

ABSTRACT
New or emerging infectious diseases are commonly caused by pathogens that cannot be readily manipulated or studied under common laboratory conditions. These limitations hinder standard experimental approaches and our abilities to define the fundamental molecular mechanisms underlying pathogenesis. The advance of capped small RNA sequencing (csRNA-seq) now enables genome-wide mapping of actively initiated transcripts from genes and other regulatory transcribed start regions (TSRs) such as enhancers at a precise moment from total RNA. As RNA is nonpathogenic and can be readily isolated from inactivated infectious samples, csRNA-seq can detect acute changes in gene regulation within or in response to a pathogen with remarkable sensitivity under common laboratory conditions. Studying valley fever (coccidioidomycosis), an emerging endemic fungal infection that increasingly impacts livestock, pet, and human health, we show how csRNA-seq can unravel transcriptional programs driving pathogenesis. Performing csRNA-seq on RNA isolated from different stages of the valley fever pathogen Coccidioides immitis revealed alternative promoter usage, connected cis-regulatory domains, and a WOPR family transcription factor, which are known regulators of virulence in other fungi, as being critical for pathogenic growth. We further demonstrate that a C. immitis WOPR homologue, CIMG_02671, activates transcription in a WOPR motif-dependent manner. Collectively, these findings provide novel insights into valley fever pathogenesis and provide a proof of principle for csRNA-seq as a powerful means to determine the genes, regulatory mechanisms, and transcription factors that control the pathogenesis of highly infectious agents.
IMPORTANCE Infectious pathogens like airborne viruses or fungal spores are difficult to study; they require high-containment facilities, special equipment, and expertise. As such, establishing approaches such as genome editing or other means to identify the factors and mechanisms underlying caused diseases, and, thus, promising drug targets, is costly and time-intensive. These obstacles particularly hinder the analysis of new, emerging, or rare infectious diseases. We recently developed a method termed capped small RNA sequencing (csRNA-seq) that enables capturing acute changes in active gene expression from total RNA. Prior to csRNA-seq, such an analysis was possible only by using living cells or nuclei, in which pathogens are highly infectious. The process of RNA purification, however, inactivates pathogens and thus enables the analysis of gene expression during disease progression under standard laboratory conditions. As a proof of principle, here, we use csRNA-seq to unravel the gene regulatory programs and factors likely critical for the pathogenesis of valley fever, an emerging endemic fungal infection that increasingly impacts livestock, pet, and human health.
KEYWORDS: Coccidioides immitis, coccidioidomycosis, valley fever, WOPR, csRNA-seq, gene regulation, genomics, phase transition, transcription factors, transcriptomics
INTRODUCTION
Coccidioides immitis is one of many emerging pathogens (https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens). It is a fungus that causes valley fever (coccidioidomycosis), which increasingly impacts wild mammals, livestock, pets, and humans (1–4). C. immitis grows as mycelia in the soil (vegetative) and forms spores called arthroconidia that are easily aerosolized. Once inhaled, arthroconidia transition to the spherule/endospore phase that can cause life-threatening pneumonia or disseminated disease (3, 5). In 2018, 15,611 clinically significant human cases were reported to the CDC, but this underestimates the true disease incidence as many infected individuals do not present to medical care, are misdiagnosed, or are not reported (6, 7) (https://www.cdc.gov/fungal/diseases/coccidioidomycosis/statistics.html). The economic impact of valley fever in 2015 was estimated to be around $3.9 billion (4).
To combat this alarming trend and identify drug targets and novel avenues for the diagnosis and treatment of this difficult-to-treat infection, it is critical to better understand the fundamental gene regulatory mechanisms underlying the complex life cycle and pathogenesis of C. immitis. However, similar to other highly infectious pathogens, propagating C. immitis requires high-containment practices, equipment, facilities, and training (8). As such, standard laboratory procedures like genome editing or mutagenesis screens are often cost-prohibitive and sometimes simply insurmountable. These obstacles call for alternative strategies and approaches, particularly those that enable a detailed readout of pathogenic states under standard laboratory conditions. Purified RNA is nonpathogenic and can be readily isolated from inactivated infectious samples and subsequently shipped and analyzed under standard laboratory conditions. Consecutive RNA sequencing (RNA-seq) analysis enables the identification of all RNAs present in a given sample. However, many transcripts such as enhancer RNAs (eRNAs) or primary microRNAs (pri-miRNAs) are unstable and, thus, poorly detected by RNA-seq. RNA-seq also cannot distinguish actively expressed genes from those that are inactive but have stable transcripts that reside in the cell. The method is thus limited in its ability to identify regulatory mechanisms. Therefore, although RNA-seq has been used to capture changes in gene expression in C. immitis phase transition (9–11) as well as other highly pathogenic samples (12, 13), the underlying gene regulatory mechanisms and, thus, pathogenesis are not understood.
To decode gene regulatory mechanisms, it is important to capture all regulatory elements and transcription start sites (TSSs) active at a specific moment in time. Such efforts require nascent transcription approaches that capture ongoing transcription independent of transcript stability (14–16) rather than steady-state approaches such as RNA-seq, which capture the sum of stable but not necessarily actively transcribed RNAs present in the cell. Capturing nascent TSSs allows the identification of DNA motifs of key activating transcription factors (TFs) with remarkable sensitivity (17–20). As such, nascent transcription approaches have revolutionized our understanding of gene regulation, but methodological constraints have limited their application. Most nascent methods, including global run-on sequencing (GRO-seq) (14) and precision run-on sequencing (PRO-seq) (21), require rather advanced expertise and millions of purified live nuclei. These prerequisites make the application of such run-on methods challenging, particularly in organisms with cell walls, including plants (22) and fungi, and largely obstruct the analysis of highly infectious samples. To overcome these limitations, we recently developed capped small RNA sequencing (csRNA-seq), which accurately detects the TSSs of actively transcribed stable and unstable RNAs at single-nucleotide resolution from total RNA (15). Here, we demonstrate the feasibility of using csRNA-seq to unravel the gene regulatory mechanisms in highly infectious samples. Given the pressing need to learn more about valley fever pathogenesis as well as the challenges of analyzing highly infectious fungal spores that are rich in secondary metabolites and protected by thick cell walls (23), we focused this study on C. immitis. Performing csRNA-seq on RNA isolated from different stages of the valley fever pathogen revealed alternative promoter usage, connected cis-regulatory domains, and WOPR family TFs as likely critical for infection. WOPR TFs are known regulators of virulence in other fungi (24–27), and we further demonstrate that a C. immitis WOPR homologue, CIMG_02671, activates transcription in a WOPR motif-dependent manner. These findings provide a foundation to explore avenues for the diagnosis and treatment of valley fever and a proof of principle for csRNA-seq as a powerful means to identify genes, regulatory mechanisms, and transcription factors that control pathogenesis.
RESULTS
Functional transcriptome annotation of the BSL3 pathogen C. immitis.
With the aim to further our understanding of the genes and transcription factors driving C. immitis phase transition and, thus, pathogenesis, we grew C. immitis RS mycelia and young, 48-h-old, and mature, 8-day-old, spherules under biosafety level 3 (BSL3) conditions. Young spherules (48 h old) differ from mature ones (8 days old) in that the former do not contain endospores (Fig. 1A) (28, 29). Samples were inactivated using QIAzol (Qiagen) and ZR BashingBead lysis at 50 Hz for 25 min, and total RNA was extracted under common laboratory conditions. This RNA was then used to capture the steady-state transcriptome by ribosome-depleted paired-end RNA-seq as well as ongoing transcription by csRNA-seq (15) (Fig. 1B). All assays were performed on duplicate samples that were prepared on separate days for all three stages, and the generated data were highly reproducible and correlated across methods (see Fig. S1 in the supplemental material). Combined, they reveal the functional transcriptome of C. immitis at these three stages at unprecedented resolution: RNA-seq accurately quantifies RNAs present in the sample, while csRNA-seq captures actively initiating RNAs and, thus, the transcription start sites (TSSs) of both stable and unstable transcripts (Fig. 1C).
FIG 1.

Functional transcriptome annotation of the BSL3 pathogen C. immitis. (A) Life cycle of C. immitis. The dimorphic fungal pathogen grows as mycelia in the soil but upon inhalation transitions to a parasitic spherule phase as alternating spherules and progeny endospores. (B) RNA from three selected life stages. Mycelia as well as young (48-h-old) and mature (8-day-old) spherules were isolated, cytosolic steady-state RNAs were captured by total RNA-seq, and the transcription start sites of actively transcribed stable and unstable transcripts were captured by csRNA-seq. (C) Example browser shot of a bidirectionally transcribed region in C. immitis mycelia. (D) Overview of the regions where identified transcription start regions (TSRs) mapped in C. immitis mycelia and spherules. (E) RNA-seq reads associated with csRNA-seq-defined TSRs in different locations as a proxy for transcript stability. (F) Number and stability of TSRs captured across diverse fungal species and humans.
Reproducibility and correlation of generated biological replicates. (A and B) Scatterplots of replicates for RNA-seq (A) and csRNA-seq (B). (C) Comparison of csRNA-seq reads at gene promoters to RNA-seq reads across genes demonstrated that changes in initiation and gene expression were correlated across phases (r = 0.63). Download FIG S1, TIF file, 2.7 MB (2.7MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
As accurate transcriptome annotations have become an integral part of research (30, 31), we first exploited our data to annotate genes and regulatory elements in C. immitis (Fig. 1C). Using StringTie to assemble transcripts directly from our RNA-seq data (32), we identified 7,163 genes (10,069 isoforms in total) in mycelia and 9,669 genes (14,639 isoforms) in spherules (Fig. S2A). Unlike in mycelia, where only 393 novel putative genes were annotated, 2,240 (23.2%) putative genes that are not represented in the official reference annotation GTF file (Ensembl) were identified in spherules, underscoring the need for studying the pathogenic, in addition to the vegetative mycelial, stage. Using csRNA-seq, we identified in mycelia 11,728 promoter, putative enhancer, and other transcribed regulatory regions that we jointly refer to as transcription start regions (TSRs) here. Most TSRs found in mycelia were in promoters (±500 bp and either sense or antisense relative to the annotated 5′ ends of genes [TSSs]) (Fig. S2B). In 48-h- and 8-day-old spherules, 21,284 and 19,771 TSRs were identified, respectively. Most TSRs started transcription bidirectionally (91.04%) from the same regulatory regions and initiated from several dispersed individual TSSs rather than a single dominant TSS (Fig. 1C; Fig. S2C and D). Interestingly, TSRs in spherules, but not mycelia, were frequently promoter distal and initiated unstable RNAs (∼49% of TSRs are >500 bp from reference annotated TSSs) (Fig. 1D; Fig. S3A). On average, TSRs at annotated promoters had higher levels of associated RNA reads than promoter-distal loci (Fig. 1E). Similarly, intragenic TSRs had higher levels of associated RNA than intergenic TSRs (Fig. 1E).
Features of C. immitis mycelium transcription start regions (TSRs). (A) Overview of the genes and TSRs annotated in mycelia, young (48-h-old) spherules, and old (8-day-old) spherules. (B) Alignment of the major transcription start site (TSS) of mycelial TSRs as defined by csRNA-seq with the C. immitis genome annotation. (C) The average focus ratio of C. immitis mycelial TSRs as defined by the fraction of reads initiating within 5 bp of the primary TSS in the region versus the surrounding ±75-bp region as a whole suggests widespread dispersed transcription in C. immitis. (D) Example of a focused TSR that initiates on a single or several closely spaced nucleotides and a dispersed TSR with multiple TSSs. Download FIG S2, EPS file, 1.2 MB (1.2MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Features of C. immitis transcription start regions (TSRs) across mycelial and spherule stages. (A) Comparison of the percentages of TSRs with stable transcripts reveals a decrease in the percentage of TSRs associated with stable transcripts in spherules. (B) The directionality of transcription initiation in C. immitis TSRs is predominantly bidirectional. (C) Comparison of the focused ratio as defined by the fraction of reads initiating within 5 bp of the primary transcription start site (TSS) in the region versus the surrounding ±75-bp region as a whole suggests widespread dispersed transcription across all stages of C. immitis. (D) The percentage of promoter-distal TSRs highlights the relative enrichment of distal unstable transcripts in the spherules over mycelia. (E) Overview of TSR features across diverse species. Download FIG S3, EPS file, 0.9 MB (971.6KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To place these findings into a broader concept, we next profiled other fungi, Saccharomyces cerevisiae, Schizosaccharomyces pombe, and champignon mushroom (Agaricus bisporus), and integrated published data from the mold Neurospora crassa and humans (15). Integrated, these data reveal that the patterns of transcription initiation in Coccidioides mycelia share many properties with those of other fungi, including bidirectionally transcribed regulatory elements, multiple initiation sites per promoter, and limited promoter-distal initiation (Fig. 1E and F; Fig. S3). The transition to Coccidioides spherules, on the other hand, is associated with a massive increase in the numbers of TSRs and unstable RNAs and a higher percentage of TSRs located at promoter-distal locations.
Phase transition in C. immitis is accompanied by large changes in transcription programs.
To gain insights into the genes differentially expressed during phase transition, we next compared RNA-seq data from mycelia to those from spherules, which identified 1,930 downregulated and 1,750 upregulated genes in mature spherules (8 days old) and 1,699 downregulated and 1,183 upregulated genes in young spherules (48 h old) (log2 fold change [Log2FC], >1; false discovery rate [FDR], <5%) (Fig. 2A). The majority of differentially regulated genes in spherules were identified at both 48 h and 8 days (Fig. 2A). Similarly, the transition from mycelia to spherules was associated with massive changes in transcription initiation, as determined by csRNA-seq. In 48-h- or 8-day-old spherules, 11,768 or 12,752 TSRs were differentially upregulated and 5,536 or 5,573 TSRs were downregulated compared with mycelia, respectively (Log2FC, >1; FDR, <0.05) (Fig. 2B), and the majority (>71%) of differentially regulated TSRs were shared among both spherule stages. Together, these findings suggest a sharp transition in transcriptional regulation from mycelia to spherules, while changes are less profound as spherules develop endospores.
FIG 2.

Phase transition in C. immitis is accompanied by large changes in transcription programs and connected cis-regulatory domains. (A and B) Scatterplot and quantitative bar graph of differentially expressed (Log2FC, >1; FDR, <0.05) nonredundant transcripts as captured by total RNA-seq (A) and csRNA-seq (B). Note the difference in scale between RNA-seq and csRNA-seq. (C) Example of a part of a connected cis-regulatory domain (20 kb) activated in spherules with novel intergenic transcription start regions (TSRs) of both stable (*) and unstable (#) transcripts. (D) Genome-wide overview of TSRs or repeats per 50 kb showing an increase in spherule-specific cis-regulatory domains in regions that lack genes in the current annotation (ASM14933v2) and often are enriched for repetitive DNA. C. immitis RS supercontig (SC) 3.1-3.6 are shown.
Spherule-specific promoter-distal TSR activity is concentrated in specific genomic regions.
To obtain more insights into the phase-specific gene regulation that drives pathogenesis, we first inspected the ∼28% of mycelial TSRs that are distal of currently annotated promoters (annotation ASM14933v2). Many of these TSRs are associated with transcripts detected by RNA-seq, suggesting that they may mark novel gene loci. In contrast, 49% of spherule TSRs are promoter distal. While some of these distal TSRs also associate with RNA-seq and are thus likely novel gene loci (Fig. 2C, “*”), 34%, or 3,316, of the distal intergenic TSRs have low levels of, or no, associated stable RNA (Fig. 2C, “#”). These promoter-distal elements have some features that resemble those of unstable enhancer RNAs (eRNAs) that are produced at metazoan enhancers (33). Furthermore, these intergenic TSRs are three times more likely to be located in the vicinity of genes upregulated in spherules (based on the distance to the closest promoter) than in the vicinity of those upregulated in mycelia (640 versus 220, >2-fold change; FDR < 0.05). To determine the locations where TSR activity changed between the mycelial and spherule stages, we compared the genomic locations of all annotated TSSs with those of differentially regulated TSRs. In contrast to mycelium-specific TSRs that were most commonly found at or near annotated TSSs, spherule-specific TSRs were frequently found in regions that lack gene annotations (Fig. 2D). In addition to being “gene-poor” regions, these locations were also enriched for repetitive elements, primarily long terminal repeat (LTR) retrotransposons (Copia and Gypsy types) (Fig. 2D) (34). The morphological transition to spherules is thus associated with a significant increase in total TSRs, the majority of which are promoter distal and give rise to rapidly degraded RNAs. Intriguingly, these “enhancer-like” TSRs appear to be coregulated and form cis-regulatory domains with spherule-specific genes, similar to a phenomenon thus far found largely in mammals (35, 36).
Phase-specific TSR switching as a potential means to alter gene expression.
csRNA-seq defines the start sites of transcripts at single-nucleotide resolution and can thus reveal genes that are expressed in both mycelia and spherules but initiate from different TSRs, leading to distinct 5′ mRNA isoforms and occasionally alternative exon usage (Fig. S4A). As such, we identified 99 transcribed annotated genes where the primary TSR in mycelia and 8-day-old spherules was separated by more than 100 bp (∼1% of annotated transcripts) (Fig. S4B). Alternative TSRs were usually specific to phase transition: only seven 5′ TSRs were different among the 48-h- and 8-day-old spherules (Fig. S4C). Shifting the TSR upstream from mycelia to spherules but not vice versa led to a significant loss of transcription, suggesting a role for TSR switching in gene regulation (Fig. S4D and E), as previously reported for Histoplasma capsulatum (37). These findings propose alternative promoter usage as a notable regulatory mechanism during spherule formation.
Phase-specific transcription start region (TSR) switching is associated with changes in gene expression. (A) Example browser shots for TSR switching among different morphological states of C. immitis. (B) Overview of TSR shifts that moved upstream in mycelia or spherules in percentages. These switches were further subgrouped into those that just extend the 5′ untranslated region (UTR) and those that lead to a gain of one or more exons (Exon). (C) Bar plot of the numbers of genes expressed that switch their TSRs upstream or downstream among mycelia or young and old spherules. (D) Comparison of gene expression levels of TSRs that moved upstream or downstream in mycelia or spherules revealed a significant reduction in RNA expression levels upon 5′-UTR extension in spherules but not mycelia. (E) Model for how TSR switching impacts the expression of many but not all genes. Download FIG S4, TIF file, 1.9 MB (1.9MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
cis-regulatory sequences and transcription initiation in C. immitis.
While our refined analysis of the C. immitis transcriptome is fundamental to research, disease diagnostics and, eventually, new treatments require molecular targets and the identification of the pathways critical for pathogenesis. We therefore next investigated enriched sequence patterns proximal to csRNA-seq-defined TSSs. As a quality control, we first assessed the nucleotide frequencies in proximity to transcribed TSSs that, consistent with most other eukaryotic species, revealed a strong preference for the initiator motif “YCA(+1)” (Fig. 3A) (38, 39). The lack of a clear TATA box signature suggested that C. immitis, in agreement with its phylogeny (40), utilizes the mode of “scanning initiation” that is specific to some yeasts and has been elegantly characterized (41, 42) (Fig. S5A).
FIG 3.

HSE and WOPR are phase-specific TF motifs in mycelia and spherules. (A) Sequence logo and nucleotide frequency plot of the primary transcription start site of all TSRs common to mycelia and spherules. (B) A motif resembling the HSE motif is particularly enriched in mycelium-specific genes. (C) A motif resembling the WOPR motif is especially enriched in spherule- but not mycelium-specific genes.
cis-regulatory sequences mediating general and phase-specific transcriptional regulation in C. immitis. (A) The position of the TATA box and other top 10 motifs most strongly enriched in active TSRs across evolution suggests that the assembly of the RNA polymerase II complex and the site of transcription initiation can be spatially separated in C. immitis, a mode of transcription that is commonly referred to as “scanning initiation.” (B) Overview of the top six DNA sequence elements and their predicted transcription factors enriched in TSRs active in both mycelia and spherules. Their relative positions and abundances are differentially plotted for TSRs shared among mycelia and spherules and those specific to either stage. (C) The TATA box is enriched in both mycelium and spherule stage-specific TSRs over ubiquitously expressed ones. (D) Coenrichment analysis of the 11 most prominent TSR motifs in C. immitis reveals that WOR1 frequently cooccurs with the TATA box but is mutually exclusive with the mycelium-specific HSE motif. Download FIG S5, EPS file, 1.7 MB (1.7MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To further identify putative regulatory sequences in an unbiased manner, we next applied de novo motif discovery (HOMER [43]) to TSRs from positions −150 to +50 relative to the primary TSS. Using sequence-content-matched regions of random genomic sequence as a control, our analysis revealed six motifs that were most prevalent in TSRs active in both mycelia and spherules, including the E-box motif (basic helix-loop-helix [bHLH]), CRE/TRE (bZIP), and the yeast family-specific RIM101 and STP3 TFs (Fig. S5B). Together, these motifs are likely critical for maintaining gene expression programs in both morphological states of C. immitis.
cis-regulatory sequences mediating phase-specific transcriptional regulation in C. immitis.
To probe gene regulatory differences in mycelia and the pathogenetic spherule stages, we next probed the stage-specific TSRs for enriched motifs. Consistent with previous findings that report the TATA box as being linked to regulated rather than constitutively active genes (44, 45), we found that the TATA box was enriched in stage-specific TSRs and depleted at ubiquitous TSRs in both mycelia and spherules (Fig. S5C). Furthermore, we identified two DNA motifs that were highly enriched at either mycelium- or spherule-specific TSRs. The single mycelium-specific motif was a heat shock response element-like (HSE) motif (Fig. 3B) (46). The single motif that was highly enriched in spherule-specific but not mycelial TSRs was the WOPR motif (Fig. 3C). Consistently, the HSE and WOPR motifs were largely mutually exclusive (Fig. S5D). Intriguingly, the WOPR motif was enriched from positions −100 to −50 relative to the TSS, resembling the distribution of an activator, and WOPR transcription factors are known fungus-specific master regulators of morphological changes and virulence (24, 25, 47).
A C. immitis Ryp1 orthologue can drive gene expression using the WOPR motif.
To further explore the mechanisms of spherule stage-specific gene expression, we searched the C. immitis genome for putative WOPR TFs. Protein BLAST analysis of white-opaque regulator (Wor1) from Candida albicans (CaWor1) identified C. immitis CIMG_02671 as the likely homologue (NCBI BLASTp E value of 7e−33). Given the limitations associated with C. immitis as a BSL3 pathogen that hinder experimental approaches such as genome editing, we next modeled the CIMG_02671 structure and superimposed it onto available crystal structures from 2 WOPR family members, Wor1 from C. albicans and YHR177w from S. cerevisiae (ScYHR177w) (24, 48). This approach demonstrated similar structures and positionings of key amino acids, including those critical for WOPR DNA motif binding (Fig. 4A and B; Fig. S6A and B). Furthermore, the amino acids reported to be required for Wor1-dependent white-to-opaque morphology switching in C. albicans or mutations in YHR177w that disrupt its binding to DNA containing the WOPR motif were highly conserved across WOPR TFs and CIMG_02671 (Fig. 4C). We therefore next cloned CIMG_02671 and tested its ability to activate transcription in a WOPR motif-dependent manner using a reporter system in S. cerevisiae. An intact or mutated WOPR motif was cloned upstream of the Cyc1 promoter (Pcyc1) that lacked an upstream activating site (UAS-less), driving a β-galactosidase (LacZ) reporter (Fig. 4D). We then cotransformed this Pcyc1 LacZ reporter vector with plasmids expressing the positive control H. capsulatum Ryp1 (HcRyp1) or CIMG_02671 into S. cerevisiae and measured reporter expression. As reported previously and as shown in Fig. 4E, Ryp1 potently induced β-galactosidase expression when the WOPR motif was intact but not when it was mutated (25). Similarly, CIMG_02671 induced β-galactosidase expression in a WOPR motif-dependent manner (Fig. 4E). Collectively, these results demonstrate that CIMG_02671 resembles other WOPR TFs at conserved and functionally important amino acid residues and can activate transcription in a WOPR motif-dependent manner.
FIG 4.
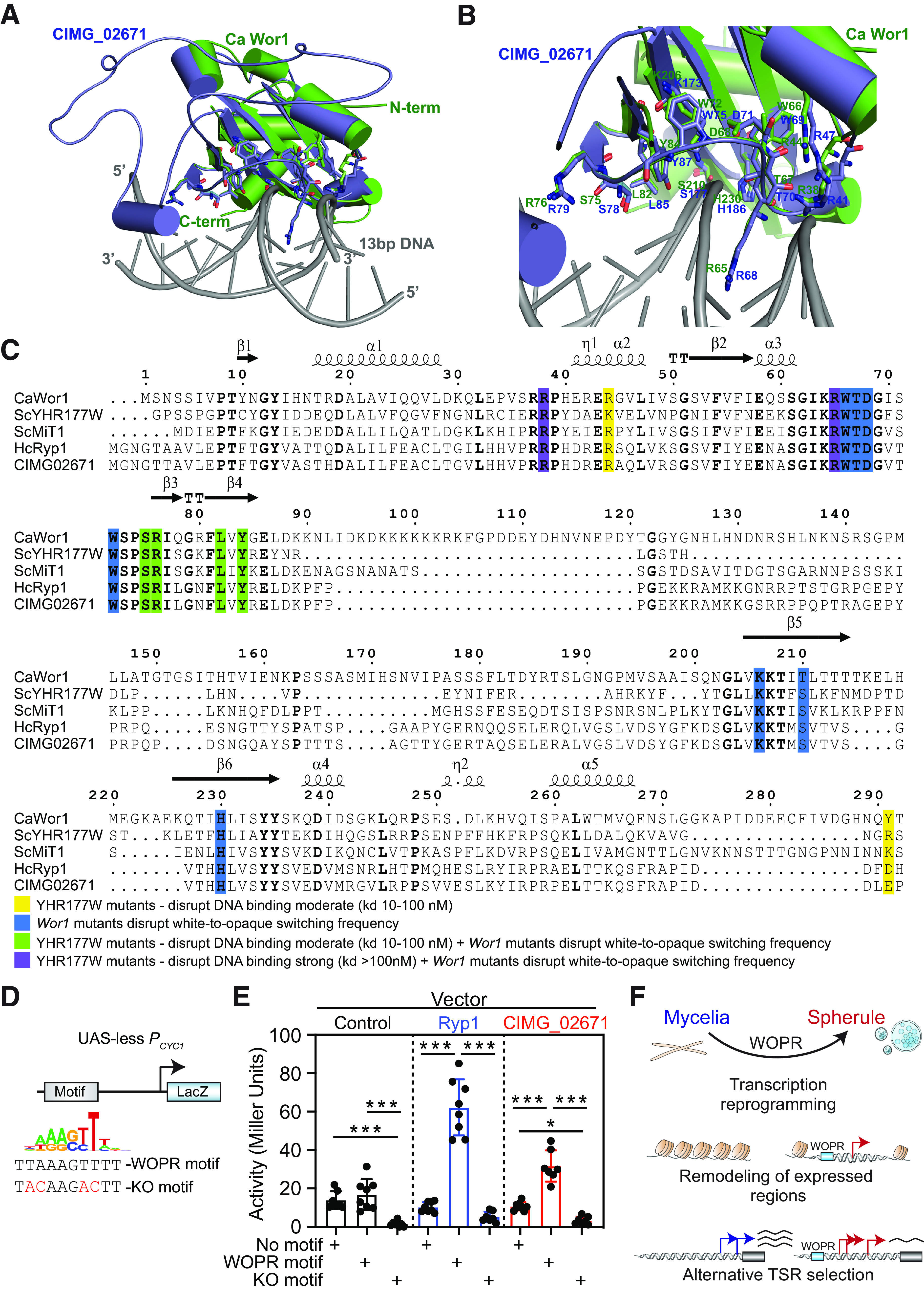
C. immitis CIMG_02671 has WOPR TF-like activity. (A and B) Superimposed model of CIMG_02671 (blue) with the Wor1 crystal structure from C. albicans (green) complexed with 13-bp DNA (gray) containing the WOPR motif (A) and detailed view of the DNA binding domain identifying amino acids that when mutated disrupt white-to-opaque morphology switching in C. albicans (B). (C) Structure-based sequence alignment of the WOPR TFs Wor1 from C. albicans, YHR177w from S. cerevisiae, Mit1 from S. cerevisiae, Ryp1 from H. capsulatum, and CIMG_02671 revealed conservation of the key amino acids required for WOPR TF DNA binding or white-to-opaque morphology switching in C. albicans. The amino acid numbering and the secondary structures of CaWor1 are marked at the top of the alignment. Analogous functionally relevant residues are highlighted in shaded boxes of different colors, as described at the bottom of the sequence alignment. kd, dissociation constant. (D) Overview of the reporter plasmid with the Cyc1 promoter (Pcyc1) driving a β-galactosidase (LacZ) and the wild-type and mutant WOPR (knockout [KO] motif) motifs utilized. (E) Reporter activity of the plasmid from panel D with variations of no motif (control DNA), the WOPR motif, or the mutant WOPR motif (KO motif) cotransfected into S. cerevisiae with either a control vector (empty), H. capsulatum Ryp1, or CIMG_02671 revealed WOPR motif-dependent activation by both WOPR TFs Ryp1 and CIMG_02671. (F) Model for the transcriptional regulatory mechanisms underlying C. immitis phase transition.
The DNA binding domain of WOPR transcription factors is highly conserved across fungi. Shown are superimposed crystal structures (A) with a detailed view of the DNA binding domain of the two WOPR family members, Wor1 from C. albicans and YHR177w from S. cerevisiae (24, 48) (B). Download FIG S6, TIF file, 2.6 MB (2.6MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
Here, we exploit the advance of csRNA-seq to capture active (nascent) TSSs from >500 ng of total RNA instead of intact nuclei or cells as required by other nascent methods (16). This key advantage makes csRNA-seq readily applicable to pathogenic or biohazardous samples, including fungi and others where morphological constraints like cell walls can hinder nucleus isolation. The use of total RNA as the input also greatly facilitates the analysis of pathogens that replicate or reside in the cytoplasm (49). As such, the approach used here to deepen our understanding of the fundamental transcription regulatory mechanisms of C. immitis, the causative agent of valley fever, should also be directly translatable to other infectious diseases.
Using inactivated BSL3 samples from the mycelial and spherule phases of C. immitis and performing csRNA-seq on the isolated RNA identified the WOPR motif as being highly enriched in the promoters and putative enhancer elements of spherule-specific genes. Using homology, we identified CIMG_02671 and showed that it can activate transcription in a WOPR motif-dependent manner. These findings comprise the first comprehensive description and a model for the gene regulatory programs underlying phase transition in the valley fever pathogen (Fig. 4F).
csRNA-seq identified >11,000 TSRs in C. immitis mycelia that were primarily promoter proximal. In contrast, spherule transition nearly doubled the number of TSRs and substantially increased the fraction of promoter-distal TSRs, many of which were associated with rapidly degraded transcripts. These findings were consistent among independent biological replicates. In some cases, spherule-specific transcription activation was found spanning a genomic region that included both putative novel genes and enhancer-like TSRs rather than individual genes. This finding implies a role of alternative regulatory mechanisms beyond cis-regulatory promoters in spherule maturation. Eukaryotic chromosomes exhibit domains of correlated gene expression where gene positioning influences the activation or silencing of transcription (35, 36). Ascomycota fungi, of which Coccidioides spp. are members, are known to cluster into functionally related gene families (50). In our data, the spherule-specific activation of promoter-distal TSRs was enriched near spherule-specific genes. Thus, the transcriptional activation of spherule-specific genomic domains, rather than individual gene promoters, may be a key mechanism by which genes involved in spherule maturation are coordinately regulated.
Spherule-specific TSRs were further highly enriched for the WOPR motif. WOPR TFs regulate morphological changes and pathogenesis in diverse fungal species, including the human pathogens C. albicans (24) and H. capsulatum (26). We further found CIMG_02671, which contains a conserved WOPR binding domain, to activate transcription in a WOPR motif-dependent manner. Intriguingly, unlike both RYP1 and WOR1 WOPR TFs that are preferentially expressed with their activity in yeast or opaque cells, respectively (26, 27), CIMG_02671 is not transcriptionally upregulated in 48-h- or 8-day-old spherules compared to mycelia. CIMG_02671 activity is thus likely differently regulated compared to WOR1 and RYP1. One potential mechanism could include TSR switching. We found that significantly more TSRs in spherules shifted 5′, which resulted in transcription and, potentially, translation activation (37, 51). These more active, alternative TSRs commonly contained a WOPR motif and invite speculation that alternative TSR selection and, thus, mRNAs could be a means to regulate gene expression between fungal differentiation states. In support of the importance of CIMG_02671 in valley fever pathogenesis, a recent preprint found that knocking out Coccidioides posadasii CPSG_00528, which has 100% identity to CIMG_02671, blocked spherule maturation under spherule-promoting conditions and was avirulent in the mouse model of coccidioidomycosis (52). Collectively, these findings propose CIMG_02671 as a WOPR TF that is important for spherule morphogenesis and C. immitis pathogenesis.
In conclusion, our findings provide insights into the fundamental transcriptional programs underlying the phase transition of C. immitis and highlight regulatory domains, TSR switching, and WOPR transcription factors as being central to valley fever pathogenesis. We hope that these findings and the data generated will provide a resource and stepping-stone to combat the increasing disease incidence and expanding geographic range of valley fever. On a broader scope, this study also provides a proof of principle for the utility of csRNA-seq as a novel approach to reveal key candidate transcription factors and regulatory programs underlying the pathogenesis of infectious or biohazardous agents under standard laboratory conditions.
MATERIALS AND METHODS
Culture conditions.
The C. immitis RS strain was grown as mycelia or spherules as previously described (28). To grow mycelia, 2 × 106 arthroconidia/mL were incubated in 250-mL flat-bottom Erlenmeyer flasks (Corning) in 50 mL glucose-yeast extract (GYE) medium. Flasks were cultured in a 30°C incubator without shaking for 5 days. To grow spherules, arthroconidia were washed 2 times in modified Converse medium (53). The spores were inoculated at 4 × 106 arthroconidia/mL into a 250-mL baffled Erlenmeyer flask containing 50 mL of modified Converse medium. Flasks were set up and grown on a shaker at 160 rpm in 14% CO2 at 42°C. Four flasks were harvested 2 days after inoculation, and the remaining four flasks were harvested after 8 days. Fresh Converse medium was not added. The spherules did not rupture and release endospores within that time in this culture system. Saccharomyces cerevisiae (strain RYH2863) was grown as described previously (54). Schizosaccharomyces pombe (strain TH972) was generously provided by Tony Hunter (Salk Institute for Biological Sciences) and grown in yeast extract with supplements (YES) (55). White and brown ecotypes of Agaricus bisporus, better known as “champignon mushroom,” common mushroom, or “crimini mushroom,” were kindly provided by Monterey Mushroom farms.
RNA extraction and purification.
C. immitis mycelium and spherule samples were stored in QIAzol (Qiagen) at −70°C and processed as previously described (9). Samples were added to a 2 mL ZR BashingBead lysis tube with 0.5 mm beads (Zymo Research), and tubes were arranged in a precooled TissueLyser II adapter (Qiagen) and disrupted by shaking at 50 Hz for 25 min. QIAzol samples were spun at 21,000 × g for 5 min at 4°C, and the supernatant was transferred to a fresh tube. Total RNA was purified from mycelium and spherule samples (2 replicates/condition) using chloroform extraction and isopropanol precipitation and quantified using a Qubit 3.0 fluorometer (Invitrogen). RNA from white and brown Agaricus bisporus mushrooms was isolated as described previously (22), using TRIzol LS extraction following tissue homogenization. Libraries were generated for each ecotype separately and then pooled for the analysis. RNA from S. cerevisiae and S. pombe was isolated by resuspending a pelleted culture in TRIzol LS. Next, silica beads were added, and cells were lysed with a multivortexor, with 1 min on and 1 min off, on ice for a total lysis time of 6 min. Samples were spun at 21,000 × g for 5 min at 4°C, and the supernatant was transferred to a fresh tube, followed by TRIzol LS RNA purification as described by the manufacturer.
RNA and csRNA sequencing.
For RNA-seq, strand-specific, paired-end libraries were prepared from total RNA by ribosomal depletion using the yeast Ribo-Zero rRNA removal kit (Illumina) and then using the TruSeq stranded total RNA-seq kit (Illumina) according to the manufacturer’s instructions. Next, 100 bases were sequenced from both ends using a NovaSeq 6000 system according to the manufacturer’s instructions (Illumina).
csRNA-seq was performed as described previously (15). Small RNAs (sRNAs) of ∼20 to 60 nucleotides (nt) were size selected from 0.4 to 2 μg of total RNA by denaturing gel electrophoresis. A 10% input sample was taken aside, and the remainder was enriched for 5′-capped RNAs. Monophosphorylated RNAs were selectively degraded by Terminator 5′-phosphate-dependent exonuclease (Lucigen), and RNAs were 5′ dephosphorylated by quickCIP (New England BioLabs [NEB]). Input (sRNA) and csRNA-seq libraries were prepared as described previously (22) using RppH (NEB) and the NEBNext small RNA library prep kit, amplified for 14 cycles, and sequenced for single end 75 base pair reads (SE75) on the Illumina NextSeq 500 system.
WOPR transcription factor structure-based sequence alignment and modeling.
Structure-based sequence alignment was performed with CIMG_02671 and known WOPR family transcriptional regulators such as Candida albicans white-opaque regulator 1 (Wor1) (CaWor1), ScYHR177w, ScMiT1, and HcRyp1 using ESPript (56). To understand the DNA binding properties of CIMG_02671, we performed automated protein structure homology modeling using SWISS-MODEL (57). To perform protein homology modeling, we provided a truncated input sequence (positions 1 to 270) of CIMG_02671 (UniProt accession number J3KLV5_COCIM J3KLV5 Camp-independent regulatory protein; https://www.uniprot.org/uniprot/J3KLV5) and used the crystal structure of the WOPR family member YHR177w of S. cerevisiae with the 19-bp double-stranded DNA (dsDNA) (PDB accession number 4M8B) as the template. This was one of the top-ranked template searches for the resulting model of CIMG_02671, with a top global model quality estimation (GMQE) value of 0.4. Model building was carried out using the ScYHR177w crystal structure (PDB accession number 4M8B) as the template, and a three-dimensional (3D) model was automatically generated by the target-template alignment. The quality of the generated model was evaluated by global and local quality estimates, by Ramachandran plots, and by the qualitative model energy analysis (QMEAN) value for the different geometric properties for a single model. The final CIMG_02671 model contains amino acids 11 to 223. To analyze its potential DNA binding properties, the CIMG homology model was aligned with the crystal structure of the complex of CaWor1–13-bp DNA using PyMOL (http://www.pymol.org).
Data analysis. (i) RNA-seq.
Sequencing reads were aligned to the genome using STAR with default parameters (58). Genomes and their gene annotation files (GTFs) were downloaded from Ensembl and include C. immitis (ASM14933v2), S. cerevisiae (R64-1-1/sacCer3), S. pombe (ASM294v2), A. bisporus (gca_000300555/Agabi_varbur_1), N. crassa (NC12), Drosophila melanogaster (BDGP6), and human (GRCh38/hg38). Reference-guided transcript assembly was performed using StringTie2 (32) with the additional parameters “-m 100 –rf.” The assembled transcripts were compared to the existing C. immitis annotation using cuffcompare from the Cufflinks suite (59). Gene expression was determined by counting the number of overlapping reads per gene using HOMER’s analyzeRepeats.pl tool, considering only reads with a single, unique alignment (mapping quality score [MAPQ] value of ≥10) for all downstream analyses. DESeq2 was used to identify differentially expressed genes (60).
(ii) csRNA-seq.
Sequencing reads were trimmed for 3′ adapter sequences using HOMER (“homerTools trim -3 AGATCGGAAGAGCACACGTCT -mis 2 -minMatchLength 4 -min 20”) and aligned to the appropriate genome using STAR with default parameters (58). Only reads with a single, unique alignment (MAPQ value of ≥10) were considered in the downstream analysis. Furthermore, reads with spliced or soft-clipped alignments were discarded. Transcription start regions (TSRs), representing loci with significant transcription initiation activity (i.e., “peaks” in csRNA-seq), were defined using HOMER’s findcsRNATSS.pl tool, which uses short input RNA-seq, traditional total RNA-seq, and annotated gene locations to eliminate loci with csRNA-seq signals arising from noninitiating, high-abundance RNAs that nonetheless are captured and sequenced by the method (see reference 15 for more details). Replicate experiments were first pooled to form meta-experiments for each condition prior to identifying TSRs. Annotation information, including gene assignments and promoter-distal, stable transcript, and bidirectional annotations, are provided by findcsRNATSS.pl. Stable TSSs were defined as TSS clusters containing at least 2 per 107 RNA-seq reads within positions −100 to +500 relative to the TSS (15). To identify differentially regulated TSRs, TSRs identified under each condition were first pooled (union) to identify a combined set of TSRs represented in the data set using HOMER’s mergePeaks tool, using the option “-strand.” The resulting combined TSRs were then quantified across all individual replicate samples by counting the 5′ ends of reads aligned at each TSR on the correct strand. The raw read count table was then analyzed using DESeq2 to identify differentially regulated TSRs (60). Normalized genome browser visualization tracks were generated using HOMER and visualized using IGV (61).
(iii) Sequence/motif analysis.
Known motif enrichment and de novo motif discovery were performed using HOMER’s (43) findMotifsGenome.pl tool with default parameters using TSR sequences from positions −150 to +50 relative to the primary TSS. When performing de novo motif discovery, TSR sequences were compared to a background set of 50,000 random genomic regions matched for overall GC content. Nucleotide frequency and motif density plots were created using HOMER’s annotatePeaks.pl tool (43).
(iv) In vivo transcriptional assays.
For the construction of plasmids used in the in vivo transcriptional assay, CIMG_02671 was amplified from C. immitis RS genomic DNA (gDNA) using oligonucleotides 5′-GCACTAGTATGGGTAACGGCACTACAGC-3′ and 5′-GCGTCGACCTATTGCCCTCCGTAGCTTCC-3′. The amplified fragment was digested with SpeI and SalI and ligated into a similarly digested p414TEF vector (62). The resulting plasmid (pSB474) was maintained in the Escherichia coli DH5α strain and sequenced to ensure that no mutation was introduced during the cloning process. pSB474, p414TEF (empty vector), and the previously generated plasmid pSB94 (carrying H. capsulatum Ryp1) (25) were transformed into previously constructed S. cerevisiae Δmit1 Δyhr177w strains carrying p228 (PCYC1-ΔUAS-lacZ::WOPR motif [5′-AAAAATTAAAGTTTTTTTAT-3′]), p230 (PCYC1-ΔUAS-lacZ::WOPR knockout [5′-AAAAATACAAGACTTTTTAT-3′]), and an empty vector control (PCYC1-ΔUAS-lacZ) (25, 63). Four independent isolates of each S. cerevisiae strain were stored and used in β-galactosidase assays, which were performed as previously described (64). Each isolate was assayed in duplicate at least three independent times.
(v) 5′ switch analysis.
To identify genes with major changes in isoform usage due to changes in TSR promoter usage, we first merged the de novo-assembled transcripts found using StringTie2 with cuffmerge into a single transcript set. We then identified the TSR from spherules (8 days old) or mycelia with the most reads that overlapped on the correct strand in each gene locus (allowing the TSR to be up to 200 bp upstream of the 5′ end). The difference in the positions of the top spherule and mycelial TSRs was recorded for each gene, and the mature transcript level expressed in each stage was estimated using the RNA-seq data by counting the fragments per kilobase per million (FPKM) levels from the upstream TSR to the downstream TSR (uFPKM) and then from the downstream TSR to the end of the gene (dFPKM). These values were then filtered by the distance between TSRs (100 bp to 1,000 bp), by the level of expression (FPKM of >5 in mycelia or spherules), and to ensure basal expression in both groups (FPKM of >2 in both mycelia and spherules). To identify alternative TSRs leading to differential starting positions in RNA-seq, the log2 ratios of uFPKM and dFPKM were calculated for both mycelia and spherules with the addition of a small pseudocount {log2[(uFPKM + 3)/(dFPKM + 3)]}. Genes with log2 fold changes of greater than 1 or less than −1 between spherule and mycelial upstream-to-downstream ratios were identified (n = 159 comparing mycelia with 8-day-old spherules). Each of these loci was visually evaluated using the IGV browser to confirm 2 distinct TSRs and different RNA-seq starting positions associated with the alternative TSRs (n = 99 visually confirmed).
Data availability.
All raw and processed csRNA-seq data generated for this study can be accessed at the NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE179468. Previously published Neurospora crassa RNA-seq and csRNA-seq data can be accessed under GEO accession number GSE135498. Previously published Coccidioides immitis RNA-seq data can be accessed under GEO accession number GSE171286.
Overview of generated and analyzed data (GEO submission). Download Table S1, DOCX file, 0.02 MB (17.2KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
ACKNOWLEDGMENTS
This work was supported by NIH grant K08AI130381 and a career award for medical scientists from the Burroughs Wellcome Fund to A.F.C. as well as NIH grants K99GM135515 to S.D., R01AI137418 to S.B., and U19AI135972 and GM134366 to C.B. These studies were also supported by NIH grant 1R35GM133565-01 and Pew Biomedical Award to S.N. A.F.C., S.V., J.F., J.E.S., and T.N.K. were supported by VFR-19-633952, Investigating Fundamental Gaps in Valley Fever Knowledge. S.V., J.F., J.E.S., and T.N.K. were supported by the MRP-17-454959, UC Valley Fever Research Initiative. J.E.S. is a CIFAR fellow in the program Fungal Kingdom: Threats & Opportunities. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 system that was purchased with funding from a National Institutes of Health SIG grant (number S10 OD026929).
We are grateful to Tony Hunter (Salk Institute) for the generous donation of Schizosaccharomyces pombe, Randy Hampton (UC San Diego) for Saccharomyces cerevisiae, and the Monterey Mushroom farms for champignons (Agaricus bisporus).
We declare no competing interests.
Contributor Information
Aaron F. Carlin, Email: acarlin@health.ucsd.edu.
Sergio Baranzini, University of California, San Francisco.
REFERENCES
- 1.Graupmann-Kuzma A, Valentine BA, Shubitz LF, Dial SM, Watrous B, Tornquist SJ. 2008. Coccidioidomycosis in dogs and cats: a review. J Am Anim Hosp Assoc 44:226–235. doi: 10.5326/0440226. [DOI] [PubMed] [Google Scholar]
- 2.Hector RF, Laniado-Laborin R. 2005. Coccidioidomycosis—a fungal disease of the Americas. PLoS Med 2:e2. doi: 10.1371/journal.pmed.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen C, Barker BM, Hoover S, Nix DE, Ampel NM, Frelinger JA, Orbach MJ, Galgiani JN. 2013. Recent advances in our understanding of the environmental, epidemiological, immunological, and clinical dimensions of coccidioidomycosis. Clin Microbiol Rev 26:505–525. doi: 10.1128/CMR.00005-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorris ME, Neumann JE, Kinney PL, Sheahan M, Sarofim MC. 2021. Economic valuation of coccidioidomycosis (valley fever) projections in the United States in response to climate change. Weather Clim Soc 13:107–123. doi: 10.1175/WCAS-D-20-0036.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirkland TN, Fierer J. 2018. Coccidioides immitis and posadasii; a review of their biology, genomics, pathogenesis, and host immunity. Virulence 9:1426–1435. doi: 10.1080/21505594.2018.1509667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang DC, Anderson S, Wannemuehler K, Engelthaler DM, Erhart L, Sunenshine RH, Burwell LA, Park BJ. 2008. Testing for coccidioidomycosis among patients with community-acquired pneumonia. Emerg Infect Dis 14:1053–1059. doi: 10.3201/eid1407.070832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCotter OZ, Benedict K, Engelthaler DM, Komatsu K, Lucas KD, Mohle-Boetani JC, Oltean H, Vugia D, Chiller TM, Sondermeyer Cooksey GL, Nguyen A, Roe CC, Wheeler C, Sunenshine R. 2019. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol 57:S30–S40. doi: 10.1093/mmy/myy095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaki AN. 2010. Biosafety and biosecurity measures: management of biosafety level 3 facilities. Int J Antimicrob Agents 36(Suppl 1):S70–S74. doi: 10.1016/j.ijantimicag.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 9.Carlin AF, Beyhan S, Peña JF, Stajich JE, Viriyakosol S, Fierer J, Kirkland TN. 2021. Transcriptional analysis of Coccidioides immitis mycelia and spherules by RNA sequencing. J Fungi (Basel) 7:366. doi: 10.3390/jof7050366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whiston E, Zhang Wise H, Sharpton TJ, Jui G, Cole GT, Taylor JW. 2012. Comparative transcriptomics of the saprobic and parasitic growth phases in Coccidioides spp. PLoS One 7:e41034. doi: 10.1371/journal.pone.0041034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mead HL, Roe CC, Higgins Keppler EA, Van Dyke MCC, Laux KL, Funke AL, Miller KJ, Bean HD, Sahl JW, Barker BM. 2020. Defining critical genes during spherule remodeling and endospore development in the fungal pathogen, Coccidioides posadasii. Front Genet 11:483. doi: 10.3389/fgene.2020.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanley JP, Tu HA, Dragon JA, Dickson DM, Rio-Guerra RD, Tighe SW, Eckstrom KM, Selig N, Scarpino SV, Whitehead SS, Durbin AP, Pierce KK, Kirkpatrick BD, Rizzo DM, Frietze S, Diehl SA. 2021. Immunotranscriptomic profiling the acute and clearance phases of a human challenge dengue virus serotype 2 infection model. Nat Commun 12:3054. doi: 10.1038/s41467-021-22930-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matranga CB, Andersen KG, Winnicki S, Busby M, Gladden AD, Tewhey R, Stremlau M, Berlin A, Gire SK, England E, Moses LM, Mikkelsen TS, Odia I, Ehiane PE, Folarin O, Goba A, Kahn SH, Grant DS, Honko A, Hensley L, Happi C, Garry RF, Malboeuf CM, Birren BW, Gnirke A, Levin JZ, Sabeti PC. 2014. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol 15:519. doi: 10.1186/PREACCEPT-1698056557139770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duttke SH, Chang MW, Heinz S, Benner C. 2019. Identification and dynamic quantification of regulatory elements using total RNA. Genome Res 29:1836–1846. doi: 10.1101/gr.253492.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wissink EM, Vihervaara A, Tippens ND, Lis JT. 2019. Nascent RNA analyses: tracking transcription and its regulation. Nat Rev Genet 20:705–723. doi: 10.1038/s41576-019-0159-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. 2011. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, Glass CK, Rosenfeld MG, Fu X-D. 2011. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duttke SHC, Lacadie SA, Ibrahim MM, Glass CK, Corcoran DL, Benner C, Heinz S, Kadonaga JT, Ohler U. 2015. Human promoters are intrinsically directional. Mol Cell 57:674–684. doi: 10.1016/j.molcel.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim J-Y, Duttke SH, Baker TS, Lee J, Gambino KJ, Venturini NJ, Ho JSY, Zheng S, Fstkchyan YS, Pillai V, Fajgenbaum DC, Marazzi I, Benner C, Byun M. 2021. DNMT3A haploinsufficiency causes dichotomous DNA methylation defects at enhancers in mature human immune cells. J Exp Med 218:e20202733. doi: 10.1084/jem.20202733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwak H, Fuda NJ, Core LJ, Lis JT. 2013. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339:950–953. doi: 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hetzel J, Duttke SH, Benner C, Chory J. 2016. Nascent RNA sequencing reveals distinct features in plant transcription. Proc Natl Acad Sci USA 113:12316–12321. doi: 10.1073/pnas.1603217113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Rubio R, de Oliveira HC, Rivera J, Trevijano-Contador N. 2019. The fungal cell wall: Candida, Cryptococcus, and Aspergillus species. Front Microbiol 10:2993. doi: 10.3389/fmicb.2019.02993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lohse MB, Rosenberg OS, Cox JS, Stroud RM, Finer-Moore JS, Johnson AD. 2014. Structure of a new DNA-binding domain which regulates pathogenesis in a wide variety of fungi. Proc Natl Acad Sci USA 111:10404–10410. doi: 10.1073/pnas.1410110111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beyhan S, Gutierrez M, Voorhies M, Sil A. 2013. A temperature-responsive network links cell shape and virulence traits in a primary fungal pathogen. PLoS Biol 11:e1001614. doi: 10.1371/journal.pbio.1001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen VQ, Sil A. 2008. Temperature-induced switch to the pathogenic yeast form of Histoplasma capsulatum requires Ryp1, a conserved transcriptional regulator. Proc Natl Acad Sci USA 105:4880–4885. doi: 10.1073/pnas.0710448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zordan RE, Galgoczy DJ, Johnson AD. 2006. Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc Natl Acad Sci USA 103:12807–12812. doi: 10.1073/pnas.0605138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viriyakosol S, Singhania A, Fierer J, Goldberg J, Kirkland TN, Woelk CH. 2013. Gene expression in human fungal pathogen Coccidioides immitis changes as arthroconidia differentiate into spherules and mature. BMC Microbiol 13:121. doi: 10.1186/1471-2180-13-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cole GT, Hung CY. 2001. The parasitic cell wall of Coccidioides immitis. Med Mycol 39(Suppl 1):31–40. doi: 10.1080/744118874. [DOI] [PubMed] [Google Scholar]
- 30.Shamie I, Duttke SH, Karottki KJLC, Han CZ, Hansen AH, Hefzi H, Xiong K, Li S, Roth SJ, Tao J, Lee GM, Glass CK, Kildegaard HF, Benner C, Lewis NE. 2021. A Chinese hamster transcription start site atlas that enables targeted editing of CHO cells. NAR Genom Bioinform 3:lqab061. doi: 10.1093/nargab/lqab061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steward CA, Parker APJ, Minassian BA, Sisodiya SM, Frankish A, Harrow J. 2017. Genome annotation for clinical genomic diagnostics: strengths and weaknesses. Genome Med 9:49. doi: 10.1186/s13073-017-0441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, Pertea M. 2019. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20:278. doi: 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirkland TN, Muszewska A, Stajich JE. 2018. Analysis of transposable elements in Coccidioides species. J Fungi (Basel) 4:13. doi: 10.3390/jof4010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, Fonseca GJ, Troutman TD, Spann NJ, Strid T, Sakai M, Yu M, Hu R, Fang R, Metzler D, Ren B, Glass CK. 2018. Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell 173:1796–1809.e17. doi: 10.1016/j.cell.2018.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen BA, Mitra RD, Hughes JD, Church GM. 2000. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat Genet 26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- 37.Gilmore SA, Voorhies M, Gebhart D, Sil A. 2015. Genome-wide reprogramming of transcript architecture by temperature specifies the developmental states of the human pathogen Histoplasma. PLoS Genet 11:e1005395. doi: 10.1371/journal.pgen.1005395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmoll M, Dattenböck C (ed). 2016. Gene expression systems in fungi: advancements and applications. Springer, New York, NY. [Google Scholar]
- 39.Ngoc LV, Cassidy CJ, Huang CY, Duttke SHC, Kadonaga JT. 2017. The human initiator is a distinct and abundant element that is precisely positioned in focused core promoters. Genes Dev 31:6–11. doi: 10.1101/gad.293837.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Z, Lin Z. 2021. The origin and evolution of a distinct mechanism of transcription initiation in yeasts. Genome Res 31:51–63. doi: 10.1101/gr.264325.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuehner JN, Brow DA. 2006. Quantitative analysis of in vivo initiator selection by yeast RNA polymerase II supports a scanning model. J Biol Chem 281:14119–14128. doi: 10.1074/jbc.M601937200. [DOI] [PubMed] [Google Scholar]
- 42.Fishburn J, Hahn S. 2012. Architecture of the yeast RNA polymerase II open complex and regulation of activity by TFIIF. Mol Cell Biol 32:12–25. doi: 10.1128/MCB.06242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, Stark A. 2015. Enhancer–core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518:556–559. doi: 10.1038/nature13994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basehoar AD, Zanton SJ, Pugh BF. 2004. Identification and distinct regulation of yeast TATA box-containing genes. Cell 116:699–709. doi: 10.1016/s0092-8674(04)00205-3. [DOI] [PubMed] [Google Scholar]
- 46.Rabindran SK, Giorgi G, Clos J, Wu C. 1991. Molecular cloning and expression of a human heat shock factor, HSF1. Proc Natl Acad Sci USA 88:6906–6910. doi: 10.1073/pnas.88.16.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tollot M, Assmann D, Becker C, Altmüller J, Dutheil JY, Wegner C-E, Kahmann R. 2016. The WOPR protein Ros1 is a master regulator of sporogenesis and late effector gene expression in the maize pathogen Ustilago maydis. PLoS Pathog 12:e1005697. doi: 10.1371/journal.ppat.1005697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang S, Zhang T, Yan M, Ding J, Chen J. 2014. Crystal structure of the WOPR-DNA complex and implications for Wor1 function in white-opaque switching of Candida albicans. Cell Res 24:1108–1120. doi: 10.1038/cr.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.den Boon JA, Diaz A, Ahlquist P. 2010. Cytoplasmic viral replication complexes. Cell Host Microbe 8:77–85. doi: 10.1016/j.chom.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hagee D, Abu Hardan A, Botero J, Arnone JT. 2020. Genomic clustering within functionally related gene families in fungi. Comput Struct Biotechnol J 18:3267–3277. doi: 10.1016/j.csbj.2020.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rojas-Duran MF, Gilbert WV. 2012. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA 18:2299–2305. doi: 10.1261/rna.035865.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mandel MA, Beyhan S, Voorhies M, Shubitz LF, Galgiani JN, Orbach MJ, Sil A. 2021. The WOPR family protein Ryp1 is a key regulator of gene expression, development, and virulence in the thermally dimorphic fungal pathogen Coccidioides posadasii. bioRxiv https://www.biorxiv.org/content/10.1101/2021.07.26.453804v1. [DOI] [PMC free article] [PubMed]
- 53.Converse JL. 1956. Effect of physico-chemical environment of spherulation of Coccidioides immitis in a chemically defined medium. J Bacteriol 72:784–792. doi: 10.1128/jb.72.6.784-792.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neal S, Jaeger PA, Duttke SH, Benner C, Glass CK, Ideker T, Hampton RY. 2018. The Dfm1 Derlin is required for ERAD retrotranslocation of integral membrane proteins. Mol Cell 69:915. doi: 10.1016/j.molcel.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 55.Leverson JD, Huang H-K, Forsburg SL, Hunter T. 2002. The Schizosaccharomyces pombe aurora-related kinase Ark1 interacts with the inner centromere protein Pic1 and mediates chromosome segregation and cytokinesis. Mol Biol Cell 13:1132–1143. doi: 10.1091/mbc.01-07-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gouet P, Robert X, Courcelle E. 2003. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mumberg D, Müller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 63.Lohse MB, Zordan RE, Cain CW, Johnson AD. 2010. Distinct class of DNA-binding domains is exemplified by a master regulator of phenotypic switching in Candida albicans. Proc Natl Acad Sci USA 107:14105–14110. doi: 10.1073/pnas.1005911107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reynolds A, Lundblad V, Dorris D, Keaveney M. 2001. Yeast vectors and assays for expression of cloned genes. Curr Protoc Mol Biol Chapter 13:Unit 13.6. doi: 10.1002/0471142727.mb1306s39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reproducibility and correlation of generated biological replicates. (A and B) Scatterplots of replicates for RNA-seq (A) and csRNA-seq (B). (C) Comparison of csRNA-seq reads at gene promoters to RNA-seq reads across genes demonstrated that changes in initiation and gene expression were correlated across phases (r = 0.63). Download FIG S1, TIF file, 2.7 MB (2.7MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Features of C. immitis mycelium transcription start regions (TSRs). (A) Overview of the genes and TSRs annotated in mycelia, young (48-h-old) spherules, and old (8-day-old) spherules. (B) Alignment of the major transcription start site (TSS) of mycelial TSRs as defined by csRNA-seq with the C. immitis genome annotation. (C) The average focus ratio of C. immitis mycelial TSRs as defined by the fraction of reads initiating within 5 bp of the primary TSS in the region versus the surrounding ±75-bp region as a whole suggests widespread dispersed transcription in C. immitis. (D) Example of a focused TSR that initiates on a single or several closely spaced nucleotides and a dispersed TSR with multiple TSSs. Download FIG S2, EPS file, 1.2 MB (1.2MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Features of C. immitis transcription start regions (TSRs) across mycelial and spherule stages. (A) Comparison of the percentages of TSRs with stable transcripts reveals a decrease in the percentage of TSRs associated with stable transcripts in spherules. (B) The directionality of transcription initiation in C. immitis TSRs is predominantly bidirectional. (C) Comparison of the focused ratio as defined by the fraction of reads initiating within 5 bp of the primary transcription start site (TSS) in the region versus the surrounding ±75-bp region as a whole suggests widespread dispersed transcription across all stages of C. immitis. (D) The percentage of promoter-distal TSRs highlights the relative enrichment of distal unstable transcripts in the spherules over mycelia. (E) Overview of TSR features across diverse species. Download FIG S3, EPS file, 0.9 MB (971.6KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phase-specific transcription start region (TSR) switching is associated with changes in gene expression. (A) Example browser shots for TSR switching among different morphological states of C. immitis. (B) Overview of TSR shifts that moved upstream in mycelia or spherules in percentages. These switches were further subgrouped into those that just extend the 5′ untranslated region (UTR) and those that lead to a gain of one or more exons (Exon). (C) Bar plot of the numbers of genes expressed that switch their TSRs upstream or downstream among mycelia or young and old spherules. (D) Comparison of gene expression levels of TSRs that moved upstream or downstream in mycelia or spherules revealed a significant reduction in RNA expression levels upon 5′-UTR extension in spherules but not mycelia. (E) Model for how TSR switching impacts the expression of many but not all genes. Download FIG S4, TIF file, 1.9 MB (1.9MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
cis-regulatory sequences mediating general and phase-specific transcriptional regulation in C. immitis. (A) The position of the TATA box and other top 10 motifs most strongly enriched in active TSRs across evolution suggests that the assembly of the RNA polymerase II complex and the site of transcription initiation can be spatially separated in C. immitis, a mode of transcription that is commonly referred to as “scanning initiation.” (B) Overview of the top six DNA sequence elements and their predicted transcription factors enriched in TSRs active in both mycelia and spherules. Their relative positions and abundances are differentially plotted for TSRs shared among mycelia and spherules and those specific to either stage. (C) The TATA box is enriched in both mycelium and spherule stage-specific TSRs over ubiquitously expressed ones. (D) Coenrichment analysis of the 11 most prominent TSR motifs in C. immitis reveals that WOR1 frequently cooccurs with the TATA box but is mutually exclusive with the mycelium-specific HSE motif. Download FIG S5, EPS file, 1.7 MB (1.7MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The DNA binding domain of WOPR transcription factors is highly conserved across fungi. Shown are superimposed crystal structures (A) with a detailed view of the DNA binding domain of the two WOPR family members, Wor1 from C. albicans and YHR177w from S. cerevisiae (24, 48) (B). Download FIG S6, TIF file, 2.6 MB (2.6MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Overview of generated and analyzed data (GEO submission). Download Table S1, DOCX file, 0.02 MB (17.2KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data Availability Statement
All raw and processed csRNA-seq data generated for this study can be accessed at the NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE179468. Previously published Neurospora crassa RNA-seq and csRNA-seq data can be accessed under GEO accession number GSE135498. Previously published Coccidioides immitis RNA-seq data can be accessed under GEO accession number GSE171286.
Overview of generated and analyzed data (GEO submission). Download Table S1, DOCX file, 0.02 MB (17.2KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.