hardware maintenance. However, since the service runs and stores data on the Amazon Web Services platform, it should be used with care when storing sensitive or confidential data. In addition, use of the underlying Amazon Web Services produces some costs when analyzing data; however, the cost remains lower when compared with managing computational resources locally35.
Experimental design
Our protocol emerged from the need to simultaneously and repeatedly record neural activity from distant cortical areas in head-restrained mice during perceptual tasks. This demanded combining a highly sensitive macroscope with a surgical preparation for optical access to the entire dorsal cortex. Further, it prompted the development of software for synchronizing imaging data with behavior, and the implementation of a scalable data analysis pipeline.
Macroscope assembly (Stage 1)
We based our macroscope on the design from Ratzlaff et al. that allows efficient light collection and excitation using a tandem-lens configuration36. The design uses commercially available parts, and is customizable, easy to assemble and relatively cost effective (under US$10,000, excluding the camera), which places it within reach of laboratories without previous imaging expertise or with limited resources.
A wide-field macroscope consists of two main components: the excitation path (blue and violet in Fig. 1b,c) that delivers light to the cortical surface to excite the fluorescent indicator, and the collection path (green in Fig. 1b,c) that collects light emitted by the fluorescent indicator and terminates at the camera sensor. In our design, the excitation path is optimized for imaging green fluorophores (e.g., GCaMP637) and uses a set of dichroic mirrors to direct light from the two light-emitting diodes (LEDs) to the objective lens (L1). An alternative is to provide illumination externally from the side of the objective17,26,38. Side illumination requires fewer components and allows the flexibility to use different wavelengths (e.g., when green excitation light17 or multiple wavelengths are required38); however, its implementation can be challenging when using visual stimulation assays where light shielding is important.
The sensitivity of the collection path is an important factor that determines if weaker signals can be recorded and how much light power needs to be delivered to the cortex. Light collection in our design uses two single-lens reflex camera lenses that are facing each other in an inverted tandem-lens configuration to form a low-magnification collection path36. This provides long working distance (> 40 mm, dictated by the flange focal distance of the objective lens), narrow depth of field that maximizes signal collection from cortex, and high sensitivity (numerical aperture of ~0.36 versus ~0.05 for low-magnification microscope objectives). Alternative designs, using a single camera lens26 or a microscope objective39, can also be used to reduce cost or setup size but often result in shorter working distances, lower sensitivity and wider depth of field.
For the tandem-lens configuration, the objective lens is inverted (i.e., the lens’ camera mount faces the brain surface) to project near-collimated light to a second lens that focuses the light on the camera sensor. The ratio of the focal lengths (f) of two lenses, L1 and L2 in Fig. 1b, determines the magnification. We chose L1f = 105 mm for the imaging (top) lens and L2f = 85 mm for the objective (bottom lens), which results in a magnification of 1.24 (L2f/L1f). To increase or decrease the magnification factor, one can use lenses with different focal lengths. It is important to consider that the effective field of view depends on the magnification factor and the size of the camera sensor. We use a camera with a 16.6 × 14 mm sensor, which results in a field of view of 13.4 × 11.3 mm with a 1.24 magnification factor. Because both lenses include zoom optics, it is possible to adjust the field of view, without replacing the lenses, between 17.1 × 14.4 mm and 12.5 × 10.5 mm, which is ideal for imaging the whole cortex of adult mice.
To obtain high signal sensitivity and a large field of view, we use a camera (PCO Edge 5.5) with a scientific CMOS sensor that provides high-quantum efficiency (~60% for green light), large sensor (16.6 × 14 mm), low noise, large dynamic range (16 bit), fast pixel scan rate (286 MHz) and high frame rates (100 Hz at full resolution). The camera resolution is 2,560 × 2,160 pixels (~5 μm2/pixel), which is far below the required resolution for intact skull wide-field imaging (~50 μm)20. We therefore recommend fourfold pixel binning, which combines 4 × 4 pixel arrays on the sensor (~20 μm2/merged pixel) and increases light sensitivity while reducing data size. Sufficiently high spatial resolution (at least ~80 μm2/pixel) should be maintained to resolve activity in smaller cortical areas.
The shutter mode (rolling or global shutter) of the camera is also an important consideration when choosing the camera because a rolling shutter can provide higher speed and sensitivity but also produce artifacts when acquiring fast-changing signals. We further discuss how to image in rolling shutter mode in the ‘data acquisition’ stage below. It is also possible to use cameras with lower sensitivity, depending on the experimental constraints (brightness of the fluorescent indicator and required sensitivity). In earlier versions of the setup, we used the MV-D1024E-160-CL-12 camera from Photonfocus with satisfactory results; recent studies even demonstrated cortex-wide imaging using a lower-cost Raspberry Pi camera module26,31.
To maximize the amount of collected light, we use a 60-mm cage system (hollow cube where optical components, i.e., lenses, mirrors and filters, are mounted) in the collection path. The ratio between the focal length and the diameter of the entrance pupil (effective aperture) is the f-stop (or f-number) and is used in photography as a measure of the aperture in different lenses. Our objective lens (L1) has an f-stop of 1.4 and a focal length of 85 mm. The entrance pupil is therefore 85 (mm)/1.4 = 60.7 mm. Using a 60 mm, and not a 30 mm, cage system is an important design consideration because it allows more light to be collected by the imaging lens (L2). Another consideration is that the light exiting the L1 lens is slightly divergent, and therefore the entrance pupil of L2 should be larger than the exit pupil of L136. In our design, the exit pupil of L1 has a 50 mm aperture (the size of optical mounts in our cage system) and the L2 lens has entrance pupil of 105 (mm)/2 = 52.5 mm.
The near-collimated light path between the objective (L1) and the imaging (L2) lenses can accommodate optics specific to fluorescence imaging (Fig. 1b,c). We use a dichroic mirror to reflect blue (470 nm) and violet light (405 nm) on the sample while collecting the emitted, longer wavelength (>495 nm) light through a green bandpass filter (525 ± 25 nm) (Fig. 1b,c). These optics are optimized for imaging green fluorophores (e.g., GCaMP6) but can easily be exchanged to record with other activity indicators, e.g., red-shifted indicators.
The acquired fluorescence is a combination of neuronal signals from the activity indicator but also nonneuronal signals from other sources, such as flavoprotein- and hemodynamic-related fluorescence changes38,40. To isolate neuronal signals, we alternate the excitation light from frame to frame between 470 nm (blue light) and 405 nm (violet light)4,5,40. While blue light excites GCaMP to emit green light depending on the binding of calcium, violet light is at the isosbestic point for GCaMP, which means that the emitted fluorescence is largely independent of the calcium binding. Since fluorescence at both wavelengths contains nonneural signals but only blue-excited fluorescence additionally contains neural signals, we can rescale violet-excited fluorescence to match blue-excited fluorescence and then subtract it to remove nonneural signals4. This effectively removes intrinsic signal contributions and has been validated in single neurons40 and GFP-expressing control mice4. Nonetheless, a consensus on the optimal strategy for hemodynamic correction has not yet been reached. Efforts to further quantify different components of the hemodynamic response, its relation to GCaMP fluorescence and the development of indicators that are less prone to contamination are important to find novel ways of accurately estimating calcium fluorescence38,41. An alternative approach, which may reduce photobleaching, is to use green instead of violet illumination17. A second alternative is to use combinations of blue, green and red illumination to individually quantify hemodynamic signals, such as fluorescence changes due to oxygenation of hemoglobin and local blood volume38. The approaches discussed above require alternating the excitation light from frame to frame between different wavelengths; a related approach is to use continuous illumination with two wavelengths and separate the emitted light using an RGB camera31, or building a spatial model of the hemodynamic response using a separate cohort of non-GCaMP mice41.
In some cases, the hemodynamic correction can also be omitted2,18,42. For example, when imaging from densely labeled neuronal populations, using bright indicators such as GcaMP6s, the neural signal can be strong enough to not be confounded by nonneural signals2,42,43. However, since intrinsic signal contamination strongly increases the more excitation and emission photons travel through brain tissue38, the correction is particularly important when imaging from deeper cortical layers even when using bright neural indicators.
Animal preparation (Stage 2)
Imaging cortical activity requires an animal preparation that ensures optical access to the cortex for the duration of the experiment. A specific procedure, often referred to as ‘skull clearing’19, allows the maintaining of optical clarity similar to that observed when covering the skull with saline over months without chemically altering the bone structure20. We favor preparing the skull with cyanoacrylate19 over invasive procedures44,45 because of the low risk of tissue damage, straightforward surgical procedure and long-term implant stability. The cyanocrylate we use was selected for four reasons: it prevents the formation of opaque spots that are due to locking in small air bubbles in the bone, it has a medium viscosity that allows even distribution across the skull, is very clear after curing and it remains stable to allow repeated imaging for at least 5 months (Fig. 1d).
When imaging from specific cell populations, brightness and expression of the indicator in the neuronal population of interest is critical. For imaging excitatory neurons in cortex, the transgenic tetO-GCaMP6s/CaMK2a-tTA mouse line17 provides very high signal strength and even expression across cortical areas46. In contrast, some GCaMP6-expressing mouse lines, such as the Ai93D and Ai95D lines, have weaker fluorescence signal and can exhibit epileptiform activity patterns47.
Data acquisition (Stage 3)
Several considerations are important regarding data acquisition when imaging repeatedly from transgenic mice. First, most high-speed CMOS cameras operate in ‘rolling shutter’ mode. However, this can cause artifacts when neural dynamics change quickly because not all lines are exposing at the same time. To prevent such artifacts, we restrict the excitation light to times when all lines are exposing. To do this, the camera common exposure trigger is connected to a fast microcontroller (Teensy) that triggers the LEDs in an alternating fashion and only during the exposure time common to all lines (Fig. 2a,b). It is therefore important to prevent contamination of the imaging signals with ambient light because some lines are partially exposing during the time that the excitation light is turned off. For single-wavelength illumination, the camera common exposure trigger can be connected to the LED directly. Note that this is only critical for cameras with ‘rolling shutter’ acquisition but does not apply to cameras that operate in ‘global shutter’ mode.
Second, imaging for long durations can induce photobleaching, the photochemical degradation of the fluorophore, which reduces the signal amplitude over time. In addition to optimizing the light sensitivity so that less power is required, and triggering LED illumination with the camera, we devised a trial-based acquisition system in which the LEDs are extinguished during the period between trials. Together, this strategy greatly reduces the net power delivered to cortex by restricting light exposure periods and thus enabling chronic imaging across months without signal decay. We usually use 10–25 mW of alternating blue/violet light when imaging at 30 Hz. The required light power depends on the brightness, expression and labeling density of the indicator and therefore varies from mouse to mouse. We observed that light powers of ≥50 mW can induce photobleaching within 5–10 min of imaging (Extended Data Fig. 1a). Photobleaching is reversible because of the regeneration of indicator protein over time, but bleaching effects can also accumulate when imaging daily (Extended Data Fig. 1b). If bleaching occurs within a session, waiting several days between imaging sessions can avoid such accumulation.
Third, recording behavioral events and stimulus triggers alongside imaging data is critical for the accurate interpretation of neural activity. We use a data acquisition board (DAQ) to acquire the exposure for each frame and the LED triggers, and to monitor behavior variables (e.g., from a treadmill or from sensors in the behavioral setup). An off-line algorithm then asserts proper alignment of behavioral and imaging data. The DAQ also controls the microcontroller (Fig. 2c) that controls the excitation light for multiple wavelength illumination and is used to recover the wavelength that was used for each frame. An alternative approach is to use a camera that supports ‘general purpose input–output’ to record the triggers directly in the camera frames or trigger the camera to acquire a predetermined number of frames when the trial structure is known a priori.
Lastly, wide-field imaging produces very large datasets (on the order of ~150 GB/h when imaging continuously at 60 Hz, depending on the resolution) that might be prohibitively expensive to store. However, since behavioral tasks often have periods during which data are not critical, we established an approach to discard the intertrial periods even when the subject itself initiates the trials in the assay. This is a challenge because in self-initiated tasks, the baseline needs to be captured before the trial is initiated. To solve this problem, we implemented a sliding window recording method that allows us to discard the totality of the intertrial interval with the exception of the baseline period and effectively relaxes the storage requirements (effectively cutting the length of the dataset by a factor of ~3, depending on the behavioral task).
To tackle all the aforementioned considerations, we developed a custom MATLAB-based acquisition software available at https://github.com/churchlandlab/WidefieldImager. The software aims to provide user-friendly control of the camera acquisition through a graphical user interface and to integrate with different behavior assays or stimulation paradigms (Fig. 2d). We also recently developed acquisition software written in Python with similar functionality (https://bitbucket.org/jpcouto/labcams). Furthermore, on our GitHub page, we provide code to evaluate the amount of bleaching that can be useful for repeated measurements from the same mouse (checkSession-Bleaching.m).
Data analysis (Stage 4)
A key challege for implementing the technique is the data analysis. Chronic wide-field imaging of transgenic mice is a high-throughput technique that produces massive datasets. Specific analysis algorithms may depend on the scientific question or experiment at hand. Here we dicuss some common processing steps to extract neuronal activity from the acquired frames. The first step is to correct for artifacts resulting from motion of the skull in relation to the objective. These can be corrected by aligning each frame to a time-averaged reference image. We obtained better results with rigid body registration algorithms that consider both translation and rotation artifacts. If there are multiple channels, it is essential that motion is corrected for all channels because movement artifacts can be amplified when subtracting activity across channels. The next step is to compute relative fluorescence changes (ΔF/F) for each pixel. This is important to reveal small changes in fluorescence due to cortical activity. Computing ΔF/F is done by subtracting and dividing the value at each frame by the baseline fluorescence. The baseline can be computed by taking the average of a number of frames before trial onset or by taking the average of all frames in the session if the former is not possible.
The third step is to denoise and compress the dataset to improve the signal-to-noise ratio and separate the dataset into temporal and spatial components of reduced size. This simplifies subsequent analysis (e.g., linear decoding) because that can be performed on temporal components instead of individual pixels. Denoising aims to isolate the signal to boost the signal-to-noise ratio, whereas compression reduces the size of the dataset and is critical for the computational efficiency of downstream analyses. Here we discuss two related methods for denoising and compression. The standard method is to use singular value decomposition, and keep the components that carry the most variance. In our experience, 200 components are sufficient to capture >90% of the variance in the dataset and reduce the dataset size by a factor of 1504,22. A more specialized and efficient method for denoising and compressing wide-field recordings is penalized matrix decomposition (PMD), which uses assumptions on the structure of noise for denoising and can achieve higher compression rates (~300 times reduction in size), with faster computation time22.
The fourth step is to perform hemodynamic correction to remove nonneuronal-related activity using measurements with violet light excitation (Fig. 3). Before correcting, data are filtered with a 0.1 Hz temporal high-pass filter. An additional low-pass filter can be applied to remove fast transients in the violet channel since the nonneural signals are usually slower than neural signals. Frames acquired with violet illumination are then extrapolated to account for differences in acquisition time, rescaled to the amplitude of the frames acquired with blue illumination and subtracted from the frames with blue illumination. The result is dramatic for frames with high hemodynamic response where the blood vessels were clearly visible in frames acquired with violet and blue illumination but not in the corrected frames (Fig. 3a,b). Frames with no hemodynamic response are unaffected because the subtracted component is close to zero.
Fig. 3 |. Correction of hemodynamic artifacts with alternating violet and blue illumination.
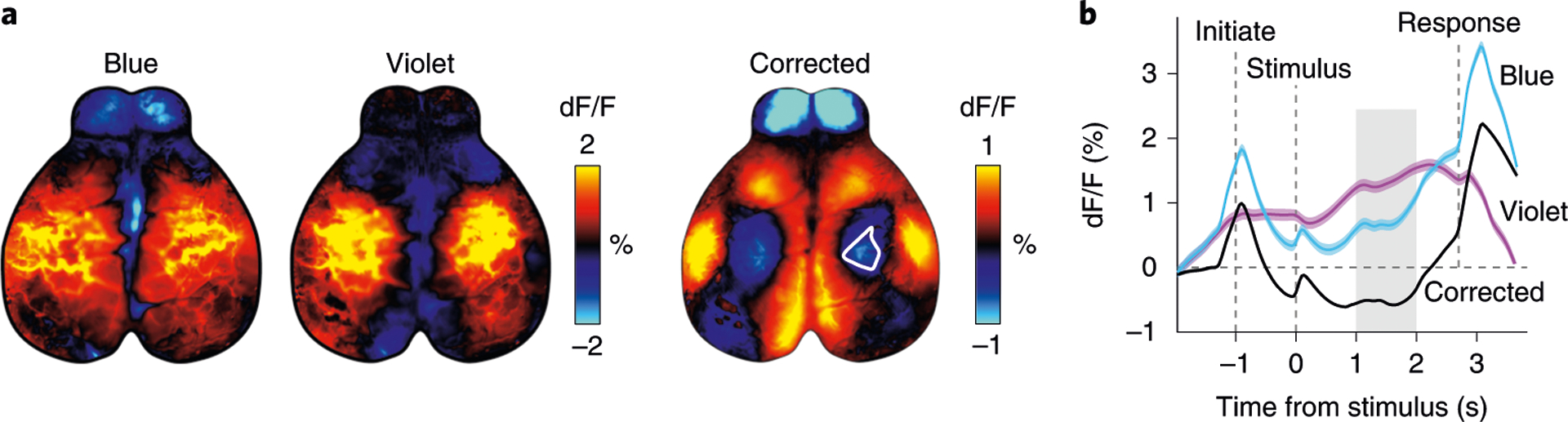
a, Trial-averaged fluorescence with either blue or violet excitation (left) during a behavioral task4 (n = 402 trials). Hemodynamic corrected trial average (right) shows reduced activity in the hind-limb area (white outline). b, Hemodynamic correction recovers the temporal dynamics of neural signals. Traces show averaged fluorescence from the somatosensory hind-limb area (white outline in a), which appears to be above baseline when averaging activity acquired with blue excitation. However, the hemodynamic corrected trace (black) shows that activity is below baseline. Gray shading shows the time from which maps in a were averaged. Dashed lines show the time of different events in the task. Animal experiments followed NIH guidelines and were approved by the Institutional Animal Care and Use Committee of Cold Spring Harbor Laboratory.
The last step is to interpret the neural data, for example, by comparing the activity in different cortical areas in response to a sensory stimulus or behavioral event. To achieve this, the data can be aligned to a brain atlas4,5, such as the Allen Common Coordinate Framework48, and then averaged over all pixels in an area of interest. However, averaging is problematic because it unselectively collapses different activity patterns within larger areas or potentially confuses the activity in small, neighboring areas because of individual differences in brain anatomy or imperfect atlas alignment. To resolve this issue and reliably extract all activity patterns from known brain areas, we developed a new analysis method called LocaNMF23. Using area locations from a brain atlas as a prior, LocaNMF obtains interpretable components that represent the activity in specific cortical regions and is also capable of isolating overlapping activity patterns in the same cortical area (see fig. 4a in Saxena et al., 2020). This approach is highly useful to transform the low-dimensional data from singular value decomposition or PMD into components that can be readily interpreted as different activity patterns from known cortical areas.
Fig. 4 |. Detailed build instructions for the wide-field macroscope.
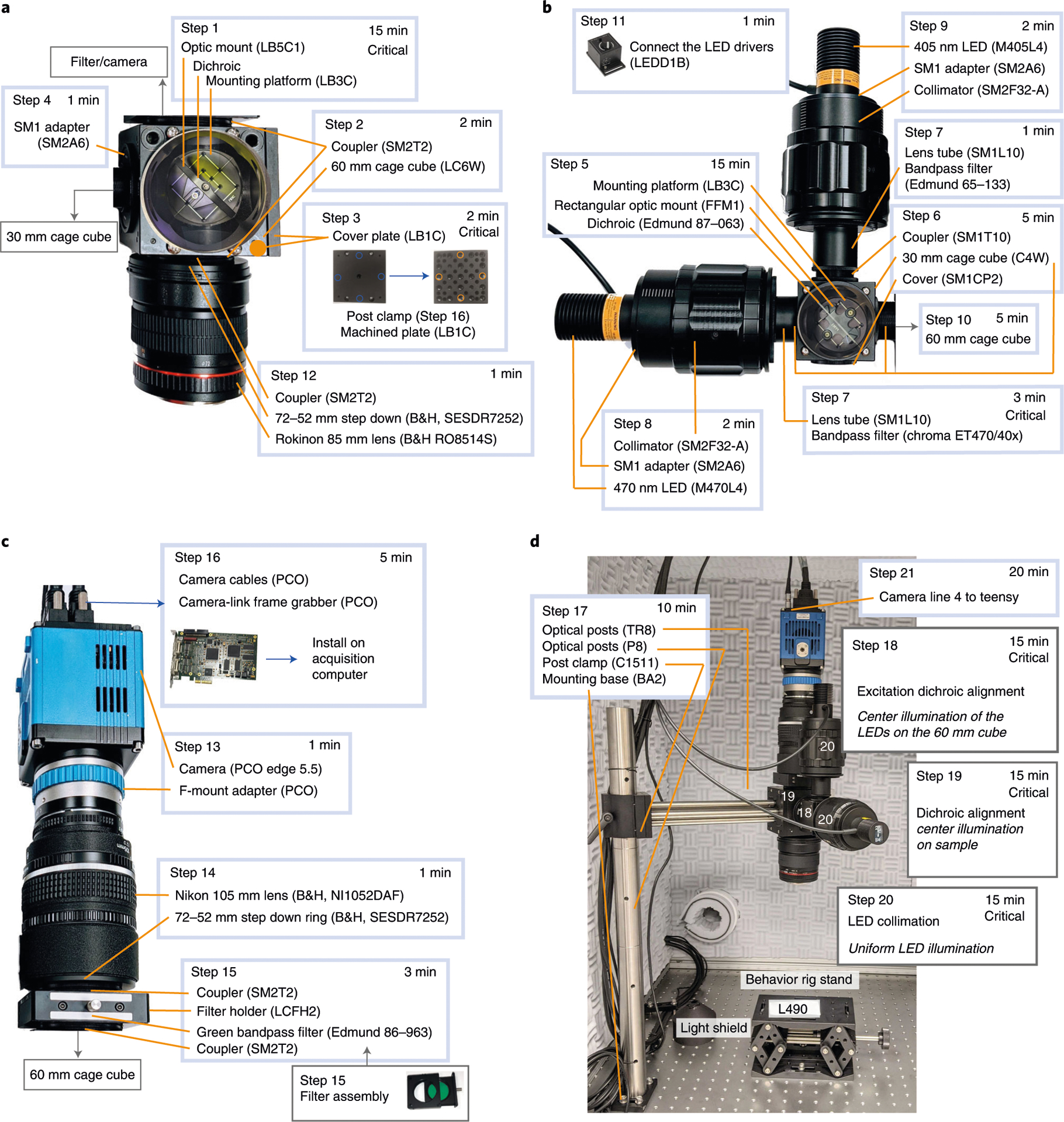
a, Steps for assembling the main cage cube and objective module. b, Steps for assembling the excitation cage cube and light collimation module. c, Details for assembling the light collection module. d, Example setup with a rig stand and highlighting the steps for calibration.
To efficiently apply these analyses, we developed tools for both Python (https://github.com/churchlandlab/wfield) and MATLAB (https://github.com/churchlandlab/WidefieldImager/tree/master/Analysis, also used in ref.4) that are compatible with the aforementioned acquisition software, optimized to use limited computational resources and flexible to also accommodate other imaging formats. This also includes example applications and test datasets to create stimulus-triggered averages or visual phase maps (https://github.com/churchlandlab/WidefieldImager/tree/master/Analysis).
Because longitudinal experiments generate large datasets that require a potentially expensive infrastructure for efficient data storage and processing, we also present an analysis pipeline that combines all the steps described above on the cloud-based analysis platform NeuroCAAS (http://neurocaas.org), which allows reproducible and streamlined analysis35. One of the major advantages of this platform is that it deploys well-established, state-of-the-art algorithms and runs on optimized hardware without local infrastructure and maintenance costs; users simply drag-and-drop datasets onto the system and then download the denoised, compressed and demixed output. Importantly, sophisticated analysis algorithms often take advantage of specific libraries or hardware that are often not trivial to implement locally but are already implemented in the cloud. Using a cloud platform therefore streamlines data analysis, enables access to standardized high-performance algorithms, reduces processing time, eliminates hardware cost and reduces the challenges of implementing processing pipelines on local premises. To simplify the interaction with the online platform, we created a graphical user interface, dedicated to launching analysis and retrieving results from the NeuroCAAS platform (https://github.com/churchlandlab/wfield).
Materials
Biological materials
Transgenic mice expressing GCaMP6 in a neuronal population of interest (Charles River or Jackson Laboratories) ! CAUTION Experiments involving animals must be conducted in accordance with relevant institutional and governmental guidelines and regulations. Experiments for developing this protocol conformed to guidelines established by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of Cold Spring Harbor Laboratory.
Reagents
Skull clearing reagents
Cyanoacrylate glue (Pacer Technologies, cat. no. ZAP-A-GAP CA+) ! CAUTION Cyanoacrylate is toxic. Use gloves when handling the cyanoacrylate.
Drugs
Isoflurane (Covetrus, cat. no. 1169567761)
Ophthalmic ointment (Dechra, NDC 17033-211-38)
Ketamine, 10% (wt/vol) (Dechra, cat. no. 17033-100-10)
Dexmedetomidine hydrochloride, 0.5 mg/mL (Zoetis, cat. no. 1-000-2752)
Antisedan, 5 mg/mL (Zoetis, cat. no. 10000-449)
Lidocaine, 1% (vol/vol) (Bichsel, cat. no. 2057659)
Meloxicam, 5 mg/mL (Böhringer Ingelheim, cat. no. 136327)
Other
Ethanol (EtOH), 70% (vol/vol) (VWR, cat. no. 97065-058)
Isotonic saline solution: 0.9% NaCl (wt/vol) (B. Braun, cat. no. L8001)
Betadine (Henry Schein, cat. no. 6906950)
Black dental acrylic, Ortho-Jet (Lang Dental cat. nos. 1520BLK and 1503AMB), for bonding the headpost to the skull and for light shielding
Vetbond (3M, cat. no. 70200742529)
Equipment
Surgical equipment
Leica M60 stereomicroscope (Leica Microsystems, Leica M60)
Small rodent stereotaxic frame (Kopf, model 900LS)
Fiber optic lamp with gooseneck (Schott, cat. no. SCHOTT-ACE-GOOSENECK)
Isoflurane vaporizer and anesthesia system (Kent Scientific, cat. no. VetFlo-1205S-M)
Feedback-controlled thermal blanket (Kent Scientific, cat. no. PhysioSuite-PS-02)
Surgical tools
Scalpel (Fine Science Tools, cat. no. 10003-12)
Forceps (Fine Science Tools, cat. no. 11049-10)
Scissors (Fine Science Tools, cat. no. 14040-10)
Vannas scissors (World Precision Instrument, cat. no. 501777)
Micro probe, angled (Fine Science Tools, cat. no. 10032-13)
Delicate bone scraper (Fine Science Tools, cat. no. 10075-16)
Fine forceps (Fine Science Tools, cat. nos. 11254-20, 11251-35)
High-speed dental drill (Foredom, cat. no. K.1070), can be used to removed particles trapped in the cement
Consumable supplies
Insuline syringes 29G (BD, cat. no. 324704)
Paper wipers (Kimberly-Clark, cat. no. 34155)
Cotton swabs (Thermo Fisher, cat. no. 22-363-160)
Sterile drape (Thermo Fisher Scientific, cat. no. 19-160-140)
Gauze (Thermo Fisher Scientific, cat. no. 22-415-469)
Gloves (Thermo Fisher Scientific, cat. no. 19-041-171)
Surgical mask (Thermo Fisher Scientific, cat. no. 12-888-001)
Hair net (VWR, cat. no. 10135-738)
Lens cleaning paper (Thorlabs, cat. no. MC-5)
Macroscope camera and optics
PCO edge 5.5 camera sCMOS, 2560 × 2160 with cameralink interface (PCO AG, cat. no. PCO.EDGE 5.5 RS AIR), highly sensitive, low-noise camera
Nikon AF DC-NIKKOR 105 mm f/2D lens (B&H, cat. no. NI1052DAF), i.e., the imaging lens (L2)
Rokinon 85 mm f/1.4 AS IF UMC lens for Sony A (B&H, cat. no. RO8514S), i.e., objective lens (L1)
Blue excitation bandpass filter, 470 nm with 40 nm bandwidth (Chroma, cat. no. ET470/40x), to restrict the wavelength of the blue LED to excite GCaMP
Violet excitation bandpass filter, 405 nm with 10 nm bandwidth (Edmund optics, cat. no. 65–133, or ET405/10× from Chroma), to restrict the wavelength of the hemodynamics correction LED
GFP bandpass filter, 525 nm with 45 nm bandwidth (Edmund optics, cat. no. 86–963, or ET525/36m from Chroma), for restricting the light that reaches the camera sensor
Dichroic mirror, R: 325–425, T: 444–850, 435 nm cutoff, 25.2 mm × 35.6 mm, 1.05 mm thick (Edmund Optics, cat. no. 87–063, or T425lpxr from Chroma), combines the excitation light, lets pass blue and reflects violet 495 nm long-pass dichroic mirror, R: 450–490, T: 500–575 and 600–700 nm, 50 mm diameter, 1 mm thick (Chroma, cat. no. T495lpxr, ring mounted, use T495LPXR-UF2 if a rectangular filter is needed as when using the DMF2), to reflect blue and violet light to the sample and let green fluorescence pass to the camera sensor
Thread adapters
Nikon F to C-mount adapter (B&H, cat. no. VELACNF)
2× Sensei 72–52 mm step-down ring (B&H, cat. no. SESDR7252)
3× Adapter with external SM2 threads and internal SM1 threads (Thorlabs, cat. no. SM2A6)
2× Coupler with external threads, 1″ long (Thorlabs, cat. no. SM1T10)
4× Coupler with external threads, 1/2″ long (Thorlabs, cat. no. SM2T2)
2× SM1 lens tube, 1″ thread depth with one retaining ring (Thorlabs, cat. no. SM1L10)
Macroscope frame and assembly
▲ CRITICAL Parts numbers are for an imperial system; an alternative setup using metric parts is possible.
60-mm cage cube (Thorlabs, cat. no. LC6W or DMF2 for easier assembly)
Mounting platform for 60-mm cage cube, Imperial taps (Thorlabs, cat. no. LB3C)
2″ Optic mount for 60-mm cage cube with setscrew optic retention (Thorlabs, cat. no. LB5C1)
2× Light tight blank cover plate (Thorlabs, cat. no. LB1C)
60-mm cage system removable filter holder (Thorlabs, cat. no. LCFH2 or omit if the filter is mounted directly on the DMF2)
30-mm cage cube (Thorlabs, cat. no. C4W or DFM1 for easier assembly)
Fixed cage cube platform for C4W/C6W (Thorlabs, cat. no. B3C)
30-mm cage compatible rectangular filter mount (Thorlabs, cat. no. FFM1)
Lens tube spacer, 1″ long (Thorlabs, cat. no. SM1S10)
Adapter plate for 1.5″ post mounting clamp (Thorlabs, cat. no. C1520)
Post mounting clamp (Thorlabs, cat. no. C1511)
Mounting base (Thorlabs, cat. no. BA2)
3× Mounting posts (Thorlabs, cat. no. P8)
4× Optical posts (Thorlabs, cat. no. TR8)
Optical breadboard 36″ × 72″ × 2.28″ with 1/4″ 20 mounting holes (Thorlabs, cat. no. B3672FX). Alternatively for a smaller setup: optical breadboard, 36″ × 48″ × 2.28″ with 1/4″−20 mounting holes (Thorlabs, cat. no. B3648FX)
High rigid frame 900 × 1,500 mm (3′ × 5′) (Thorlabs, cat. no. PFR90150-8). Alternatively for 36″ × 48″ breadboard: high rigid frame 750 × 900 mm (2.5′ × 3′) (Thorlabs, cat. no. PFR7590–8)
Lab Jack (Thorlabs, cat. no. L490) to adjust the height of the behavioral setup. Alternatively, the macroscope height can be adjusted with a focusing module (Thorlabs, cat. no. ZFM1020)
Macroscope excitation light and wavelength alternation
GCaMP excitation LED, 470 nm (Thorlabs, cat. no. M470L4)
Hemodynamic correction LED, 405 nm (Thorlabs, cat. no. M405L4)
2× Adjustable collimation adapter (Thorlabs, cat. no. SM2F32-A)
2× T-cube LED driver (Thorlabs, cat. no. LEDD1B)
Teensy 3.2 (PJRC, cat. no. TEENSY32)
SMA female RF coaxial adapter PCB mount plug (Amazon, ASIN B06Y5WZ1TK)
USB to micro USB, 6 ft (Digikey, cat. No. AE10342-ND)
Prototyping perforated breadboard 2″ × 3.2″ (Adafruit, cat. no. 1609)
BNC connector jack, female socket 75 Ohm panel mount (Digikey, cat. no. A97562-ND)
Adapter coaxial connector, female socket to BNC jack (Digikey, cat. no. ARFX1069-ND)
Coaxial BNC to BNC male to male RG-58 29.53″, 750.00 mm (Digikey, cat. no. J10339-ND)
Coaxial SMA to SMA male to male RG-316 5.906″, 150.00 mm (Digikey, cat. no. J10284-ND)
BNC, male plug to wire lead 72.0″, 1,828.80 mm (Digikey, cat. no. 501–2556-ND)
Other
Data acquisition device to synchronize with behavior setups (National Instruments, USB-6001, cat. no. 782604-01)
Power meter (Thorlabs, cat. no. PM100D)
Photodiode power sensor, 400–1,100 nm (Thorlabs, cat. no. S120C)
3D-printed light-shielding cone (custom piece printed from Shapeways (https://www.shapeways.com) in opaque plastic (e.g., Multi Jet Fusion PA 12, design files in https://github.com/churchlandlab/WidefieldImager/blob/master/CAD/LightCone/LargerWidefieldCone_v3.stl)
Data acquisition computer, at least 4-core CPU with 32 GB RAM, with a full-length PCI slot for the frame grabber and a separate solid state drive for data acquisition
Software
MATLAB 2016b or newer, image/data acquisition toolboxes (https://www.mathworks.com/)
PCO frame grabber drivers (https://www.pco.de/fileadmin/user_upload/pco-driver/PCO_SISOINSTALL_5.7.0_0002.zip)
PCO Camware (https://www.pco.de/fileadmin/user_upload/pco-software/SW_CAMWAREWIN64_410_0001.zip)
PCO SDK (https://www.pco.de/fileadmin/user_upload/pco-software/SW_PCOSDKWIN_125.zip)
PCO MATLAB toolkit (https://www.pco.de/fileadmin/user_upload/pco-software/SW_PCOMATLAB_V1.1.2.zip)
Recording software: https://github.com/churchlandlab/WidefieldImager ▲ CRITICAL A fully functioning Python alternative can be found here: https://bitbucket.org/jpcouto/labcams
Preprocessing and graphical interface: https://github.com/churchlandlab/wfield
NeuroCAAS computing platform: http://neurocaas.org
Reagent setup
! CAUTION Prepare reagents using aseptic techniques. Some of the reagents are controlled substances.
Ketamine/dexmedetomidine anesthetic
Mix 8.4 mL of 0.9% (wt/vol) NaCl with 0.6 mL of ketamine (from a 100 mg/mL stock) and 1 mL of dexmedetomidine (from a 0.5 mg/mL stock) in a sterile vial. This solution can be stored at room temperature (20–25 °C) and protected from light for no longer than 30 d. Inject 0.3 mL for a 30 g mouse to deliver a dose of 60 ketamine/0.5 dexmedetomidine (mg/kg).
Meloxicam
Mix 9 mL of 0.9% (wt/vol) NaCl and 1 mL of Meloxicam (5 mg/mL stock) into a sterile vial. This solution can be stored for several months at room temperature (20–25 °C).
Equipment setup
Implants for head fixation
The implants for head fixation (headpost) are laser cut from a 0.05″ titanium sheet. An aluminum custom-machined holder secures the headpost during imaging (CAD files in https://github.com/churchlandlab/WidefieldImager).
Procedure
Macroscope assembly ● Timing 2–3 h
▲ CRITICAL Clear the optical table, and gather the tools and parts for the build.
-
1
Assembling the 60-mm cage cube (Steps 1–4; Fig. 4a): attach the optic mount (Thorlabs, LB5C1) to the mounting platform (Thorlabs, LB3C). Place the 495-nm long-pass dichroic mirror (Chroma, T495lpxr) in the optic mount.
▲ CRITICAL STEP Orient dichroic correctly. The correct orientation is in the datasheet that comes from the manufacturer. When handling optics, use gloves and lens paper, and be careful not to scratch the optics.
-
2
Tighten two couplers (Thorlabs, SM2T2) to the cage cube (Thorlabs, LC6W).
-
3
Drill four holes (slightly larger than the 8–32 screws) on a cover plate (Thorlabs, LB1C); these will attach to the post clamp (Thorlabs, C1511), as shown in Fig. 4a. Use a workshop (e.g., from your institute) if possible. Cover the side of the cube (that will be used for supporting the macroscope, see Fig. 4d) with the machined cover plate (Thorlabs, LB1C). Attach an additional cover plate (Thorlabs, LB1C), orthogonal to the machined cover plate, on the cage cube. Note that mounting holes are already included when using a DFM2 instead of a LC6W cage cube.
-
4
Attach the SM1 to SM2 adapter (Thorlabs, SM2A6) to the side directly facing the dichroic.
-
5
Assembling the 30-mm cage cube (Steps 5–10; Fig. 4b): attach the rectangular optic mount (Thorlabs, FFM1) to the mounting platform (Thorlabs, LB3C). Secure the excitation dichroic mirror (Edmund, 87–063) on the optic mount.
-
6
Attach two SM1 couplers (Thorlabs, SM1T10) to the cage cube (Thorlabs, C4W). Attach an SM1 end cap (Thorlabs, SM1CP2) to the bottom of the cube. Attach the assembly from Step 5 to the cage cube. Seal the cage cube with a blanking cover plate (Thorlabs, B1C) on the side opposing the dichroic mirror.
-
7
Secure the violet bandpass filter (Edmund, 65–133) to a lens tube using the retainer rings (Thorlabs, SM1L10). Attach the lens tube to the top of the cage cube. Repeat for the blue bandpass filter (Chroma ET, 470/40x) and connect the assembly to the side of the cage cube.
▲ CRITICAL STEP Follow the filter manufacturer’s instructions to orient the filter correctly. Install the LEDs so that violet light is reflected and blue light passes through the dichroic mirror.
-
8
Connect the 470 nm excitation LED (Thorlabs, M470L4) to one of the collimators (Thorlabs, SM2F32-A). Attach the whole assembly to the blue filter lens tube using an SM2 adapter (Thorlabs, SM2A6).
-
9
Similar to Step 8, connect the 405 nm LED (Thorlabs, M405L4).
-
10
Connect the 30-mm cube assembly to the larger 60-mm cube, by connecting a lens tube spacer (SM1S10) to the SM2-adaptor ring (Thorlabs, SM2A6).
-
11
Connect the LEDs to the T-cube LED drivers (Thorlabs, LEDD1B), and set the drivers current limit to the maximum (arrow on the side of the driver) using a screwdriver.
▲ CRITICAL STEP The maximum driver current is 1.2 A, but the LEDs are rated for 1 A. In our experience, using 1.2 A works and provides more power but might reduce LED lifetime.
-
12
Connecting the lenses, camera, and green filter (Steps 12–16; Fig. 4c): attach a 72–52 mm step-down ring (B&H, SESDR7252) to the filter threading of the Rokinon 85 mm lens (B&H, RO8514S). Use a coupler (SM2T2) to secure the step-down ring and lens to the bottom of the cage cube (Fig. 4a). The lens must be attached in an inverted position (with the lens’ camera mount facing away from the cage cube).
▲ CRITICAL STEP Using a lens with different threading may require a different step-down ring.
-
13
Attach the Nikon F to C-mount adapter (B&H, VELACNF) to the PCO Edge camera.
-
14
Secure a 72–52 mm step-down ring (B&H, SESDR7252) to the Nikon 105 mm lens (B&H, NI1052DAF). Attach the Nikon 105 mm lens (B&H, NI1052DAF) to the f-mount adapter and lock it in place.
▲ CRITICAL STEP Like for Step 12, a different lens may require a different step-down ring.
-
15
Prepare the filter assembly. Secure the green bandpass filter (Edmund, 86–963) to the filter holder (Thorlabs, LCFH2). Attach two couplers (Thorlabs, SM2T2) to the filter holder cage. Connect the holder to the step-down ring on the camera assembly and to the top of the 60-mm cage cube (Fig. 4a).
-
16
Install the frame grabber in the acquisition computer. Turn the computer on and install the camera drivers and software following the camera manufacturer instructions (PCO frame grabber drivers, PCO Camware and PCO SDK). Connect the cables from the camera to the computer. Connect the power cable of the camera.
-
17
Securing the wide-field assembly to the optical table (Fig. 4d): Combine three large optical posts (Thorlabs, P8), attach them to the mounting base (Thorlabs, BA2) and secure the assembly to the optical table. Attach four optical posts (Thorlabs, TR8) to the machined cover plate (from Step 3). Secure the optical posts to the post clamp (Thorlabs, C1511). Slide the full assembly on the 1.5″ optical posts (Thorlabs, P8).
? TROUBLESHOOTING
-
18
Calibrating the excitation light path (Steps 18–20; Fig. 4d): loosen the setscrew on the collimators (from Step 9 and 8). Remove the dichroic on the 60-mm cage cube (from Step 1). Loosen the cover on the 30-mm cage cube (from Step 5) enough to rotate the mirror. Turn on the blue and violet LEDs, using a moderate power level to be able to see both. Adjust the collimators to obtain a small square image of each LED on the back of the 60-mm cube. Rotate the dichroic mirror (Step 5, Extended Data Fig. 2a) to center the violet light spot on the blue. Secure the dichroic in place when the violet and blue light spots are directly on top of each other. Put the dichroic back in the 60-mm cage cube.
▲ CRITICAL STEP Alignment between the two excitation LEDs is a critical step that if done improperly, will impair the hemodynamic correction. This step is not required if using a DFM1 cage cube because the mirror is fixed at 45° in the kinematic filter mount.
? TROUBLESHOOTING
-
19
Turn the camera on, and open the Camware software in the computer. Click the preview button. Activate the grid in the software using the dropdown menu (right click the image). Open the aperture of both lenses completely (rotate clockwise). Place a white paper under the Rokinon lens (i.e., the objective) at focus height. Loosen the dichroic in the 60-mm cage cube. Turn on the blue LED until it almost saturates the center of the preview image (Extended Data Fig. 2b). Rotate the dichroic to center the light in the camera image. Secure the dichroic in place by tightening the screws.
▲ CRITICAL STEP This step is essential to illuminate the sample evenly. This step is not required if using a DFM2 cage cube because the mirror is fixed at 45° in the kinematic filter mount.
? TROUBLESHOOTING
-
20
In the Camware software, activate the line profiler on the dropdown menu. Draw a line that spans the camera sensor (Extended Data Fig. 2c). Adjust the collimators for each LED until the light is uniformly covering the entire preview image and the profile histogram is flat on both channels (Extended Data Fig. 2c). Use a power meter (Thorlabs, PM100D; probe S120C) to measure the power at the sample (470 nm, ~160 mW with 1.2 A; 405 nm, ~44 mW with 1.2 A).
▲ CRITICAL STEP This step is important to achieve even illumination of the sample.
? TROUBLESHOOTING
-
21
Synchronizing the illumination and the camera (Steps 21–24): Connect line 4 of the camera (all lines exposure signal) to the microcontroller (PJRC, Teensy 3.2) circuit (pin 3). Connect the LED drivers to pin 5 (470 nm LED) and 6 (405 nm LED) (Fig. 2b,c). Connect the microcontroller to the computer and load the Teensy firmware (LEDswitcher from the WidefieldImager repository) using the Arduino IDE.
-
22
Connect the DAQ (National Instruments; USB-6001) to the acquisition computer. Connect the Teensy microcontroller and the digital and analog lines as shown in Fig. 2c.
▲ CRITICAL STEP The trigger signals control the start and end of each trial. Analog input ports are used to synchronize with behavior offline.
-
23
Set the LED drivers to triggered mode (switch in the middle position). Configure camera output sync in the Camware software: go to Features | Hardware IO control | Status Expos | Signal: Status Expos; Timing: Show common time of all lines; Status: ON; Signal: High.
-
24
Clone or download the github repository https://github.com/churchlandlab/WidefieldImager, and add it to the MATLAB path. Install the pco.matlab adaptor package (PCO, check materials). Type ‘imaqhwinfo’ in MATLAB. The camera should appear as ‘pcocameraadaptor’. Type ‘WidefieldImager’ in the command window, and click ‘start preview’ to see a video stream from the camera. Click ‘Light OFF’ to switch on the blue LED.
? TROUBLESHOOTING
Surgical preparation to image through the intact skull ● Timing 1–2 h
▲ CRITICAL Apply aseptic techniques during Steps 25–40.
-
25
Place the animal in the induction chamber, prefilled with isoflurane (3% in 100% oxygen 0.5–1 L/min) for 20–60 s. This step reduces stress and facilitates handling.
! CAUTION All experiments involving animals must be performed in accordance with relevant institutional and governmental guidelines and regulations.
-
26
Induce anesthesia with an intraperitoneal injection of ketamine–dexmedetomidine (60/0.5 mg/kg in saline). Let the anesthesia take full effect (usually 5–10 min). Confirm the anesthesia depth with the absence of toe-pinch reflexes. The surgical procedures can only start when the mouse is deeply anesthetized.
-
27
Place the animal in the stereotaxic frame, making sure that the earbars and the incisors are firmly holding the skull. Make sure that the feedback-controlled heating blanket is operating and maintaining body temperature.
-
28
Inject meloxicam (1–2 mg/kg) subcutaneously in the back of the animal for systemic analgesia. Apply lidocaine on the scalp for topical analgesia.
-
29
Cover the eyes with ophthalmic ointment to prevent drying.
-
30
Trim the hair in a region starting 2 mm in front of the eyes until the middle of the ears (anterior-posterior) and below the level of the eyes (lateral). Use a pair of scissors or alternatively an electric trimmer. Apply a hair removal product (e.g., Nair) if needed to remove all hair from the surgical area.
-
31
Clean the area using cotton swabs alternating between 70% (vol/vol) EtOH and betadine.
-
32
Make four small incisions using a fine pair of scissors in the locations highlighted in Fig. 5a. Remove the scalp using the scissors to cut between the incisions and a pair of forceps.
-
33
Use the number 11 scalpel blade or a pair of fine forceps to push the periosteum to the edges of the skull. Scrape any remaining periosteum until the skull is clean.
▲ CRITICAL STEP This step is essential for proper optical clearance. It is critical that there are no scratches or damage to the skull.
-
34
Retract the temporal muscles slightly using a pair of closed forceps. Use the same approach to carefully retract the muscles posterior to the lambda suture.
-
35
Apply Vetbond to the exposed skin 1 mm around the cuts (Fig. 5b).
-
36
Apply dental acrylic to cuts and in the temporal areas. The acrylic should form a flat surface to which the headpost can be attached (Fig. 5c).
-
37
Place the headpost, and hold it in place with Vetbond and dental cement (Fig. 5d).
-
38
After the cement is cured, apply cyanoacrylate glue to the skull (Fig. 5e). Start at the sutures, and cover the entire skull. Only apply a small amount of cyanoacrylate, and form a thin layer over the skull. Then wait for the layer to cure and repeat this step four to five times. Use a toothpick to distribute the cyanoacrylate.
▲ CRITICAL STEP The cyanoacrylate cures spontaneously within minutes. We found that repeated application of four to five layers of cyanoacrylate produces the best results. It takes ~5 min for the first layer to cure. Subsequent layers tend to cure more quickly. Be careful because it can cure faster it makes contact with the dental cement. Too fast curing from the sides can cause heat, which is harmful to the clearing. To avoid this, build the first layer from the center and avoid touching the cement walls. Fill gaps on the sides when adding subsequent layers.
-
39
Wait until the glue is dry. Skull clearing should set in gradually, and brain vessels should be clearly visible after 5–10 min. Inject Antisedan (0.05 mg/kg) to reverse anesthesia, and monitor the animal as it recovers.
? TROUBLESHOOTING
-
40
Monitor the animal recovery for at least 48 h, and inject analgesia as needed (inject meloxicam (2 mg/kg) in saline every 24 h).
▲ CRITICAL STEP While optical clarity settles less than 1 h after the procedure, allow enough time for recovery and habituation before imaging to minimize animal stress.
? TROUBLESHOOTING
Fig. 5 |. Overview of the surgical procedures.
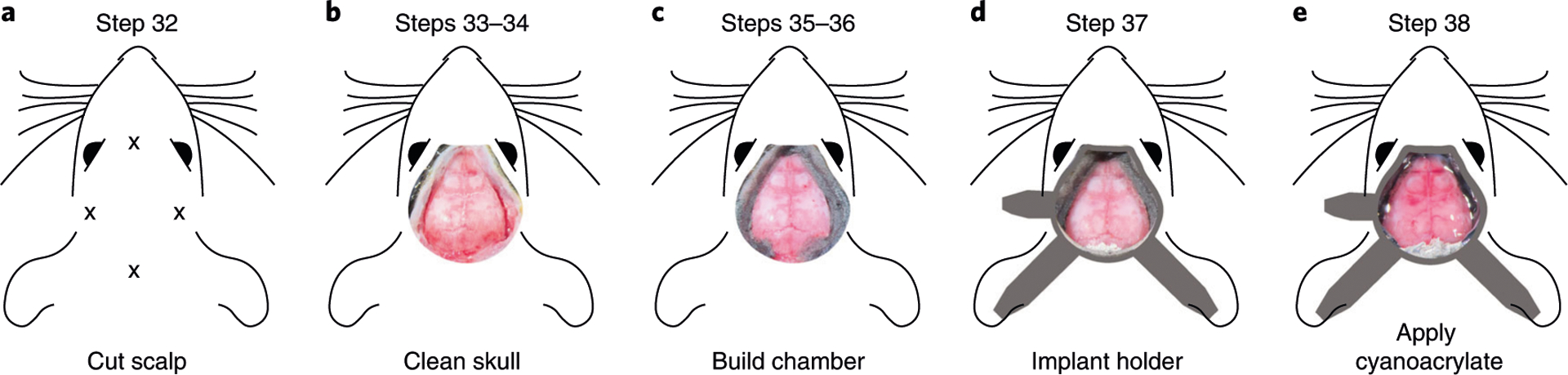
a, Location of scalp cuts in Step 32. b, Clean skull preparation with temporal muscles retracted from the skull in Steps 33–34. c, Formation of the dental cement chamber in Steps 35–36. d, Placement of the headpost implant for head restraint (Step 37). e, Completed skull preparation and headpost at the end of the surgery (Step 38). Animal experiments followed NIH guidelines and were approved by the Institutional Animal Care and Use Committee of Cold Spring Harbor Laboratory.
Imaging ● Timing 1–2 h
-
41
Open the Camware software. Go to the Camera settings tab and select I/O Signals. On Line 4 set Signal to Status Expos. Click the clock icon and select ‘Show common time of “All Lines”‘.
▲ CRITICAL STEP This step is critical to avoid artifacts due to the moving shutter. The camera setting ensures that the exposure trigger is active only when all lines of the camera are exposing. Cameras with global shutter allow all lines to be read simultaneously but usually do not allow high sampling rates.
-
42
Type the command ‘WidefieldImager’ in the MATLAB command prompt to start the acquisition software (Fig. 2d). Click ‘New animal’ to create a new animal ID, and enter a name for the current experiment in the pop-up dialog. Make sure other settings, such as the trial duration and file path are correct. A detailed, step-by-step instruction manual that includes descriptions of all features and further details for how to perform an imaging session with the software is at https://github.com/churchlandlab/WidefieldImager.
-
43
Attach the animal to the behavior apparatus. Clean the implant with a cotton swab dipped in 70% (vol/vol) EtOH. Center the animal under the objective, and switch on the blue light. Position the light-shielding cone over the implant.
▲ CRITICAL STEP Make sure the shielding blocks all the light but does not push on the animals’ eyes or occludes the field of view. The light shield is essential because the camera operates in rolling shutter mode. It can be omitted, for experiments in absolute darkness. However, there might be activation in visual cortex upon light onset because mice will be able to see the excitation light.
-
44
Open the aperture of the imaging lens completely (Fig. 1c), and adjust the height of the setup until the most lateral brain vessels are clearly in focus. Due to the brain curvature, the center of the implant will appear blurry (Fig. 2d). Adjust the light power of the blue and violet LED.
▲ CRITICAL STEP More excitation light results in stronger fluorescence signals, but too much light will saturate the camera or cause photobleaching of the indicator (Extended Data Fig. 1). We found that light power between 10 and 25 mW is an appropriate range, but the optimal light intensity varies strongly, depending on the localization, expression level and brightness of the indicator. To avoid saturation, we recommend setting the light levels so that the brightest pixels remain below 70% of the cameras full dynamic range. Pixels above this threshold are shown in red in the camera preview window (Fig. 2d). To avoid photobleaching, the light intensity should remain below 50 mW (Extended Data Fig. 1). For dim samples, reduce the framerate to increase the exposure time and therefore the measured signal.
? TROUBLESHOOTING
-
45
Click ‘Take Snapshot’ to save a vessel image to disc for later reference. The depth of field can be extended momentarily to obtain a sharper image of the blood vessels by closing the aperture. To ensure that a blood vessel image is acquired in every session, the recording cannot be started before an image is taken. Make sure that the aperture is fully open during imaging. Set the excitation light to ‘Mixed light’ and click the ‘Wait for trigger’ button. The software is now ready to acquire imaging data and waiting for a ‘trial start’ trigger. The trigger needs to be sent to the NI-DAQ to start acquisition (Fig. 2b). If the DAQ is not connected, you can mimic triggers in software by clicking the ‘Trial Trigger’ button.
-
46
Start the stimulation or behavioral paradigm and send a ‘trial start’ TTL trigger to the DAQ. The software starts acquiring frames and waits for a ‘stimulus trigger’ TTL. When a stimulus trigger is received, the software will record the baseline frames and acquire frames until the end of the poststimulus period. If no stimulus trigger is received within the ‘wait’ period, no data are recorded, the trial is stopped and a new ‘trial start’ trigger needs to be provided. An optional ‘trial end’ trigger can be provided to stop a trial at any time. Monitor the MATLAB command window to ensure that the correct number of frames was saved in each trial. To stop the recording, unlock the recording panel by clicking the ‘Locked’ button, and end the current session by clicking the ‘Wait for Trigger’ button.
? TROUBLESHOOTING
Data preprocessing ● Timing 20 min to 4 h
-
47
Follow the instructions to install the processing tools as described in our GitHub repository: https://github.com/churchlandlab/wfield#installation.
-
48
(Optional) In the terminal run: wfield open_raw <folder name>. To roughly match each imaging session to the Allen Common Coordinate Framework, drag the points from the left, center and right olfactory bulb as well as the base of the retrosplenial cortex to the respective landmarks (Fig. 6a). Press the ‘save’ button to store the results. This step generates a transformation matrix to align the imaging data to the Allen Common Coordinate Framework and is required for some algorithms/analysis (e.g., locaNMF).
-
49Run the analysis through NeuroCAAS (Fig. 6b) either using the graphical user interface (Option A) or uploading data to the website (http://neurocaas.org/, Option B). The website allows the user to run only PMD or LocaNMF, whereas the graphical interface allows the running of all the analysis steps, including also motion correction and hemodynamics compensation.
- Running NeuroCAAS using the web interface
- To run the analysis from the web interface, go to http://neurocaas.org and create user credentials (use group code EF1E04 when creating an account to have access to the test datasets from the website).
- Log in and navigate to the ‘Available analysis’ page.
- Click on ‘View analysis’ followed by ‘Start analysis’.
- Select a dataset by clicking the checkbox next to the files, or drag and drop your own dataset to upload.
- Tune analysis parameters in the configuration file by downloading, changing the parameters value with a text editor and reuploading to the website. Click on a config file to select the parameters.
- Start the analysis by pressing the ‘Submit’ button. A dialog will appear when the analysis completes (in under 15 min for the example datasets) after which the results can be downloaded.
-
Running NeuroCAAS using the GUI▲ CRITICAL To run the entire analysis pipeline with NeuroCAAS, use the graphical user interface. This includes tools for motion correction, PMD, hemodynamic compensation and LocaNMF. Instructions for operating the graphical interface are here: https://github.com/churchlandlab/wfield/blob/master/usecases.md.
-
Open a terminal and type the command ‘wfield ncaas’. The first time, it will ask for the credentials. Close the dialog window after inserting the neurocaas.org credentials.▲ CRITICAL STEP An account on NeuroCAAS is required to run the analysis. Request access to the cshl-wfield-locanmf and wfield-preprocess analysis after creating an account. Test datasets are available in the repository http://labshare.cshl.edu/shares/library/repository/38599/.
- Drag the data folder from the local computer browser (on the left) to the cloud browser window (on the top left).
-
On the dialog window, select the analyses to run and the analysis parameters and select the files to upload.▲ CRITICAL STEP For the compression step, the values for block height and width depend on the frame size and must allow dividing the frame in equal chunks (e.g., for a frame height of 540, we use block height 90; and for frame width 640, block width 80).
- Press ‘Submit’ to close the dialog and add the analysis to the transfer queue.
- To upload data and actually run the analysis press the ‘Submit to NeuroCAAS’ button. You can monitor progress by clicking the log files. The results of preprocessing, i.e., the motion-corrected, denoised, compressed and demixed data, are automatically copied back to the local computer when the analysis completes.
-
Fig. 6 |. Diagram of the data processing pipeline.
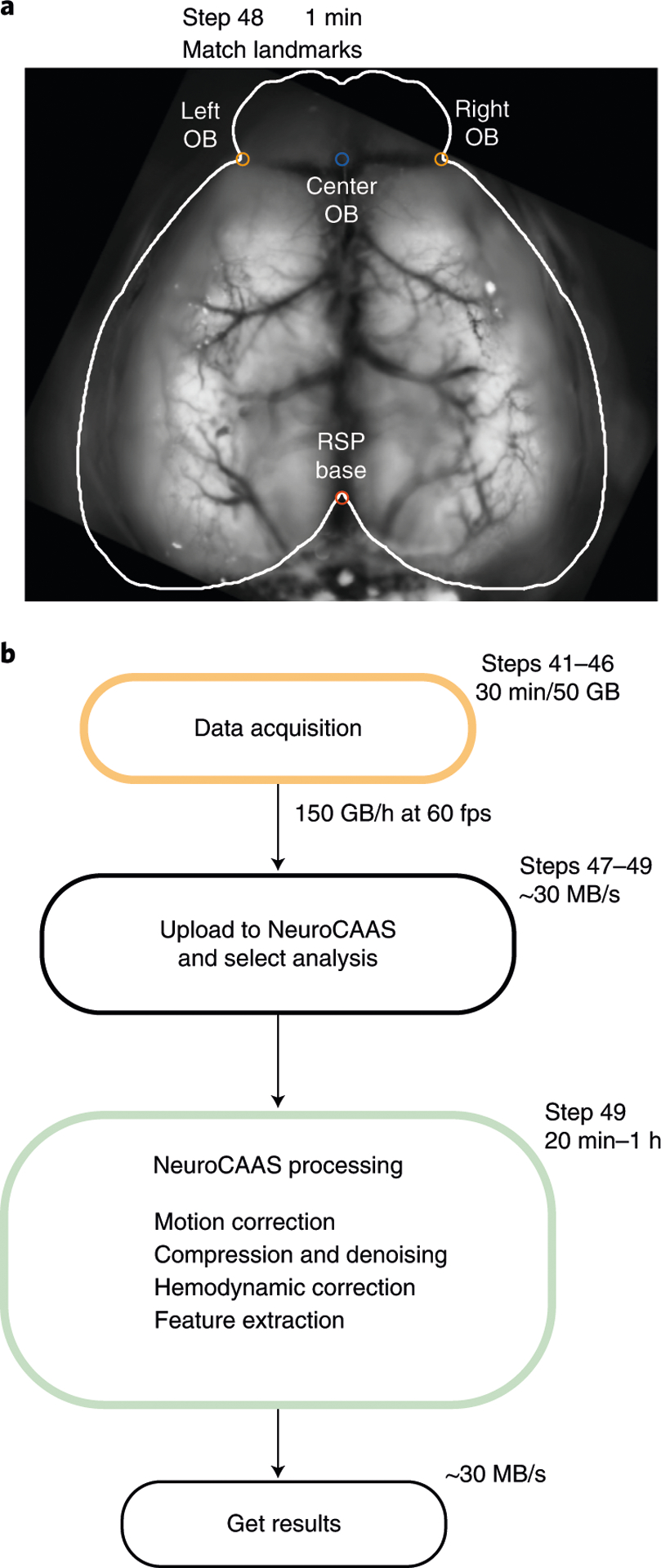
a, Following data acquisition, data from all trials are concatenated (Step 47) and uploaded to the cloud platform NeuroCAAS (Step 49). Preprocessing steps are performed in the cloud (Step 49), and data are retrieved for further analysis (Step 50). b, Landmarks for roughly matching the data to the Allen Common Coordinate Framework (Step 48). This step is required for using locaNMF.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 17 | Lenses wobble | Couplers are not tight | Readjust couplers in Steps 13–15 |
| 18 | Illumination is not even across channels | Improper alignment of the dichroic in the excitation cube | Repeat the alignment Step 18 |
| 19 | No light out of the objective | Misaligned dichroics, incorrect cube orientation | Check the orientation of the filters in Step 7 and the dichroics in Steps 1 and 5 |
| 20 | The object is not fully illuminated | The aperture of the objective (Rokinon lens) is partially closed or the LEDs not collimated | Rotate the aperture of the objective clockwise. Repeat Step 20 |
| 24 | Imager cannot find ‘Dev1’ | DAQ numbering not correct | Use the command daqlist(‘ni’) in MATLAB to discover the name of the USB DAQ device. Edit the daqName in line 63 of WidefieldImager.m |
| 39 | Cyanoacrylate is uneven or there are air bubbles | Too much glue or application too fast | Apply thin layers of cyanoacrylate and allow time to cure |
| Skull clearing is uneven or seems opaque | Glue applied before letting the cement cure | Let the cement cure before applying the cyanoacrylate. If the glue contacts the liquid phase of the cement, it can create uneven patterns | |
| 40 | Mouse is stressed in the setup or has poor task performance | Headpost too close to the eye or light shield is misplaced | Make sure that the implant is at least 3 mm from the eye by creating a structure from dental cement before securing the headpost (Steps 31–34) |
| Slow recovery after surgery, the animal seems irritated by the implant | Excessive dental cement around the implant | Use less dental cement on the sides of the implant, avoid sharp edges or open pockets that might cause skin irritation or infection | |
| 44 | Dirt trapped in the cyanoacrylate after clearing, or dents or cracks appear after weeks | Short curation time | Allow more time for the cyanoacrylate to cure before finishing the procedure. Use a dental drill to remove the debris and reapply the cyanoacrylate |
| No signal after repeated imaging | Photobleaching | Increase the interval between consecutive imaging sessions, reduce LED power and/or decrease the acquisition rate | |
| The imager does not show a preview of the camera | The software could not connect to the camera | Make sure the camera is accessible through Camware. Close Camware and attempt restarting the imager. Restart the computer and check the driver installation if the problem persists | |
| 46 | Error in the number of frames collected | The acquisition settings are incorrect | Check the ‘framerate’, ‘baseline’ and ‘poststim’ duration settings in the imager |
Timing
Steps 1–24, macroscope assembly and alignment: 2–3 h
Steps 1–5, assembling the 60-mm cage cube: 20 min
Steps 5–10, assembling the 30-mm cage cube: 30 min
Steps 12–16, connecting the lenses, camera, and green filter: 10 min
Step 17, securing the wide-field assembly to the optical table: 10 min
Steps 18–20, calibrating the excitation light path: 45 min
Steps 21 and 24, synchronizing the illumination and the camera: 25 min
Steps 25–40, surgical preparation to image through the intact skull: 1–2 h
Steps 41–46, imaging: 1–2 h per session
Steps 47–49, data preprocessing: 15 min to 3 h, depending on the dataset and the analysis
Anticipated results
This procedure allows functional imaging of the entire dorsal cortex through the intact skull of awake mice. There are many possible applications for functional imaging: mapping experiments can identify the location of many sensory areas, based on their neural response to repeated visual or somatosensory stimulation (Fig. 7a,b). By careful quantification of animal behavior, a similar approach can be used to identify areas that are related to specific animal movements (Fig. 7c). Responses to sensory stimulation or movements can also be extracted when mice perform a behavioral task. For example, a linear encoding model can be used to isolate cortical activity patterns that are most related to different task or movement events4,8,46 (Fig. 7d). Because the procedure is minimally invasive and leaves the skull intact, mice can be imaged for long timespans (we confirmed stable implants up to 1 year). This makes wide-field imaging ideal to observe changes in cortical activity during task learning49 (Fig. 7e) or following long-term changes due to perturbations such as a stroke28 or tumor growth50. Highly specific transgenic mouse lines enable imaging genetically defined neuronal subpopulations25 and provide exciting new possibilities when combined with whole-cortex wide-field imaging (Fig. 7f).
Fig. 7 |. Wide-field imaging applications.
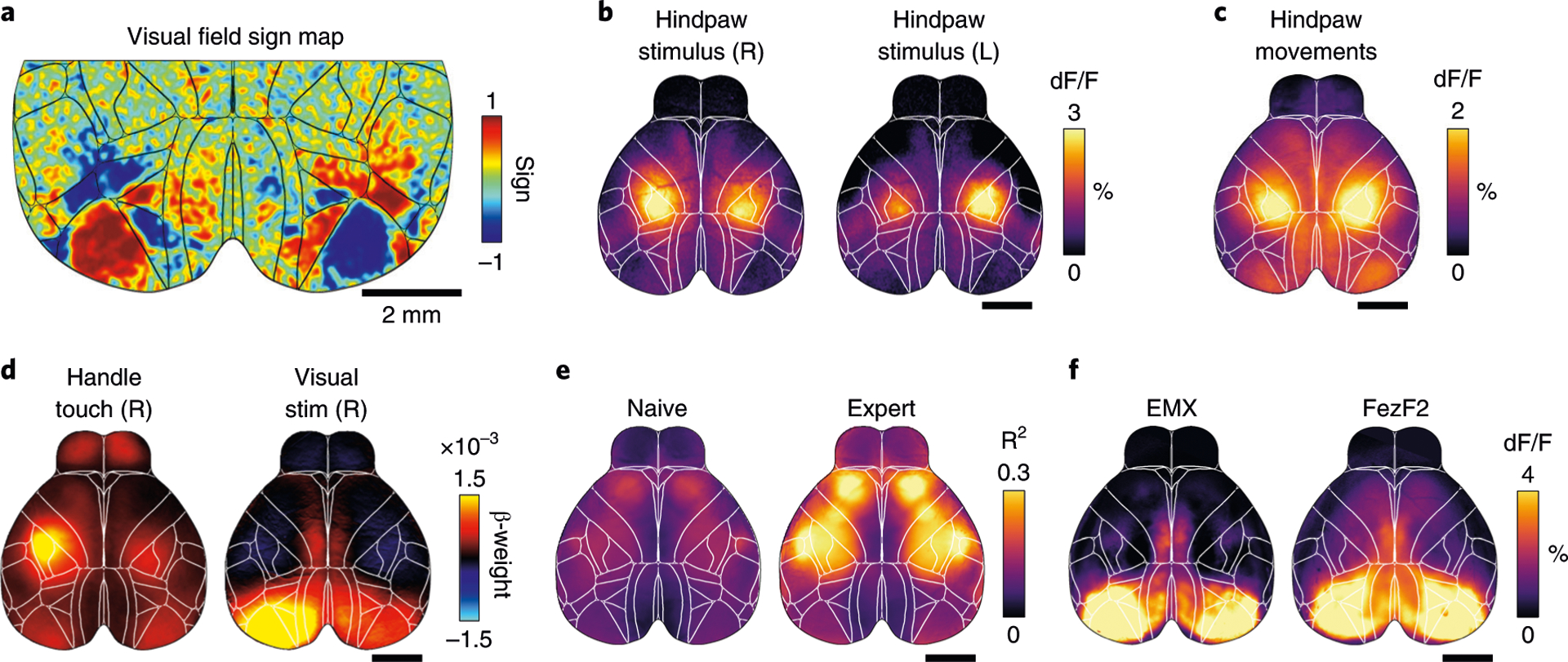
a, Retinotopic mapping reveals the location of primary and secondary visual areas in both hemispheres. b, Somatosensory hindpaw stimulation shows robust and localized activation of somatosensory cortex (scale bar is 2 mm). c, Analyzing data from a pressure sensor can be used to infer hindpaw movements. Cortical activation strongly resembles sensory stimulation data in b. d, Model weights from linear regression analysis can show cortical response patterns to different variables, such as touching a handle or visual stimulation. e, Example data from two recordings in the same mouse, 6 weeks apart. Imaging quality is stable, and task-related cortical activity strongly increases with learning. f, Imaging specific cell populations using transgenic mouse lines. Average dF/F during visual stimulus presentation for a mouse with expression in all pyramidal neurons (controlled by the emx1 gene) or restricted to a subpopulation of layer 5 pyramidal neurons (FezF2). Despite expression being restricted to deep layers, the response to the visual stimuli in the FezF2 mouse line is comparable to the activity recorded in an Emx1 line. Animal experiments followed NIH guidelines and were approved by the Institutional Animal Care and Use Committee of Cold Spring Harbor Laboratory.
Taken together, this protocol puts within reach of many investigators a powerful platform for collecting, processing and analyzing cortex-wide responses in awake animals. Compared with other imaging approaches, the reduced cost, streamlined assembly and integrated analysis tools that define our platform will equip diverse investigators to make new discoveries about brain function.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Photobleaching due to repeated or high-power imaging.
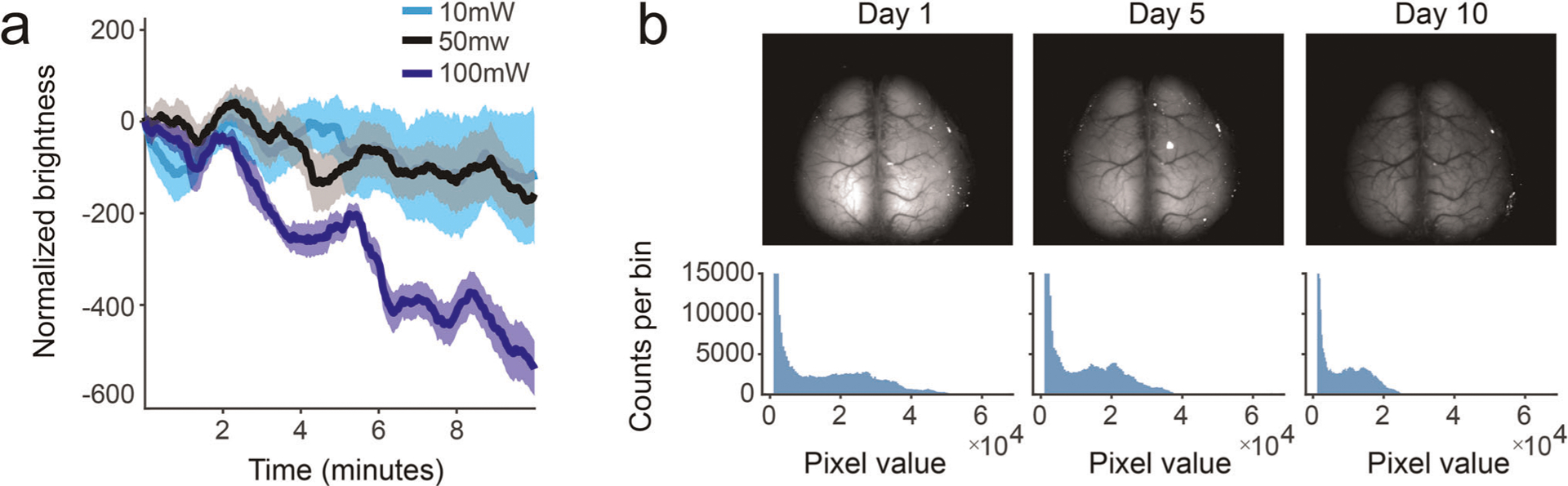
a, Photochemical degradation of the fluorophore can occur over days because of repeated imaging. Decrease in fluorescence can be observed after 10 min when imaging at more than 50 mW of blue light (blue trace); signals are more stable at lower intensities (black, cyan traces). b, Top: images from a mouse expressing GCaMP6 in a subpopulation of cortical excitatory projection neurons. Visible decrease in overall fluorescence is evident after daily imaging at day 5 and 10. Bottom: histogram of pixel intensities corresponding to the images above.
Extended Data Fig. 2 |. Steps for setup calibration.
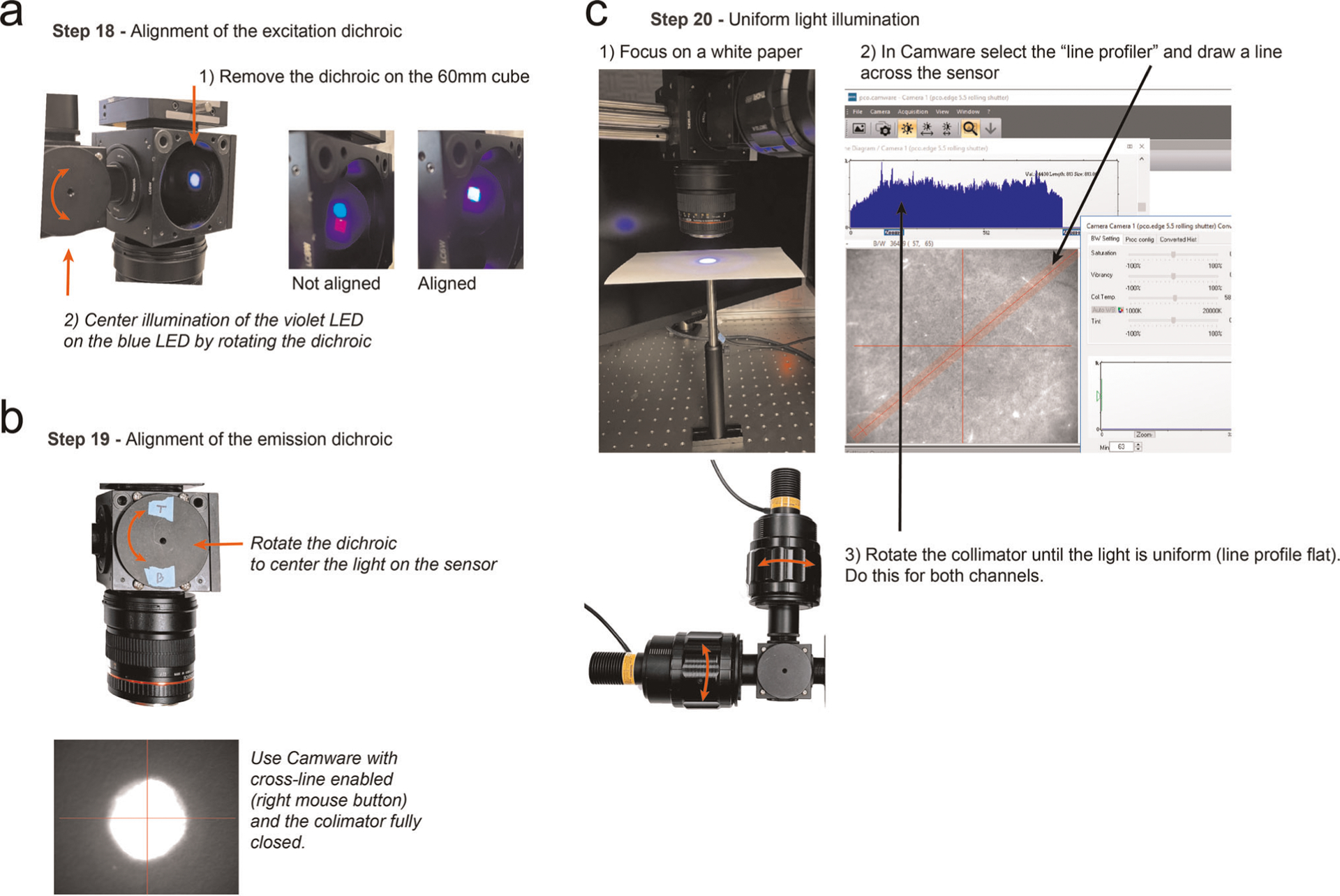
a, Alignment of the excitation dichroic. b, Alignment of the emission dichroic using Camware. c, Procedure for obtaining uniform illumination with the Camware software and the line profiler. The WidefieldImager also has a calibration mode with similar functionality and supporting cameras from multiple vendors.
Acknowledgements
We thank M. Kaufman and K. Odoemene for help with developing early versions of the protocol; P. Gupta, F. Albeanu and J. Wekselblatt for technical advice; N. Steinmetz, M. Pachitariu and K. Harris for help with wide-field analysis; and Z. Josh Huang for providing FezF2 mice. Financial support was received from the Swiss National Science foundation (S.M., grant no. P2ZHP3_161770), the Deutsche Forschungsgemeinschaft (German Research Foundation, DFG - 368482240/GRK2416), the NIH (grant no. EY R01EY022979 and BRAIN initiative 5R01EB026949) and the Army Research Office under contract no. W911NF-16-1-0368 as part of the collaboration between the US DOD, the UK MOD and the UK Engineering and Physical Research Council under the Multidisciplinary University Research Initiative (A.K.C.). X.R.S. was supported by the NINDS BRAIN Initiative of the National Institutes of Health under award number F32MH120888. T.A. was supported by NIH training grant 2T32NS064929-11. S.S. was supported by the Swiss National Science Foundation P400P2 186759 and NIH 5U19NS104649. J.P.C. was supported by Simons 542963 and the McKnight Foundation. L.P. was funded by IARPA MICRONS D16PC00003, NIH 5U01NS103489, 5U19NS104649, 5U19NS107613, 1UF1NS107696, 1UF1NS108213, 1RF1MH120680, DARPA NESD N66001-17-C-4002 and Simons Foundation 543023. L.P. and J.P.C. were supported by NSF Neuronex Award DBI-1707398.
Footnotes
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41596-021-00527-z.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-021-00527-z.
Code availability
Source code used in this protocol is available in the online repositories without access restrictions under a general public license at https://github.com/churchlandlab. The code and NeuroCAAS platform will remain available for the foreseeable future.
Data availability
The raw datasets used to generate the visual sign, stimuli triggered averages and linear regression analysis maps are available in a public repository, maintained by Cold Spring Harbor Laboratory with https://doi.org/10.14224/1.38599. Example datasets to test the analysis pipeline are at http://labshare.cshl.edu/shares/library/repository/38599/2021-01-20-Update/.
References
- 1.Stringer C et al. Spontaneous behaviors drive multidimensional, brainwide activity. Science 364, 255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanni MP, Chan AW, Balbi M, Silasi G & Murphy TH Mesoscale mapping of mouse cortex reveals frequency-dependent cycling between distinct macroscale functional modules. J. Neurosci 37, 7513–7533 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clancy KB, Orsolic I & Mrsic-Flogel TD Locomotion-dependent remapping of distributed cortical networks. Nat. Neurosci 22, 778–786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Musall S, Kaufman MT, Juavinett AL, Gluf S & Churchland AK Single-trial neural dynamics are dominated by richly varied movements. Nat. Neurosci 22, 1677–1686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen WE et al. Global representations of goal-directed behavior in distinct cell types of mouse neocortex. Neuron 94, 891–907.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinto L et al. Task-dependent changes in the large-scale dynamics and necessity of cortical regions. Neuron 104, 810–824.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zatka-Haas P, Steinmetz NA, Carandini M & Harris KD A perceptual decision requires sensory but not action coding in mouse cortex. Preprint at bioRxiv 10.1101/501627 (2020). [DOI] [Google Scholar]
- 8.Salkoff DB, Zagha E, McCarthy E & McCormick DA Movement and performance explain widespread cortical activity in a visual detection task. Cereb. Cortex 30, 421–437 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grinvald A, Lieke E, Frostig RD, Gilbert CD & Wiesel TN Functional architecture of cortex revealed by optical imaging of intrinsic signals. Nature 324, 361–364 (1986). [DOI] [PubMed] [Google Scholar]
- 10.Frostig RD, Lieke EE, Ts’o DY & Grinvald A Cortical functional architecture and local coupling between neuronal activity and the microcirculation revealed by in vivo high-resolution optical imaging of intrinsic signals. Proc. Natl Acad. Sci. USA 87, 6082–6086 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shmuel A & Grinvald A Functional organization for direction of motion and its relationship to orientation maps in cat area 18. J. Neurosci 16, 6945–6964 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrett ME, Nauhaus I, Marshel JH & Callaway EM Topography and areal organization of mouse visual cortex. J. Neurosci 34, 12587–12600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andermann ML, Kerlin AM, Roumis DK, Glickfeld LL & Reid RC Functional specialization of mouse higher visual cortical areas. Neuron 72, 1025–1039 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juavinett AL, Nauhaus I, Garrett ME, Zhuang J & Callaway EM Automated identification of mouse visual areas with intrinsic signal imaging. Nat. Protoc 12, 32–43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferezou I et al. Spatiotemporal dynamics of cortical sensorimotor integration in behaving mice. Neuron 56, 907–923 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Akemann W, Mutoh H, Perron A, Rossier J & Knöpfel T Imaging brain electric signals with genetically targeted voltage-sensitive fluorescent proteins. Nat. Methods 7, 643–649 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Wekselblatt JB, Flister ED, Piscopo DM & Niell CM Large-scale imaging of cortical dynamics during sensory perception and behavior. J. Neurophysiol 115, 2852–2866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilad A, Gallero-Salas Y, Groos D & Helmchen F Behavioral strategy determines frontal or posterior location of short-term memory in neocortex. Neuron 99, 814–828.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Guo ZV et al. Flow of cortical activity underlying a tactile decision in mice. Neuron 81, 179–194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silasi G, Xiao D, Vanni MP, Chen ACN & Murphy TH Intact skull chronic windows for mesoscopic wide-field imaging in awake mice. J Neurosci Methods 267, 141–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunningham JP & Yu BM Dimensionality reduction for large-scale neural recordings. Nat. Neurosci 17, 1500–1509 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchanan EK et al. Penalized matrix decomposition for denoising, compression, and improved demixing of functional imaging data Preprint at https://arxiv.org/abs/1807.06203 (2018).
- 23.Saxena S et al. Localized semi-nonnegative matrix factorization (LocaNMF) of widefield calcium imaging data. PLoS Comput. Biol 16, e1007791 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim EJ, Juavinett AL, Kyubwa EM, Jacobs MW & Callaway EM Three types of cortical layer 5 neurons that differ in brain-wide connectivity and function. Neuron 88, 1253–1267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matho KS et al. Genetic dissection of glutamatergic neuron subpopulations and developmental trajectories in the cerebral cortex. Preprint at bioRxiv 10.1101/2020.04.22.054064 (2020). [DOI] [Google Scholar]
- 26.Murphy TH et al. High-throughput automated home-cage mesoscopic functional imaging of mouse cortex. Nat. Commun 7, 11611 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy TH et al. Automated task training and longitudinal monitoring of mouse mesoscale cortical circuits using home cages. eLife 9, e55964 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cramer JV et al. In vivo widefield calcium imaging of the mouse cortex for analysis of network connectivity in health and brain disease. NeuroImage 199, 570–584 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Shimaoka D, Harris KD & Carandini M Effects of arousal on mouse sensory cortex depend on modality. Cell Rep 25, 3230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dana H et al. Sensitive red protein calcium indicators for imaging neural activity. eLife 5, e12727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao D et al. Mapping cortical mesoscopic networks of single spiking cortical or sub-cortical neurons. eLife 6, e19976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deffieux T, Demene C, Pernot M & Tanter M Functional ultrasound neuroimaging: a review of the preclinical and clinical state of the art. Curr. Opin. Neurobiol 50, 128–135 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Lake EMR et al. Simultaneous cortex-wide fluorescence Ca 2+ imaging and whole-brain fMRI. Nat. Methods 17, 1262–1271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shemesh OA et al. Precision calcium imaging of dense neural populations via a cell body-targeted calcium indicator. Preprint at bioRxiv 10.1101/773069 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abe T, Kinsella I, Saxena S, Paninski L & Cunningham JP Neuroscience cloud analysis as a service. Preprint at bioRxiv 10.1101/2020.06.11.146746 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ratzlaff EH & Grinvald A A tandem-lens epifluorescence macroscope: hundred-fold brightness advantage for wide-field imaging. J. Neurosci. Methods 36, 127–137 (1991). [DOI] [PubMed] [Google Scholar]
- 37.Chen T-W et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma Y et al. Wide-field optical mapping of neural activity and brain haemodynamics: considerations and novel approaches. Phil. Trans. R. Soc. B 371, 20150360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Musall S, Haiss F, Weber B & von der Behrens W Deviant processing in the primary somatosensory cortex. Cereb. Cortex 27, 863–876 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Lerner TN et al. Intact-brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell 162, 635–647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valley MT et al. Separation of hemodynamic signals from GCaMP fluorescence measured with wide-field imaging. J. Neurophysiol 123, 356–366 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Vanni MP & Murphy TH Mesoscale transcranial spontaneous activity mapping in GCaMP3 transgenic mice reveals extensive reciprocal connections between areas of somatomotor cortex. J. Neurosci 34, 15931–15946 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao D et al. Mapping cortical mesoscopic networks of single spiking cortical or sub-cortical neurons. eLife 6, e19976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim TH et al. Long-term optical access to an estimated one million neurons in the live mouse cortex. Cell Rep 17, 3385–3394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghanbari L et al. Cortex-wide neural interfacing via transparent polymer skulls. Nat. Commun 10, 1500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinmetz NA, Zatka-Haas P, Carandini M & Harris KD Distributed coding of choice, action and engagement across the mouse brain. Nature 576, 266–273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steinmetz NA et al. Aberrant cortical activity in multiple GCaMP6-expressing transgenic mouse lines. eNeuro 10.1523/ENEURO.0207-17.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oh SW et al. A mesoscale connectome of the mouse brain. Nature 508, 207–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilad A & Helmchen F Spatiotemporal refinement of signal flow through association cortex during learning. Nat. Commun 11, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montgomery MK et al. Glioma-induced alterations in neuronal activity and neurovascular coupling during disease progression. Cell Rep 31, 107500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
Related links
Key reference using this protocol
Musall, S. et al. Nat. Neurosci. 22, 1677–1686 (2019): https://doi.org/10.1038/s41593-019-0502-4
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw datasets used to generate the visual sign, stimuli triggered averages and linear regression analysis maps are available in a public repository, maintained by Cold Spring Harbor Laboratory with https://doi.org/10.14224/1.38599. Example datasets to test the analysis pipeline are at http://labshare.cshl.edu/shares/library/repository/38599/2021-01-20-Update/.