

Abstract
Growing evidence suggests that plasmin is involved in a number of physiological processes in addition to its key role in fibrin cleavage. Plasmin inhibition is critical in preventing adverse consequences arising from plasmin overactivity, e.g., blood loss that may follow cardiac surgery. Aprotinin was widely used as an antifibrinolytic drug before its discontinuation in 2008. Tranexamic acid and ε-aminocaproic acid, two small molecule plasmin inhibitors, are currently used in the clinic. Several molecules have been designed utilizing covalent, but reversible, chemistry relying on reactive cyclohexanones, nitrile warheads, and reactive aldehyde peptidomimetics. Other major classes of plasmin inhibitors include the cyclic peptidomimetics and polypeptides of the Kunitz and Kazal-type. Allosteric inhibitors of plasmin have also been designed including small molecule lysine analogs that bind to plasmin’s kringle domain(s) and sulfated glycosaminoglycan mimetics that bind to plasmin’s catalytic domain. Plasmin inhibitors have also been explored for resolving other disease states including cell metastasis, cell proliferation, angiogenesis, and embryo implantation. This review highlights functional and structural aspects of plasmin inhibitors with the goal of advancing their design.
Keywords: plasmin(ogen), serine proteases antifibrinolytics, tranexamic acid, aprotinin, cyclic peptidomimetics, glycosaminoglycan mimetics, allosteric inhibition
1. INTRODUCTION
Given the key role of plasmin in fibrinolysis, plasmin inhibitors are used in the clinic to treat hyperfibrinolysis-associated bleeding events and adverse consequences. Hyperfibrinolysis-associated bleeding occurs in many major surgeries that require blood transfusion.1 Approximately 55% of cardiac surgery patients receive blood transfusions.2 Such transfusions are associated with longer hospital stay, multi-organ dysfunction and increased mortality.3 The estimated cost of transfusing one unit of blood is $700–$1200 in United States,4 which justifies the use of relatively inexpensive antifibrinolytic agents. Nearly 70% of cardiac surgeries performed in United States utilize antifibrinolytics to reduce the economical burden.5 Antifibrinolytics can also be used as adjunct therapy to control hemorrhage in some cases of disseminated intravascular coagulation (DIC), which is a hyperfibrinolysis-associated secondary disorder.6, 7 DIC, in particular, occurs in 40% of patients with sepsis leading to a mortality rate of 50–75%.7 DIC patients with a primary hyperfibrinolytic state and having severe bleeding can be treated with lysine analogs, a group of interesting plasmin inhibitors.6 In addition, hemophilia,8 menorrhagia,9 von Willebrand syndrome,10 and thrombolytics-induced bleeding can be fully or partially managed by the antifibrinolytic activity of plasmin inhibitors.11
Hemostasis concludes with thrombin cleavage of fibrinogen to generate fibrin monomers that are rapidly crosslinked by factor XIIIa (Fig. 1).12 Under normal physiologic conditions, the fibrin-rich clot is dissolved by plasmin, which is produced following activation of plasminogen. This activation process takes place on the surface of fibrin. The C-terminal region of fibrin monomers that are rich in lysine residues facilitate binding to the lysine-binding sites (LBSs) on both plasminogen and tPA13-16 resulting in the formation of fibrin-plasminogen-tPA ternary complex, which initiates the plasminogen activation process. The plasmin so formed remains bound to fibrin. Plasmin that disengages from fibrin is rapidly neutralized by α2-antiplasmin, which is present in plasma at high concentrations. This helps localize plasmin’s proteolytic activity.17, 18 Fibrinolysis can also be regulated by thrombin-activatable fibrinolysis inhibitor,19 plasminogen activator inhibitor-1 and -2,20, 21 or α2-macroglobulin.22 In a clinical setting, fibrinolysis can be enhanced by using thrombolytics such as the fibrin-cleaving preparations of streptokinase, urokinase, or tissue plasminogen activator (tPA).23
Figure 1.
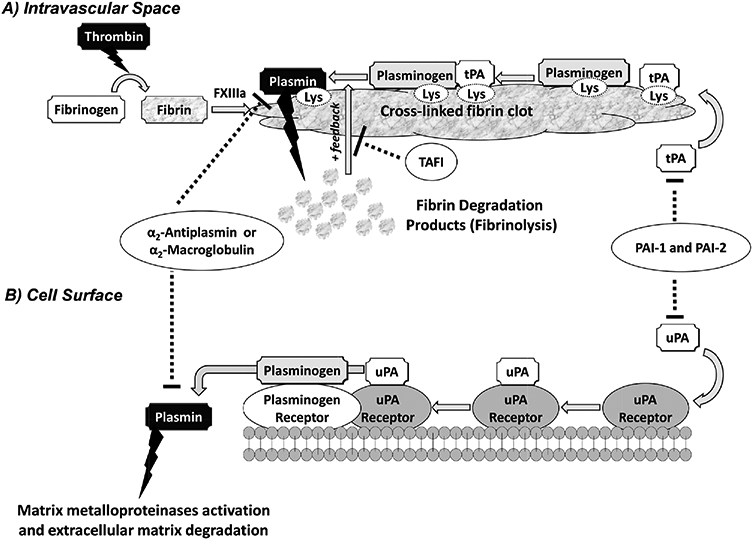
A simplified schematic representation of the plasminogen-plasmin system. Plasminogen is activated in the intravascular space by tissue plasminogen activator (tPA) (A) or at the cell surface by urokinase plasminogen activator (uPA) (B). (A) During coagulation, thrombin converts fibrinogen into soluble fibrin monomers, which cross-link by the action of factor XIIIa resulting in formation of insoluble cross-linked fibrin clot. If the fibrinolysis is to be initiated, plasminogen and tPA bind to fibrin through their lysine-binding sites (LBSs) present on the kringle domains. Formation of such ternary complex activates plasminogen and releases plasmin, which hydrolyzes fibrin. In a positive feedback mechanism, plasmin promotes its own formation by exposing more C-terminal lysine residues of fibrin. Four physiologic inhibitors regulate fibrinolysis including plasminogen-activator inhibitor −1 and −2 (PAI-1 and PAI-2), which inhibit tPA and uPA, and α2-antiplasmin and α2-macroglobulin, which inactivate any unbound plasmin. In addition, activated thrombin-activatable fibrinolysis inhibitor, which upon activation by thrombin removes the C-terminal lysine residues of fibrin, also prevents plasmin generation. (B) Activator uPA binds to its receptor at the cell surface and activates plasminogen that is bound to its receptor nearby. This releases plasmin into the extracellular matrix. Plasmin generated at the cell surface is primarily regulated by the action of α2-antiplasmin, PAI-1, and -2. Plasmin generated at the cell surface plays key roles in MMP activation and ECM degradation.
Plasmin is a serine protease that exhibits trypsin-like broad specificity, particularly when plasminogen is activated by urokinase plasminogen activator (uPA) in a cell surface receptor-mediated process.24, 25 Plasmin generated at the cell surface plays a critical role in degradation of extracellular matrix (ECM) resulting in modulation of several processes including tissue remodeling,26, 27 cell invasion and metastasis,28, 29 chemotaxis,29, 30 wound healing and tissue repair,31-33 neuritogenesis,34, 35 ovulation and embryo implantation,36, 37 and others.38 Plasmin itself can degrade components in the ECM such as laminin,39 fibronectin,40 and collagens.41 Plasmin also contributes indirectly to ECM degradation by its activation of matrix metalloproteinases (MMPs), which degrade other ECM components.38, 42-44 In fact, potential benefits of plasmin inhibition have been projected for angioedema,45, 46 chronic inflammatory responses,47, 48 embryogenesis,37, 49 and lymphoid malignancies.50, 51 Thus, plasmin inhibitors may also be of value as anticancer and antiinflammatory agents in addition to their well-known therapeutic use as antifibrinolytic agents.45-51
In this review, the plasminogen-plasmin system and its central physiological role in the fibrinolytic pathway will be discussed in an attempt to understand fundamental rationale for plasmin inhibitors design and use. Recent advances made on the design, discovery, and development of plasmin inhibitors as antifibrinolytic agents will be more elaborately discussed. Current plasmin inhibitors in use or development belong to either the orthosteric (competitive) or the allosteric (noncompetitive) class of inhibitors. Structurally, the inhibitors belong to one of three chemical classes including peptidomimetics, natural and engineered polypeptides, and polymeric sulfated glycosaminoglycan (GAG) mimetics. The broad categories of plasmin inhibitors reported in the literature imply considerably diverse structures and mechanisms. Finally, studies on the role of plasmin in other pathophysiological events and the potential benefits of plasmin inhibitors under these conditions will also be outlined.
2. THE PLASMINOGEN-PLASMIN SYSTEM
Human plasmin is known to be the key player in the fibrinolytic system (Fig. 1). It catalyzes the proteolytic cleavage of blood clots. Plasmin is derived from its zymogen, plasminogen, which is a single-chain polypeptide biosynthesized as 810 amino acid residues. The mature form of this zymogen is a glycoprotein comprising 791 residues arising from the cleavage of a 19-residue leader peptide upon secretion.27, 52-54 Although plasminogen is primarily biosynthesized in liver, its messenger RNA has been detected in other tissues.55 Plasminogen is physiologically activated by the action of either tPA or uPA, of which the former appears to be the primary activator for intravascular events, whereas the latter is mainly important for extravascular cell surface events. The plasminogen/plasmin structural architecture is relatively complex as depicted in Figure 2.27, 52-54
Figure 2.

A schematic depiction of the plasminogen structure. Plasminogen possesses an N-terminal plasmin-apple-nematode (PAN) domain (1–77), five kringle domains K1–K5 (residues 162–542), and a catalytic domain (562–791). K77-K78 and R561-V562 are two cleavage sites. Cleavage at the R561-V562 scissile bond by tPA and other activators produces full-length active plasmin. Cleavage at the K77-K78 bond can produce a shorter zymogen called Lys-plasminogen. Ligand- or substrate-binding sites are also shown (see legend). Competitive inhibitors such as the polypeptides aprotinin, DX-1000, KDI-L17R, and the cyclic peptidomimetics 39 and 40 bind to the active site, whereas lysine analogs such asTXAand EACA bind to the kringle domains. GAG mimetics bind to the putative heparin-binding site on the catalytic domain.
Structurally, plasmin has two polypeptide chains linked by two disulfide linkages. The N-terminal heavy chain (Glu1-Arg561) comprises five sequential kringle (K1–K5) domains each of which is a triple disulfide-linked polypeptide comprising approximately 80 residues.27, 53 Each kringle domain contains an LBS characterized by anionic and cationic centers interrupted by a hydrophobic channel. For example, the K1 domain contains anionic and cationic centers made up of two Asp and two Arg residues, respectively, and also a hydrophobic groove that is lined with three Tyr residues.56, 57 The LBSs facilitate binding of small and large molecules, such as Lys-like ligands,58-62 inorganic chloride,63 fibrin(ogen),64, 65 bacterial proteins,66, 67 mammalian cell surfaces,68, 69 and the main physiological inhibitor α2-antiplasmin.17, 18 Interestingly, despite similarity among the five LBSs, structural differences do exist among them leading to differential recognition of a ligand by the five domains. For example, the affinity of antifibrinolytic drug 6-aminohexanoic acid increases in the order K1 > K3 > K2 > K5 > K4.27 Likewise, although the K3 domain possesses a dysfunctional LBS, a single Lys311Asp mutation results in enhanced function.70 The other structural feature of plasmin is the C-terminal light chain (Val562-Asn791), which is the trypsin-like catalytic domain having the triad of His603, Asp646, and Ser741 residues (His57, Asp102, and Ser195 in chymotrypsinogen numbering).71 The catalytic domain appears to bind to α2-macroglobulin, a physiological inhibitor of plasmin.22, 72
A very important component of the plasminogen-plasmin system is the 77-residue activation domain, named as plasmin-apple-nematode domain.73-75 Physiologic activators cleave the scissile bond Arg561-Val562 in full-length plasminogen (Glu1-plasminogen) between the heavy chain (K5) and the light chain (catalytic domain) to produce the active plasmin (Glu1-plasmin or full-length plasmin).76 Another scissile bond that has been identified is Lys77-Lys78, cleavage of which appears to be self-catalyzed leading to the formation of a shorter length plasminogen (Lys78-plasminogen) or a shorter length plasmin (Lys78-plasmin). A fundamental difference between the two forms is the conformation of the corresponding protein. For example, Glu1-protein exists in more tight conformation that is favored by the chloride ion, whereas Lys78-protein exists in a more open conformation that appears to be favored by Lys-like ligands.53 Crystal structures shedding light on the structural aspects of plasminogen have been published recently.74, 75
Although plasmin is a trypsin-like protease with significant similarity to other enzymes in the superfamily, structural differences between these enzymes do exist and these differences have aided the design of selective active site inhibitors. Of particular interest is the absence of segment 95–100 in plasmin(ogen), which led to the discovery of the most selective plasmin inhibitor belonging to the cyclic peptidomimetics class.11 Furthermore, several approaches including positional-scanning synthetic combinatorial library screening77-79 and noncombinatorial sparse matrix peptide library screening80 have been performed to deduce the plasmin active site specificity. Although a general consensus on plasmin specificity has not been derived as of yet, some understanding has been deduced.81 Using macromolecule protein substrates, it was suggested that the P1 specificity of plasmin includes basic amino acids Lys and Arg with a slight preference for Arg; P2 residues can be Leu or Ser, and to a lesser extent Pro, Ala, or Phe; P3 preference is for Arg, followed by Ser, Gln, or Gly; and P4 specificity is for Pro and Ala, and to a lesser extent Arg. On the C-terminal side of the scissile bond, Ser and Ala are overwhelmingly favored for the P1’ position, Arg, Ser, or Val are preferred at the P2’ position, Ser and Pro are favored at the P3’ position, and Gly, Pro, or Leu are favored at the P4’ position. In studies with smaller substrates having P4–P1 domains, plasmin was found to prefer Lys at the P1 position (to a lesser extent Arg), and Trp, Phe, and Tyr at the P2 position. However, there was significant uncertainty with respect to P3 and P4 positions suggesting that most amino acids may be tolerated in the two subsites.77-81
3. PLASMIN INHIBITORS AS ANTIFIBRINOLYTICS
A. Peptidomimetic Inhibitors
1. Lysine Analogs
ε-Aminocaproic acid (EACA or 6-aminohexanoic acid, 1) and tranexamic acid (TXA, 2) are synthetic derivatives of the amino acid lysine (Fig. 3) and are antifibrinolytic agents used in the clinic today.4, 6, 82-85 EACA and TXA were discovered by Okamoto et al. in the 1950s.86-88 The two agents do not bind in the active site of plasmin, rather, they bind to the LBSs of the kringle domains on plasmin(ogen) and prevent tPA/uPA-induced activation of plasminogen activation to plasmin.4, 6, 82-85 TXA is approximately tenfold more potent per unit dose than EACA.89, 90 TXA produces higher and sustained antifibrinolytic activity than EACA. Structurally, TXA is the trans-stereoisomer of 4-aminomethyl-cyclohexane carboxylic acid. Initial studies to assess its antifibrinolytic activity were performed using a mixture of cis- and trans-isomers. However, Okamoto et al. concluded later that the trans-isomer is the only active species in the mixture.87, 89, 90 The two analogs demonstrate different pharmacokinetic profiles. EACA is orally well absorbed and reaches a maximal plasma level of 30 mg/L but shows a shorter half life (60 min). Nearly 80% of EACA is excreted in urine in the unmetabolized form. In contrast, TXA shows a half life of 80 min, is recovered in the urine in an unchanged form, and requires a therapeutic concentration of 5–10 mg/L. TXA shows a higher volume of distribution and crosses the blood–brain barrier.90 Complications associated with the use of either derivative include renal failure (more with EACA),91 seizures (more with TXA),91, 92 and rhabdomylosis.5
Figure 3.
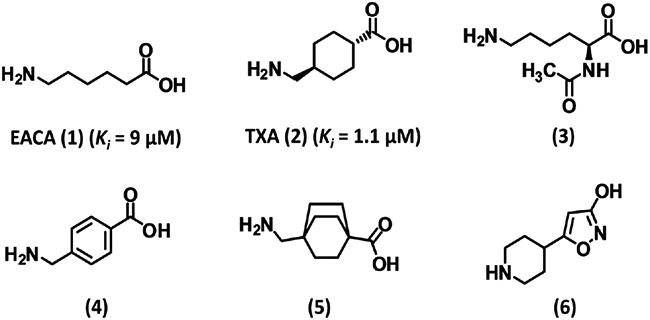
Structures of lysine analogs 1–6. EACA (1) and TXA (2) are the most widely used antifibrinolytic agents. These agents are noncompetitive inhibitors of plasmin binding to kringle domains except inhibitor (3), which is a competitive inhibitor. Inhibitor 6 was recently identified by computational chemistry and exhibited fourfold better potency than TXA (2). Ki is the inhibition constant for plasmin.
Several studies have implicated the use of lysine analogs to reduce perioperative and postoperative blood loss in cardiac surgeries,1, 2, 5 orthotopic liver transplantation,82, 84 menorrhea,9, 93 DIC,6, 7 von Willebrand syndrome,10, 85 and some forms of hemophilia.8 For example, TXA saves an average of 300 mL of blood per patient during cardiac surgery and reduces blood transfusion by 32%.1 It reduces blood loss by 40%, thereby reducing number of transfusions in liver transplantation.87 Both analogs have been exhaustively studied to establish their safety profiles and have shown a superior safety margin, particularly in cardiac procedures, with less than 1% of cases associated with severe complications. Both analogs effect significant cost-savings relative to other antifibrinolytics.5
Several biochemical studies have shed light on the mechanism of TXA/EACA. The two lysine analogs bind to kringle domains of plasmin(ogen).72 Kringle domain 1 (K1) appears to be the primary target of TXA (1.1 μM) and EACA (9.0 μM), while their affinities are intermediate for K4 and low for K2, K3, and K5.72 Both TXA and EACA do not affect the amidolytic activity of plasmin, miniplasmin, and streptokinase-plasmin complex but inhibit plasmin and miniplasmin-mediated fibrinolysis. At the molecular level, miniplasmin contains only the K5 domain (instead of the five kringles present in plasmin) and the catalytic domain, which implies that the two lysine derivatives inhibit fibrinolysis by binding to K5.72 However, this does not imply that other kringles are not involved. TXA and EACA reduce the rate of inhibition of plasmin by α2-antiplasmin, which utilizes K1–K3.72 The two lysine derivatives do not affect activated partial thromboplastin time (APTT) or prothrombin time (PT) but prolong euglobulin clot lysis time at 10 mM concentration.72 These studies suggest that TXA and EACA are allosteric inhibitors.
Structure-activity relationship (SAR) studies have been performed as to enhance the moderate efficacy of TXA and EACA and to minimize their side effects. A lysine ester, Nα-acetyl-L-Lys-methyl ester (3, Fig. 3), was found to inhibit the amidolytic activity of plasmin variants, in contrast to TXA and EACA, suggesting competitive inhibition. Inhibitor 3 did not inhibit plasmin-mediated fibrinolysis and also did not reduce plasmin inhibition by α2-antiplasmin at 100 μM. Inhibitor 3 did not affect APTT and PT as well as euglobulin clot lysis time, which was in contrast to TXA and EACA.72 This suggested that allosteric inhibition through the kringle domains required carboxylic acid and a free amine on a Lys analog. Other SAR studies also confirmed the role of free amine and carboxylic acid groups for antifibrinolytic activity.89 Studies on homologous aminocarboxylic acids showed that the antifibrinolytic activity depended critically on the distance between the free amine and carboxylic acid groups (~7 Å).89 Yet interestingly, replacing the C2-C5 segment with a benzene ring (presumably only 4.5 Å in length), as in 4 (Fig. 3), was found to increase potency by fivefold. Saturating the benzene ring to produce cis- and trans-isomers led to the discovery of trans-TXA (2), which is tenfold more potent than EACA and is most often used nowadays. Several derivatives of trans-TXA were synthesized including 4-aminomethyl-bicyclo-2,2,2-octane carboxylic acid (5, Fig. 3), however none displayed superior potency.
The crystal structure of human plasminogen K1 domain with TXA and EACA shows that the amine of the two ligands recognizes K1 Asp54 and Asp56 residues, while the carboxyl moiety binds to Arg70 and Arg34. A hydrogen bond between the carboxyl group and the phenolic group of Tyr63 appears to contribute to binding affinity. The methylene groups of the ligands are stabilized by van der Waals contacts with the side-chain atoms of Trp61 and Tyr71.57
To optimize EACA’s pharmacokinetic/dynamic profiles, several short peptides based on the EACA moiety were synthesized and evaluated.94-100 Analogs containing residues such as L-Lys, L-Leu, L-Nle, or L-Cys demonstrated variable antifibrinolytic activity relative to the parent EACA. Of note was the L-Lys derivative that exhibited tenfold better antifibrinolytic potency (IC50 < 200 μM).94
More recently, Boström et al. reported the use of a computational technique coupled with a low-throughput screening to find 6 (Fig. 3), which displays an electrostatic potential similar to TXA.101 Inhibitor 6 was found to be about four times as potent as TXA with an IC50 for plasma clot lysis of 0.8 μM. Inhibitor 6 is predicted to exhibit GABA-A activity, which leads to convulsions, similar to that associated with TXA. Yet, 6 has been proposed as a good lead for further development.101
Finally, the low bioavailability of TXA (~34%)93 was sought to be resolved through its maleamic acid102 and acyloxyalkyl carbamate derivatives.103 In November 2009, Lysteda™, a novel TXA formulation, was approved by the FDA to treat heavy menstrual bleeding.93 This oral formulation was designed to minimize gastrointestinal adverse effects. The formulation requires less frequent dosing because of its higher per-tablet dosage relative to the immediate-release preparation.93
2. Trans-4-aminomethylcyclohexanecarbonyl Conjugated Inhibitors
Okada et al. and others have reported that plasmin’s chromogenic substrate D-Ile-Phe-Lys-pNA (7, Fig. 4) has a low Km (20 μM) as well as mild inhibitory action (IC50 30 μM).104 Hence, this substrate was used as a template to help design more potent active site inhibitors. Replacing the p-nitroanilide group with more hydrophobic moieties such as p-benzoylanilide (BZA, 8), p-acetylanilide or 3-(p-dimethylaminobenzoyl) anilide increased the IC50 some 6–9-fold as measured by plasmin fibrinolysis.104 Yet, the analogs were better inhibitors than 7 as these molecules were minimally hydrolyzed by plasmin.104 Same investigators attempted to replace the P3-P2 domain of substrate 7. Introducing a tosyl group, instead of the D-Ile-Phe motif as in Tos-Lys-pNA, displayed an IC50 of 700 μM (against S-2251) and 780 μM (against fibrin).104 The above two efforts were combined to design Tos-Lys-BZA (9, Fig. 4), which inhibited plasmin better (IC50 140–150 μM).104
Figure 4.
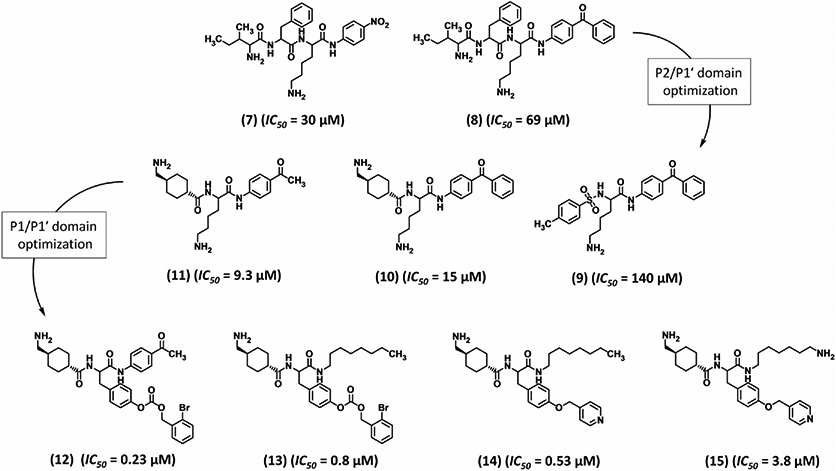
Structures of trans-4-aminomethylcyclohexanecarbonyl-conjugated inhibitors 7–15. These inhibitors are active site inhibitors. The development of inhibitor 15 was accomplished through two stages of P2/P1′ optimization followed by P1/P1′ optimization. IC50 refers to inhibition of plasmin amidolysis.
These initial studies were exploited to develop irreversible inhibitors containing the chloromethyl ketone moiety. Replacing P1′ (BZA) inhibitor 8 (Fig. 4) with chloromethyl ketone moiety led to an inhibitor with an excellent Kd of 1.75 μM and a catalytic efficiency (k2/Ki) of 77,000 M−1s−1. But the inhibitor lacked selectivity as it also inhibited thrombin, plasma kallikrein, factor Xa, and trypsin.105 An analog, D-Ile-Phe-Arg-CH2Cl, synthesized on the basis of inhibitor 8, was actually found to be a more potent inhibitor of plasma kallikrein.105
Teno et al. have reported Tra-Lys-BZA (10, Fig. 4) as a moderately potent, active site-directed plasmin inhibitor displaying IC50 of 15 and 6.1 μM against chromogenic substrate S-2251 and fibrin, respectively.106 This inhibitor was intuitively designed from trans-TXA, which replaced the D-Ile-Phe domain of inhibitor 8.106 Interestingly, 10 inhibited fibrinogenolysis too with an IC50 of 13 μM.106
Dissecting the inhibitor Tra-Lys-BZA (10) into three structural domains gave some interesting structure-function insights. It was found that L-Lys is an optimal central part and replacing it with ornithine or D-Lys abolished inhibition of plasmin. Substituting the terminal Tra domain with racemic 4-aminocyclohexanecarbonyl or 3-aminocyclohexanecarbonyl groups resulted in essentially inactive analogs. Likewise, the cis-isomer of Tra diminished the activity toward chromogenic substrate and fibrin nearly 27- and 43-fold, respectively. Likewise, replacing the Tra group with the tosyl group led to 10–25-fold decrease in inhibitory effect on plasmin amidolysis, fibrinolysis, and fibrinogenolysis.106 However, introducing 5-aminopentoyl or 6-aminohexanoyl (EACA) group in the place of Tra group maintained inhibition potency against amidolysis (IC50 12–16 μM), fibrinolysis (IC50 10–17 μM), and fibrinogenolysis (IC50 36 μM). Replacement of the third domain, the BZA group, with substituted piperidineamide, p-nitroanilide, 5,6,7,8-tetrahydro-2-aminoronaphthyl amide, or p-methoxycarbonylanilide resulted in compounds that did not inhibit plasmin amidolytic or fibrinolytic activity at concentrations as high as 500 μM. But substituting only the 4-benzoyl group of BZA with aceyl group resulted in moderately potent fibrinolysis inhibitor 11 (IC50 9.3 μM).106 Last, replacing the central amide linker with an ester also reduced the overall inhibitor potency.106 Thus, 10 was arguably the best inhibitor in the series.
Mechanistically, 10 is a competitive inhibitor that bound in the active site of plasmin. Plasmin’s S1 subsite interacts with the basic side chain of Lys or Arg residues. Using this information, two derivatives were synthesized to gain insights on 10’s mode of binding. The free amino groups of Lys or Tra moieties in 10 were individually protected by the benzyloxycarbonyl (Cbz) group. While the former derivative retained inhibitory activity, the latter derivative was significantly less active.106 This implied that the free amine of Tra, and not that of Lys, interacted with the S1 Asp of plasmin.
The observation that the Tra group when conjugated to appropriate amino acids induces potent plasmin inhibition led to the design of new peptidomimetics with three domains (P1-P1′-P2′) in which P1 is the Tra group.107-109 The most potent plasmin inhibitor in the new set had 2-bromobenzyloxycarbonyltyrosine as P1′ group and p-acetylanilide as P2′ group. This compound 12 inhibited plasmin amidolysis with an IC50 of 0.23 μM. But 12 also inhibited plasma kallikrein, urokinase, and thrombin with IC50 of 0.37, 43, and 63 μM, respectively.107 An organic chemistry-driven approach was developed to improve the three domain peptidomimetic. The P2′ domain was modified with branched and unbranched alkylamides, p-alkylanilides, pyridineanilides, pyridinealkylanilides, or a Tra group.108 Each of these displayed plasmin inhibition potency comparable to the parent inhibitor 12. Their specificity over thrombin and urokinase was significantly improved but not against kallikrein or trypsin. This suggested that the S2′ subsites in both plasmin and kallikrein tolerate bulky hydrophobic groups as opposed to thrombin and urokinase. Thus, inhibitor 13 was designed with a P2′ octylamide and contained P1 and P1′ groups of 12.108 Compound 13 inhibited plasmin with an IC50 of 0.8 μM (amidolysis) or 0.23 μM (fibrinolysis) as well as plasma kallikrein (IC50 16 μM) and trypsin (IC50 1.6 μM) but not urokinase or thrombin (both IC50 > 50 μM).108 Further replacement of the protecting group on P1′ Tyr from 2-bromobenzyloxycarbonyl to 4-pyrdinylmethyl group produced inhibitor 14 (Fig. 4), which displayed better plasmin inhibition (IC50 0.53 μM) and 60-fold higher selectivity over plasma kallikrein (IC50 30 μM). However, 14 lost specificity against urokinase (Ki 5.3 μM).109 To restore selectivity against urokinase, the P2′ octylamide was modified to contain a terminal amine as in 15, which improved the selectivity index eightfold against plasma kallikrein, urokinase, and thrombin.109, 110 To capitalize on these gains, a set of four domain peptidomimetics (P2-P1-P1′-P2′) were synthesized based on 12. The Tra group at P1 was modified to 4-aminocyclohexylalanine, P1′ and P2′ groups were retained as in 12 and a tosyl group was replaced for P2. These changes neither improved potency nor enhanced specificity of inhibition.107
3. Cyclohexanone-Based Inhibitors
Seto and colleagues were the first to report that 4-heterocyclohexanone moiety gives reversible, covalent, active site inhibitors of several serine and cysteine proteases.111 The inhibitor’s ketone moiety is attacked by nucleophilic active site Ser or Cys of the protease to form a reversible hemiketal adduct. Steric relationship between the heteroatom and the carbonyl group was proposed as key to enhance the reactivity of the molecule.112, 113 Compound 16 (Fig. 5) was the first to be designed for plasmin.111 It relied on three interactions including those of the S1 and S3 subsites with the two hexylamine chains. Inhibitor 16 displayed a Ki of 400 μM against plasmin with approximately threefold selectivity over trypsin and more than 25-fold selectivity over thrombin and kallikrein.111 To increase its potency, one hexylamine was replaced with D-Ile-L-Phe, a favorable P3–P2 plasmin motif derived earlier, to give 17 (Fig. 5). One diastereomer of 17 displayed threefold better inhibition of plasmin (Ki 50 μM) than the other diastereomer and good selectivity over trypsin, thrombin, and kallikrein (34-, 15-, and 13-fold, respectively). Lineweaver-Burk analysis confirmed that inhibitor 17 was a reversible competitive inhibitor of plasmin. The importance of 17’s hexylamine chain was realized when replacing it with a hydrogen eliminated plasmin inhibition potential (18, Ki 9–16 mM, Fig. 5). Likewise, replacement of the heteroatom with a hydrogen, as in 19, also lost potency by approximately threefold.111 Finally, 20 was found to be about 12- and fivefold less potent than inhibitors 17 and 19, respectively suggesting the importance of covalent hemiketal adduct formation in inhibition.111
Figure 5.
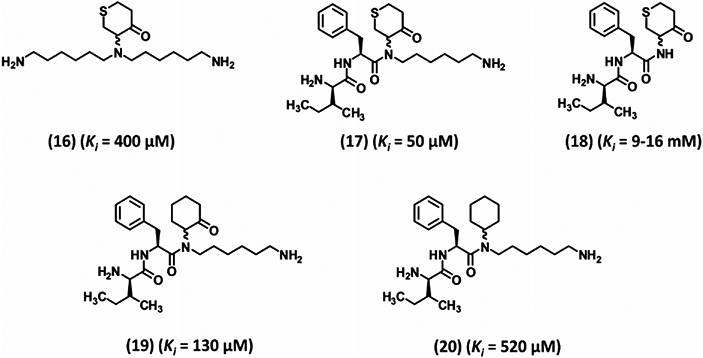
Early development of cyclohexanone-based peptidomimetic inhibitors 16–20. These inhibitors are reversible, covalent inhibitors (except inhibitor 20) targeting the active site of human plasmin. Inhibition involves formation of hemiketal tetrahedral complex with the Ser residue of the catalytic triad. Ki is the inhibition constant for plasmin.
Abato et al. exploited another cyclohexanone core structure to design the next generation of bidirectional plasmin inhibitors.114 A combinatorial chemistry approach was developed to synthesize some 400 potential inhibitors, each of which contained two variations at positions 2 and 6 of the cyclohexanone core that fit into the S2 and S2′ subsites (Fig. 6). Based on an initial screening, four inhibitors (21–24, Fig. 6) were chosen for further analysis. Inhibitor 21 having two Trp residues demonstrated the highest potency in the group with a Ki of 5–10 μM and at least 38-fold selectivity over the cysteine protease papain. Phe-containing inhibitors were less potent (Ki > 100 μM) and less selective.114 To improve upon the potency, a hexylamine chain was added so as to engage the S1 subsite of plasmin. However, the resulting diastereomeric mixture (25, Fig. 6) did not show much improvement.114, 115 A follow-up study developed an extended bidirectional cyclohexanone inhibitor comprising five domains corresponding to P3-P2-cyclohexanone-P1′-P2′-P3′.116 A combinatorial chemistry approach synthesized 400 compounds, of which 26 displayed an IC50 of 2.7 μM against plasmin and a selectivity index of > 150-fold over kallikrein, thrombin, and trypsin. This suggested a preference for Trp at the P3, P2, and P2′ sites. Likewise, Tyr was found to be favored at the P3′ site as other residues displayed slightly weaker inhibition.116
Figure 6.
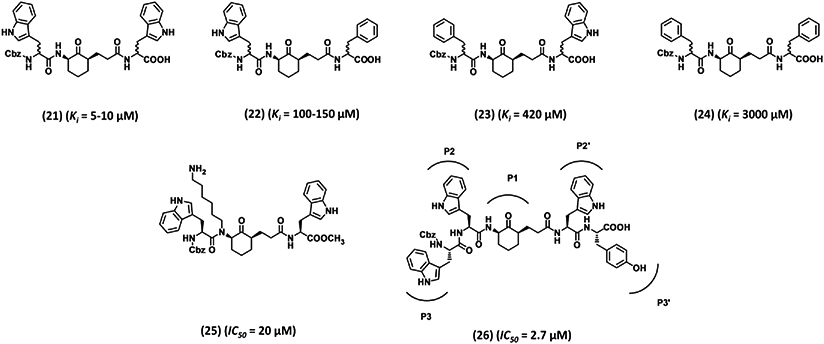
Advanced cyclohexanone-based peptidomimetic inhibitors 21–26. These inhibitors were developed by combinatorial chemistry approach to reversibly inhibit plasmin. Inhibitor 26 has six domains spaning plasmin active site from subsite S3-S3′. Ki is the inhibition constant for plasmin. IC50 refers to inhibition of plasmin amidolysis.
The success of the ketone–hemiacetal trapping strategy led Seto and co-workers to further optimize the type and size of the cyclic ketone core structure, the residue at the P2 domain, the amine cation at the P1 domain, and the N-terminus of a four-domain inhibitor P4-P3-P2-P1.117 Varying the P2 domain to either Phe (as in inhibitor 27) or Trp did not affect inhibition potency (~22 μM). In contrast, Cbz removal from the N-terminus (as in inhibitor 28) diminished the activity ~17-fold suggesting its significance in binding to the S4 subsite of plasmin. Replacing the nuclear oxygen of the heterocyclic ring of 27 with a methylene or a protected amine reduced the potency by 45- or 16-fold, respectively,117 suggesting its probable role in enhancing electrophilicity of the ketone. Supporting this hypothesis was the observation that a sulfone moiety in the place of the nuclear oxygen, as in inhibitor 29, enhanced the potency by approximately twofold. Likewise, expansion to a six-membered ring essentially eliminated inhibitory potential (IC50 1000 μM). With regard to the P1 domain, butylamine, pentylamine, heptylamine, or 4-aminocyclohexylmethyl containing molecules were less active than inhibitor 27 (Fig. 7), except for the hexylamine containing derivative 30 that displayed twofold improved IC50. Combining these results with that of the sulfone derivative 29 led to inhibitor 31, which showed an IC50 of 5.7 μM, the most potent inhibitor in this series.117 No selectivity data have been published for this set of plasmin inhibitors.
Figure 7.
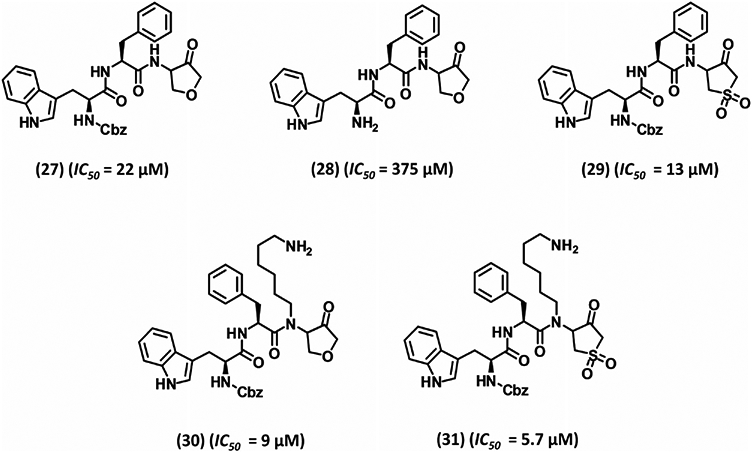
Development of five membered heterocycle-based peptidomimtic inhibitors 27–31. Inhibitors were developed by divalent classical bioisosterism replacement of position 4 methylene unit in previously designed inhibitors with either ether or sulfone moiety. IC50 refers to inhibition of plasmin amidolysis.
Overall, the designed reversible, covalent, hemiketal-based competitive inhibitors of plasmin demonstrate moderate inhibitory potency (low μM) with the 26 being the best in this class. Unfortunately, further biochemical or crystallographic studies have not been reported on this inhibitor which has stymied development of new directions. Also, no in vivo studies have been published. It is possible that the size and the peptidic nature of this inhibitor is a detriment for such a translation. Nevertheless, it is important to recognize that this chemical class has led to three important observations on plasmin specificity and cooperativity. Plasmin strongly prefers Lys or Arg at the P1 position and extended hydrophobic and aromatic residues, particularly Trp, at the P2 position. Plasmin also favors hydrophobic residues (Ile, Phe, and Tyr) at P2′. Furthermore, the inhibition studies show that cooperative binding in the S1 and S2 subsites of plasmin is important for enhancing specificity. A positively charged P1 group that binds in the S1 subsite indiscriminately positions any P2 aromatic group (Trp, Tyr, or Phe) to bind in the S2 pocket. In contrast, inhibitors that do not bind in the S1 subsite prefer Trp at P2, perhaps due to favorable van der Waals or hydrogen-bonding interactions with the S2 subsite.115 Thus, the S2 substrate specificity can be altered depending on whether S1 is occupied or not. S3 and S3′ subsites prefer to bind hydrophobic and/or aromatic residues such as Trp, Phe, Tyr, or Leu. Less hydrophobic residues, such as Ala, Ile, or Val, show good affinity for plasmin, whereas polar amino acids, such as Orn and His, and conformationally constrained residues, such as Pro and hydroxyPro, are detrimental for binding to the S3′ subsite.115 For this class of inhibitors, it was found that Trp is the preferred residue at P2, P3, and P2′, whereas Tyr is preferred at the P3′ site. These results are consistent with previous reports on the specificity of plasmin substrates and inhibitors.77, 78, 118
4. Cyclic Peptidomimetic Inhibitors
Cyclization of known peptide inhibitors to form macrocylic peptidomimetics has recently emerged as a viable drug design strategy.119, 120 An important feature that has helped inspire this protocol is that substrate-like inhibitors are likely to bind in the form of an antiparallel β-strand to the active site of trypsin-like serine proteases. The conformational constraint introduced by cyclization should have two special advantages over the corresponding linear peptides. First, cyclization is expected to reduce the entropic penalty for binding by decreasing flexibility. Second, cyclization reduces proteolytic processing by physiologic peptidases, which can improve bioavailability.121-123
To put this concept to work for plasmin, Xue and Seto synthesized cyclic analogs by treating the free N-terminus of a Tyr-containing analog of inhibitor 28 with appropriate bromoalkanoyl chloride followed by NaI/acetone-mediated cyclization.117 Inhibitors 32, 33, and 34, containing macrocyles with varying atoms (Fig. 8) showed reasonable improvement in the inhibition potency relative to that of parent molecule 28. The best analog 32 contained a 19-atom macrocycle and showed ~15-fold improvement over 28.117 However, 33 and 34 did not exhibit more than 20% inhibition at a concentration as high as 250 μM.124
Figure 8.
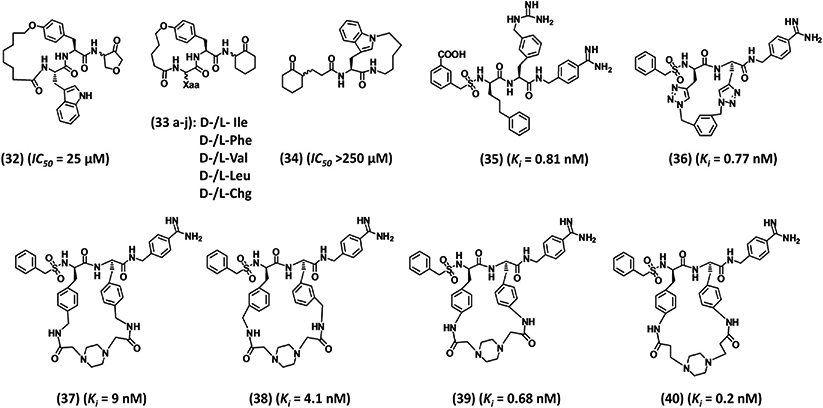
Structures of various macrocyclic inhibitors 32–40. Inhibitors 39 and 40 represent the most potent and selective small peptidomimetics reported in the literature. Ki is the inhibition constant for plasmin. IC50 refers to inhibition of plasmin amidolysis.
These results do not appear to demonstrate much potential arising from the cyclization strategy. One reason could be the absence of optimal elements for plasmin inhibition in the acyclic precursors. This led Saupe and Steinmetzer to prepare a series of macrocyclic peptidomimetics by simple amidation, metathesis, or click chemistry so as to link the P2 and P3 chains of inhibitor 35.125 The designed molecule 36 (Fig. 8) was a potent plasmin inhibitor with a Ki of 0.77 nM. This inhibitor exhibited a selectivity index of 12324-, 268-, and 3896-fold over thrombin, factor Xa, and activated protein C, respectively.125 However, 36 inhibited plasma kallikrein with high potency too (Ki 2.4 nM).
The success of the Saupe and Steinmetzer strategy appears to reside in the starting acyclic lead 35 (Fig. 8), which inhibited plasmin (Ki 0.81 nM), plasma kallikrein (Ki 0.075 nM), thrombin (Ki 667 nM), factor Xa (Ki 42 nM), and activated protein C (Ki 0.61 nM). The nonselective inhibitor 35 contained 4-amidinobenzylamide as an Arg mimetic at the P1 position, 3-guanidinomethylphenylalanine at the P2 position, D-phenylpropylglycine at the P3 position, and benzylsulfonyl moiety at the P4 position. A comparison of the crystal structures of several serine proteases shows that plasmin’s active site is devoid of the 95–100 segment, the so-called 94-shunt. This allows continuity of proximal (S1 and S2 subsites) and distal (S3 and S4 subsites) binding pockets rendering the active site wide and open. This accounts for the relative nonspecificity of plasmin, which can cleave many different substrates in a fashion similar to trypsin. Saupe and Steinmetzer exploited this feature as to devise cyclic analogs having bulky group in the vicinity of the 99 residue. The cyclic loop of the designed inhibitors was expected to fit well into plasmin active site but generate steric clash with other serine proteases that contain this segment, as is the case of thrombin, factor Xa, factor IXa, factor XIa, factor VIIa, activated protein C and uPA. They used the ‘i to i+1’ cyclization strategy as opposed to the ‘i to i+2’ strategy utilized by Xue and Seto.124 The shorter cyclization strategy increased bulkiness and rigidity in the macrocycles and enhanced their selectivity over other homologous proteases.
The same study explored the SAR further through 11 additional cyclic inhibitors containing 1,3- or 1,4-disubstituted benzene, 1,4-disubstituted piperazine, 1,3-disubstituted pyridine or N-oxide pyridine, n-butyl, or butylene linkers. Owing to rigidity, inhibitors with smaller rings (17-membered) generally exhibited better potency (Ki < 3 nM) and selectivity relative to inhibitors with larger rings (24 to 26-membered rings).125 A plausible mode of binding and selectivity was explained through molecular modeling, which suggested that the benzamidine group of inhibitor 36 fits into the S1 subsite, as expected, and hydrogen bonds to both the carbonyl of Gly219 as well as the side chain oxygen of Ser190. Also the benzylsulfonyl group of 36 seems to fit into the S4 subsite and the sulfonyl group hydrogen bonds to the side chain of Gln192, which is also involved in interacting with the P2 carbonyl group. The two triazole rings with the 1,3-substituted aromatic linker were hypothesized to be directed toward the large open binding pocket. Superimposition of thrombin, factor Xa and activated protein C structures onto the docked plasmin-36 complex showed that the triazole-containing linker would sterically clash with the 95–100 segment of the related proteases.125 At the same time, steric clash is greatly reduced for plasma kallikrein because of its small and flexible S2 subsite residue (Gly99), which explained the observed comparable potency. Overall, the modeling results corroborated with the observed selectivity profile of inhibitor 36.
To enhance the selectivity of inhibitor 36, especially against kallikrein, and to improve its aqueous solubility, additional cyclic peptidomimetics were synthesized.11 Inhibitor 37, containing a 26-membered, piperazine linker-based macrocycle, was found to display better selectivity profile in comparison to inhibitor 38, which had a 24-atom macrocycle (Fig. 8).125 This suggested that the 1,4-disubstituted piperazine linker was a better lead for further optimization. In a new set, the −CH2–of 4-aminomethylene-Phe present in 37 was eliminated to yield 39, which inhibited plasmin with 13-fold (Ki 0.68 nM) better potency than 37. Inhibitor 39 was 100-fold more selective over trypsin, 470-fold over kallikrein, and > 15,000-fold selective over thrombin, factor Xa, and activated protein C.11 Replacing the 1,4-diacetylpiperazine linker with the 1,4-dipropionylpiperazine linker to give a 26-membered macrocycle 40 (Fig. 8) further enhanced the potency (Ki plasmin 0.2 nM) as well as selectivity over kallikrein, thrombin, factor Xa, and activated protein C. But its selectivity over trypsin was reduced (40 38.3 nM).11 Additional modifications of piperazine ring had marginal impact on plasmin inhibition.11 Inhibitors 39 and 40 also did not affect many other proteases including the enzymes of the coagulation cascade (Table I) and possessed excellent solubility in 0.9% saline. Both inhibitors efficiently inhibit the tPA-induced fibrinolysis in human plasma. Clot lysis time was doubled at 280 nM for inhibitor 39 and 180 nM for inhibitor 40. Both inhibitors showed no anticoagulant activity in plasma as demonstrated by APTT, PT, and thrombin time (TT) assays as well as insignificant binding to hERG, natrium, and L-type calcium ion channels at 10 μM concentration. Incubation with human liver microsomes revealed not much metabolism for both inhibitors as the IC50 for the inhibition of various Cyp-P450 enzymes was found to be > 10 μM.11
Table I.
Equilibrium Inhibition Constants (Ki) in Nanomolar for Selected Plasmin Inhibitors, Their Effect on APTT, PT, and Fibrinolysis
| Protease | (40)a | CU-2010b | Aprotininc | Textilinin-1d | DX-1000e | KD1-L17Rf | CDSO3g |
|---|---|---|---|---|---|---|---|
| Plasmin | 0.2 | 2.2 | 0.18 | 0.11–3.5 | 0.087 | 0.9 | 242 |
| Trypsin | 38.3 | ND | 0.02 | 0.76 | ND | ND | >10,000 |
| Chymotrypsin | ND | ND | 1.3 | ND | ~10,000 | ND | >10,000 |
| Plasma kallikrein | 1000 | 0.019 | 13.4 | 4830 | 250 | >3000 | >10,000 |
| Pancreatic kallikrein | ND | ND | 0.023 | ND | ND | ND | ND |
| Tissue kallikrein | ND | ND | 0.004 | 12,900 | ND | ND | ND |
| Urinary kallikrein | ~10,000 | ND | ND | ND | ND | ND | ND |
| Factor IXa | >5000 | ND | >5000 | ND | ND | ND | 3380 |
| Factor Xa | 25,000 | 45 | >30,000 | NI | ~1300 | ND | 34 |
| Factor Xia | 3370 | 18 | 288 | ND | ND | >3000 | 22 |
| Factor XIIa | >20,000 | 5200 | 6800 | NI | ND | ND | >15,000 |
| Factor VIIa/tissue factor | >50,000 | ND | >1000 | NI | ND | >3000 | >29,000 |
| Factor IIa | 26,420 | 1700 | >3000 | NI | >>100,000 | >3000 | 18 |
| Factor IIa/thrombomodulin | ND | ND | ~3000 | ND | ND | >3000 | ND |
| Activated protein C | >15,000 | ND | ~2000 | >100,000 | ND | >3000 | >12,000 |
| Tissue plasminogen activator | >40,000 | ND | >3000 | >500,000 | ND | >3000 | ND |
| Urokinase | ND | ND | ~20,000 | >100,000 | >100,000 | ND | ND |
| Cathepsin G | ND | ND | ~4000 | ND | ND | ND | 232 |
| Leukocyte elastase | ND | ND | ~3500 | >100,000 | 800 | ND | 11 |
| Pancreatic elastase | ND | ND | >2,000,000 | ND | ND | ND | >10,000 |
| Clotting assay | 2 × clotting time (μM) | ||||||
| APTT | >100 | 1.4 | 11.9 | >12 | >0.7 | >6 | 2.9 |
| PT | >100 | 9.1 | >100 | >12 | >5.6 | >20 | 13.1 |
| Fibrinolysis inhibition | ’Concentration (μM) | ||||||
| EC200/IC50 | 0.18 | 0.32 | 0.33 | 2—5 | ~0.56 | 0.36 | ND |
Data from reference [11].
Data from references [197–199]. Factor VIIa data were reported without tissue factor. Concentration of fibrinolysis assay induces ~100% inhibition.
Data from reference [155].
Data from references [249, 250-251]. Factor VIIa data were reported without tissue factor and povine pancreatic elastase was used.
ND, not determined; NI, no significant inhibition.
To get atomistic insight on the interactions of 39 and 40 within the active site of human plasmin, molecular modeling was employed. Computational docking revealed that the amidino group of the P1 domain fits well into S1 subsite and hydrogen bonds to the carbonyl group of Gly219 as well as the side chain oxygen of Ser190.11 The amide NH of the P1 residue binds to the carbonyl oxygen of Ser214. A hydrogen bond was predicted between the P2 carbonyl oxygen of the two inhibitors and the side chain amide of Gln192. Sulfonyl oxygens were involved in hydrogen bonding to the NH of Gly219. A comparison of the two inhibitor conformations suggested better fit of 40 in plasmin’s active site because of stronger van der Waals interactions. Interestingly, no hydrogen bond between the linker segment and plasmin was identified. Finally, a steric clash between the 95–100 loop of kallikrein, thrombin, factor Xa, activated protein C and trypsin and the piperazine linker of inhibitors 39 and 40 was predicted by overlaying the models, thus corroborating the observed high selectivity profile.11
Overall, the cyclization strategy was highly successful in designing potent and selective substrate-like inhibitors of plasmin. It puts forward two inhibitors 39 and 40 as clinically relevant candidates, although the molecules appear to have been not tested in animal models.
5. Miscellaneous Agents
Nitrile warheaded inhibitors
An attractive strategy in the design of covalent enzyme inhibitors is that of transforming a peptidic substrate into a nitrile warhead-based inhibitor. The strategy has been reported for several serine proteases with reasonable success.126, 127 Teno et al. utilized tripeptide D-Ile-Phe-Lys-pNA (7, Fig. 4) to design a new class of covalent inhibitors of plasmin utilizing the nitrile warhead concept. In this strategy, the scissile bond of 7 was replaced by a nitrile warhead,128 which was found to selectively inhibit plasmin with a nearly fourfold improved IC50 (78 μM). This initial success inspired design of new nonpeptidic, nitrile warhead-containing inhibitors (Fig. 9). Molecular modeling studies predicted optimal positioning of the nitrile group of a lead molecule 41 with regard to the active site Ser. In addition, the lysyl side chain would fit into the S1 subsite, the meta-substituent would occupy the S2 subsite, and the para-picolyl group would bind to the S2 subsite. Thus, seven analogs of 41 containing various aromatic substituents at the P3 domain were synthesized. Of these, 42 (Fig. 9), in which the P3 domain is N-(4-fluorobenzyl)-3-indole propionamide, was found to be most potent (IC50 140 μM) and selective over plasma kallikrein and urokinase (IC50 > 1000 μM). It is worth mentioning that replacing the picolyl moiety of 42 with a methyl group abolished plasmin inhibition, but retained plasma kallikrein inhibition.128
Figure 9.
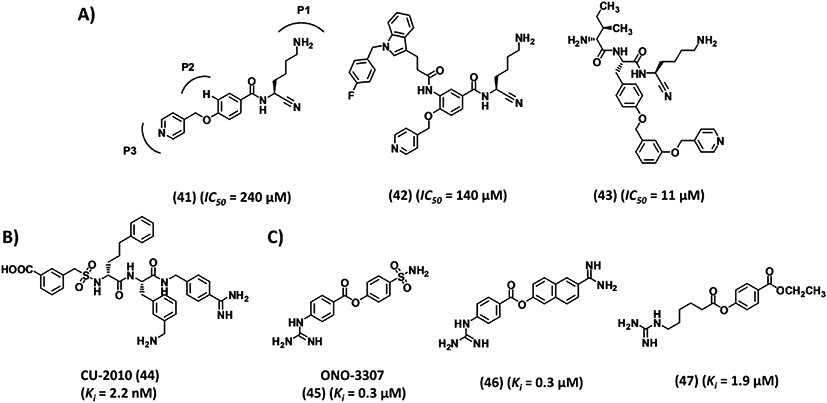
Structures of diverse group of human plasmin inhibitors 41–49. (A) Examples of nitrile warhead-based covalent inhibitors. (B) CU-2010 is amidinobenzyl-based peptidomimetic inhibitor that showed high promise, but its development was recently stopped. (C) Nonspecific inhibitors that are commonly used in complex pathologies such as DIC and pancreatitis. Ki is the inhibition constant for plasmin. IC50 refers to inhibition of plasmin amidolysis.
In a parallel study, five different tripeptidic nitrile warheads of D-Ile-Phe-Lys-CN type were studied containing variations at the Phe-containing P2 domain.129 The lead tripeptide showed a plasmin IC50 of 78 μM and 13-fold selectivity over plasma kallikrein and urokinase. Introducing O-benzyl, O-picolyl or O-picolyloxybenzyl Tyr (43, Fig. 9) in the place of Phe enhanced the potency against plasmin by —sevenfold to tenfold. However, this decreased the selectivity against plasma kallikrein (IC50 210–320 μM). P1 domain modifications to imidazole or triazole ethylene, or butyl side chains rendered the compounds inactive. Likewise, substitution of the P3 D-Ile with Ile, Gly, acetyl, or pivaloyl significantly diminished plasmin inhibition suggesting that the S3 subsite of plasmin prefers a branched hydrophobic residue, which was consistent with previous reports.129
Aldehyde-based peptidomimetic inhibitors
In the manner of the nitrile group as a warhead, the aldehyde group can also be used to replace the scissile bond and develop a warhead that covalently nullifies the activity of active site Ser of enzymes. Swedberg and Harris identified two tetrapeptide aldehydes from a library as potent active site inhibitors of plasmin.80 Tetrapeptides Ac-Lys-Met(sulfone)-Tyr-Arg-H and Ac-Arg-Met(sulfone)-Tyr-Arg-H inhibited plasmin with Ki of 3.1 and 9.9 nM, respectively. The former also inhibited trypsin and plasma kallikrein with IC50 of 95 and 366 nM, respectively. In addition, the molecules inhibited fibrinogenolysis by plasmin at 250 nM. Molecular modeling studies of the tetrapeptide binding to the catalytic plasmin unit showed that the aldehyde group is likely to be well accommodated in S1 subsite and multiple hydrogen bonds between the two interacting partners are likely to form. In addition, Tyr of the tetrapeptide was likely to interact with His603 (His57 in chymotrypsinogen numbering) of the active site through pi–pi interactions.80 It is important to mention that this study’s results are at variance with previous residue level specificity conclusions.77-79 For example, substrates with Arg, rather than Lys, were cleaved more efficiently by plasmin. There was a slight preference for Tyr at P2 in contrast to previous studies showing preference for Trp or Phe. There was no clear evidence that plasmin prefers a basic residue at P4, rather preference for Val or Phe was observed. Finally, the high in vitro potency and selectivity of the tetrapeptide aldehyde inhibitors has not been translated to in vivo evaluation as yet.
CU-2010
CU-2010 (renamed MDCO2010 (44), Fig. 9) is a peptidomimetic inhibitor.130, 131 The inhibitor has a benzamidinomethylamine moiety at the C-terminus mimicking a P1 motif. Inhibitor 44 displays a Ki of 2.2 nM against plasmin, but also good potency against other proteases including factors Xa (Ki 45 nM), XIa (Ki 18 nM), XIIa (Ki 5200 nM), and IIa (Ki 1700 nM) (Table I). It is especially more potent against plasma kallikrein (Ki 0.019 nM).130 Human whole blood clot lysis was suppressed by inhibitor 44 at 150 nM, which is 2- and 18-fold better than that for aprotinin and TXA, respectively.130 CU-2010 had a significant effect on APTT and a weak effect on PT, while aprotinin prolonged only the APTT at comparable concentration.
Considering its interesting inhibition profile, CU-2010 half-life was studied and found to be 20 min in dogs and rats following intravenous administration, which increases to 45 min for its pegylated analog CU-2020.132 But pegylation had variable effect on inhibition potencies of factor XIIa (4.5-fold increase), plasmin (fourfold loss), kallikrein (fourfold decrease), and factor XIa (31-fold decrease), while retaining factor Xa and thrombin inhibition potential.132 Both CU-2010 and CU-2020 reduced blood loss by approximately threefold relative to the vehicle (149 ± 24 mL) after cardiopulmonary bypass surgery in a canine model, which was similar to that observed for aprotinin.132 Despite the controversial lack of specificity, CU-2010 not only dose-dependently reduced postoperative blood loss, similar to aprotinin, but also improved the postischemia recovery of myocardial and endothelial functions in a canine model of cardiac surgery. This probably arises from its kallikrein inhibition that results in an antiinflammatory effect.133
Surprisingly, Phase 2b study of CU-2010 was stopped because of some evidence of enhanced risk.134 This could be because of CU-2010’s prolongation of activated clotting time arising from inhibition of coagulation factors Xa, Xia, and activated protein C. Activated clotting time is widely used for monitoring heparin anticoagulation in patients during cardiac surgery and its prolongation by CU-2010 may interfere with the evaluation of a patient’s anticoagulation state leading to underdosing of heparin.135
ONO-3307, nafamostat, and gabexate
ONO-3307 (45), nafamostat (46), and gabexate (47) (Fig. 9) are guanidine/amidine-based, highly nonselective serine proteases inhibitors. The first two molecules inhibit plasmin with a Ki of 0.31 μM, whereas 47 inhibits with sixfold lower potency.136 They also inhibit trypsin, thrombin, and kallikrein competitively (Ki < 5 μM) and their effects can be reversed by dialysis.136 ONO-3307 and gabexate were shown to completely inhibit the deposition of labeled fibrinogen in kidney, lung, and liver in experimental animal model of t-AMCHA-induced thrombosis in rats at 10 mg/kg/hr. In addition, 100 μM ONO-3307 was shown to inhibit elastase release from stimulated human leukocytes. Likewise, the three inhibitors inhibited thromboplastin release from stimulated leukocytes at ~10 μM dosage.136 Due to the combined effect on thrombin (coagulation) and plasmin (fibrinolysis), ONO-3307 was tested as a treatment for DIC137 and pancreatitis.138-140 Yoshikawa et al. found a protective effect against endotoxin-induced DIC in rats with 10 or 100 μg/kg/h of ONO-3307 as measured by fibrin degradation products, number of renal glomeruli with fibrin deposition, and other effects.137 Likewise, Hirano and others observed beneficial effects of ONO-3307 in rat acute pancreatitis.138-140
Both nafamostat and gabexate have been approved for clinical use in Japan and Italy. Reports suggest that prophylactic intravenous nafamostat mesilate reduces the frequency of postendoscopic retrograde cholangiopancreatography pancreatitis.141 Nafamostat is also useful as an anticoagulant during continuous venous hemodialysis142 and it attenuates postreperfusion syndrome during liver transplantation.143 Likewise, gabexate appears to be pharmacologically useful in pancreatitis,144 liver transplantation,145 and hemodialysis anticoagulation.146 Both drugs are also potentially beneficial in the treatment of DIC.147, 148
B. Polypeptide Inhibitors of Plasmin–Natural and Engineered
Plasmin can be functionally regulated by several polypeptide inhibitors of the Kunitz or Kazal type. Despite their structural similarity to plasmin substrates, the polypeptides act as extremely potent inhibitors. The polypeptides bind to the active site of plasmin in a substrate-like fashion utilizing the enzyme’s catalytic triad,149, 150 but the products of hydrolysis remain associated due to extremely slow hydrolysis, which favors resynthesis of the peptide bond.149-152 This inhibition mechanism is widely known as the Finkenstadt and Laskowski mechanism.151, 152 Several reasons have been proposed to explain such an inhibitory mechanism including (1) the extreme rigidity of the protease–polypeptide complex stemming from the internal network of stabilizing hydrogen and disulfide bonds in the binding loops of the protease inhibitors, (2) the poor orientation of the reactive groups resulting in a nonproductive complex, or (3) the orientation of the leaving group in the acyl-enzyme complex, which favors the reverse reaction to regenerate the Michaelis complex.149, 150
A brief description of the loop sequences for each natural polypeptide inhibitors of plasmin along with their Ki’s is provided in Table II. The Schechter and Berger nomenclature is used to describe the inhibitory peptide sequence.153 Each polypeptide is a competitive inhibitor, which blocks the active site without inducing any major conformational change and forms an antiparallel β sheet in the active site of proteases. The inhibitors typically display nanomolar affinity but suffer from poor selectivity. Nearly all sequences are natural, except DX-1000154 and KD1-L17R,155 which have been engineered. Of these, bovine pancreatic trypsin inhibitor (BPTI) has been therapeutically used as antifibrinolytic agent.156
Table II.
Polypeptide Inhibitor’s Human Plasmin and Plasma Kallikrein Inhibition Constants (Ki, in nM) and Their Recognition Sequences
| Ki (nM) |
Recognition sequence |
References | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Human plasmin | Plasma kallikrein | P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ | ||
| Natural Kunitz-type | |||||||||||
| Aprotinin (BPTI) | 0.18 | 13.4 | Gly | Pro | Cys | Lys | Ala | Arg | Ile | Ile | 160-165 |
| KD APPH | 81 | 86 | Gly | Pro | Cys | Arg | Ala | Val | Met | Pro | 188 |
| KD APP (KPI PN2) | 42 | 82 | Gly | Pro | Cys | Arg | Ala | Met | Ile | Ser | 188 |
| KD TFPI-2 | 10 | ND | Gly | Pro | Cys | Arg | Ala | Leu | Leu | Leu | 190 |
| KD1 rTFPI-1 | 26 | ND | Gly | Pro | Cys | Lys | Ala | Ile | Met | Lys | 191 |
| Placental bikunin | 0.5–1.0 | ~0.7 | Gly | Pro | Cys | Arg | Ala | Phe | Ile | Gln | 192, 193 |
| L1TI | 0.32 | 6.3 | Ser | Pro | Tyr | Arg | Leu | Gly | Ser | Asn | 194 |
| TdPI | 55 | ND | Gly | Leu | Cys | Lys | Ala | Arg | Phe | Tyr | 195 |
| Textilinin-1 | 0.11–3.5 | 4830 | Gly | Pro | Cys | Arg | Val | Arg | Phe | Pro | 196-199 |
| AvKTI | 4.9 | ND | Gly | Pro | Cys | Lys | Ala | Ser | Leu | Tyr | 203 |
| DrKIn-II | 0.2 | ND | Gly | Arg | Cys | Arg | Ala | His | Leu | Arg | 204 |
| Bt-KTI | 2.01 | ND | Gly | Thr | Cys | Arg | Gly | Tyr | Phe | Pro | 205, 206 |
| Tryptogalinin | 5.83 | NI | Gly | Pro | Cys | Lys | Ala | Met | Phe | Arg | 207 |
| Engineered Kunitz-type | |||||||||||
| DX-1000 | 0.087 | 250 | Gly | Pro | Cys | Arg | Ala | Arg | Phe | Asp | 154, 224 |
| 4PEG DX-1000 | 0.232 | ND | Gly | Pro | Cys | Arg | Ala | Arg | Phe | Asp | 225 |
| KD1-L17R | 0.9 | >3000 | Gly | Pro | Cys | Arg | Ala | Arg | Leu | Leu | 155 |
| KPI PN2-R15K/M17K | 8 | NI | Gly | Pro | Cys | Lys | Ala | Lys | Ile | Ser | 189 |
| Natural Kazal-type | |||||||||||
| Infestin-4 | 2.1 | ND | Ala | Cys | Phe | Arg | Asn | Tyr | Val | Pro | 210-214 |
| Bdellin-KL | 8.64 | ND | Val | Cys | Thr | Lys | Glu | Leu | Leu | Arg | 215 |
| clTI-1 | 83 | ND | Gly | Cys | Pro | Arg | Asp | Tyr | Ser | Pro | 216, 217 |
| Bikazin | 2000 | ND | Ala | Cys | Pro | Arg | Leu | His | Gln | Pro | 217 |
| AaTI | 3.8 | NI | Ala | Cys | Pro | Arg | Ile | Tyr | Met | Pro | 218, 219 |
| Natural Subtilisin-type | |||||||||||
| Plasminostreptin | 0.49 | NI | Ala | Cys | Thr | Lys | Gln | Phe | Asp | Pro | 220 |
ND, not determined or reported; NI, no inhibition at the highest concentration tested.
1. Natural Kunitz-Type Inhibitors
Bovine pancreatic trypsin inhibitor (BPTI, also called aprotinin)
BPTI is a Kunitz-type serine protease inhibitor of bovine pancreatic trypsin that was introduced independently by Kraut et al.157 and Kunitz and Northrop in the 1930s.158 Structurally, it is a single polypeptide chain that is crosslinked by three disulfide bridges. It can be obtained from the pancreas, parotid gland, and the lung of cows, however, only the lung form is clinically used. It is a 58 residue basic protein containing six Arg residues at positions 1, 17, 20, 39, 42, and 53 as well as four Lys residues at positions 15, 26, 41, and 46.156 To identify the P1 residue, chemical labeling with N-carboxy-DL-alanine anhydride in the presence and absence of trypsin was employed. Lys15 was the only basic residue that remained unmodified in the presence of trypsin suggesting its binding in the S1 anionic pocket. This was further confirmed through sequencing of the tryptic digest that identified Lys15-Ala16 as the scissile bond.159
Aprotinin targets several serine proteases (Tables I and II).156 The broad specificity of wild-type aprotinin can be altered through site-specific mutagenesis.160-165 For example, Lys15Arg mutation did not impact plasmin inhibition, but increased antifactor Xa (~484-fold) and antithrombin activity (~17-fold).164 Likewise, Lys15Val aprotinin exhibited higher potency against leukocyte elastase.160
Aprotinin was widely used in antifibrinolytic therapy for reducing perioperative bleeding and the need for blood transfusions during organ transplantation, and orthopedic, cardiac, and thoracic surgeries.1, 2, 5, 82-84, 90, 91 Aprotinin reduced the high risk of allergic reactions and transmission of infections associated with multiple blood transfusions.3, 4, 83, 166, 167 In addition, the polypeptide has other beneficial effects due to its activity against serine proteases of the coagulation and inflammatory systems that help reduce some adverse consequences during cardiopulmonary bypass.168-170 Likewise, the coagulation and inflammatory complications arising from contact with the artificial surfaces of heart and lung devices also tend to be lower because of aprotinin.52 Evaluation of aprotinin in different in vitro and in vivo models of fibrinolysis, coagulation, and thrombus formation has been reported.171 Fibrinolysis was inhibited by aprotinin with an IC50 of 0.16 μM. Aprotinin also inhibited in vivo thrombus formation and reduced rat tail bleeding time in a dose-dependent manner. It appears that aprotinin reduced blood loss and the need of blood transfusion by an average of 45–60%.1, 172
Aprotinin is monomeric in solution, but exists as a decamer at high-salt concentrations. Inorganic sulfate neutralizes its basic amino acids and stabilizes the decameric form. A clinical counterpart of such sulfate–aprotinin interaction is the heparin–aprotinin interaction, for which a model has been proposed.173 Heparin is a mixture of highly sulfated polysaccharide chains that is biosynthesized in the mast cells along with aprotinin.174 This implies that bovine heparin may carry aprotinin, which may impact the heparin anticoagulant therapy.175 Aprotinin is known to interfere with heparin binding to platelets and appears to reduce peri- and postoperative blood loss resulting from heparin usage.176, 177
Despite the many advantageous effects of aprotinin, results from the BART clinical trial led to suspension of aprotinin’s clinical use in May 2008.178, 179 This large multicenter study found that even though the risk of bleeding was lowest in the aprotinin group for patients undergoing high-risk cardiac surgery in comparison to TXA and EACA groups, mortality was significantly higher, which led to early termination of the trial. A recent Cochrane review also concluded that the risk of death was higher with aprotinin.180 Aprotinin has been also reported to increase the incidences of renal failure,178, 181, 182 myocardial infarction,178 vein graft hypercoagulation,183 anaphylactic shock,184 and mortality.179, 181, 182 However, several subsequent studies have shown the BART results to be controversial.185, 186 Health Canada published a safety review of aprotinin in September 2011, which suggested that the benefit of using aprotinin in cardiac surgery might offset the risk.83, 187 Accordingly, aprotinin was made available again in Canada for restricted use in isolated coronary bypass graft surgery. The European Medicines Agency also recommended lifting the suspension of aprotinin in February 2012 after a review of the risks and benefits of antifibrinolytic drugs.83
2. Other Natural Kunitz-Type Inhibitors
Alzheimer amyloid precursor protein and its homolog
The Kunitz domain of Alzheimer amyloid precursor protein homolog (KD APPH) was found to inhibit plasmin with a Ki of 81 nM. KD APPH inhibited several other proteases too including trypsin (Ki 0.02 nM), plasma kallikrein (Ki 86 nM), glandular kallikrein (Ki 8.8 nM), chymotrypsin (Ki 78 nM), and factor XIa (Ki 14 nM).188 Likewise, the Kunitz domain of amyloid precursor protein (KD APP), also reported in the literature as Kunitz protease inhibitor of protease nexin-2 (KPI PN2), potently inhibits trypsin, chymotrypsin, and factor XIa with Ki of 0.02, 6.0, and 0.7 nM, respectively, and moderately inhibits plasmin, plasma kallikrein, and glandular kallikrein (Ki 42–82 nM).188 Mutagenesis at specific sites in the recognition sequence of KPI PN2, especially P1 Arg→Lys and P2′ Met→Lys/Arg (Table II), fine tuned activity against plasmin (Ki 8 nM) at the cost of activity against factor XIa and kallikrein.189 Although wild-type KPI PN2 was potently antithrombotic, the double mutants were antifibrinolytic without displaying antithrombotic complications.189
Tissue factor pathway inhibitor-1 (TFPI-1) and TFPI-2
The first Kunitz domain of human TFPI-2 (KD TFPI-2) inhibited plasmin (Ki 10 nM), while also inhibiting trypsin (Ki 7 nM).190 In contrast, recombinant domain 1 of hTFPI-1 inhibited several enzymes including factor VIIa/tissue factor (Ki 250 nM), cathepsin G (Ki 200 nM), and plasmin (Ki 26 nM). Its recombinant domain 2 inhibited factor Xa (Ki 90 nM), trypsin (Ki 0.1 nM), and chymotrypsin (Ki 0.75 nM), while Kunitz domain 3 appeared to have no inhibitory function.191
Bikunin
Placental bikunin is a 170-amino acid human serine protease inhibitor containing two Kunitz-type inhibitory domains, the N-terminal(7–64) Kunitz domain, and the C-terminal(102–159) Kunitz domain.192, 193 The two domains can function independently and can also direct ternary complexation with selected serine proteases.192, 193 Both domains inhibited several enzymes of the intrinsic coagulation and fibrinolytic pathways including plasmin (Ki 0.5–1.0 nM), trypsin (Ki ~0.03 nM), chymotrypsin (Ki ~2 nM), plasma kallikrein (Ki ~0.7 nM), and pancreatic kallikrein (Ki ~0.5 nM). But there were some interesting differences too. Tissue kallikrein was more potently inhibited by C-terminal(102–159) Kunitz domain (Ki 0.13 nM). However, the high potency could not be easily exploited because of considerable cross reactivity.
L1T1
A 174-residue polypeptide L1TI was isolated from Leucaena leucocephala seeds and found to inhibit human plasmin with a Ki of 0.32 nM.194 Other enzymes (plasma kallikrein, trypsin, and chymotrypsin) were inhibited with at least 20-fold lower potency. L1TI prolonged clotting time in the APTT assay, but not in PT or TT assays, suggesting its primary effect on the contact activation pathway. L1TI also inhibited kinin release from high molecular weight kininogen, decreased carrageenin-induced edema, and lowered bradykinin suggesting an antiinflammatory effect.194
Tick-derived protease inhibitor
Another natural plasmin inhibitor (Ki 55 nM) is tick-derived protease inhibitor (TdPI), which is a 97-amino acid polypeptide isolated from Rhipicephalus appendiculatus. The polypeptide also inhibited human tryptase (Ki ~1.5 nM) and trypsin (Ki 5.6 nM),195 but it does not affect urokinase, thrombin, factor Xa, factor XIIa, elastases, kallikreins, cathespsin G, granzyme B, chymase, and chymotrypsin.195
Textilinin-1 and -2
Textilinin-1 and -2 are Kunitz-type serine protease inhibitors isolated from the Australian snake Pseudonaja textilis having 59 residues and ~45% and 43% identity to aprotinin, respectively.196 Both inhibitors contain six conserved cysteines common to all Kunitz-type inhibitors and bind tightly to plasmin with Ki of ~0.11–3.5 nM.197 Both inhibitors reduced blood loss by a substantial 60% in a murine tail vein bleeding model.196 Although textilinin-1 appears to be a more specific plasmin inhibitor than aprotinin, the latter was able to inhibit clot lysis better.198 Textilinin-1 was several-fold less potent than aprotinin in inhibiting kallikrein (plasma and tissue), trypsin and plasmin (Tables I and II). But it was more specific than aprotinin with respect to direct inhibition of tPA, urokinase, activated protein C, and elastase.199 The crystal structure of free textilinin-1 has just been reported and found to be similar to that of aprotinin.200 Also, the crystal structure of textilinin-1–microplasmin complex has been solved.201 The narrower specificity of textilinin-1 most probably arises from its bulkier P1′ Val in comparison to the P1’ Ala present in aprotinin.201 Textilinin-1 appears to be worth investigating further as an antifibrinolytic agent.202
AvKT1, DrKln-II, and Bt-KTI
A plasmin inhibitor was isolated from Araneus ventricosus spider and found to be a Kunitz-type protease inhibitor (AvKTI).203 Recombinant AvKTI having a 57-amino acid Kunitz domain inhibited plasmin (Ki 4.9 nM), neutrophil elastase (Ki 169 nM), trypsin (Ki 7.3 nM), and chymotrypsin (Ki 37.8 nM).203 Likewise, recently a slow and tight-binding inhibitor of plasmin (Ki 0.2 nM) was isolated from Russell’s viper (Daboia russelii) venom and named as Kunitz-type protease inhibitor (DrKIn-II).204 It appeared to inhibit plasmin more than most other proteases screened. It prolonged APTT but not PT. DrKIn-II demonstrated antifibrinolytic activity in fibrin plate assay and prolonged the clot lysis time. Finally, DrKIn-II inhibited formation of fibrin/fibrinogen degradation product in a coagulation-stimulated mice model and diminished murine tail bleeding time.204 The third Kunitz-type polypetide Bt-KTI is a 58-residue inhibitor from bumblebee Bombus terrestris venom.205, 206 Bt-KTI is antifibrinolytic owing to its potent plasmin inhibition activity.
Tryptogalinin
Tryptogalinin is a tick-derived Kunitz-type serine protease inhibitor isolated from Ixodes scapularis.207 Besides its potent inhibition of β-tryptase (Ki 0.01 nM), tryptogalinin potently inhibited plasmin, trypsin, and chymotrypsin with Ki values of 5.8, 0.5, and 0.4 nM, respectively. It also moderately inhibited matriptase and elastase (Ki ~14 and 19 nM, respectively). This inhibitor did not inhibit coagulation enzymes (thrombin, factors Xa, XIa, or Xlla), chymase, kallikren, or plasminogen activators indicating its special specificity features.
3. Natural Kazal-Type Inhibitors
Kazal-type serine protease inhibitors are similar to the Kunitz-type inhibitors in terms of their size (40–60 residues) and the number of the conserved disulfide bridges. Yet, the difference arises from the combination of the six conserved cysteines in forming the disulfide bridges. Although cysteines 1 and 5, 2 and 4, and 3 and 6208 combine for Kazal-type inhibitors, it is 1 with 6, 2 with 4 and 3 with 5 for Kunitz-type polypeptides.209 This results in their characteristic three-dimensional structures that are different. Five natural Kazal-type plasmin inhibitors are described below.
Infestins
Infestins are Kazal-type serine protease inhibitors found in the midgut of the Chagas’ disease vector, Triatoma infestans.210 Infestins are composed of seven Kazal domains. Although native infestin did not show plasmin inhibition, recombinant infestin 1–4 (fourth to seventh Kazal domains), infestin 3–4, and infestin 4 inhibited plasmin potently with Ki of 1.1, 0.4 and 2.1 nM, respectively.210-214 These Kazal domains also inhibited other serine proteases including thrombin, trypsin, factor XIIIa, and factor Xa, although not all were inhibited by each polypeptide. Structural characterization of the seven Kazal domains revealed that domains second to fifth share high-sequence homology and thus, inhibitory properties.211 However, the data appears to indicate that infestin’s antiplasmin activity stems from the seventh Kazal domain (infestin-4). The plasmin inhibitory activity of infestins has not been exploited for antifibrinolytic purposes. Instead, infestins are being studied in molecular imaging.214
Bdellin-KL
Bdellin-KL is a trypsin (Ki 3.6 nM) and plasmin inhibitor (Ki 8.6 nM) from the leech Hirudo nipponia and comprises a 48-amino acid Kazal-type protease inhibitor domain at the N-terminal (fragment 19–66) sequence.215 The intact protein (fragment 19–155) displayed similar inhibitory profile. Interestingly, its plasmin inhibitory activity remained intact up to 90°C at pH 1 and up to 50°C at pH 2 or 12.
clTI-1
Recently, Kubiak et al. identified a Kazal-type trypsin inhibitor, named as clTI-1 from the liver of Mleagris gallopavo chicken.216 This inhibitor avidly binds to human plasmin (Kd 83 nM) and different forms of trypsin (Kd 0.08–2.2 nM). Interestingly, the P1–P1’ sequence was found to be ArgP1–AspP1′.216, 217 Not much additional information is available on this molecule.
4. Other Kazal-Type Inhibitors
Other inhibitors belonging to the Kazal-type inhibitor family include the bikazin salivary inhibitor isolated from Canis familiaris dog submandibular glands and a trypsin inhibitor from Aedes aegypti (AaTI). The former has two structural domains, of which domain I inhibits trypsin and plasmin (Ki 2 μM) and domain II inhibits chymotrypsin, elastase, and subtilisin.217 The recombinant form of the latter inhibitor inhibited human plasmin, trypsin, and thrombin with Ki of 3.8, 0.15, and 320 nM, respectively.218, 219
5. Natural non-Kunitz-Type or Non-Kazal-Type Inhibitors
Plasminostreptin has been reported as a plasmin inhibitor isolated from Streptomyces antifibrinolyticus microbe.220 It does not belong to either Kunitz-type or Kazal-type inhibitor class. Plasminostreptin inhibited human plasmin (ID50 8 μg), bovine trypsin (ID50 1 μg) and several microbial alkaline proteases (e.g., subtilisin (ID50 2.5 μg)), but not thrombin, elastase or kallikrein.221 Plasminostreptin inhibited plasmin by forming a stoichiometric complex of 1:1 and doubled the clot lysis time at 8 μg dose.
6. Engineered Kunitz-Type Protease Inhibitors
Kunitz domains appear to be attractive platforms to design novel therapeutic proteins.154, 155, 222-226 Several reasons contribute to this characteristic. Structurally, Kunitz domains are relatively small peptides of about 60 amino acids that exhibit high stability because of the presence of three disulfide linkages.227 They can be engineered to possess high stability to inactivation by oxidants, high temperatures, and extreme acidity or basicity. Their expression in yeast occurs with high efficiency and yields.154, 155 Finally, literature reports their relatively safe use in humans, although concerns about their immunogenicity, bioavailability, and plasma half-life have to be kept in mind. We describe here two promising engineered Kunitz domains that have been developed for potential use as antifibrinolytics to substitute the recently withdrawn aprotinin.
DX-1000
Markland et al. reported the iterative use of phage display to generate small libraries of Kunitz domains having nearly human sequences and possessing high affinity and specificity for human plasmin.154 A series of libraries having variants of the first Kunitz domain of hTFPI-1 were iteratively generated through variations at 13 positions in the P1 region (residues 10–21) and in the second loop (residues 31–39). These two regions comprise the end of the Kunitz domain, which interacts with the serine protease target. One particular engineered protein (EPI-P302 or DX-1000) was identified to have interesting antifibrinolytic profile comparable to aprotinin.
DX-1000 binds tightly to human plasmin (Ki 0.087 nM), which reflects about 300-fold increase in the affinity relative to the parent domain KD hTFPI-1 (Table I). It was also several thousand-fold selective over plasma kallikrein (Ki ~250 nM), factor Xa (Ki 1.3 μM), human chymotrypsin (Ki 10 μM), urokinase (Ki > 100 μM), and human neutrophil elastase (Ki ~800 nM). Interestingly, DX-1000 did not inhibit human thrombin at the highest concentration tested (Ki >> 100 μM).154 Comparison of DX-1000 sequence with its parent KD hTFPI-1 revealed seven differences including position 10 (E vs. D), 11 (T vs. D), 15 (R vs. K), 17 (R vs. I), 18 (F vs. M), 19 (D vs. K), and 21 (W vs. F).154 Interestingly, the plasmin inhibitory activity of DX-1000 remained essentially unaffected by temperatures as high as 85°C, or by prolonged incubation at pH 10, or in the presence of chloramine T, a very strong oxidant.154 DX-1000 was recently evaluated in hemostasis and coagulation studies.226 In these studies, DX-1000 inhibited plasmin with a high affinity (Ki 0.1 nM) and did not inhibit human tPA or uPA (Ki > 10 μM). Despite the good antiplasmin activity, DX-1000 was evaluated as an antineoplastic agent226 rather than as an antifibrinolytic agent.
KD1-L17R
An interesting example of an engineered Kunitz-type plasmin inhibitor is KD1-L17R. It is the first Kunitz domain of hTFPI-2 in which the P2′ residue was mutated to Arg.155 Human TFPI-2 contains three Kunitz domains, of which the first domain was found to inhibit several enzymes of the digestive system, coagulation pathways, and the fibrinolytic pathway. The second Kunitz domain was found to inhibit factor Xa and the third domain has no known inhibitory function.191, 228, 229 Taking these selectivities in consideration, a rational structure-based approach was developed that led to the design of KD1-L17R, which selectively inhibited fibrinolysis but not coagulation.
The reason why a single mutation was sufficient to engineer high selectivity is that digestive and coagulation proteases prefer hydrophobic residues at the P2′ position in their substrates and inhibitors, whereas plasmin favors a basic residue. The cationic P2’ in the engineered inhibitor was expected to interact with the strongly electronegative Glu73 and Glu143 present in the S2’ subsite of plasmin.230 In contrast, the S2′ subsite in plasma kallikrein, factor XIa and factor VIIa comprises a hydrophobic pocket that prefers nonpolar residues. Biochemical studies indicated that KD1-L17R had a Ki of 0.9 nM against plasmin and a Ki of at least 3000 nM against other serine proteases (Table I). This was in contrast to KD1-WT, which was less potent against plasmin (Ki 6 nM).155
KD1-WT prolonged the clotting time in APTT and PT assays. In contrast, KD1-L17R did not prolong the clotting times as expected of its higher selectivity. In the fibrinolysis assay, KD1-L17R inhibited tPA-induced plasma clot fibrinolysis with an IC50 of 0.36 μM, which was at least fivefold better than KD1-WT and tranexamic acid. Mouse liver laceration animal model was also exploited to assess the effect of KD1-L17R on the amount of blood loss. It was found that the amount of blood loss was reduced by ~84% by KD1-L17R, ~70% by aprotinin, ~52% by TXA, and ~10% by KD1-WT. These results suggest that KD1-L17R is very effective in inhibiting plasma clot fibrinolysis and in reducing blood loss in animal models in a manner comparable to clinically used aprotinin.155
Based on the fact that KD1-L17R and aprotinin share similar size, and that the dose at which each satisfies the required therapeutic outcome is also similar, it was proposed that they both share similar pharmacokinetics. KD1-L17R did not induce renal toxicity in mice when used at doses higher than that of aprotinin and resulted in no detectable histopathologic changes in major organs including heart, lungs, brain, and liver.155 KD1-L17R also did not cause any seizures as observed for TXA.231 These results suggested that KD1-L17R was likely to have a better safety profile than existing antifibrinolytic agents. At this point, the potential of this engineered protein is being further studied.
It is important to mention here that several reports indicate the value of direct inhibition of plasmin in reducing blood loss during on-pump cardiothoracic surgery.225, 232, 233 For example, DX 88 is a mutated version of the Kunitz domain from human lipoprotein-associated coagulation inhibitor domain 1. It was studied for two major indications; hereditary angioedema, for which it was approved in 2009,232 and reduction of blood loss during on-pump cardiothoracic surgery, in which it failed relative to TXA.225, 233 Likewise, DXX 88 is a potent kallikrein inhibitor (Ki 25 pM). But it also inhibits plasmin (Ki 29 nM). The inhibitor has good safety and tolerability profiles.217, 232, 233 Hence, the failure for the second indication does not necessarily implicate any dangerous risk, yet it does emphasize the importance of direct inhibition of plasmin to reduce blood loss over kallikrein inhibition.
C. Sulfated Polymers as Plasmin Inhibitors
1. Heparin and Heparan Sulfate
Heparin is a variably sulfated, highly heterogeneous and polydisperse mixture of linear sulfated GAGs. Structurally, heparin is composed of repeating disaccharide units of glucosamine and uronic acid (Fig. 11A).234 Heparin has long been clinically used as an anticoagulant to prevent and treat thromboembolic diseases. Heparin’s anticoagulant activity stems from its binding to antithrombin (AT), which enhances inactivation of coagulation enzymes, particularly thrombin and factor Xa.12
Figure 11.
Structures of glycosaminoglycan (GAG) mimetics. (A) Heparin is a polysaccharide that consists of variably sulfated glucosamine and uronic acid units. (B) Sulfated LMWLs are liginin-based polymers that have variable level of sulfation and hydrophobicity. (C) PVA copolymers. (D) Chemically modified dextran sulfates.
In addition to its effect on the coagulation system, heparin was also found to affect the fibrinolytic system. Yet, how heparin interacts with different components of the fibrinolytic pathway remains controversial.235 The reason for this is that many of the experimental conditions used were not physiological. Nevertheless, the heparin–plasmin interaction has received significant attention, particularly to explore a route to novel antifibrinolytic agents. Even here, it is important to recognize that heparin’s impact on plasmin activity, direct or indirect, seems to be limited at NaCl concentrations higher than 25 mM.
Plasmin inhibition can be brought about by other plasma inhibitors including α2-antiplasmin, α1-antitrypsin, C1 esterase inhibitor, α2-macroglobulin, and AT. Highsmith et al. studied the interaction of AT and plasmin.236 AT was found to be a time-dependent inhibitor of plasmin’s proteolytic activities. Sodium dodecylsulfate gel electrophoresis indicated that AT forms a stoichiometric covalent complex, which is stable in the presence of denaturing or reducing agents. The presence of heparin accelerated plasmin inhibition by AT by about 60-fold. However, increasing the plasmin concentration relative to heparin reduced the rate.237
Heparin bound directly to plasmin with a KD of 10 nM238-240 and induced a conformational change in its active site.241 Interestingly, heparin enhanced hydrolysis of several chromogenic substrates through an increase in kCAT (KM unchanged). The polysaccharide also enhanced the rate of enzyme inactivation by N-α-tosyl-lysine-chloromethylketone, an active site histidine targeting agent, but not by phenyl-methylsulfonyl fluoride, an active site serine probe. Likewise, heparin increased the heat sensitivity (denaturation) of plasmin when a P1-Lys chromogenic substrate was used, but did not affect enzyme activity when P1-Arg substrates were used. These results suggest that heparin induced microscopic environmental changes in the active site of plasmin by binding to an allosteric site.241 In fact, kinetic studies using chromogenic substrate S-2251 demonstrated that heparin is a noncompetitive inhibitor of plasmin with a Ki of 4.9 μM.240
Recently, Chander et al. reported plasmin inhibition under physiological conditions by a covalent complex of AT and heparin.235 Results revealed that the increase in plasmin inactivation is brought about by inducing a conformational change in the enzyme. The study also found that plasmin inhibition was decreased in the presence of fibrin, fibrinogen, and EACA.235
Earlier studies have shown that oleic acid exhibits a contradictory effect on the amidolytic and fibrinolytic activity of plasmin. Although oleic acid stimulated plasmin amidolysis of S-2251, it inhibited cleavage of the physiological substrate fibrin.242, 243 Competitive studies with lysine analogs indicated that oleic acid bound to the LBSs of plasmin and induced a conformational change to bring about such contradictory effects. The effect of oleic acid on plasmin was later exploited to enhance the inhibitory action of heparin. Combining oleic acid and heparin in a single molecule, as in N-oleoyl heparin, yielded a more potent inhibitor of plasmin (IC50 16 nM).244 N-oleoyl heparin was also devoid of antifactor Xa activity, which means that it may carry only antifibrinolytic activity.244 Other GAGs including hyaluronic acid, chondroitin sulfate, dermatan sulfate, and heparan sulfate did not inhibit the peptidolytic activity of plasmin. Yet, oversulfation of chondroitin sulfate generated an antiplasmin molecule (IC50 60 nM), especially when four sulfates were present per disaccharide unit.244 Plasmin inhibition by N-oleoyl heparin was highly dependent on salt and detergent concentration suggesting a role for both ionic and hydrophobic interactions. Lineweaver-Burk analysis indicated that N-oleoyl heparin is a noncompetitive inhibitor with a Ki of 8 nM.244
Plasmin’s heparin-binding site(s) is(are) yet to be determined. The site(s) is(are) most probably located in the catalytic domain, but could also be in the kringle domains or both. Several researchers have exploited these putative heparin-binding site(s) to introduce heparin mimetics that are capable of potently inhibiting plasmin.245-247 Three classes of heparin mimetics functioning as plasmin inhibitors are described in the following sections.
2. Sulfated Low Molecular Weight Lignins (LMWLs)
Sulfated LMWLs have been developed as polymeric, nonsaccharide mimetics of heparin that utilize dual ionic and hydrophobic interactions in binding to proteins.248 Three sulfated LMWLs, CDSO3, FDSO3, and SDSO3 (Fig. 11B), were synthesized and found to be potent inhibitors of human plasmin as well as thrombin.245, 249 The polymers were synthesized in two steps involving a chemo-enzymatic process in which horseradish peroxidase-catalyzed oxidative coupling of 4-hydroxycinnamic acid monomers was followed by sulfation using triethylamine-sulfur trioxide complex. The enzymatic process relies on a radical coupling reaction, which results in polymers containing multiple inter-residue linkages, such as β-O-4 and β-5, and heterogeneity and polydispersity resembling low molecular weight heparins. The three sulfated LMWLs displayed molecular weights in the range of 3000–4000 Da, chain lengths in the range of 5–15 units and 0.3–0.4 sulfate groups per unit.
Chromogenic substrate hydrolysis assays indicated that CDSO3 potently inhibited the amidolytic activity of human plasmin with an IC50 value of 0.24 μM and efficacy of nearly 100%. FDSO3 and SDSO3 were threefold and fivefold less potent. Indirect AT-mediated inhibition of plasmin was also evaluated and found to have limited contribution to the overall inhibition process.245 Among the three polymers, FDSO3 was tested for the inhibition of fibrin cleavage using an in vitro transmittance assay, which revealed good reduction in fibrinolysis over 0–6.4 μM concentration. Mechanistically, sulfated LMWLs were found to disrupt the plasmin’s catalytic apparatus as revealed by Michaelis–Menten kinetics in presence of CDSO3. Binding of CDSO3 to plasmin affected both KM and VMAX of the hydrolysis suggesting a mixed inhibition mechanism. Competitive binding studies indicated that FDSO3 competed well with 30 nM and 300 nM full-length heparin, which implied binding to the heparin-binding site on plasmin.245
Although sulfated LMWLs are good inhibitors of plasmin, they inhibit several other serine proteases,250, 251 including thrombin, factor Xa, factor IXa, factor XIa, leuckocyte elastase, and cathepsin G (Table I).250, 251 Especially for thrombin, extensive biochemical studies and site-directed mutagenesis studies have revealed that sulfated LMWLs bind in exosite 2, which is the heparin-binding site in thrombin.252 Thus, superimposition of the thrombin and plasmin structures helped identify the corresponding residues in plasmin that may recognize sulfated LMWLs.245, 252 The authors hypothesized that Arg644, Lys645, Arg637, Arg776, and Arg779 of plasmin may be important (Fig. 12).
Figure 12.

A snapshot of the active site of human microplasmin (PDB ID: 3UIR) and the adjacent putative heparin-binding site. The pose shows basic amino acids that are likely to be targeted by GAGs mimetics of the sulfated LMWLs type. The putative heparin-binding site includes Arg637, Arg644, Lys645, Lys651, Arg776, and Arg779 in addition to other hydrophobic amino acid (see text). The catalytic triad of His603, Asp646, and Ser741 (His57, Asp102, and Ser195 in chymotrypsinogen numbering) is also shown. Colors used are gray for the protein backbone, cyan for carbons of the catalytic triad, yellow for the carbons of the putative heparin-binding site, red for oxygen, and blue for nitrogen atoms. The figure was generated in PyMOL (http://www.pymol.org/).
Overall, sulfated LMWLs can be considered as nonspecific inhibitors that may be useful in preventing and/or treating complex pathologies. Clinically, several disordered conditions are known in which a cluster of serine proteases are active including pancreatitis, DIC, and cancer.253 Thrombin and plasmin are the two critical enzymes in DIC, in which coagulation and fibrinolysis processes are aberrant. Extensive clotting due to thrombin action in DIC is followed by bleeding due to plasmin action.6, 7, 137, 148 Hence, the dual inhibitory function of LMWLs may lead to a novel therapy for DIC. Also, inhibiting thrombin and plasmin could offer an efficient strategy to treat thrombotic cancers.26, 27, 42, 48, 243 Plasmin also activates MMPs, which play important roles in tissue remodeling and have been recently indicated to contribute to emphysema.254 Thus, direct inhibition of neutrophil elastase and plasmin-mediated indirect inhibition of MMPs could represent a novel therapy for chronic obstructive pulmonary diseases. This was recently demonstrated by Saluja et al. in an in vitro antiemphysema study of sulfated LMWLs.255
3. Chemically Modified Dextran Sulfate Derivatives
Considering plasmin’s biological roles in tissue remodeling (in addition to fibrinolysis), Ledoux reported synthetic dextran sulfates (DS) that mimic heparin’s antiplasmin activity.246 Four chemically modified DSs were synthesized from dextran T40. This included DSs that were carboxymethylated (RG1100), O-sulfonated (RG1003), carboxymethylated and O-sulfonated (RG1503), and carboxymethylated, amidated, and O-sulfonated (RG1192). The resulting polymers of 40–140 kDa size had variable levels of hydrophobic/hydrophilic substitution (Fig. 11D).246
S-2251 hydrolysis assay indicated that RG1100 and RG1003 were inactive at pH 7.4 and 37°C, but the dual carboxylated and O-sulfonated derivative, RG1503, inhibited human plasmin with an IC50 of 20 nM and a moderate efficacy of 30%. The most potent activity was derived upon introducing aromatic hydrophobic elements (benzyl groups) through amidation on the carboxylated polymer. The resulting triple-modified dextran derivative RG1192 was found to potently inhibit plasmin with an IC50 of 2 nM and efficacy of 80%. Interestingly, RG1192 inhibited tPA also (IC50 34 nM, efficacy 70%) but did not affect trypsin, chymotrypsin, and uPA.246
Mechanistically, both RG1503 and RG1192 were found to be reversible tight binding, noncompetitive inhibitors of human plasmin and displayed a stoichiometry of 1:6 and 1:20, respectively. The derivatives relied significantly on their sulfate groups for inhibiting plasmin. Using physiological salt concentration, only RG1192 retained an inhibitory activity toward plasmin (efficacy 70%, Ki 0.31 μM). Competitive studies in presence of EACA, kringle 1–3, kringle 4, or kringle 5 indicated that RG1192 preferred to bind to the LBSs on kringles 1, 4, and 5 with variable affinites but not to the active site. RG1192 bound to plasminogen also with high affinity (Kd 0.03 μM). SDS-PAGE analysis revealed that RG1192 renders the Arg561-Val562 scissile bond of plasminogen inaccessible and/or unrecognizable by uPA. This implies that RG1192 could contribute to the regulation of plasmin activity by three plausible mechanisms; (1) inhibition of plasmin generation due to plasminogen binding; (2) inhibition of tPA so as to prevent plasmin generation; and (3) direct and noncompetitive plasmin inhibition. Analysis by SDS-PAGE further revealed that plasmin-catalyzed degradation of fibronectin and laminin was inhibited by RG1192 (1–2.5 μM).246 In addition, the DSs protected various heparin-binding growth factors against proteolytic degradation in vitro and thus enhanced their bioavailability.256, 257 The derivatives also inhibited the enzymatic activity of neutrophil elastase.258 The inhibitory profile of DSs partially accounted for their roles in in vivo models of tissue repair and wound healing.256-262 Despite this success, reports on their antifibrinolytic activity to reduce blood loss have not been published as yet.
4. Sulfated Polyvinylalcohol-Acrylate Copolymers
Four sulfated polyvinylalcohol-acrylic acid (PVA) copolymers were prepared as heparin mimetics having varying number of negative charges (40.5–73.5%) and molecular weights (5600–8800 Da, Fig. 11C).247 The four copolymers were named PVA30, PVA38, PVA47, and PVA64 according to their sulfate content. Each copolymer, except for PVA30, inhibited small molecule hydrolysis by plasmin (IC50 80–110 nM) in pH 7.4 buffer containing 150 mM NaCl. Miniplasmin (kringles 1–4 absent) and microplasmin (kringles 1–5 absent) were similarly inhibited (IC50 28–31 nM and 120–140 nM, respectively).247 These results suggested that the binding site(s) for sulfated PVA polymers is(are) likely to be in the catalytic domain of plasmin. Under similar conditions, heparin had no effect on plasmin activity at concentrations as high as 2.2 μM, although it did inhibit plasmin at low ionic strengths of buffer.247 A change in the intrinsic fluorescence of plasmin was observed for all four copolymers indicating a possible change in conformation upon binding. PVA38, PVA47, and PVA64 also inhibited the fibrinolytic activity of plasmin with an IC50 of 1.6–2.2 μM, whereas heparin was not effective under similar conditions.247 PVA47 decreased the fibrin dissolution when co-added with plasminogen and tPA but had no effect on fibrinolysis when co-added with fibrin-bound plasminogen and tPA. This probably implied that the polymers are not effective when plasmin was generated from plasminogen that is attached to the fibrin but inhibit plasmin in its free, solution form.247 Earlier, these copolymers had been reported to inhibit thrombin with potencies similar to that against plasmin.263
4. PLASMIN INHIBITORS FOR OTHER DISEASES
The above discussion was focused on structural and mechanistic aspects of plasmin inhibitors with respect to their antifibrinolytic activity so as to treat hyperfibrinolysis-associated bleeding consequences. However, plasmin inhibitors can potentially serve several other roles. We highlight other applications of plasmin inhibitors, especially as antimetastatic, antiproliferative, antiangiogenesis agents as well as in embryo implantation.
A. PASI-535
Wanaka et al. reported that PASI-535 (48, Fig. 10) potently inhibited plasmin-mediated fibrinolysis (IC50 2.9 μM) and fibrinogenolysis (IC50 4.5 μM). Interestingly, PASI-535 suppressed ascites retention and growth of tumor cells in sarcoma-180 bearing mice following its subcutaneous injection for 5 days at 30–50 mg/kg/day.264 This activity was nearly 40 times better than that for TXA, which reduced both ascites retention and tumor growth at ~2 g/kg/day.264
Figure 10.
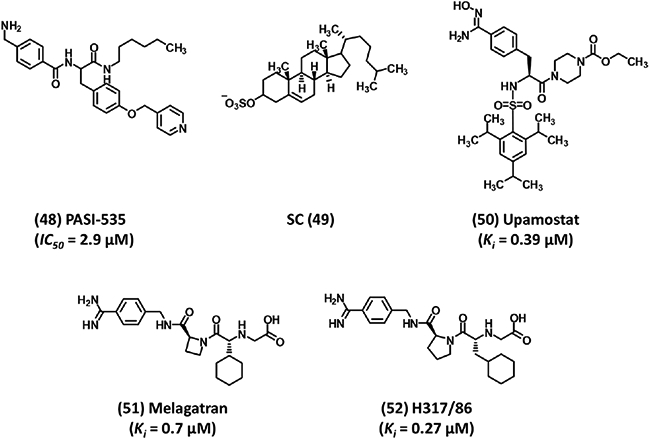
Structures of plasmin inhibitors that show activities other than antifibrinolytic activity. Inhibitor 48 exhibits antitumor potential; SC 49 has been implicated in embryo implantation; Upamostat 50 is in pancreatic cancer Phase II studies; melagatran 51 was approved as a direct thrombin inhibitor; and H317/86 52, a benzamidine derivative related to upamostat, also inhibits plasmin. IC50 refers to inhibition of plasmin fibrinolysis.
B. YO-2
Another interesting discovery was that of a selective plasmin inhibitor (14, Fig. 4). This agent not only inhibited plasmin-mediated fibrinolysis (IC50 0.36 μM), but also induced apoptosis of both M1 melanoma and HT29 colon carcinoma cell lines within 24 hr.50 Mechanistic studies revealed that inhibitor 14 activated caspase-3 at low doses (10–20 μg/mL).50 The pro-apoptotic activity rose from a combination of intranucleosomal DNA fragmentation and enhancement in capsase-3, capsase-8, and capsase-9-like activities.265, 266 Further, inhibition of tumor growth by 14 was examined using HT29 human colon carcinoma, HT18 human melanoma, and HT58 human B-cell lymphoma inoculated as xenografts into immuno-deprived mice.267 Its antimetastatic activity was also investigated in the B16 mouse melanoma muscle-lung model. Results indicated that 14 inhibited the growth of all xenografts by 40–50%, when administered at a dose of 0.4–2.0 mg/kg. It also effectively decreased the number of lung metastasis found in mice inoculated with B16 melanoma at 4 mg/kg.267 Recently, 14 was shown to reduce T-cell lymphoid tumor growth by suppressing MMP-9-dependent CD11b+ F4/80+ myeloid cell recruitment.51
C. Pegylated DX-1000
Recently, engineered polypeptide DX-1000 was modified using methoxy-PEG-succinimidyl propionate (mPEG-SPA, Mr 5000 Da) at its three lysines (positions 10, 54, and 55) and the amino terminus to produce pegylated DX-1000 (4PEG DX-1000).226 Pegylation increased the Ki for plasmin by 2.5-fold to 0.23 nM. Both DX-1000 and 4PEGDX-1000 were found to efficiently inhibit plasmin-mediated activation of pro-MMP-9 in HL60 cells as well as inhibit tube formation in HUVEC (IC50 1.4 nM for DX-1000, 8.3 nM for 4PEG DX-1000) and LEII cells (IC50 16.6 nM for DX-1000,15.8 nM for 4PEG DX-1000).226 DX-1000 was rapidly cleared from plasma in mice and rabbits, however, 4PEG DX-1000 exhibited an extended plasma half-life and pharmacological profile.226 4PEG DX-1000 was found to block human breast cancer growth and tumor metastasis in vivo at a dose of 10 mg/kg. Furthermore, 4PEG DX-1000 treatment also led to a significant inhibition of tumor cell proliferation and a decrease of tumor vessel hot spots. Mechanistically, 4PEG DX-1000 inhibitory effects were found to be mediated by activating mitogen-activated protein kinase signaling pathways.226 Interestingly, 4PEG GDX-1000 treatment also reduced uPA and plasminogen expression by 55% and 63%, respectively. Overall, 4PEG DX-1000 was found to have antiproliferative, antimetastatic, and antiangiogenic effects in vivo with improved bioavailability.
D. Sulfated Cholesterol (SC)
An important function in which plasmin is involved as a key enzyme is embryo implantation into maternal endometrium during pregnancy. The implantation process relies on the invasive capacity of the trophoblast cells, which is attributed to plasmin as well as MMPs. The proteases are required for the degradation of the matrix so that pregnancy can be established. While MMPs contribute to the process directly, plasmin contributes indirectly by converting the pro-MMP latent form to the active form. Matrix degradation is necessary but excessive degradation is detrimental because the damage can prevent or terminate pregnancy. Thus, regulation of plasmin proteolytic activities is important for embryo implantation.
It was shown that sulfated cholesterol (SC) content drastically increases in rabbit endometrium during the implantation period.37 Koizumi et al. studied the inhibition of these enzymes by SC (49, Fig. 10).37 In a concentration-dependent fashion, SC was found to reduce plasmin and MMP-3 activities to 14% and 26%, respectively, at a concentration of 30 μM in culture media of the extracellular matrix. Western blot analysis demonstrated that SC inhibited the plasmin-mediated conversion of MMP-3’s latent form to its active form. Gelatin zymography analysis indicated inhibition of plasmin-mediated activation of MMP-9 by 10 μM SC.37 In another study, Nakae et al. showed that SC directly inhibited plasmin activity in a noncompetitive manner, which led to reduction of invasion of a trophoblast cell line.49 In combination, the results appear to indicate that SC inhibition of plasmin may be essential for regulating the process of embryo implantation.
E. EACA, TXA, Aprotinin, Nafamostat, and Gabexate
Several reports present the potential benefit of using EACA (1), TXA (2), or aprotinin in managing, treating, and preventing disorders such as hereditary angioedema,45, 46 traumatic bleeding,268 chronic inflammation,47, 48 and lymphoid malignancies.50, 51 In particular, data from the Clinical Randomization of an Antifibrinolytic in Significant Hemorrhage 2 (CRASH-2) study revealed that early administration of TXA reduced the risk of death in trauma patients in comparison to the placebo (4.9% vs. 5.7%, respectively; p = 0.0077).269 Also, aprotinin’s anticoagulant and antiinflammatory effects could further aid cardiopulmonary bypass success.168, 170
Recently, plasmin inhibitors have been reported to prevent leukocyte accumulation and remodeling events in the postischemic microvasculature,270 which highlights their potential in the prevention of ischemia-reperfusion injury. Moreover, although not fully understood, TXA has been implicated in the treatment of melasma, prevention of UV-induced pigmentation, and the recovery of skin barrier.270, 271 Finally, nafamostat and gabexate have been found useful for complex pathologies such as DIC,7, 137, 148 pancreatitis,138-144 and liver diseases.272
F. Upamostat (Mesupron)
Upamostat, inhibitor 50 (Fig. 10), is a hydroxyamidino prodrug of WX-UK1, a nonspecific inhibitor of several serine proteases.273, 274 WX-UK1 was found to inhibit uPA (Ki 0.41 μM), plasmin (Ki 0.39 μM), thrombin (Ki 0.49 μM), factor Xa (Ki 1.7 μM), activated protein C (Ki 2.3 μM), plasma kallikrein (Ki 7.2 μM), trypsin (Ki 0.037 μM), and tryptase (Ki 6.3 μM). It appears that its direct and indirect inhibition of plasmin contributes to its activity against primary tumor growth and metastasis formation.275, 276 Currently, upamostat is in phase II studies for patients with special forms of pancreatic cancer or breast cancer.277 Interestingly, melagatran (51) and inhibitor H317/86 (52) (Fig. 10), related benzamidine derivatives to upamostat, were also found to inhibit plasmin with Ki values of 0.7 and 0.27 μM, respectively. Melagatran was approved as an anticoagulant direct thrombin inhibitor, yet it was withdrawn later from market due to hepatotoxicity.278
5. CONCLUSIONS
Plasmin is intravascularly formed upon activation of plasminogen by tPA, whereas uPA appears to be the major extravascular activator of plasminogen.27, 52, 54 The resulting plasmin plays diverse physiological and pathophysiological roles, of which fibrinolysis is the key role for intravascular plasmin. Cell surface-derived plasmin is more involved in tissue remodeling, matrix degradation, and cell migration.26-28 A couple of plasmin inhibitors are used clinically today including EACA (1) and TXA (2), which are lysine analogs. Despite their success in reducing blood loss associated with major surgeries, the two antifibrinolytics suffer from severe lack of efficacy. Large quantities need to be administrated and the therapeutic regimen has to be adjusted to account for every patient.171, 279-281 Further, specificity is also an issue as these lysine analogs may bind a nontarget protein possessing an appropriate negatively charged domain. An example of this is seizure attacks in some patients, which likely happen because of their action on GABA receptors.91, 92, 231, 280, 282 A plasmin inhibitor in limited use in some countries is aprotinin. The controversy surrounding this polypeptide is an ongoing debate. Its disadvantage is its lack of specificity, which is the probable cause of associated morbidity and mortality.83, 178-187
The similarity of plasmin active site with several other serine proteases, e.g., coagulation enzymes, introduces considerable challenges in developing new potent and selective plasmin inhibitors. This problem is further compounded by the limited number of plasmin-ligand co-crystal structures resulting in considerable lack of understanding of interactions at the atomic level. This hampers progress in the direction of establishing key structural elements that enhance potency as well as selectivity. A thorough understanding of the spatiotemporal properties of extravascular and intravascular plasmin action is also required to develop novel plasmin inhibitors that selectively target fibrinolysis or tissue remodeling and cell migration processes.
Despite these difficulties, several novel plasmin inhibitors are emerging as antifibrinolytics. The molecules being developed are highly diverse spanning a wide range of size, scaffold, and physicochemical properties. Mechanistically, the group comprises competitive as well as noncompetitive inhibitors. Some exciting antiplasmin agents include textilinin-1,196-202 KDI-L17R,155 pegylated DX-1000,154, 226 and the cyclic peptidomimetics 39 and 40.11 Majority of these molecules enjoy high affinity for plasmin. Yet, their specificity is not high enough and there is a possibility that the molecules may suffer unexpected consequences as they advance through the various stages of drug development.
An emerging trend in targeting plasmin is nonsaccharide GAG mimetics (NSGMs). Sulfated LMWLs, chemically modified sulfated dextrans, and sulfated PVA polymers are sulfated NSGMs that functionally mimic heparin/heparan sulfate and inhibit plasmin in an allosteric manner. This is worth investigating in detail. Allosteric inhibitors usually convey two advantages over orthosteric inhibitors. One, allosteric sites are typically less conserved among proteins of the same family leading to an enhanced specificity for molecules targeting these sites. Two, allosteric sites in principle afford the possibility of controlled reduction in enzyme function (<100% maximal inhibition), which may become critical for enzymes involved in multiple pathophysiological roles, such as plasmin.
The discovery of polymeric NSGMs as allosteric inhibitors may lead to the design of small NSGMs that specifically target plasmin. This is a relatively new concept and is in rapid development. For example, NSGMs of a specific type, tetrasulfated tetrahydroisoquinolines, were rationally designed to target the pentasaccharide-binding site of AT.283 Likewise, several NSGMs including disulfated quinazolinone dimers284 and polysulfated pentagalloyl glucosides285 have been developed to target factor XIa, while monosulfated benzofuran dimers286 and trimers287 were designed to target exosite 2 of thrombin. Small NSGMs agents offer many advantages including (1) high water solubility, which is expected to help antifibrinolytic use during surgeries, (2) low cellular and central nervous system toxicity arising from their highly charged nature, (3) good chemical stability, and (4) ease of chemical synthesis.288 Yet, major efforts are needed to develop NSGMs as inhibitors of plasmin for clinical use.
ACKNOWLEDGMENTS
This work was supported by a Mid-Atlantic Affiliate postdoctoral fellowship to R.A.A.H. from the American Heart Association (grant 12POST10930004) and grants HL090586 and HL107152 from the National Institutes of Health to U.R.D.
6. ABBREVIATIONS
- AaTI
Aedes aegypti thrombin inhibitor
- APTT
activated partial thromboplastin times
- AT
antithrombin
- AvKTI
Araneus ventricosus spider is Kunitz-type serine protease inhibitor
- BPTI
bovine pancreatic trypsin inhibitor
- Bt-KTI
Bombus terrestris Kunitz-type serine protease inhibitor
- BZA
p-Benzoylanilide
- Cbz
benzyloxycarbonyl
- clTI-1
chicken liver trypsin inhibitor-1
- DIC
disseminated intravascular coagulation
- DrKIn-II
Daboia russelii Kunitz Inhibitor-II
- EACA
ε-Aminocaproic acid
- GAGs
glycosaminoglycans
- hTFPI-1
human tissue factor pathway inhibitor-1
- K
kringle domain
- KD APP
Kunitz domain of amyloid precursor protein
- KD APPH
Kunitz domain of Alzheimer amyloid precursor protein homolog
- KD hTFPI-1
Kunitz domain of human tissue factor pathway inhibitor-1
- KD TFPI-2
Kunitz domain of tissue factor pathway inhibitor-2
- KPI PN2
Kunitz protease inhibitor domain of protease nexin 2
- LBS
lysine-binding site
- LMWLs
low molecular weight lignins
- MMPs
matrix metalloproteinases
- NSGMs
nonsaccharide glycosaminoglycan mimetics
- PT
prothrombin time
- PVA
polyvinyl-acrylate
- SAR
structure-activity relationship
- SC
sulfated cholesterol
- TdPI
tick-derived protease inhibitor
- Tos
tosyl
- tPA
tissue plasminogen activator
- Tra
Nα-trans-4-aminomethylcyclohexanecarbonyl
- TT
thrombin time
- TXA
tranexamic acid
- uPA
urokinase plasminogen activator
Biography
Rami A. Al-Horani is an American Heart Association senior postdoctoral associate in the laboratory of Dr. Desai in Department of Medicinal Chemistry at Virginia Commonwealth University (VCU), Richmond, Virginia, USA. He obtained his first degree in pharmacy from University of Jordan, Amman, Jordan. He was recently awarded his Ph.D. degree from Department of Medicinal Chemistry at VCU under the supervision of Dr. Desai. He obtained the Fulbright scholarship sponsored by the US Department of State during his graduate studies. He received the Charles T. Rector and Thomas W. Rorrer, Jr. Dean’s Award, and the J. Doyle Smith Award from School of Pharmacy at VCU in 2012.
Umesh R. Desai is a Professor of Medicinal Chemistry in the School of Pharmacy at Virginia Commonwealth University, Richmond, VA. He received his Ph.D. from the Indian Institute of Technology, Bombay, India, following which he did postdoctoral work with Professors Linhardt (Iowa) and Klibanov (MIT), and an AHA research fellowship with Professor Olson (UIC). Since 1998, he has been at VCU and works on rational design of small and large mechanism-based anticoagulants. He received an Established Investigator Award from the AHA in 2006; Faculty Research and Teaching Excellence Awards from the School of Pharmacy in 2003 and 2010, respectively; and Distinguished Scholarship Award from VCU in 2013.
Footnotes
CONFLICT OF INTEREST
Authors declare no competing financial conflict of interest.
REFERENCES
- 1.Levy JH. Pharmacologic methods to reduce perioperative bleeding. Transfusion 2008;48:31S–38S. [DOI] [PubMed] [Google Scholar]
- 2.Dhir A Antifibrinolytics in cardiac surgery. Ann Card Anaesth 2013;16:117–125. [DOI] [PubMed] [Google Scholar]
- 3.Spahn DR, Goodnough LT. Alternatives to blood transfusion. Lancet 2013;381:1855–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shander A, Hofmann A, Ozawa S, Theusinger OM, Gombotz H, Spahn DR. Activity-based costs of blood transfusions in surgical patients at four hospitals. Transfusion 2010;50:753–765. [DOI] [PubMed] [Google Scholar]
- 5.Trudell J, McMurdy N. Current antifibrinolytic therapy for coronary artery revascularization. AANA 2008;76:121–124. [PubMed] [Google Scholar]
- 6.Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33. [DOI] [PubMed] [Google Scholar]
- 7.Toh CH, Dennis M. Disseminated intravascular coagulation: Old disease, new hope. BMJ 2003;327:974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hvas AM, Sorensen HT, Norengaard L, Christiansen K, Ingerslev J, Sorensen B. Tranexamic acid combined with recombinant factor VIII increases clot resistance to accelerated fibrinolysis in severe hemophilia A. J Thromb Haemost 2007;5:2408–2414. [DOI] [PubMed] [Google Scholar]
- 9.Philipp CS. Antifibrinolytics in women with menorrhagia. Thromb Res 2011;127:S113–S115. [DOI] [PubMed] [Google Scholar]
- 10.Tiede A, Rand JH, Budde U, Ganser A, Federici AB. How I treat the acquired von Willebrand syndrome. Blood 2011;117:6777–6785. [DOI] [PubMed] [Google Scholar]
- 11.Saupe SM, Leubner S, Betz M, Klebe G, Steinmetzer T. Development of new cyclic plasmin inhibitors with excellent potency and selectivity. J Med Chem 2013;56:820–831. [DOI] [PubMed] [Google Scholar]
- 12.Henry BL, Desai UR. Anticoagulants: Drug discovery and development. In: Rotella D, Abraham DJ, Eds. Burger’s Medicinal Chemistry, 7th ed. New York: John Wiley and Sons; 2010. p 365–408. [Google Scholar]
- 13.Anglés-Cano E Overview on fibrinolysis: Plasminogen activation pathways on fibrin and cell surfaces. Chem Phys Lipids 1994;67–68:353–362. [DOI] [PubMed] [Google Scholar]
- 14.Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem 1982;257:2912–2919. [PubMed] [Google Scholar]
- 15.Lerch PG, Rickli EE, Lergier W, Gillessen D. Localization of individual lysine-binding regions in human plasminogen and investigations on their complex-forming properties. Eur J Biochem 1980;107:7–13. [DOI] [PubMed] [Google Scholar]
- 16.Lijnen HR, Van Hoef B, Collen D. On the molecular interactions between fibrin, tissue-type plasminogen activator and plasminogen. Thromb Res Suppl 1990;10:45–54. [DOI] [PubMed] [Google Scholar]
- 17.Lu BG, Sofian T, Law RH, Coughlin PB, Horvath AJ. Contribution of conserved lysine residues in the alpha2-antiplasmin C terminus to plasmin binding and inhibition. J Biol Chem 2011;286:24544–24552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsurupa G, Yakovlev S, McKee P, Medved L. Noncovalent interaction of alpha(2)-antiplasmin with fibrin(ogen): localization of alpha(2)-antiplasmin-binding sites. Biochemistry 2010;49:7643–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foley JH, Kim PY, Mutch NJ, Gils A. Insights into thrombin activatable fibrinolysis inhibitor function and regulation. J Thromb Haemost 2013;(Suppl 1)11:306–315. [DOI] [PubMed] [Google Scholar]
- 20.Nicholl SM, Roztocil E, Davies MG. Plasminogen activator system and vascular disease. Curr Vasc Pharmacol 2006;4:101–116. [DOI] [PubMed] [Google Scholar]
- 21.Binder BR, Christ G, Gruber F, Grubic N, Hufnagl P, Krebs M, Mihaly J, Prager GW. Plasminogen activator inhibitor 1: Physiological and pathophysiological roles. News Physiol Sci 2002;17:56–61. [DOI] [PubMed] [Google Scholar]
- 22.Steiner JP, Migliorini M, Strickland DK. Characterization of the reaction of plasmin with alpha 2-macroglobulin: Effect of antifibrinolytic agents. Biochemistry 1987;26:8487–8495. [DOI] [PubMed] [Google Scholar]
- 23.Kunadian V, Gibson CM. Thrombolytics and myocardial infarction. Cardiovasc Ther 2012;30:e81–e88. [DOI] [PubMed] [Google Scholar]
- 24.Ceruti P, Principe M, Capello M, Cappello P, Novelli F. Three are better than one: Plasminogen receptors as cancer theranostic targets. Exp Hematol Oncol 2013;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwaan HC, McMahon B. The role of plasminogen-plasmin system in cancer. Cancer Treat Res 2009;148:43–66. [DOI] [PubMed] [Google Scholar]
- 26.Deryugina EI, Quigley JP. Cell surface remodeling by plasmin: A new function for an old enzyme. J Biomed Biotechnol 2012;2012:564259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaller J, Gerber SS. The plasmin-antiplasmin system: Structural and functional aspects. Cell Mol Life Sci 2011;68:785–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang L, Han X. The urokinase plasminogen activator system in breast cancer invasion and metastasis. Biomed Pharmacother 2013;67:179–182. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Syrovets T, Genze F, Pitterle K, Oberhuber A, Orend KH, Simmet T. Plasmin triggers chemotaxis of monocyte-derived dendritic cells through an Akt2-dependent pathway and promotes a T-helper type-1 response. Arterioscler. Thromb Vasc Biol 2010;30:582–590. [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Laumonnier Y, Syrovets T, Simmet T. Recruitment of CCR6-expressing Th17 cells by CCL20 secreted from plasmin-stimulated macrophages. Acta Biochim Biophys Sin (Shanghai) 2013;45:593–600. [DOI] [PubMed] [Google Scholar]
- 31.Rønø B, Engelholm LH, Lund LR, Hald A. Gender affects skin wound healing in plasminogen deficient mice. PLoS ONE 2013;8:e59942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawao N, Nagai N, Okada K, Okumoto K, Ueshima S, Matsuo O. Role of plasminogen in macrophage accumulation during liver repair. Thromb Res 2010;125:e214–e221. [DOI] [PubMed] [Google Scholar]
- 33.Wilkins-Port CE, Higgins SP, Higgins CE, Kobori-Hotchkiss I, Higgins PJ. Complex regulation of the pericellular proteolytic microenvironment during tumor progression and wound repair: Functional interactions between the serine protease and matrix metalloproteinase cascades. Biochem Res Int 2012;2012:454368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutiérrez-Fernández A, Gingles NA, Bai H, Castellino FJ, Parmer RJ, Miles LA. Plasminogen enhances neuritogenesis on laminin-1. J Neurosci 2009;29:12393–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacovina AT, Zhong F, Khazanova E, Lev E, Deora AB, Hajjar KA. Neuritogenesis and the nerve growth factor-induced differentiation of PC-12 cells requires annexin II-mediated plasmin generation. J Biol Chem 2001;276:49350–49358. [DOI] [PubMed] [Google Scholar]
- 36.Ny A, Leonardsson G, Hägglund AC, Hägglöf P, Ploplis VA, Carmeliet P, Ny T. Ovulation in plasminogen-deficient mice. Endocrinology 1999;140:5030–5035. [DOI] [PubMed] [Google Scholar]
- 37.Koizumi M, Momoeda M, Hiroi H, Nakazawa F, Nakae H, Ohno T, Yano T, Taketani Y. Inhibition of proteases involved in embryo implantation by cholesterol sulfate. Hum Reprod 2010;25:192–197. [DOI] [PubMed] [Google Scholar]
- 38.Syrovets T, Simmet T. Novel aspects and new roles for the serine protease plasmin. Cell Mol Life Sci 2004;61:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakagami Y, Abe K, Nishiyama N, Matsuki N. Laminin degradation by plasmin regulates long-term potentiation. J Neurosci 2000;20:2003–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res 2000;98:323–332. [DOI] [PubMed] [Google Scholar]
- 41.Netzel-Arnett S, Mitola DJ, Yamada SS, Chrysovergis K, Holmbeck K, Birkedal-Hansen H, Bugge TH. Collagen dissolution by keratinocytes requires cell surface plasminogen activation and matrix metalloproteinase activity. J Biol Chem 2002;277:45154–45161. [DOI] [PubMed] [Google Scholar]
- 42.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost 2001;86:324–333. [PubMed] [Google Scholar]
- 43.Kwaan HC, McMahon B. The role of plasminogen-plasmin system in cancer. Cancer Treat Res 2009;148:43–66. [DOI] [PubMed] [Google Scholar]
- 44.Danø K, Behrendt N, Høyer-Hansen G, Johnsen M, Lund LR, Ploug M, Rømer J. Plasminogen activation and cancer. Thromb Haemost 2005;93:676–681. [DOI] [PubMed] [Google Scholar]
- 45.Ritchie BC. Protease inhibitors in the treatment of hereditary angioedema. Transfus Apheresis Sci 2003;29:259–267. [DOI] [PubMed] [Google Scholar]
- 46.Du-Thanh A, Raison-Peyron N, Drouet C, Guillot B. Efficacy of tranexamic acid in sporadic idiopathic bradykinin angioedema. Allergy 2010;65:793–795. [DOI] [PubMed] [Google Scholar]
- 47.Syrovets T, Lunov O, Simmet T. Plasmin as a proinflammatory cell activator. J Leukocyte Biol 2012;92:509–519. [DOI] [PubMed] [Google Scholar]
- 48.Schuliga M, Westall G, Xia Y, Stewart AG. The plasminogen activation system: New targets in lung inflammation and remodeling. Curr Opin Pharmacol 2013;13:386–393. [DOI] [PubMed] [Google Scholar]
- 49.Nakae H, Hiroi H, Momoeda M, Koizumi M, Iwamori M, Taketani Y. Inhibition of cell invasion and protease activity by cholesterol sulfate. Fertil Steril 2010;94:2455–2457. [DOI] [PubMed] [Google Scholar]
- 50.Okada Y, Tsuda Y, Wanaka K, Tada M, Okamoto U, Okamoto S, Hijikata-Okunomiya A, Bokonyi G, Szende B, Keri G. Development of plasmin and plasma kallikrein selective inhibitors and their effect on M1 (melanoma) and HT29 cell lines. Bioorg Med Chem Lett 2000;10:2217–2221. [DOI] [PubMed] [Google Scholar]
- 51.Ishihara M, Nishida C, Tashiro Y, Gritli I, Rosenkvist J, Koizumi M, Okaji Y, Yamamoto R, Yagita H, Okumura K, Nishikori M, Wanaka K, Tsuda Y, Okada Y, Nakauchi H, Heissig B, Hattori K. Plasmin inhibitor reduces T-cell lymphoid tumor growth by suppressing matrix metalloproteinase-9-dependent CD11b(+)/F4/80(+) myeloid cell recruitment. Leukemia 2012;26:332–339. [DOI] [PubMed] [Google Scholar]
- 52.Swedberg JE, Harris JM. Natural and engineered plasmin inhibitors: Applications and design strategies. Chembiochem 2012;13:336–348. [DOI] [PubMed] [Google Scholar]
- 53.Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thromb Haemost 2005;93:647–654. [DOI] [PubMed] [Google Scholar]
- 54.Novokhatny V. Structure and activity of plasmin and other direct thrombolytic agents. Thromb Res 2008;122:53–58. [DOI] [PubMed] [Google Scholar]
- 55.Zhang L, Seiffert D, Fowler BJ, Jenkins GR, Thinnes TC, Loskutoff DJ, Parmer RJ, Miles LA. Plasminogen has a broad extrahepatic distribution. Thromb Haemost 2002;87:493–501. [PubMed] [Google Scholar]
- 56.Ramesh V, Petros AM, Llinás M, Tulinsky A, Park CH. Proton magnetic resonance study of lysine-binding to the kringle 4 domain of human plasminogen. The structure of the binding site. J Mol Biol 1987;198:481–498. [DOI] [PubMed] [Google Scholar]
- 57.Mathews II, Vanderhoff-Hanaver P, Castellino FJ, Tulinsky A. Crystal structures of the recombinant kringle 1 domain of human plasminogen in complexes with the ligands epsilon-aminocaproic acid and trans-4-(aminomethyl)cyclohexane-1-carboxylic Acid. Biochemistry 1996;35:2567–2576. [DOI] [PubMed] [Google Scholar]
- 58.Sehl LC, Castellino FJ. Thermodynamic properties of the binding of alpha-, omega-amino acids to the isolated kringle 4 region of human plasminogen as determined by high sensitivity titration calorimetry. J Biol Chem 1990;265:5482–5486. [PubMed] [Google Scholar]
- 59.Menhart N, Sehl LC, Kelley RF, Castellino FJ. Construction, expression and purification of recombinant kringle 1 of human plasminogen and analysis of its interaction with omega-amino acids. Biochemistry 1991;30:1948–1957. [DOI] [PubMed] [Google Scholar]
- 60.Menhart N, McCance SG, Sehl LC, Castellino FJ. Functional independence of the kringle 4 and kringle 5 regions of human plasminogen. Biochemistry 1993;32:8799–8806. [DOI] [PubMed] [Google Scholar]
- 61.Menhart N, Castellino FJ. The importance of the hydrophobic components of the binding energies in the interaction of omega-amino acid ligands with isolated kringle polypeptide domains of human plasminogen. Int J Pept Protein Res 1995;46:464–470. [DOI] [PubMed] [Google Scholar]
- 62.Marti DN, Schaller J, Llinas M. Solution structure and dynamics of the plasminogen kringle 2-AMCHA complex: 3(1)-Helix in homologous domains. Biochemistry 1999;38:15741–15755. [DOI] [PubMed] [Google Scholar]
- 63.Urano T, Chibber BAK, Castellino FJ. The reciprocal effects of ε-aminohexanoic acid and chloride ion on the activation of human [Glu1]plasminogen by human urokinase. Proc Natl Acad Sci USA 1987;84:4031–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lucas MA, Fretto LJ, McKee PA. The binding of human plasminogen to fibrin and fibrinogen. J Biol Chem 1983;258:4249–4256. [PubMed] [Google Scholar]
- 65.Kim PY, Tieu LD, Stafford AR, Fredenburgh JC, Weitz JI. A high affinity interaction of plasminogen with fibrin is not essential for efficient activation by tissue-type plasminogen activator. J Biol Chem 2012;287:4652–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanderson-Smith ML, Dowton M, Ranson M, Walker MJ. The plasminogen-binding group A streptococcal M protein-related protein Prp binds plasminogen via arginine and histidine residues. J Bacteriol 2007;189:1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fulde M, Rohde M, Polok A, Preissner KT, Chhatwal GS, Bergmann S. Cooperative plasminogen recruitment to the surface of Streptococcus canis via M protein and enolase enhances bacterial survival. MBio 2013;4:e00629–e00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang H, Doll JA, Jiang K, Cundiff DL, Czarnecki JS, Wilson M, Ridge KM, Soff GA. Differential binding of plasminogen, plasmin, and angiostatin4.5 to cell surface beta-actin: Implications for cancer-mediated angiogenesis. Cancer Res 2006;66:7211–7215. [DOI] [PubMed] [Google Scholar]
- 69.Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL. The cell biology of the plasminogen system. FASEB J 1995;9:939–945. [DOI] [PubMed] [Google Scholar]
- 70.Bürgin J, Schaller J. Expression, isolation and characterization of a mutated human plasminogen kringle 3 with a functional lysine binding site. Cell Mol Life Sci 1999;55:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, Lin X, Loy JA, Tang J, Zhang XC. Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science 1998, 281:1662–1665. [DOI] [PubMed] [Google Scholar]
- 72.Anonick PK, Vasudevan J, Gonias SL. Antifibrinolytic activities of alpha-N-acetyl-L-lysine methyl ester, epsilon-aminocaproic acid, and tranexamic acid. Importance of kringle interactions and active site inhibition. Arterioscler Thromb 1992;12:708–716. [DOI] [PubMed] [Google Scholar]
- 73.Tordai H, Bányai L, Patthy L. The PAN module: The N-terminal domains of plasminogen and hepatocyte growth factor are homologous with the apple domains of the prekallikrein family and with a novel domain found in numerous nematode proteins. FEBS Lett 1999;461:63–67. [DOI] [PubMed] [Google Scholar]
- 74.Xue Y, Bodin C, Olsson K. Crystal structure of the native plasminogen reveals an activation-resistant compact conformation. J Thromb Haemost 2012;10:1385–1396. [DOI] [PubMed] [Google Scholar]
- 75.Law RH, Caradoc-Davies T, Cowieson N, Horvath AJ, Quek AJ, Encarnacao JA, Steer D, Cowan A, Zhang Q, Lu BG, Pike RN, Smith AI, Coughlin PB, Whisstock JC. The X-ray crystal structure of full-length human plasminogen. Cell Rep 2012;1:185–190. [DOI] [PubMed] [Google Scholar]
- 76.Robbins KC, Summaria L, Hsieh B, Shah RJ. The peptide chains of human plasmin. Mechanism of activation of human plasminogen to plasmin. J Biol Chem 1967;242:2333–2342. [PubMed] [Google Scholar]
- 77.Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci USA 2000;97:7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Backes BJ, Harris JL, Leonetti F, Craik CS, Ellman JA. Synthesis of positional-scanning libraries of fluorogenic peptide substrates to define the extended substrate specificity of plasmin and thrombin. Nat Biotechnol 2000;18:187–193. [DOI] [PubMed] [Google Scholar]
- 79.Gosalia DN, Salisbury CM, Maly DJ, Ellman JA, Diamond SL. Profiling serine protease substrate specificity with solution phase fluorogenic peptide microarrays. Proteomics 2005;5:1292–1298. [DOI] [PubMed] [Google Scholar]
- 80.Swedberg JE, Harris JM. Plasmin substrate binding site cooperativity guides the design of potent peptide aldehyde inhibitors. Biochemistry 2011;50:8454–8462. [DOI] [PubMed] [Google Scholar]
- 81.Rawlings ND, Barrett AJ, Bateman A. MEROPS: The peptidase database. Nucleic Acids Res 2010;38:D227–D233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xia VW, Steadman RH. Antifibrinolytics in orthotopic liver transplantation: current status and controversies. Liver Transpl 2005;11:10–18. [DOI] [PubMed] [Google Scholar]
- 83.Ortmann E, Besser MW, Klein AA. Antifibrinolytic agents in current anaesthetic practice. Br J Anaesth 2013;111:549–563. [DOI] [PubMed] [Google Scholar]
- 84.Alkozai EM, Lisman T, Porte RJ. Bleeding in liver surgery: Prevention and treatment. Clin Liver Dis 2009;13:145–154. [DOI] [PubMed] [Google Scholar]
- 85.Tiede A. Diagnosis and treatment of acquired von Willebrand syndrome. Thromb Res 2012;130(Suppl 2):S2–S6. [DOI] [PubMed] [Google Scholar]
- 86.Okamoto S. Plasmin and antiplasmin: Their pathologic physiology. Keio J Med 1959;8:211–217. [Google Scholar]
- 87.Okamoto S, Sato S, Takada Y, Okamoto U. An active stereo-isomer (trans-form) of AMCHA and its antifibrinolytic (antiplasminic) action in vitro and in vivo. Keio J Med 1964;13:177–185. [DOI] [PubMed] [Google Scholar]
- 88.Okamoto S, Hijikata A. Rational approach to proteinase inhibitors. In: Ariens EJ, Ed. Medicinal Chemistry: A Series of Monographs, Vol. 11. Drug Design: Parts 1–10. MA, USA: Academic Press, Inc; 1975. p 143–169. [Google Scholar]
- 89.Markwardt F Synthetic inhibitors of fibrinolysis. In: Markwardt F, Ed. Handbook of Experimental Pahramcology: Fibrinolytics and Anti-fibrinolytics, Vol. 46. Berlin: Springer; 1978. p 511–577. [Google Scholar]
- 90.Royston D Blood-sparing drugs: Aprotinin, tranexamic acid, and epsilon-aminocaproic acid. Int Anesthesiol Clin 1995;33:155–179. [PubMed] [Google Scholar]
- 91.Makhija N, Sarupria A, Kumar Choudhary S, Das S, Lakshmy R, Kiran U. Comparison of epsilon aminocaproic acid and tranexamic acid in thoracic aortic surgery: Clinical efficacy and safety. J Cardiothorac Vasc Anesth 2013;27:1201–1207. [DOI] [PubMed] [Google Scholar]
- 92.Lecker I, Wang DS, Romaschin AD, Peterson M, Mazer CD, Orser BA. Tranexamic acid concentrations associated with human seizures inhibit glycine receptors. J Clin Invest 2012;122:4654–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lukes AS, Kouides PA, Moore KA. Tranexamic acid: A novel oral formulation for the treatment of heavy menstrual bleeding. Womens Health (Lond Engl) 2011;7:151–158. [DOI] [PubMed] [Google Scholar]
- 94.Midura-Nowaczek K, Purwin M, Markowska A, Drozdowska D, Bruzgo M. Effect of short peptides containing lysine and epsilon-aminocaproic acid on fibrinolytic activity of plasmin and topoisomerase II action on supercoiled DNA. Acta Pol Pharm 2013;70:431–434. [PubMed] [Google Scholar]
- 95.Purwin M, Bruzgo I, Markowska A, Midura-Nowaczek K. Short peptides containing L-lysine and epsilon-aminocaproic acid as potential plasmin inhibitors. Pharmazie 2009;64:765–767. [PubMed] [Google Scholar]
- 96.Midura-Nowaczek K, Lepietuszko I, Bruzgo I. Synthesis of alkylamides of dipeptides as potential plasmin inhibitors. Acta Pol Pharm 2006;63:33–37. [PubMed] [Google Scholar]
- 97.Midura-Nowaczek K, Roszkowska-Jakimiec W, Lepietuszko I, Bruzgo I. Synthesis of benzylamides of dipeptides as potential inhibitors of plasmin. Pharmazie 2003;58:687–689. [PubMed] [Google Scholar]
- 98.Midura-Nowaczek K, Bruzgo I, Roszkowska-Jakimiec W, Worowski K. Synthesis and activity of dipeptides containing epsilon-aminocaproic acid. Acta Pol Pharm 1994;51:499–504. [PubMed] [Google Scholar]
- 99.Midura-Nowaczek K, Bruzgo I, Roszkowska-Jakimiec W, Worowski K. Synthesis and activity of N epsilon-aminocaproyl-L-norleucine derivatives. Acta Pol Pharm 1996;53:221–223. [PubMed] [Google Scholar]
- 100.Bruzgo I, Midura-Nowaczek K, Bruzgo M, Kaczyńska J, Roszkowska-Jakimiec W. Effect of epsilon-aminocaproylamino acids on fibrin formation. Acta Pol Pharm 2006;63:149–152. [PubMed] [Google Scholar]
- 101.Boström J, Grant JA, Fjellström O, Thelin A, Gustafsson D. Potent fibrinolysis inhibitor discovered by shape and electrostatic complementarity to the drug tranexamic acid. J Med Chem 2013;56:3273–3280. [DOI] [PubMed] [Google Scholar]
- 102.Karaman R, Ghareeb H, Dajani KK, Scrano L, Hallak H, Abu-Lafi S, Mecca G, Bufo SA. Design, synthesis and in vitro kinetic study of tranexamic acid prodrugs for the treatment of bleeding conditions. J Comput Aided Mol Des 2013;27:615–635. [DOI] [PubMed] [Google Scholar]
- 103.Zerangue N, Jandeleit B, Li Y, Gallop MA. Acyloxyalkyl carbamate prodrugs of tranexamic acid, methods of synthesis and use. US Patent Application US20100036148 A1,2010.
- 104.Okada Y, Tsuda Y, Teno N, Wanaka K, Bohgaki M, Hijikata-Okunomiya A, Naito T, Okamoto S. Synthesis of active center-directed peptide inhibitors of plasmin. Chem Pharm Bull (Tokyo) 1988;36:1289–1297. [DOI] [PubMed] [Google Scholar]
- 105.Tsuda Y, Teno N, Okada Y, Wanaka K, Bohgaki M, Hijikata-Okunomiya A, Okamoto U, Naito T, Okamoto S. Synthesis of tripeptide chloromethyl ketones and examination of their inhibitory effects on plasmin and plasma kallikrein. Chem Pharm Bull (Tokyo) 1989;37:3108–3111. [DOI] [PubMed] [Google Scholar]
- 106.Teno N, Wanaka K, Okada Y, Tsuda Y, Okamoto U, Hijikata-Okunomiya A, Naito T, Okamoto S. Development of active center-directed inhibitors against plasmin. Chem Pharm Bull (Tokyo) 1991;39:2340–2346. [DOI] [PubMed] [Google Scholar]
- 107.Teno N, Wanaka K, Okada Y, Taguchi H, Okamoto U, Hijikata-Okunomiya A, Okamoto S. Development of active center-directed plasmin and plasma kallikrein inhibitors and studies on the structure-inhibitory activity relationship. Chem Pharm Bull (Tokyo) 1993;41:1079–1090. [DOI] [PubMed] [Google Scholar]
- 108.Okada Y, Matsumoto Y, Tsuda Y, Tada M, Wanaka K, Hijikata-Okunomiya A, Okamoto S. Development of plasmin-selective inhibitors and studies of their structure-activity relationship. Chem Pharm Bull (Tokyo) 2000;48:184–193. [DOI] [PubMed] [Google Scholar]
- 109.Okada Y, Tsuda Y, Tada M, Wanaka K, Okamoto U, Hijikata-Okunomiya A, Okamoto S. Development of potent and selective plasmin and plasma kallikrein inhibitors and studies on the structure-activity relationship. Chem Pharm Bull (Tokyo) 2000;48:1964–1972. [DOI] [PubMed] [Google Scholar]
- 110.Tsuda Y, Tada M, Wanaka K, Okamoto U, Hijikata-Okunomiya A, Okamoto S, Okad Y. Structure-inhibitory activity relationship of plasmin and plasma kallikrein inhibitors. Chem Pharm Bull (Tokyo) 2001;49:1457–1463. [DOI] [PubMed] [Google Scholar]
- 111.Sanders TC, Seto CT. 4-Heterocyclohexanone-based inhibitors of the serine protease plasmin. J Med Chem 1999;42:2969–2976. [DOI] [PubMed] [Google Scholar]
- 112.Conroy JL, Sanders TC, Seto CT. Using the electrostatic field effect to design a new class of inhibitors for cysteine proteases. J Am Chem Soc 1997;119:4285–4291. [Google Scholar]
- 113.Xue F, Seto CT. A comparison of cyclohexanone and tetrahydro-4H-thiopyran-4-one 1,1-dioxide as pharmacophores for the design of peptide-based inhibitors of the serine protease plasmin. J Org Chem 2005;70:8309–8321. [DOI] [PubMed] [Google Scholar]
- 114.Abato P, Conroy JL, Seto CT. Combinatorial library of serine and cysteine protease inhibitors that interact with both the S and S’ binding sites. J Med Chem 1999;42:4001–4009. [DOI] [PubMed] [Google Scholar]
- 115.Abato P, Yuen CM, Cubanski JY, Seto CT. Inhibitors of plasmin that extend into both the S and S’ binding sites: Cooperative interactions between S1 and S2. J Org Chem 2002;67:1184–1191. [DOI] [PubMed] [Google Scholar]
- 116.Xue F, Seto CT. Selective inhibitors of the serine protease plasmin: Probing the S3 and S3’ subsites using a combinatorial library. J Med Chem 2005;48:6908–6917. [DOI] [PubMed] [Google Scholar]
- 117.Xue F, Seto CT. Structure-activity studies of cyclic ketone inhibitors of the serine protease plasmin: Design, synthesis, and biological activity. Bioorg Med Chem 2006;14:8467–8487. [DOI] [PubMed] [Google Scholar]
- 118.Hervio LS, Coombs GS, Bergstrom RC, Trivedi K, Corey DR, Madison EL. Negative selectivity and the evolution of protease cascades: The specificity of plasmin for peptide and protein substrates. Chem Biol 2000;7:443–453. [DOI] [PubMed] [Google Scholar]
- 119.Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery-an underexploited structural class. Nat Rev Drug Discov 2008;7:608–624. [DOI] [PubMed] [Google Scholar]
- 120.Hamada Y, Shioiri T. Recent progress of the synthetic studies of biologically active marine cyclic peptides and depsipeptides. Chem Rev 2005;105:4441–4482. [DOI] [PubMed] [Google Scholar]
- 121.Tyndall JD, Nall T, Fairlie DP. Proteases universally recognize beta strands in their active sites. Chem Rev 2005;105:973–999. [DOI] [PubMed] [Google Scholar]
- 122.Loughlin WA, Tyndall JD, Glenn MP, Fairlie DP. Beta-strand mimetics. Chem Rev 2004;104:6085–6117. [DOI] [PubMed] [Google Scholar]
- 123.Tyndall JD, Fairlie DP. Macrocycles mimic the extended peptide conformation recognized by aspartic, serine, cysteine and metallo proteases. Curr Med Chem 2001;8:893–907. [DOI] [PubMed] [Google Scholar]
- 124.Xue F, Seto CT. Macrocyclic inhibitors for the serine protease plasmin. J Enzyme Inhib Med Chem 2009;24:779–794. [DOI] [PubMed] [Google Scholar]
- 125.Saupe SM, Steinmetzer T. A new strategy for the development of highly potent and selective plasmin inhibitors. J Med Chem 2012;55:1171–1180. [DOI] [PubMed] [Google Scholar]
- 126.Lecaille F, Kaleta J, Brömme D. Human and parasitic papain-like cysteine proteases: Their role in physiology and pathology and recent developments in inhibitor design. Chem Rev 2002;102:4459–4488. [DOI] [PubMed] [Google Scholar]
- 127.Hedstrom L Serine protease mechanism and specificity. Chem Rev 2002;102:4501–4524. [DOI] [PubMed] [Google Scholar]
- 128.Teno N, Gohda K, Wanaka K, Sueda T, Tsuda Y. Identification of novel plasmin inhibitors possessing nitrile moiety as warhead. Bioorg Med Chem Lett 2011;21:6305–6309. [DOI] [PubMed] [Google Scholar]
- 129.Teno N, Otsubo T, Gohda K, Wanaka K, Sueda T, Ikeda K, Hijikata-Okunomiya A, Tsuda Y. Synthesis and evaluation of tripeptidic plasmin inhibitors with nitrile as warhead. J Pept Sci 2012;18:620–625. [DOI] [PubMed] [Google Scholar]
- 130.Dietrich W, Nicklisch S, Koster A, Spannagl M, Giersiefen H, van de Locht A. CU-2010-a novel small molecule protease inhibitor with antifibrinolytic and anticoagulant properties. Anesthesiology 2009;110:123–130. [DOI] [PubMed] [Google Scholar]
- 131.Steinmetzer T, Schweinitz A, Stuerzebecher J, Steinmetzer P, Soffing A, Van de Locht A, Nicklisch S, Reichelt C, Ludwig F-A, Schulze A, Daghisch M, Heinicke J. Trypsin-like serine protease inhibitors, and their preparation and use. US Patent Application 20100022781, 2010.
- 132.Szabó G, Veres G, Radovits T, Haider H, Krieger N, Bährle S, Miesel-Gröschel C, Niklisch S, Karck M, van de Locht A. Effects of novel synthetic serine protease inhibitors on postoperative blood loss, coagulation parameters, and vascular relaxation after cardiac surgery. J Thorac Cardiovasc Surg 2010;139:181–188. [DOI] [PubMed] [Google Scholar]
- 133.Szabó G, Veres G, Radovits T, Haider H, Krieger N, Bährle S, Niklisch S, Miesel-Gröschel C, van de Locht A, Karck M. The novel synthetic serine protease inhibitor CU-2010 dose-dependently reduces postoperative blood loss and improves postischemic recovery after cardiac surgery in a canine model. J Thorac Cardiovasc Surg 2010;139:732–740. [DOI] [PubMed] [Google Scholar]
- 134.The Medicines Company Discontinues Phase 2b Trial of MDCO-2010. http://ir.themedicinescompany.com/phoenix.zhtml?c=122204&p=irol-newsArticle&ID=1741990&highlight=. (Accessed on 09/25/2013).
- 135.Kim H, Szlam F, Tanaka KA, van de Locht A, Ogawa S, Levy JH. The effects of MDCO-2010, a serine protease inhibitor, on activated clotting time in blood obtained from volunteers and cardiac surgical patients. Anesth Analg 2012;115:244–252. [DOI] [PubMed] [Google Scholar]
- 136.Matsuoka S, Futagami M, Ohno H, Imaki K, Okegawa T, Kawasaki A. Inhibitory effects of ONO-3307 on various proteases and tissue thromboplastin in vitro and on experimental thrombosis in vivo. Jpn J Pharmacol 1989;51:455–463. [DOI] [PubMed] [Google Scholar]
- 137.Yoshikawa T, Takeda S, Naito Y, Kondo M. Protective effects of ONO-3307, a new synthetic protease inhibitor against experimental disseminated intravascular coagulation in rats. Thromb Res 1990;60:1–7. [DOI] [PubMed] [Google Scholar]
- 138.Sobajima H, Hayakawa T, Kondo T, Shibata T, Kitagawa M, Sakai Y, Ishiguro H, Tanikawa M, Nakae Y. Effect of a new synthetic trypsin inhibitor on taurocholate-induced acute pancreatitis in rats. Pancreas 1993;8:240–277. [DOI] [PubMed] [Google Scholar]
- 139.Hirano T, Manabe T, Tobe T. Cytoprotective effects of prostaglandins and a new potent protease inhibitor in acute pancreatitis. Am J Med Sci 1992;304:154–163. [DOI] [PubMed] [Google Scholar]
- 140.Hirano T, Manabe T, Tobe T. Protection by a new synthetic protease inhibitor, ONO3307, of the rat exocrine pancreas during acute edematous pancreatitis induced by a supramaximal dose of caerulein in comparison with FOY007. Pharmacology 1992;45:107–116. [DOI] [PubMed] [Google Scholar]
- 141.Yoo KS, Huh KR, Kim YJ, Kim KO, Park CH, Hahn T, Park SH, Kim JH, Park CK, Kwon YJ, Lehman GA. Nafamostat mesilate for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis: A prospective, randomized, double-blind, controlled trial. Pancreas 2011;40:181–186. [DOI] [PubMed] [Google Scholar]
- 142.Maruyama Y, Yoshida H, Uchino S, Yokoyama K, Yamamoto H, Takinami M, Hosoya T. Nafamostat mesilate as an anticoagulant during continuous veno-venous hemodialysis: A three-year retrospective cohort study. Int J Artif Organs 2011;34:571–576. [DOI] [PubMed] [Google Scholar]
- 143.Ryu HG, Jung CW, Lee CS, Lee J. Nafamostat mesilate attenuates postreperfusion syndrome during liver transplantation. Am J Transplant 2011;11:977–983. [DOI] [PubMed] [Google Scholar]
- 144.Werner J, Hartwig W, Hackert T, Kaiser H, Schmidt J, Gebhard MM, Büchler MW, Klar E. Multidrug strategies are effective in the treatment of severe experimental pancreatitis. Surgery 2012;151:372–381. [DOI] [PubMed] [Google Scholar]
- 145.Miyagi S, Kawagishi N, Nakanishi W, Fujio A, Miyazawa K, Maida K, Kashiwadate T, Hara Y, Sekiguchi S, Ohuchi N, Satomi S. Risk factors for hepatic artery thrombosis after microsurgical vascular reconstruction in liver transplantation. Transplant Proc 2013;45:1994–1996. [DOI] [PubMed] [Google Scholar]
- 146.Taenaka N, Shimada Y, Hirata T, Nishijima M, Yoshiya I. New approach to regional anticoagulation in hemodialysis using gabexate mesilate (FOY). Crit Care Med 1982;10:773–775. [DOI] [PubMed] [Google Scholar]
- 147.Takahashi T, Suzukawa M, Akiyama M, Hatao K, Nakamura Y. Systemic AL amyloidosis with disseminated intravascular coagulation associated with hyperfibrinolysis. Int J Hematol 2008;87:371–374. [DOI] [PubMed] [Google Scholar]
- 148.Nishiyama T, Kohno Y, Koishi K. Effects of antithrombin and gabexate mesilate on disseminated intravascular coagulation: A preliminary study. Am J Emerg Med 2012;30:1219–1223. [DOI] [PubMed] [Google Scholar]
- 149.Zakharova E, Horvath MP, Goldenberg DP. Structure of a serine protease poised to resynthesize a peptide bond. Proc Natl Acad Sci USA 2009;106:11034–11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Radisky ES, Koshland DE Jr. A clogged gutter mechanism for protease inhibitors. Proc Natl Acad Sci USA 2002;99:10316–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Laskowski M Jr, Kato I. Protein inhibitors of proteinases. Annu Rev Biochem 1980;49:593–626. [DOI] [PubMed] [Google Scholar]
- 152.Laskowski M, Qasim MA. What can the structures of enzyme-inhibitor complexes tell us about the structures of enzyme substrate complexes? Biochim Biophys Acta 2000;1477:324–337. [DOI] [PubMed] [Google Scholar]
- 153.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 1967;27:157–162. [DOI] [PubMed] [Google Scholar]
- 154.Markland W, Ley AC, Lee SW, Ladner RC. Iterative optimization of high-affinity proteases inhibitors using phage display. 1. Plasmin. Biochemistry 1996;35:8045–8057. [DOI] [PubMed] [Google Scholar]
- 155.Bajaj MS, Ogueli GI, Kumar Y, Vadivel K, Lawson G, Shanker S, Schmidt AE, Bajaj SP. Engineering kunitz domain 1 (KD1) of human tissue factor pathway inhibitor-2 to selectively inhibit fibrinolysis: Properties of KD1-L17R variant. J Biol Chem 2011;286:4329–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ascenzi P, Bocedi A, Bolognesi M, Spallarossa A, Coletta M, De Cristofaro R, Menegatti E. The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor): A milestone protein. Curr Protein Pept Sci 2003;4:231–251. [DOI] [PubMed] [Google Scholar]
- 157.Kraut E, Frey EK, Werle E. About the inactivation of Kallikrein. (In German: Hoppe-Seyler Zeitschr; ). Physiol Chem 1930;192:1–21. [Google Scholar]
- 158.Kunitz M, Northrop JH. Isolation from beef pancreas of crystalline trypsinogen, trypsin, a trypsin inhibitor, and an inhibitor-trypsin compound. J Gen Physiol 1936;19:991–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chauvet J, Acher R. The reactive site of the basic trypsin in hibitor of pancreas. Role of lysine 15. J Biol Chem 1967;242:4274–4275. [PubMed] [Google Scholar]
- 160.Beckmann J, Mehlich A, Schröder W, Wenzel HR, Tschesche H. Preparation of chemically ‘mutated’ aprotinin homologues by semisynthesis. P1 substitutions change inhibitory specificity. Eur J Biochem 1988;176:675–682. [DOI] [PubMed] [Google Scholar]
- 161.Beckmann J, Mehlich A, Schröder W, Wenzel HR, Tschesche H. Semisynthesis of Arg15, Glu15, Met15, and Nle15-aprotinin involving enzymatic peptide bond resynthesis. J Protein Chem 1989;8:101–113. [DOI] [PubMed] [Google Scholar]
- 162.Groeger C, Wenzel HR, Tschesche H. Enzymatic semisynthesis of aprotinin homologues mutated in P’ positions. J Protein Chem 1991;10:245–251. [DOI] [PubMed] [Google Scholar]
- 163.Krowarsch D, Zakrzewska M, Smalas AO, Otlewski J. Structure-function relationships in serine protease-bovine pancreatic trypsin inhibitor interaction. Protein Pept Lett 2005;12:403–407. [DOI] [PubMed] [Google Scholar]
- 164.Grzesiak A, Krokoszynska I, Krowarsch D, Buczek O, Dadlez M, Otlewski J. Inhibition of six serine proteinases of the human coagulation system by mutants of bovine pancreatic trypsin inhibitor. J Biol Chem 2000;275:33346–33352. [DOI] [PubMed] [Google Scholar]
- 165.Otlewski J, Jaskólski M, Buczek O, Cierpicki T, Czapińska H, Krowarsch D, Smalas AO, Stachowiak D, Szpineta A, Dadlez M. Structure-function relationship of serine protease-protein inhibitor interaction. Acta Biochim Pol 2001;48:419–428. [PubMed] [Google Scholar]
- 166.Porte RJ, Leebeek FW. Pharmacological strategies to decrease transfusion requirements in patients undergoing surgery. Drugs 2002;62:2193–2211. [DOI] [PubMed] [Google Scholar]
- 167.Hendrickson JE, Hillyer CD. Noninfectious serious hazards of transfusion. Anesth Analg 2009;108:759–769. [DOI] [PubMed] [Google Scholar]
- 168.Landis RC, Asimakopoulos G, Poullis M, Haskard DO, Taylor KM. The antithrombotic and antiinflammatory mechanisms of action of aprotinin. Ann Thorac Surg 2001;72:2169–2175. [DOI] [PubMed] [Google Scholar]
- 169.Poston RS, White C, Gu J, Brown J, Gammie J, Pierson RN, Lee A, Connerney I, Avari T, Christenson R, Tandry U, Griffith BP. Aprotinin shows both hemostatic and antithrombotic effects during off-pump coronary artery bypass grafting. Ann Thorac Surg 2006;81:104–110. [DOI] [PubMed] [Google Scholar]
- 170.Asimakopoulos G, Thompson R, Nourshargh S, Lidington EA, Mason JC, Ratnatunga CP, Haskard DO, Taylor KM, Landis RC. An anti-inflammatory property of aprotinin detected at the level of leukocyte extravasation. J Thorac Cardiovasc Surg 2000;120:361–369. [DOI] [PubMed] [Google Scholar]
- 171.Sperzel M, Huetter J. Evaluation of aprotinin and tranexamic acid in different in vitro and in vivo models of fibrinolysis, coagulation and thrombus formation. J Thromb Haemost 2007;5:2113–2118. [DOI] [PubMed] [Google Scholar]
- 172.Ray M, Hatcher S, Whitehouse SL, Crawford S, Crawford R. Aprotinin and epsilon aminocaproic acid are effective in reducing blood loss after primary total hip arthroplasty-a prospective randomized double-blind placebo-controlled study. J Thromb Haemost 2005;3:1421–1427. [DOI] [PubMed] [Google Scholar]
- 173.Yang IS, Kim TG, Park BS, Cho KJ, Lee JH, Park Y, Kim KH. Crystal structures of aprotinin and its complex with sucrose octasulfate reveal multiple modes of interactions with implications for heparin binding. Biochem Biophys Res Commun 2010;397:429–435. [DOI] [PubMed] [Google Scholar]
- 174.Lindahl U, Feingold DS, Roden L. Biosynthesis of heparin. Trends Biochem Sci 1986;11:221–225. [Google Scholar]
- 175.Rivera V, Levieux A, Levieux D. Characterisation of Ag1, the major speciesspecific contaminant of bovine crude heparin, and its identification as an aprotinin/heparin complex. J Pharm Biomed Anal 2002;29:443–458. [DOI] [PubMed] [Google Scholar]
- 176.Kestin AS, Valeri CR, Khuri SF, Loscalzo J, Ellis PA, MacGregor H, Birjiniuk V, Ouimet H, Pasche B, Nelson MJ. The platelet function defect of cardiopulmonary bypass. Blood 1993;82:107–117. [PubMed] [Google Scholar]
- 177.John LC, Rees GM, Kovacs IB. Reduction of heparin binding to and inhibition of platelets by aprotinin. Ann Thorac Surg 1993;55:1175–1179. [DOI] [PubMed] [Google Scholar]
- 178.Mangano DT, Tudor IC, Dietzel C. The risk associated with aprotinin in cardiac surgery. New Engl J Med 2006;354:353–365. [DOI] [PubMed] [Google Scholar]
- 179.Schneeweiss S, Seeger JD, Landon J, Walker AM. Aprotinin during coronary-artery bypass grafting and risk of death. New Engl J Med 2008;358:771–783. [DOI] [PubMed] [Google Scholar]
- 180.Henry DA, Carless PA, Moxey AJ, O’Connell D, Stokes BJ, Fergusson DA, Ker K. Antifibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Syst Rev 2011;3:CD001886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mangano DT, Miao Y, Vuylsteke A, Tudor IC, Juneja R, Filipescu D, Hoeft A, Fontes ML, Hillel Z, Ott E, Titov T, Dietzel C, Levin J. Investigators of the multicenter study of perioperative ischemia research group; Ischemia Research and Education Foundation. Mortality associated with aprotinin during 5 years following coronary artery bypass graft surgery. JAMA 2007;97:471–479. [DOI] [PubMed] [Google Scholar]
- 182.Shaw AD, Stafford-Smith M, White WD, Phillips-Bute B, Swaminathan M, Milano C, Welsby IJ, Aronson S, Mathew JP, Peterson ED, Newman MF. The effect of aprotinin on outcome after coronary-artery bypass grafting. N Engl J Med 2008;358:784–793. [DOI] [PubMed] [Google Scholar]
- 183.Cosgrove DM 3rd, Heric B, Lytle BW, Taylor PC, Novoa R, Golding LA, Stewart RW, McCarthy PM, Loop FD. Aprotinin therapy for reoperative myocardial revascularization: a placebo-controlled study. Ann Thorac Surg 1992;54:1031–1036. [DOI] [PubMed] [Google Scholar]
- 184.Dietrich W, Ebell A, Busley R, Boulesteix AL. Aprotinin and anaphylaxis: analysis of 12,403 exposures to aprotinin in cardiac surgery. Ann Thorac Surg 2007;84:1144–1150. [DOI] [PubMed] [Google Scholar]
- 185.Howell N, Senanayake E, Freemantle N, Pagano D. Putting the record straight on aprotinin as safe and effective: Results from a mixed treatment meta-analysis of trials of aprotinin. J Thorac Cardiovasc Surg 2013;145:234–240. [DOI] [PubMed] [Google Scholar]
- 186.Beckerman Z, Shopen Y, Alon H, Cohen O, Nir RR, Adler Z, Bolotin G. Coronary artery bypass grafting after aprotinin: Are we doing better? J Thorac Cardiovasc Surg 2013;145:243–248. [DOI] [PubMed] [Google Scholar]
- 187.Sniecinski RM, Karkouti K, Levy JH. Managing clotting: A North American perspective. Curr Opin Anaesthesiol 2012;25:74–79. [DOI] [PubMed] [Google Scholar]
- 188.Petersen LC, Bjørn SE, Norris F, Norris K, Sprecher C, Foster DC. Expression, purification and characterization of a Kunitz-type protease inhibitor domain from human amyloid precursor protein homolog. FEBS Lett 1994;338:53–57. [DOI] [PubMed] [Google Scholar]
- 189.Navaneetham D, Wu W, Li H, Sinha D, Tuma RF, Walsh PN. P1 and P2’ site mutations convert protease nexin-2 from a factor XIa inhibitor to a plasmin inhibitor. J Biochem 2013;153:221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kong D, Ma D, Bai H, Guo H, Cai X, Mo W, Tang Q, Song H. Expression and characterization of the first kunitz domain of human tissue factor pathway inhibitor-2. Biochem Biophys Res Commun 2004;324:1179–1185. [DOI] [PubMed] [Google Scholar]
- 191.Petersen LC, Bjørn SE, Olsen OH, Nordfang O, Norris F, Norris K. Inhibitory properties of separate recombinant Kunitz-type-protease-inhibitor domains from tissue-factor-pathway inhibitor. Eur J Biochem 1996;235:310–316. [DOI] [PubMed] [Google Scholar]
- 192.Marlor CW, Delaria KA, Davis G, Muller DK, Greve JM, Tamburini PP. Identification and cloning of human placental bikunin, a novel serine protease inhibitor containing two Kunitz domains. J Biol Chem 1997;272:12202–12208. [DOI] [PubMed] [Google Scholar]
- 193.Delaria KA, Muller DK, Marlor CW, Brown JE, Das RC, Roczniak SO, Tamburini PP. Characterization of placental bikunin, a novel human serine protease inhibitor. J Biol Chem 1997;272:12209–12214. [DOI] [PubMed] [Google Scholar]
- 194.Oliva ML, Souza-Pinto JC, Batista IF, Araujo MS, Silveira VF, Auerswald EA, Mentele R, Eckerskorn C, Sampaio MU, Sampaio CA. Leucaena leucocephala serine proteinase inhibitor: Primary structure and action on blood coagulation, kinin release and rat paw edema. Biochim Biophys Acta 2000;1477:64–74. [DOI] [PubMed] [Google Scholar]
- 195.Paesen GC, Siebold C, Harlos K, Peacey MF, Nuttall PA, Stuart DI. A tick protein with a modified Kunitz fold inhibits human tryptase. J Mol Biol 2007;368:1172–1186. [DOI] [PubMed] [Google Scholar]
- 196.Masci PP, Whitaker AN, Sparrow LG, de Jersey J, Winzor DJ, Watters DJ, Lavin MF, Gaffney PJ. Textilinins from Pseudonaja textilis. Characterization of two plasmin inhibitors that reduce bleeding in an animal model. Blood Coagul Fibrinolysis 2000;11:385–393. [DOI] [PubMed] [Google Scholar]
- 197.Filippovich I, Sorokina N, Masci PP, de Jersey J, Whitaker AN, Winzor DJ, Gaffney PJ, Lavin MF. A family of textilinin genes, two of which encode proteins with antihaemorrhagic properties. Br J Haematol 2002;119:376–384. [DOI] [PubMed] [Google Scholar]
- 198.Flight S, Johnson L, Trabi M, Gaffney P, Lavin M, de Jersey J, Masci P. Comparison of textilinin-1 with aprotinin as serine protease inhibitors and as antifibrinolytic agents. Pathophysiol. Haemost Thromb 2005;34:188–193. [DOI] [PubMed] [Google Scholar]
- 199.Flight SM, Johnson LA, Du QS, Warner RL, Trabi M, Gaffney PJ, Lavin MF, de Jersey J, Masci PP. Textilinin-1, an alternative anti-bleeding agent to aprotinin: Importance of plasmin inhibition in controlling blood loss. Br J Haematol 2009;145:207–211. [DOI] [PubMed] [Google Scholar]
- 200.Millers EK, Trabi M, Masci PP, Lavin MF, de Jersey J, Guddat LW. Crystal structure of textilinin-1, a Kunitz-type serine protease inhibitor from the venom of the Australian common brown snake (Pseudonaja textilis). FEBS J 2009;276:3163–3175. [DOI] [PubMed] [Google Scholar]
- 201.Millers EK, Johnson LA, Birrell GW, Masci PP, Lavin MF, de Jersey J, Guddat LW. The structure of human microplasmin in complex with textilinin-1, an aprotinin-like inhibitor from the Australian brown snake. PLoS ONE 2013;8:e54104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Earl ST, Masci PP, de Jersey J, Lavin MF, Dixon J. Drug development from Australian elapid snake venoms and the Venomics pipeline of candidates for haemostasis: Textilinin-1 (Q8008), Haempatch™ (Q8009) and CoVase™ (V0801). Toxicon 2012;59:456–463. [DOI] [PubMed] [Google Scholar]
- 203.Wan H, Lee KS, Kim BY, Zou FM, Yoon HJ, Je YH, Li J, Jin BR. A spider-derived Kunitz-type serine protease inhibitor that acts as a plasmin inhibitor and an elastase inhibitor. PLoS ONE 2013;8:e53343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cheng AC, Tsai IH. Functional characterization of a slow and tight-binding inhibitor of plasmin isolated from Russell’s viper venom. Biochim Biophys Acta 2014;1840:153–159. [DOI] [PubMed] [Google Scholar]
- 205.Qiu Y, Lee KS, Choo YM, Kong D, Yoon HJ, Jin BR. Molecular cloning and antifibrinolytic activity of a serine protease inhibitor from bumblebee (Bombus terrestris) venom. Toxicon 2013;63:1–6. [DOI] [PubMed] [Google Scholar]
- 206.Choo YM, Lee KS, Yoon HJ, Qiu Y, Wan H, Sohn MR, Sohn HD, Jin BR. Antifibrinolytic role of a bee venom serine protease inhibitor that acts as a plasmin inhibitor. PLoS ONE 2012;7:e32269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Valdés JJ, Schwarz A, Cabeza de Vaca I, Calvo E, Pedra JH, Guallar V, Kotsyfakis M. Tryptogalinin is a tick Kunitz serine protease inhibitor with a unique intrinsic disorder. PLoS ONE 2013;8:e62562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rimphanitchayakit V, Tassanakajon A. Structure and function of invertebrate Kazal-type serine proteinase inhibitors. Dev Comp Immunol 2010;34:377–386. [DOI] [PubMed] [Google Scholar]
- 209.Ranasinghe S, McManus DP. Structure and function of invertebrate Kunitz serine protease inhibitors. Dev Comp Immunol 2013;39:219–227. [DOI] [PubMed] [Google Scholar]
- 210.Campos IT, Amino R, Sampaio CA, Auerswald EA, Friedrich T, Lemaire HG, Schenkman S, Tanaka AS. Infestin, a thrombin inhibitor presents in Triatoma infestans midgut, a Chagas’ disease vector: Gene cloning, expression and characterization of the inhibitor. Insect Biochem Mol Biol 2002;32:991–997. [DOI] [PubMed] [Google Scholar]
- 211.Lovato DV, Nicolau de Campos IT, Amino R, Tanaka AS. The full-length cDNA of anticoagulant protein infestin revealed a novel releasable Kazal domain, a neutrophil elastase inhibitor lacking anticoagulant activity. Biochimie 2006;88:673–681. [DOI] [PubMed] [Google Scholar]
- 212.Campos IT, Tanaka-Azevedo AM, Tanaka AS. Identification and characterization of a novel factor XIIa inhibitor in the hematophagous insect, Triatoma infestans (Hemiptera: Reduviidae). FEBS Lett 2004;577:512–516. [DOI] [PubMed] [Google Scholar]
- 213.Campos IT, Souza TA, Torquato RJ, De Marco R, Tanaka-Azevedo AM, Tanaka AS, Barbosa JA. The Kazal-type inhibitors infestins 1 and 4 differ in specificity but are similar in three-dimensional structure. Acta Crystallogr D Biol Crystallogr 2012;68:695–702. [DOI] [PubMed] [Google Scholar]
- 214.Chen JW, Figueiredo JL, Wojtkiewicz GR, Siegel C, Iwamoto Y, Kim DE, Nolte MW, Dickneite G, Weissleder R, Nahrendorf M. Selective factor XIIa inhibition attenuates silent brain ischemia: Application of molecular imaging targeting coagulation pathway. JACC Cardiovasc Imaging 2012;5:1127–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kim YH, Choi JG, Lee GM, Kang KW. Domain and genomic sequence analysis of bdellin-KL, a leech-derived trypsin-plasmin inhibitor. J Biochem 2001;130:431–438. [DOI] [PubMed] [Google Scholar]
- 216.Kubiak A, Jakimowicz P, Polanowski A. A Kazal-type serine proteinase inhibitor from chicken liver (clTI-1): Purification, primary structure, and inhibitory properties. Int J Biol Macromol 2009;45:194–199. [DOI] [PubMed] [Google Scholar]
- 217.Lopuska A, Polanowska J, Wilusz T, Polanowski A. Purification of two low-molecular-mass serine proteinase inhibitors from chicken liver. J Chromatogr A 1999;852:207–216. [DOI] [PubMed] [Google Scholar]
- 218.Watanabe RM, Tanaka-Azevedo AM, Araujo MS, Juliano MA, Tanaka AS. Characterization of thrombin inhibitory mechanism of rAaTI, a Kazal-type inhibitor from Aedes aegypti with anticoagulant activity. Biochimie 2011;93:618–623. [DOI] [PubMed] [Google Scholar]
- 219.Watanabe RM, Soares TS, Morais-Zani K, Tanaka-Azevedo AM, Maciel C, Capurro ML, Torquato RJ, Tanaka AS. A novel trypsin Kazal-type inhibitor from Aedes aegypti with thrombin coagulant inhibitory activity. Biochimie 2010;92:933–939. [DOI] [PubMed] [Google Scholar]
- 220.Sugino H, Nakagawa S, Kakinuma A. Plasminostreptin, a protein proteinase inhibitor produced by Streptomyces antifibrinolyticus. II. Determination of the reactive site for proteinases. J Biol Chem 1978;253:1538–1545. [PubMed] [Google Scholar]
- 221.Kakinuma A, Sugino H, Moriya N, Isono M. Plasminostreptin, a protein proteinase inhibitor produced by Streptomyces antifibrinolyticus. I. Isolation and characterization. J Biol Chem 1978;253:1529–1537. [PubMed] [Google Scholar]
- 222.Wu W, Li H, Navaneetham D, Reichenbach ZW, Tuma RF, Walsh PN. The kunitz protease inhibitor domain of protease nexin-2 inhibits factor XIa and murine carotid artery and middle cerebral artery thrombosis. Blood 2012;120:671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Salameh MA, Soares AS, Hockla A, Radisky DC, Radisky ES. The P(2)’ residue is a key determinant of mesotrypsin specificity: Engineering a high-affinity inhibitor with anticancer activity. Biochem J 2011;440:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Fukushima T, Kawaguchi M, Yamasaki M, Tanaka H, Yorita K, Kataoka H. Hepatocyte growth factor activator inhibitor type 1 suppresses metastatic pulmonary colonization of pancreatic carcinoma cells. Cancer Sci 2011;102:407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lehmann A. Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery. Expert Opin Biol Ther 2008;8:1187–1199. [DOI] [PubMed] [Google Scholar]
- 226.Devy L, Rabbani SA, Stochl M, Ruskowski M, Mackie I, Naa L, Toews M, van Gool R, Chen J, Ley A, Ladner RC, Dransfield DT, Henderikx P. PEGylated DX-1000: Pharmacokinetics and antineoplastic activity of a specific plasmin inhibitor. Neoplasia 2007;9:927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ranasinghe S, McManus DP. Structure and function of invertebrate Kunitz serine protease inhibitors. Dev Comp Immunol 2013;39:219–227. [DOI] [PubMed] [Google Scholar]
- 228.Girard TJ, Warren LA, Novotny WF, Likert KM, Brown SG, Miletich JP, Broze GJ Jr. Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature 1989;338:518–520. [DOI] [PubMed] [Google Scholar]
- 229.Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP. Structure and biology of tissue factor pathway inhibitor. Thromb Haemost 2001;86:959–972. [PubMed] [Google Scholar]
- 230.Chand HS, Schmidt AE, Bajaj SP, Kisiel W. Structure-function analysis of the reactive site in the first Kunitz-type domain of human tissue factor pathway inhibitor-2. J Biol Chem 2004;279:17500–17507. [DOI] [PubMed] [Google Scholar]
- 231.Murkin JM, Falter F, Granton J, Young B, Burt C, Chu M. High-dose tranexamic acid is associated with nonischemic clinical seizures in cardiac surgical patients. Anesth Analg 2010;110:350–353. [DOI] [PubMed] [Google Scholar]
- 232.Scott CJ, Taggart CC. Biologic protease inhibitors as novel therapeutic agents. Biochimie 2010. 92:1681–1688. [DOI] [PubMed] [Google Scholar]
- 233.Bokesch PM, Szabo G, Wojdyga R, Grocott HP, Smith PK, Mazer CD, Vetticaden S, Wheeler A, Levy JH. A phase 2 prospective, randomized, double-blind trial comparing the effects of tranexamic acid with ecallantide on blood loss from high-risk cardiac surgery with cardiopulmonary bypass (CONSERV-2 Trial). J Thorac Cardiovasc Surg 2012. 143:1022–1029. [DOI] [PubMed] [Google Scholar]
- 234.Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des 2008;72:455–482. [DOI] [PubMed] [Google Scholar]
- 235.Chander A, Atkinson HM, Stevic I, Berry LR, Kim PY, Chan AK. Interactions of heparin and a covalently-linked antithrombin-heparin complex with components of the fibrinolytic system. Thromb Haemost 2013;110:1180–1188. [DOI] [PubMed] [Google Scholar]
- 236.Highsmith RF, Rosenberg RD. The inhibition of human plasmin by human antithrombin-heparin cofactor. J Biol Chem 1974;249:4335–4338. [PubMed] [Google Scholar]
- 237.Machovich R, Bauer PI, Arányi P, Kecskés E, Büki KG, Horváth I. Kinetic analysis of the heparin-enhanced plasmin-antithrombin III reaction. Apparent catalytic role of heparin. Biochem J 1981;199:521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Smith GF, Sundboom JL. Heparin and protease inhibition. II. The role of heparin in the ATIII inactivation of thrombin, plasmin, and trypsin. Thromb Res 1981;22:115–133. [DOI] [PubMed] [Google Scholar]
- 239.Jordan RE, Oosta GM, Gardner WT, Rosenberg RD. The binding of low molecular weight heparin to hemostatic enzymes. J Biol Chem 1980;255:10073–10080. [PubMed] [Google Scholar]
- 240.Yomtova VM, Stambolieva NA, Blagoev BM. Kinetic study of the effect of heparin on the amidase activity of trypsin, plasmin and urokinase. Thromb Haemost 1983;49:199–203. [PubMed] [Google Scholar]
- 241.Bauer PI, Pozsgay M, Machovich R, Elödi P, Horváth I. The interaction of heparin with human plasmin. Int J Biochem 1983;15:871–874. [DOI] [PubMed] [Google Scholar]
- 242.Higazi AA, Aziza R, Samara AA, Mayer M. Regulation of fibrinolysis by non-esterified fatty acids. Biochem J 1994;300:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Higazi AA, Finci-Yeheskel Z, Samara AA, Aziza R, Mayer M. Stimulation of plasmin activity by oleic acid. Biochem J 1992;282:863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Legras S, Diczhazi C, Moczar M. N-oleoyl heparin inhibits the amidolytic activity of plasmin and urokinase. Int J Biol Macromol 1992;14:97–99. [DOI] [PubMed] [Google Scholar]
- 245.Henry BL, Abdel Aziz M, Zhou Q, Desai UR. Sulfated, low-molecular-weight lignins are potent inhibitorsof plasmin, in addition to thrombin and factor Xa: Novel opportunity for controlling complex pathologies. Thromb Haemost 2010;103:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ledoux D, Papy-Garcia D, Escartin Q, Sagot MA, Cao Y, Barritault D, Courtois J, Hornebeck W, Caruelle JP. Human plasmin enzymatic activity is inhibited by chemically modified dextrans. J Biol Chem 2000;275:29383–29390. [DOI] [PubMed] [Google Scholar]
- 247.Vörös G, Kolev K, Csomor K, Machovich R. Inhibition of plasmin activity by sulfated polyvinylalcohol-acrylate copolymers. Thromb Res 2000;100:353–361. [DOI] [PubMed] [Google Scholar]
- 248.Monien BH, Henry BL, Raghuraman A, Hindle M, Desai UR. Novel chemo-enzymatic oligomers of cinnamic acids as direct and indirect inhibitors of coagulation proteinases. Bioorg Med Chem 2006;14:7988–7998. [DOI] [PubMed] [Google Scholar]
- 249.Henry BL, Monien BH, Bock PE, Desai UR. A novel allosteric pathway of thrombin inhibition: Exosite II mediated potent inhibition of thrombin by chemo-enzymatic, sulfated dehydropolymers of 4-hydroxycinnamic acids. J Biol Chem 2007;282:31891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Henry BL, Thakkar JN, Martin EJ, Brophy DF, Desai UR. Characterization of the plasma and blood anticoagulant potential of structurally and mechanistically novel oligomers of 4-hydroxycinnamic acids. Blood Coagul Fibrinolysis 2009;20:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Henry BL, Thakkar JN, Liang A, Desai UR. Sulfated, low molecular weight lignins inhibit a select group of heparin-binding serine proteases. Biochem Biophys Res Commun 2012;417:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Abdel Aziz MH, Mosier PD, Desai UR. Identification of the site of binding of sulfated, low molecular weight lignins on thrombin. Biochem Biophys Res Commun 2011;413:348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Snyder KM, Kessler CM. The pivotal role of thrombin in cancer biology and tumorigenesis. Semin Thromb Hemost 2008;34:734–741. [DOI] [PubMed] [Google Scholar]
- 254.Churg A, Wright JL. Proteases and emphysema. Curr Opin Pulm Med 2005;11:153–159. [DOI] [PubMed] [Google Scholar]
- 255.Saluja B, Thakkar JN, Li H, Desai UR, Sakagami M. Novel low molecular weight lignins as potential anti-emphysema agents: In vitro triple inhibitory activity against elastase, oxidation and inflammation. Pulm Pharmacol Ther 2013;26:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Meddahi A, Benoit J, Ayoub N, Sézeur A, Barritault D. Heparin-like polymers derived from dextran enhances colonic anastomosis resistance to leakage. J Biomed Mater Res 1996;31, 293–297. [DOI] [PubMed] [Google Scholar]
- 257.Tardieu M, Gamby C, Avramoglou T, Jozefonvicz J, Barritault D. Derivatized dextrans mimic heparin as stabilizers, potentiators, and protectors of acidic or basic FGF. J Cell Physiol 1992;150:194–203. [DOI] [PubMed] [Google Scholar]
- 258.Meddahi A, Lemdjabar H, Caruelle JP, Barritault D, Hornebeck W. FGF protection and inhibition of human neutrophil elastase by carboxymethyl benzylamide sulfonate dextran derivatives. Int J Biol Macromol 1996;18:141–135. [DOI] [PubMed] [Google Scholar]
- 259.Meddahi A, Blanquaert F, Saffar JL, Colombier ML, Caruelle JP, Josefonvicz J, Barritault D, New approaches to tissue regeneration and repair. Pathol Res Pract 1994;190:923–928. [DOI] [PubMed] [Google Scholar]
- 260.Blanquaert F, Saffar JL, Colombier ML, Carpentier G, Barritault D. Caruelle JP. Heparan-like molecules induce the repair of skull defects. Bone 1995;17:499–506. [DOI] [PubMed] [Google Scholar]
- 261.Fredj-Reygrobellet D, Hriskova DL, Ettaiche M, Meddahi A, Jozefonwicz J, Barritault D. CMDBS, functional analog of heparin sulfate as a new class of corneal ulcer healing agents. Ophtalmic Res 1994;26:325–331. [DOI] [PubMed] [Google Scholar]
- 262.Desgranges P, Barbaud C, Caruelle JP, Barritault D, Gautron J. A substituted dextran enhances muscle fiber survival and regeneration in ischemic and denervated rat EDL muscle. FASEB J 1999;13:761–766. [DOI] [PubMed] [Google Scholar]
- 263.Machovich R, Nagy M, Györgyi-Edelenyi J, Csomor K, Horvath I. Anticoagulant effect of sulphated polyvinylalcohol-acrylic acid copolymers. Thromb Haemost 1986;56:397–400. [PubMed] [Google Scholar]
- 264.Wanaka K, Okamoto S, Horie N, Hijikata-Okunomiya A, Okamoto U, Naito T, Ohno N, Bohgaki M, Tsuda Y, Okada Y. Use of an active center-directed plasmin inhibitor elucidates the multiplicity of plasmin actions. Thromb Res 1996;82:79–86. [DOI] [PubMed] [Google Scholar]
- 265.Lee E, Enomoto R, Takemura K, Tsuda Y, Okada Y. A selective plasmin inhibitor, transaminomethylcyclohexanecarbonyl-L-(O-picolyl)tyrosine-octylamide (YO-2), induces thymocyte apoptosis. Biochem Pharmacol 2002;63:1315–1323. [DOI] [PubMed] [Google Scholar]
- 266.Enomoto R, Sugahara C, Komai T, Suzuki C, Kinoshita N, Hosoda A, Yoshikawa A, Tsuda Y, Okada Y, Lee E. The structure-activity relationship of various YO compounds, novel plasmin inhibitors, in the apoptosis induction. Biochim Biophys Acta 2004;1674:291–298. [DOI] [PubMed] [Google Scholar]
- 267.Szende B, Okada Y, Tsuda Y, Horvath A, Bökönyi G, Okamoto S, Wanaka K, Kéri G. A novel plasmin-inhibitor inhibits the growth of human tumor xenografts and decreases metastasis number. In Vivo 2002;16:281–286. [PubMed] [Google Scholar]
- 268.Goodloe JM, Howerton DS, McAnallen D, Reed H. TXA: A difference-maker for trauma patients. JEMS 2013;38:60–65. [PubMed] [Google Scholar]
- 269.Roberts I, Shakur H, Coats T, Hunt B, Balogun E, Barnetson L, Cook L, Kawahara T, Perel P, Prieto-Merino D, Ramos M, Cairns J, Guerriero C. The CRASH-2 trial: A randomised controlled trial and economic evaluation of the effects of tranexamic acid on death, vascular occlusive events and transfusion requirement in bleeding trauma patients. Health Technol Assess 2013;17:1–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Reichel CA, Lerchenberger M, Uhl B, Rehberg M, Berberich N, Zahler S, Wymann MP, Krombach F. Plasmin inhibitors prevent leukocyte accumulation and remodeling events in the postischemic microvasculature. PLoS ONE 2011;6:e17229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Tse TW, Hui E. Tranexamic acid: An important adjuvant in the treatment of melasma. J Cosmet Dermatol 2013;12:57–66. [DOI] [PubMed] [Google Scholar]
- 272.Takahashi H, Tatewaki W, Wada K, Yoshikawa A, Shibata A. Thrombin and plasmin generation in patients with liver disease. Am J Hematol 1989;32:30–35. [DOI] [PubMed] [Google Scholar]
- 273.Stürzebecher J, Vieweg H, Steinmetzer T, Schweinitz A, Stubbs MT, Renatus M, Wikström P. 3-Amidinophenylalanine-based inhibitors of urokinase. Bioorg Med Chem Lett 1999;9:3147–3152. [DOI] [PubMed] [Google Scholar]
- 274.Froriep D, Clement B, Bittner F, Mendel RR, Reichmann D, Schmalix W, Havemeyer A. Activation of the anti-cancer agent upamostat by the mARC enzyme system. Xenobiotica 2013;43:780–784. [DOI] [PubMed] [Google Scholar]
- 275.Ertongur S, Lang S, Mack B, Wosikowski K, Muehlenweg B, Gires O. Inhibition of the invasion capacity of carcinoma cells by WX-UK1, a novel synthetic inhibitor of the urokinase-type plasminogen activator system. Int J Cancer 2004;110:815–824. [DOI] [PubMed] [Google Scholar]
- 276.Setyono-Han B, Stürzebecher J, Schmalix WA, Muehlenweg B, Sieuwerts AM, Timmermans M, Magdolen V, Schmitt M, Klijn JG, Foekens JA. Suppression of rat breast cancer metastasis and reduction of primary tumour growth by the small synthetic urokinase inhibitor WX-UK1. Thromb Haemost 2005;93:779–786. [DOI] [PubMed] [Google Scholar]
- 277.Heinemann V, Ebert MP, Laubender RP, Bevan P, Mala C, Boeck S. Phase II randomised proof-of-concept study of the urokinase inhibitor upamostat (WX-671) in combination with gemcitabine compared with gemcitabine alone in patients with non-resectable, locally advanced pancreatic cancer. Br J Cancer 2013;108:766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Gustafsson D, Antonsson T, Bylund R, Eriksson U, Gyzander E, Nilsson I, Elg M, Mattsson C, Deinum J, Pehrsson S, Karlsson O, Nilsson A, Sörensen H. Effects of melagatran, a new low-molecular-weight thrombin inhibitor, on thrombin and fibrinolytic enzymes. Thromb Haemost 1998;79:110–118. [PubMed] [Google Scholar]
- 279.Mannucci PM, Levi M. Prevention and treatment of major blood loss. New Engl J Med 2007;356:2301–2311. [DOI] [PubMed] [Google Scholar]
- 280.Koster A, Schirmer U. Re-evaluation of the role of antifibrinolytic therapy with lysine analogs during cardiac surgery in the post aprotinin era. Curr Opin Anaesthesiol 2011;24:92–97. [DOI] [PubMed] [Google Scholar]
- 281.Sander M, Spies CD, Martiny V, Rosenthal C, Wernecke KD, von Heymann C. Mortality associated with administration of high-dose tranexamic acid and aprotinin in primary open-heart procedures: A retrospective analysis. Crit Care 2010;14:R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Furtmueller R, Schlag MG, Berger M, Hopf R, Huck S, Sieghart W, Redl H. Tranexamic acid, a widely used antifibrinolytic agent, causes convulsions by a gamma-aminobutyric acid(A) receptor antagonistic effect. J Pharmacol Exp Ther 2002;301:168–173. [DOI] [PubMed] [Google Scholar]
- 283.Al-Horani RA, Liang A, Desai UR. Designing nonsaccharide, allosteric activators of antithrombin for accelerated inhibition of factor Xa. J Med Chem 2011;54:6125–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Karuturi R, Al-Horani RA, Mehta SC, Gailani D, Desai UR. Discovery of allosteric modulators of factor XIa by targeting hydrophobic domains adjacent to its heparin-binding site. J Med Chem 2013;56:2415–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Al-Horani RA, Ponnusamy P, Mehta AY, Gailani D, Desai UR. Sulfated penta-galloylglucoside is a potent, allosteric, and selective inhibitor of factor XIa. J Med Chem 2013;56:867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Abdel Aziz MH, Sidhu PS, Liang A, Kim JY, Mosier PD, Zhou Q, Farrell DH, Desai UR. Designing allosteric regulators of thrombin. Monosulfated benzofuran dimers selectively interact with Arg173 of exosite 2 to induce inhibition. J Med Chem 2012;55:6888–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Sidhu PS, Abdel Aziz MH, Sarkar A, Mehta AY, Zhou Q, Desai UR. Designing allosteric regulators of thrombin. Exosite 2 features multiple subsites that can be targeted by sulfated small molecules for inducing inhibition. J Med Chem 2013;56:5059–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Desai UR. The promise of sulfated synthetic small molecules as modulators of glycosaminoglycan function. Future Med Chem 2013;5:1363–1366. [DOI] [PubMed] [Google Scholar]