

Abstract
The influence of base type, temperature, and solvent on regioselective C(9)/C(10) “click” modifications within the tropolone ring of colchiceine (2) is investigated. New ether derivatives of 2, bearing alkyne, azide, vinyl, or halide aryl groups enable assembly of the alkaloid part with heterocycles or important biomolecules such as saccharides, geldanamycin or AZT into hybrid scaffolds by dipolar cycloaddition (CuAAC) or Heck reaction. Compared to colchicine (1) or colchiceine (2), ether congeners, as e.g. 3e [IC50s(3e) ∼ 0.9 nM], show improved or similar anticancer effects, whereby the bulkiness of the substituents and the substitution pattern of the tropolone proved to be essential. Biological studies reveal that expanding the ether arms by terminal basic heterocycles as quinoline or pyridine, decreases the toxicity in HDF cells at high anticancer potency (IC50s ∼ 1–2 nM). Docking of ether and hybrid derivatives into the colchicine pocket of αGTP/β tubulin dimers reveals a relationship between the favourable binding mode and the attractive anticancer potency.
Keywords: Colchiceine tautomers, click, heck reaction, tubulin inhibitors, anticancer
Graphical Abstract

Introduction
Colchicine (1, Figure 1), showing anticancer and other useful biological effects1,2, is a natural tropolone alkaloid, and as the other natural tropolones, is produced by autumn crocus (Colchicum autumnale)3–9. Its metabolite called colchiceine (2, Figure 1) also exhibits, albeit lower, anticancer activity, at the expense of increased antifungal properties, as compared to 110. In the structure of 2, due to a possible rotation around the single bond C(1a)–C(12a), two diastereomeric forms can exist, similarly as for 1 (Figure 1). Furthermore, H-bonding between the C=O and OH groups within the tropolone of 2 contributes to an equilibrium between the C(9)-OH and C(10)-OH keto-enol tautomeric forms (Figure 1)11. Asymmetric total syntheses of colchiceine, β-lumicolchicine and allocolchicinoid derivative were performed by Liu et al.12 In order to improve anticancer potency and to decrease the toxic effects of colchicine, its transformations were mainly performed at C(10), C(7), and C(4) or via destruction of the tropolone ring13–22. Another type of modifications of colchiceine scaffold was the formation of an extra ring, fused with the C-ring of the parent alkaloid via different approaches23–29. The synthetic challenges of regioselective functionalization of hydroxyl group within the tropolone of 2 and the other troponoid systems were undertaken in the past, however with different outcomes11,30.
Figure 1.
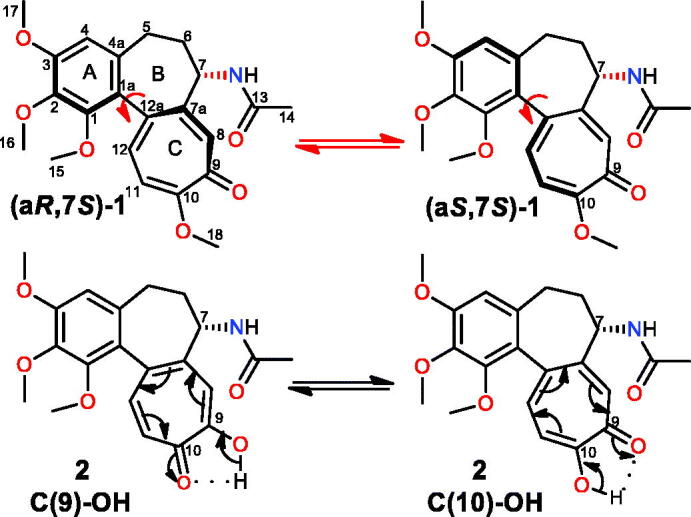
Structures of colchicine (1) and colchiceine (2) together with the atom numbering. Atropisomerization (top) and tautomerization (bottom) processes within colchicine (1) and colchiceine (2) scaffolds, respectively.
Hybrids of bioactive compounds of lower cytotoxicity towards normal cells, serve as drug delivery platforms, prodrugs, adjuvants, molecular probes, or agents active against drug-resistant cancer cell lines, parasites, or bacteria22,31–37. Colchiceine hybrids have been studied relatively rarely up to now. The amide-alkyl-ester bridge at C(7) was used to conjugate 1 with cobalamine in order to obtain tumour-targeted cytotoxin, whereas the presence of an amide linkage with a disulphide bond at C(7) enabled the formation of the thiocolchicine-podophyllotoxin hybrid38,39. Thiocolchicine conjugates bearing at C(7) long polyamide-lactone chains shown to be active in ovarian carcinoma line A2780 at IC50 ∼ 200 nM40. Dipolar cycloaddition and other modern synthetic methods as the Heck reaction yield chemically stable bonds and are worth considering at combining different bioactive blocks into a hybrid scaffold41,42. Dipolar cycloaddition reaction of CuAAC type was performed to obtain triazole-bridged hybrids of 1, but exclusively at the C(7) position43–47. Recently, new colchicine-mimicking quinoline derivatives have been obtained which showed good tubulin polymerisation inhibitory effects as well as antiproliferative potency higher than that of 148. Earlier functionalization of the 1 framework with sulphur substituents at C(10) also suggested the influence of the bulkiness of the thioether arm on the anticancer effects49.
Colchicine interacts with tubulin units mainly via H-bond and hydrophobic interactions involving rings A and C50 and hence the incorporation of new functional arms at alternative C(9) or C(10) positions within the ring C should allow designing derivatives, which fit better to the molecular target. Therefore, here we obtain functionalised colchiceine-ether intermediates at C(9) and C(10) enabling the assembly of new colchicine conjugates, using dipolar cycloaddition of CuAAC type or Heck reactions. It should be mentioned that these intermediates, bearing ether portion at alternative sites of the tropolone ring, can be helpful at designing other-type conjugates in the future. Our regioselective approach to functionalise the colchicine scaffold at C(9)/C(10) allows systematic studying of the influence of substitution pattern of tropolone ring on the anticancer activity.
Results and discussion
Chemistry studies
In order to functionalise 2 towards linkers for the construction of hybrids, we performed SN2-type etherifications of the tropolone hydroxyl group (Figure 2, Tables 1 and 1S; Supplemental Material). Regarding the tautomerization process of 2, the influence of base type, solvent, and substituent structure on the reaction course with the competitive formation of the two 3- or 4-type products (Figure 2) was tested. We focussed first on benzyl bromide as a reactant (Table 1). The most favourable conditions for the formation of the 3-type products were found using the inorganic bases NaH or K2CO3. Under those conditions the privileged formation of C(9)-ether derivative occurred (∼70%). The presence of THF as a solvent contributes to the highest ratio of C(9)/C(10) products (Table 1). The change of the inorganic base into the organic one (MTBD) in THF evokes the lack of regioselectivity because a nearly equimolar mixture of 3 with 4 was formed. A similar result was obtained when acetonitrile, acetone, and DMF were used as solvents. Favourable formation of C(10) products [ratio C(9)/C(10) was 30/70] took place when MTBD was dissolved in aromatic-type solvents (xylene or toluene). The use of other organic bases such as TMG, phosphazene-base P1-H, TMGN, or TBD yielded predominantly the 4-type product. The phosphazene base allowed to obtain a similar C(9)/C(10) ratio, as for MTBD, whereas the best regioselectivity towards the formation of the 4-type product (75%) was achieved with TMG. Thus, as indicated above, the use of the inorganic base/THF system is beneficial for the formation of C(9) analogues (of 3-type), whereas application of the organic base/toluene system alters the regioselectivity towards the favourable formation of C(10) analogues (of 4-type). In the next step, the influence of the alkyl bromide structure on the etherification site within 2 was studied (Table 1). With the NaH/THF-DMF system the highest regioselectivity was observed for cinnamyl bromide (up to 77% 3c, Table 1S; Supplemental Material). In turn, the use of propargyl bromide and ethyl bromoacetate limited the regioselectivity of the reaction. The use of the MTBD/toluene system with propargyl bromide and ethyl bromoacetate was quite beneficial (>70% of 4f, 4g) whereas the use of cinnamyl and 4-iodobenzyl bromides led to a less favourable formation of 4-type product (Table S1).
Figure 2.

Regioselective etherification of 2 with different alkyl bromides, performed at C(9) – products 3a–j and at C(10) – products 4a–j of the tropolone.
Table 1.
Comparison of SN2 reaction of 2 with benzyl bromide (BnBr) in different reaction conditions.
| SN2 reactant | Base | Solvent | Time (h) | T (°C) | Yield (%) 3a + 4a | Ratio (%) of 3a:4a |
|---|---|---|---|---|---|---|
| BnBr | NaH | DMF | 4 | 70 | 87.3 | 59.8: 40.2 |
| BnBr | NaH | THF/DMF (1:1) | 4 | 70 | 83.5 | 67.1: 32.9 |
| BnBr | NaH | THF/DMF (1:0.5) | 4 | 70 | 95.1 | 68.3: 31.7 |
| BnBr | K2CO3 | Toluene | 1 | 70 | 99.9 | 54.8: 45.2 |
| BnBr | K2CO3 | THF | 1 | 66 | 78.1 | 66.1: 33.9 |
| BnBr | K2CO3 | Acetone | 4 | 56 | 99.2 | 61.6: 38.4 |
| BnBr | K2CO3 | DMF | 1 | 115 | 97.9 | 56.6: 43.4 |
| BnBr | MTBD | Toluene | 1 | 70 | 99.5 | 28.6: 71.4 |
| BnBr | MTBD | ACN | 1 | 70 | 92.8 | 52.6: 47.4 |
| BnBr | MTBD | Acetone | 4 | 56 | 92.6 | 50.0: 50.0 |
| BnBr | MTBD | THF | 1 | 66 | 94.5 | 50.9: 49.1 |
| BnBr | MTBD | DMF | 1 | 70 | 86.4 | 51.0: 49.0 |
| BnBr | MTBD | Xylene | 1 | 115 | 99.9 | 30.4: 69.6 |
| BnBr | TMG | Toluene | 1 | 115 | 99.1 | 25.3: 74.7 |
| BnBr | P1-H | Toluene | 1 | 70 | 99.1 | 29.8: 70.2 |
| BnBr | TMGN | Toluene | 16 | 70 | 99.9 | 36.0: 64.0 |
| BnBr | TBD | Toluene | 3 | 70 | 93.4 | 37.6: 62.4 |
To explain the observed regioselectivity, DFT calculations (Figure S1) and FT-IR (Figure 2S and 3S) studies were performed. The calculated structures of complexes 2-MTBD with involvement of C(9)-O− or C(10)-O− alkoxylates (Figure 1Sa) showed that significantly enhanced electron density at one of the oxygens occurs/at O(10)−/only when C(10)-O− alkoxylate takes part in a medium strength H-bond with MTBD (distance D…A equal 2.74 Å; angle D-H…A equal 172°). In turn, the interaction between C(9)-O− alkoxylate and MTBD contributes to the almost equal negative partial charge distribution between O(9) and O(10) atoms resulting in the almost equimolar formation of the C(9)- and C(10)-ether products. Moreover, a direct comparison of the energy (E) values for complexes 2-MTBD (Figure S1) reveals that participation of C(9)-alkoxylate is less favourable than C(10)-alkoxylate in interaction with MTBD. Thus, the readily formation of C(10)-ether products 4a–j, in the presence of MTBD, is explained by an increased electron density at oxygen O(10)− within H-bonded complex 2-MTBD. DFT calculations of complexes formed between C(9)-O− or C(10)-O− alkoxylates and Na+ were performed for the octahedral coordination sphere of Na+ (Figure 1Sb). In contrast to the C(10)-O− alkoxylate complex with Na+, an analogous C(9)-O− complex is stabilised involving the oxygen of the acetamide (Figure 1Sb). Greater discrimination in the negative partial charge distribution between oxygens O(9) and O(10) was noted when O(9)− alkoxylate together with the oxygen of the acetamide is coordinated to the Na+ cation. This result explains the preferential formation of C(9)-ether products when the NaH/THF-DMF system was applied. The formation of H-bonded complex between 2 and MTBD is proved by the FT-IR spectra (Figure 2S and 3S). Protonation of the MTBD is reflected in the presence of ν(C = N+) and δ(N+-H) bands at 1624 and 1511 cm−1, respectively. In turn, the broad absorption band at ∼2750 cm−1 confirms the formation of an intermolecular N+-H…−O-C(10) H-bond between MTBD and alkoxylate of 2.
In order to demonstrate the utility of C(9)- or C(10)-ether intermediates towards the formation of conjugates, those decorated with alkyne were subjected to Huisgen dipolar cycloaddition of CuAAC type (Figure 3). Alkyne intermediates 3f and 4f were used with benzyl, saccharide, and nucleoside azides as well as with azide congener of the ansamycin antibiotic – geldanamycin to afford triazole-bridged conjugates 5a–f and 6a–f, respectively (Figure 3). Reactions were performed predominantly in THF/methanol, whereas TBA/H2O was a convenient solvent system for the synthesis of hybrids 5f and 6f. In turn, the earlier obtained 3b, 4b, 3j, and 4j ether products were used for assembling conjugates via Heck reactions (Figure 4). Structures of these hybrids were confirmed by NMR, FT-IR, and HR-MS (see Supplemental Material, exemplary 1H-13C HMBC couplings shown in Figure 4S). Irrespectively on the type of group installed at the colchicine scaffold (4-iodobenzyl, 4-vinylbenzyl) the ether-4-vinylbenzyl bridge was formed (products 7 and 8, Figure 4). When ether-allyl reactants were used, the analogous Heck reactions did not yield the expected product and we observed decomposition of 3d and 4d into compound 2.
Figure 3.

Structure of new colchiceine hybrids with arms at C(9) (5a–5f) and C(10) (6a–6f), obtained via Huisgen dipolar cycloaddition (CuAAC).
Figure 4.

Structure of new colchiceine conjugates with arms at C(9) (7a–7d) and C(10) (8a–8d), obtained via Heck reaction.
Anticancer and toxicity studies of new C(9) and C(10) ether intermediates, triazole and styryl hybrids of 2
Simple ether derivatives with substituents at C(9) 3a–3j and at C(10) 4a–j were tested in four cancer cell lines: SKBR-3, SKOV-3, PC-3, and U-87, and in healthy cell line HDF (Table 2). Analysis of the data in Table 2 shows that ether derivatives with a smaller substituent (3d–3i and 4d–4i) are more active than those containing aromatic ring at the introduced part (3a–c, 3j, and 4a–c, 4j), irrespectively on the substitution site [C(9) or C(10)]. The activities of derivatives with smaller substituents are comparable at nM level to the parent compound colchicine (1, Figure 1), despite their relatively high lipophilicities (ilogP >3, Table 2) and limited solubility in water (< 0.1 mg/mL). Compound 3e, with crotyl substituent at C(9), exhibited the highest anticancer potency (IC50 = 0.94 − 0.98 nM). The activity of compound 3e is even better than that of 1 (IC50 = 1.06 − 1.28 nM) in SKBR-3, SKOV-3, PC-3, whereas 1 and 3e show almost the same potency towards U-87 cancer cell line (IC50 (3e) = 0.95 nM; IC50 (1)= 0.94 nM). For the most biologically desired crotyl moiety at the tropolone ring, the C(9) substitution pattern was more beneficial than the C(10) one. Overall, considering ether derivatives with smaller substituents, substitution at C(9) favours better activity than substitution at C(10), except for those with the attached ester moieties (3g and 4g). It should be mentioned that for derivatives comprising aromatic rings (3a–c, 3j, and 4a–c, 4j) an analogous relationship cannot be postulated. Expanding of C(9)- and C(10)-ether arms of 3f and 4f by the formation of triazole bridges (derivatives of 5 and 6 types, Figure 3), decreases the anticancer potency when compared to 3f and 4f (Table 2). As seen from Table 2, with the increasing bulkiness of the terminal substituent at the triazole portion, the anticancer potency markedly decreases. Hence, the benzyl-triazole hybrids 5a–c and 6a–c, with less bulky ends of the arm, are the most active ones (IC50s = 2.04 − 3.97 nM) among this group derivatives. In contrast to these derivatives, combining of colchiceine with geldanamycin into one scaffold via triazole linkage, resulted in a decreased anticancer activity, when referred to 1, and enhanced potency, when referred to geldanamycin itself52. In turn, the result of the anticancer studies of 7- and 8-type derivatives is quite surprising (Table 2), taking into account the length and bulkiness of substituents introduced by the Heck reaction (Figure 4). Fusion of geldanamycin with colchiceine into one framework (7d and 8d) via E-vinyl bridge was biologically slightly more favourable than via a triazole bridge (5f and 6f). Derivatives, where the E-vinyl arms are terminated with a 2-naphthyl moiety (7a and 8a), do not show anticancer activities close to 1, similarly as was observed for the 7d and 8d. In turn, quinoline derivatives 7c and 8c, which are isostructural with 7a and 8a, showed attractive potencies (IC50 = 1.03 − 1.73 nM), close to potencies of the most active simple ether derivatives 3d–3i and 4d–4i, despite the presence of long and bulky arms at C(9) or C(10). Compound 8c showed a very good anticancer potency together with slightly decreased toxicity when referred to 1 (Table 2). Moreover, heterocyclic hybrids 7b and 8b decorated with pyridine, showed markedly higher activities than the respective naphthyl derivatives 7a and 8a, together with lower toxicities (IC50HDF(7b) = 5.11 nM; IC50HDF (8b) = 4.83 nM) than those of reference alkaloids 1 (IC50HDF(1) = 2.37 nM) or 2 (IC50HDF (2) = 3.57 nM).
Table 2.
Anticancer activities [IC50 (nM) ± SD] of 1, 2, 3a–3j, 4a–4j, 5a–5f, 6a–6f, 7a–7d and 8a–8d in SKBR-3, SKOV-3, PC-3, U-87 cells and toxicity (in Human Dermal Fibroblasts; HDF) [IC50 (nM) ± SD], and clogP, all data compared with those of other type standards as cytarabine (C), actinomycin D (ActD) and mitomycin C (MitC).
| Compd. | SKBR-3 | SKOV-3 | PC-3 | U-87 | HDF | ilogP* |
|---|---|---|---|---|---|---|
| 1 | 1.07 ± 0.05 | 1.28 ± 0.11 | 1.06 ± 0.03 | 0.94 ± 0.02 | 2.37 ± 0.15 | 3.28 |
| 2 | 1.04 ± 0.07 | 1.88 ± 0.02 | 1.37 ± 0.02 | 1.26 ± 0.04 | 3.57 ± 0.83 | 2.88 |
| 3a | 2.04 ± 0.51 | 2.19 ± 0.07 | 2.27 ± 0.04 | 2.83 ± 0.06 | 6.39 ± 1.04 | 3.67 |
| 3b | 2.55 ± 0.12 | 2.13 ± 0.40 | 2.94 ± 0.08 | 2.47 ± 0.91 | 3.94 ± 0.27 | 3.99 |
| 3c | 3.09 ± 0.02 | 3.74 ± 0.18 | 3.11 ± 0.91 | 3.38 ± 0.05 | 5.24 ± 0.26 | 4.02 |
| 3d | 1.09 ± 0.01 | 1.68 ± 0.09 | 1.05 ± 0.01 | 1.63 ± 0.04 | 2.83 ± 0.05 | 3.53 |
| 3e | 0.94 ± 0.06 | 0.98 ± 0.02 | 0.96 ± 0.01 | 0.95 ± 0.01 | 2.13 ± 0.06 | 3.82 |
| 3f | 1.37 ± 0.02 | 1.05 ± 0.07 | 1.05 ± 0.03 | 1.14 ± 0.01 | 2.83 ± 0.44 | 3.31 |
| 3g | 1.31 ± 0.08 | 1.37 ± 0.01 | 1.62 ± 0.29 | 1.41 ± 0.03 | 2.72 ± 0.03 | 3.50 |
| 3h | 1.04 ± 0.33 | 1.99 ± 0.07 | 1.53 ± 0.21 | 1.06 ± 0.03 | 1.84 ± 0.11 | 3.62 |
| 3i | 1.32 ± 0.05 | 1.05 ± 0.02 | 1.43 ± 0.02 | 1.88 ± 0.05 | 2.85 ± 0.13 | 3.90 |
| 3j | 4.19 ± 0.62 | 4.55 ± 0.07 | 4.59 ± 0.03 | 4.55 ± 0.06 | 5.38 ± 0.42 | 3.81 |
| 4a | 2.01 ± 0.02 | 1.63 ± 0.03 | 2.26 ± 0.01 | 2.08 ± 0.01 | 4.12 ± 0.06 | 4.08 |
| 4b | 2.77 ± 0.19 | 2.83 ± 0.26 | 2.41 ± 0.66 | 2.85 ± 0.03 | 4.12 ± 0.01 | 4.10 |
| 4c | 3.46 ± 0.07 | 3.05 ± 0.03 | 3.19 ± 0.01 | 3.82 ± 0.22 | 4.94 ± 0.15 | 4.22 |
| 4d | 1.73 ± 0.11 | 1.73 ± 0.07 | 1.71 ± 0.03 | 1.78 ± 0.03 | 2.39 ± 0.17 | 3.64 |
| 4e | 1.97 ± 0.04 | 1.92 ± 0.11 | 1.98 ± 0.05 | 1.93 ± 0.02 | 2.66 ± 0.03 | 3.89 |
| 4f | 1.48 ± 0.09 | 1.52 ± 0.02 | 1.46 ± 0.13 | 1.06 ± 0.04 | 1.85 ± 0.03 | 3.54 |
| 4g | 1.28 ± 0.17 | 1.06 ± 0.22 | 1.15 ± 0.03 | 1.27 ± 0.01 | 2.05 ± 0.02 | 3.61 |
| 4h | 1.69 ± 0.05 | 1.27 ± 0.16 | 1.22 ± 0.03 | 1.07 ± 0.12 | 2.71 ± 0.08 | 3.85 |
| 4i | 1.38 ± 0.03 | 1.37 ± 0.09 | 1.49 ± 0.16 | 1.19 ± 0.03 | 2.58 ± 0.12 | 3.79 |
| 4j | 3.22 ± 0.36 | 3.01 ± 0.04 | 3.05 ± 0.01 | 3.11 ± 0.03 | 5.17 ± 0.28 | 4.30 |
| 5a | 3.07 ± 0.11 | 3.28 ± 0.06 | 3.21 ± 0.15 | 3.74 ± 0.19 | 5.28 ± 0.22 | 3.65 |
| 5b | 2.07 ± 0.08 | 2.88 ± 0.47 | 2.13 ± 0.02 | 2.57 ± 0.13 | 4.92 ± 0.51 | 3.59 |
| 5c | 2.18 ± 0.02 | 2.94 ± 0.71 | 2.09 ± 0.33 | 2.04 ± 0.08 | 3.12 ± 0.41 | 5.79 |
| 5d | 6.29 ± 0.25 | 6.03 ± 0.19 | 6.39 ± 0.08 | 6.11 ± 0.26 | 7.99 ± 0.04 | 4.42 |
| 5e | 3.71 ± 0.05 | 3.74 ± 0.03 | 3.51 ± 0.09 | 3.05 ± 0.01 | 6.22 ± 0.32 | 3.49 |
| 5f | 8.91 ± 0.17 | 8.05 ± 0.58 | 8.38 ± 0.09 | 8.25 ± 0.11 | 10.36 ± 0.39 | 5.22 |
| 6a | 2.33 ± 0.09 | 2.79 ± 0.07 | 2.04 ± 0.44 | 2.78 ± 0.05 | 3.88 ± 0.14 | 3.70 |
| 6b | 3.06 ± 0.03 | 3.97 ± 0.12 | 3.15 ± 0.04 | 3.74 ± 0.06 | 5.83 ± 0.31 | 3.34 |
| 6c | 2.88 ± 0.06 | 2.42 ± 0.11 | 2.05 ± 0.03 | 2.81 ± 0.18 | 4.08 ± 0.26 | 5.79 |
| 6d | 5.21 ± 0.39 | 5.93 ± 0.12 | 5.04 ± 0.02 | 5.83 ± 0.15 | 7.29 ± 0.41 | 4.20 |
| 6e | 3.02 ± 0.31 | 3.33 ± 0.11 | 3.50 ± 0.49 | 3.84 ± 0.02 | 5.18 ± 0.18 | 3.20 |
| 6f | 17.40 ± 0.64 | 15.29 ± 0.31 | 17.84 ± 0.16 | 17.29 ± 0.35 | 18.03 ± 0.24 | 4.93 |
| 7a | 8.02 ± 0.08 | 8.54 ± 0.71 | 8.61 ± 0.38 | 8.63 ± 0.14 | 8.19 ± 0.92 | 4.86 |
| 7b | 2.09 ± 0.33 | 2.69 ± 0.02 | 2.45 ± 0.52 | 2.43 ± 0.12 | 5.11 ± 0.85 | 4.23 |
| 7c | 1.51 ± 0.02 | 1.03 ± 0.09 | 1.52 ± 0.01 | 1.73 ± 0.09 | 2.16 ± 0.60 | 4.85 |
| 7d | 7.22 ± 0.19 | 7.04 ± 0.15 | 7.49 ± 0.32 | 7.06 ± 0.44 | 9.04 ± 0.29 | 6.36 |
| 8a | 6.30 ± 0.09 | 6.49 ± 0.25 | 6.03 ± 0.26 | 6.44 ± 0.07 | 8.72 ± 0.48 | 5.31 |
| 8b | 2.77 ± 0.04 | 2.84 ± 0.03 | 2.63 ± 0.77 | 2.49 ± 0.05 | 4.83 ± 0.27 | 4.68 |
| 8c | 1.05 ± 0.06 | 1.41 ± 0.02 | 1.47 ± 0.05 | 1.65 ± 0.03 | 2.77 ± 0.25 | 5.07 |
| 8d | 11.37 ± 0.15 | 10.52 ± 0.05 | 10.97 ± 0.44 | 10.84 ± 0.74 | 17.94 ± 1.04 | 6.63 |
| C | 870 ± 10 | 990 ± 110 | 810 ± 10 | 850 ± 31 | 5940 ± 70 | 0.99 |
| ActD | 1140 ± 60 | 1140 ± 10 | 1170 ± 30 | 1610 ± 90 | 2810 ± 150 | 4.04 |
| MitC | 670 ± 10 | 610 ± 20 | 580 ± 110 | 650 ± 40 | 1380 ± 70 | 1.62 |
*-ilogP calculated by SwissADME [51]; C: cytarabine; ActD: actinomycin D; MitC: mitomycin C.
Overall, the toxicity of compounds 3–8 increases together with increasing anticancer effects (Table 2), irrespectively on the substitution pattern C(9)/C(10). Compound 3a showed the most beneficial selectivity index (SI) (e.g. SI ∼3, for SKBR-3). Taking into account the ratio of anticancer activity relative to toxicity, the most interesting derivative is heterocyclic hybrid 8c, since its potency is on the level IC50 ∼1 nM at SI ∼2.6 in SKBR-3 cells. The lowest toxic effect exhibited compound 3f, where its SIs are ∼2.7 for SKOV-3 and PC-3 cancer cell lines. In turn, compound 4h with a relatively small substituent at C(10) and attractive anticancer activity (average IC50 ∼1.5 nM), revealed the most beneficial SI ∼2.5 for U-87 cancer cell line. Unfortunately, the most potent derivative 3e showed the highest toxicity in all studied cancer cell lines at SIs ∼ 2.2. In turn, the most promising derivatives, i.e. of the lowest toxicity in HDF cells and the most anticancer activity in SKOV-3, PC-3, and SKBR-3 cancer cell lines are intermediate 3f and quinoline-based hybrid 8c.
Docking insight into SAR of colchiceine hybrids
To get a deeper insight into the SAR and in order to explain the observed differences in anticancer effects for colchiceine hybrids, docking studies into the binding site of colchicine (1), i.e. between dimeric αGTP/β tubulins, were performed (Table 2S, Figure 5, Figure 5S). Interactions between 1 and dimeric αGTP/β tubulins were also optimised (Figure 5a) for comparison. Compound 1 is stabilised between αGTP/β tubulin units (PDB 1SA0) 53, in the vicinity of the GTP binding site, mainly via hydrophobic interactions in the pocket formed by A180α, V181α, L248β, A250β, K254β, L255β, N258β, M259β, T314β, A316β, V318β, K352β and A354β and I378β and by two H-bonds with S178α (O-H) and C241β (S-H) as well as by a very weak interaction with N-H group of V181 (not marked in Figure 5a). In turn, the methoxy group at C(10) of 1 is stabilised in the binding pocket by hydrophobic interactions with T314β. Docking of the ether derivatives 3e and 4e at tubulin dimers revealed that substitution at C(9) with crotyl moiety is energetic more favourable than at C(10) (Table 2S). Furthermore, the binding energy of 3e is more beneficial than for 1 (by ∼ 10 kcal/mol), which is in line with the observed trend for their anticancer activities. The replacement of the C(10)-methoxy group (in 1) with a crotyloxy group (in 4e) was less favourable for the binding energy with the target (Table 2S). The explanation of this result is a fact that with the changed substitution pattern from C(10) to C(9) one for the crotyloxy substituent (3e), extra stabilising interactions of the ether moiety at the binding pocket are realised, i.e. π-π stacking with the carbonyl group of A180α and hydrophobic contact to T179α. Analogous binding modes and relationship as for 3e and 4e are observed for derivatives with a small substituent as propargyloxy group, whereby differences between binding energies of 3f and 4f are lower (C9ΔH°f – C10ΔH°f = ∼ 2 kcal/mol), which corresponds well with their similar anticancer activities in all studied cancer cell lines. An increase in the length of C(9) or C(10) ether substituents, as for the pairs 3a and 3j and 4a and 4j, contributes to a decrease in binding energy profit with the tubulins and could explain the lower potency of such derivatives. Thus, a general overview of the data collected in Table 2S related to 3- and 4-type derivatives suggests that compounds with less bulky substituents better fit into the pocket of αGTPβ tubulin dimers than those with bigger ether substituents, whereby the C(9) substitution pattern seems to be more energetic favourable than the C(10) one. The worse fitting into the binding pocket within dimeric tubulins is also well reflected by the decreasing distance between the acetamide group (carbon atom of the methyl group) and C(5′) carbon atom of GTP caused by the change in the substitution pattern from C(9) to C(10) in the tropolone (Figures 5,6). An increase in the length and bulkiness of C(9) or C(10) ether-triazole arms, as for compounds 5b and 6b, makes the binding energies to tubulin dimers less favourable, if compared to their synthetic precursors 3f and 4f (Table 2S). Similar results were expected for Heck reaction products of 7- and 8-types, containing lengthy and bulky arms at C(9) or C(10). This is only true for the conjugate with the naphthalene moiety 7a and 8a, for which the lowest binding energy profits and the lowest anticancer effects were together noted (Tables 2 and 2S). Surprisingly, for hybrids 7c and 8c the binding energies to αGTP/β tubulins were close to those calculated for simple ether derivatives with small substituents such as 3f and 4f. This result may explain the high anticancer potency of 7c and 8c compared to those of 3- and 4-types, in the light of similar and high lipophilicities (ilogPs >3.3, Table 2) and limited water solubilities (<0.1 mg/mL) for all of them. The reason for better stabilisation of hybrids 7c and 8c, bearing quinoline-vinylbenzyloxy arms, at the binding pocket of αGTP/β dimer, is the formation of H-bonds between the terminal and protonated quinoline and the phenol group of Y172α (Figure 6c) or the carboxylate of E183α (Figure 6d), respectively. This favourable H-bonding along with hydrophobic stabilisation of the arms between prolines P175α and P184α partially compensate the loss in binding energy profit, due to the fitting of the lengthy and bulky arms of 7c and 8c into the cavity between the tubulin units.
Figure 5.

Docking models for colchiceine and its new derivatives with small ether moieties, attached to C(9) or C(10), at the binding pocket of tubulin dimer αGTP/β (PDB 1SA0) 53: (a) 2-brown; (b) 3e – dark violet, C(9)-substitution pattern (c) 4e – dark violet, C(10)-substitution pattern; (d) 3f – orange, C(9)-substitution pattern; and (e) 4f – orange, C(10)-substitution pattern, optimised via MO-G PM6 semi-empirical method using MOZYME algorithm for huge molecules (Scigress package FJ. 2.6, 3.1.9, 2008–2019) 54. Tubulin units are distinguished by different colours: α-tubulin (green) and its key amino acids (yellow) and β-tubulin (pale blue) and its key amino acids (dark blue) whereas intermolecular interactions between new colchiceine derivatives and the key binding amino acids of αGTP/β dimer are marked by yellow dots.
Figure 6.

Docking models for colchicine (1) and colchiceine hybrids with extended ether arms, attached to C(9) or C(10), at the binding pocket of tubulin dimer αGTP/β (PDB 1SA0)53: (a) 7a – rose, C(9)-substitution pattern; (b) 8a – rose, C(10)-substitution pattern; (c) 7c – violet, C(9)-substitution pattern; and (d) 8c – violet, C(10)-substitution pattern; optimised via MO-G PM6 semi-empirical method using MOZYME algorithm for huge molecules (Scigress package 3.1.9, 2008–2019) 54. Tubulin units are distinguished by different colours: α-tubulin (green) and its key amino acids (yellow) and β-tubulin (pale blue) and its key amino acids (dark blue), intermolecular interactions between new colchiceine derivatives and the key binding amino acids of αGTP/β dimer are marked by yellow dots, distance to C5’ carbon atom of GTP is marked by the red dashed line.
Furthermore, the attractive anticancer potency of 7c and 8c may result from their basic properties reflected also in better water solubility and lower milogP in the protonated forms (milogPneutral = 5.05 and milogPprotonated = 2.51, for 7c and 8c)55. The altered arrangement of the nitrogen of the heterocyclic moiety relative to the vinyl bridge in 7b and 8b, contributes to their lower binding energy profits to the target (ΔH°f), compared to 7c and 8c (Table 2S). Less favourable binding of 7b and 8b with αGTP/β tubulin dimers is partial a result of a weaker H-bond stabilisation of the introduced arms due to unfavourable H-bond angles and distances with the proton acceptors of tubulins, i.e. with the carbonyl group of P173α (Figure 5Sg) or the carboxylate of E183α (Figure 5Sh). Thus, the decreased anticancer activities of 7b and 8b, compared to 7c and 8c, seems to result rather from a weaker H-bonding of the arms 7b and 8b with tubulins, than from their bulkiness (Table 2).
Conclusions
Using various alkyl bromides, the SN2-type etherification at C(9)/C(10) of the tropolone ring of 2 has been performed. Reactivity tests indicated that regioselectivity of etherification/C(9) or C(10)/can be altered using different bases and solvent systems. The use of the inorganic base/THF system favours the formation of C(9)-ether products, whereas application of the organic base/toluene system yielded favourably C(10)-ether products. Dipolar cycloaddition (CuAAC) and Heck reactions using C(9)-ether and C(10)-ether products were performed to obtain structurally diverse hybrids with biological relevant blocks. Among the simple ether intermediates at C(9) or C(10), those with less bulky substituents (3d–3i, 4d–4i) showed the best anticancer properties at IC50s ∼ 1–2 nM, which is a comparable or even better result than that for 1. Comparing the binding modes this-type simple ether derivatives revealed that the presence of less bulky and unsaturated substituents at C(9) increases the binding energy profit with αGTP/β dimeric tubulins due to an extra π-π stacking and hydrophobic contacts, as calculated for 3e, 4e, 3f, and 4f. Overall, the presence of lengthy and bulky substituents within most hybrids, containing triazole or vinyl-benzyl bridges (compounds of 5, 6, 7, and 8 types), destabilises binding with dimeric tubulins αGTP/β and decreases anticancer potency. Exceptions of this relationship are hybrids with basic and terminal heterocycles (7c and 8c) for which beneficial binding mode and an improved water solubility/in protonated forms/contribute to relatively high anticancer effects. It should be mentioned that the toxicities of hybrids 7b and 8b containing pyridine as an end-motif, are lower (IC50s HDF ∼ 4–5 nM) than those of the most active simple ether products and 1, at the attractive anticancer activity (IC50 ∼ 2–2.5 nM).
Supplementary Material
Acknowledgement
The authors wish to thank dr. Joanna Kurek for obtaining and purification of colchiceine samples.
Funding Statement
This work was supported by the Polish National Science Centre (NCN) under Grant Opus 13 no. UMO-2017/25/B/ST5/00291.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Gasparyan AY, Ayvazyan L, Yessirkepov M, Kitas GD.. Colchicine as an anti-inflammatory and cardioprotective agent. Expert Opin Drug Metab Toxicol 2015;11:1781–94. [DOI] [PubMed] [Google Scholar]
- 2.Dasgeb B, Kornreich D, McGuinn K, et al. Colchicine: an ancient drug with novel applications. Br J Dermatol 2018;178:350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morita H, Matsumoto K, Takeya K, Itokawa H.. Conformation of tropolone ring in antileukemic tropoloisoquinoline alkaloids. Chemical & Pharmaceutical Bulletin 1993;41:1478–80. [DOI] [PubMed] [Google Scholar]
- 4.Lee JC, Cha JK.. Total synthesis of tropoloisoquinolines: imerubrine, isoimerubrine, and grandirubrine. J Am Chem Soc 2001;123:3243–6. [DOI] [PubMed] [Google Scholar]
- 5.Shaala LA, Youssef DTA.. Identification and bioactivity of compounds from the fungus penicillium Sp. CYE-87 isolated from a marine tunicate. Mar Drugs 2015;13:1698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo H, Benndorf R, Leichnitz D, et al. Isolation, biosynthesis and chemical modifications of rubterolones A-F: rare tropolone alkaloids from actinomadura sp. 5-2. Chem Eur J 2017;23:9338–45. [DOI] [PubMed] [Google Scholar]
- 7.Carreño MC, Ortega‐Guerra M, Ribagorda M, Sanz‐Cuesta MJ.. Synthesis of 4-Aminotropones from [(Sulfinyl or Sulfonyl)Methyl]-Substituted p-Quinamines. Chem Eur J 2008;14:621–36. [DOI] [PubMed] [Google Scholar]
- 8.Trost BM, McDougall PJ.. Access to a welwitindolinone core using sequential cycloadditions. Org Lett 2009;11:3782–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan Y, Ma Y-T, Yang J, et al. Tropolone ring construction in the biosynthesis of rubrolone B, a cationic tropolone alkaloid from endophytic streptomyces. Org Lett 2016;18:1254–7. [DOI] [PubMed] [Google Scholar]
- 10.Kurek J, Kwaśniewska‐Sip P, Myszkowski K, et al. Antifungal, anticancer, and docking studies of colchiceine complexes with monovalent metal cation salts. Chem Biol Drug Design 2019;94:1930–43. [DOI] [PubMed] [Google Scholar]
- 11.Cavazza M, Pietra F.. Tautomeric selectivity towards colchicinoids in the tosylation of colchiceine on a heterogeneous, easily removable catalyst. Org Biomol Chem 2003;1:3002–3. [DOI] [PubMed] [Google Scholar]
- 12.Liu X, Hu Y-J, Chen B, et al. Asymmetric total syntheses of colchicine, β-lumicolchicine, and allocolchicinoid N-Acetylcolchinol-O-Methyl Ether (NCME). Org Lett 2017;19:4612–5. [DOI] [PubMed] [Google Scholar]
- 13.Singh B, Kumar A, Joshi P, et al. Colchicine derivatives with potent anticancer activity and reduced P-glycoprotein induction liability. Org Biomol Chem 2015;13:5674–89. [DOI] [PubMed] [Google Scholar]
- 14.Majcher U, Klejborowska G, Moshari M, et al. Antiproliferative activity and molecular docking of novel double-modified colchicine derivatives. Cells 2018;7:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghawanmeh AA, Al-Bajalan HM, Mackeen MM, et al. Recent developments on (-)-colchicine derivatives: Synthesis and structure-activity relationship. Eur J Med Chem 2020;185:111788. [DOI] [PubMed] [Google Scholar]
- 16.Czerwonka D, Sobczak S, Maj E, et al. Synthesis and antiproliferative screening of novel analogs of regioselectively demethylated colchicine and thiocolchicine. Molecules 2020;25:1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, Liu X, Hu Y-J, et al. Enantioselective total synthesis of (-)-colchicine, (+)-demecolcinone and metacolchicine: determination of the absolute configurations of the latter two alkaloids . Chem Sci 2017;8:4961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavazza M, Veracini CA, Pietra F.. Covalent reversible binding of alkoxides or thiolates to colchicinoids. J Chem Soc Perkin Trans. 2 1992;2201–4. [Google Scholar]
- 19.Czerwonka D, Sobczak S, Pędziński T, et al. Photoinduced skeletal rearrangement of N-substituted colchicine derivatives. J Org Chem 2021;86:11029–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nurieva EV, Zefirov NA, Temnyakova NS, et al. C(7)-derivatives of colchicine with guanosine and biphenyl moieties: molecular modeling, synthesis, and tubulin clustering effect in cancer cells. Russ Chem Bull 2020;69:2222–7. [Google Scholar]
- 21.Zefirov NA, Evteeva YA, Wobith B, et al. Adamantyl-substituted ligands of colchicine binding site in tubulin: different effects on microtubule network in cancer cells. Struct Chem 2019;30:465–71. [Google Scholar]
- 22.Richter M, Boldescu V, Graf D, et al. Synthesis, biological evaluation, and molecular docking of combretastatin and colchicine derivatives and their HCE1-activated prodrugs as antiviral agents. ChemMedChem 2019;14:469–83. [DOI] [PubMed] [Google Scholar]
- 23.Shchegravina ES, Svirshchevskaya EV, Combes S, et al. Discovery of dihydrofuranoallocolchicinoids – highly potent antimitotic agents with low acute toxicity. Eur J Med Chem 2020;207:112724. [DOI] [PubMed] [Google Scholar]
- 24.Shchegravina ES, Svirshchevskaya EV, Schmalz H-G, Fedorov AY.. A facile synthetic approach to nonracemic substituted pyrrolo-allocolchicinoids starting from natural colchicine. Synthesis 2019;51:1611–22. [Google Scholar]
- 25.Gracheva IA, Svirshchevskaya EV, Faerman VI, et al. Synthesis and antiproliferative properties of bifunctional allocolchicine derivatives. Synthesis 2018;50:2753–60. [Google Scholar]
- 26.Shchegravina ES, Maleev AA, Ignatov SK, et al. Synthesis and biological evaluation of novel non-racemic indole-containing allocolchicinoids. Eur J Med Chem 2017;141:51–60. [DOI] [PubMed] [Google Scholar]
- 27.Gracheva YA, Schmalz H-G, Svirshchevskaya EV, Fedorov AY.. Synthesis of new sulfur-containing derivatives of furanoallocolchicinoids. Russ J Org Chem 2016;52:1137–42. [Google Scholar]
- 28.Shchegravina ES, Knyazev DI, Beletskaya IP, et al. Synthesis of nonracemic pyrrolo-allocolchicinoids exhibiting potent cytotoxic activity: synthesis of nonracemic pyrrolo-allocolchicinoids exhibiting potent cytotoxic activity. Eur. J. Org. Chem 2016;2016:5620–3. [Google Scholar]
- 29.Tkachuk AV, Kurbatov SV, Morozov PG, Borodkin GS.. The first dipolar spirocycle based on 10-(Benzylamino) colchicine. Chem Heterocycl Comp 2015;51:948–50. [Google Scholar]
- 30.Cavazza M, Pietra F.. Fluxional sulfonyl derivatives of troponoids and colchicinoids. Tetrahedron Lett 2003;44:1895–7. [Google Scholar]
- 31.Li S, Zhao H, Fan Y, et al. Design, synthesis, and in vitro antitumor activity of a transferrin receptor-targeted peptide–doxorubicin conjugate. Chem Biol Drug Design 2020;95:58–65. [DOI] [PubMed] [Google Scholar]
- 32.Skiera I, Antoszczak M, Trynda J, et al. Antiproliferative activity of polyether antibiotic-cinchona alkaloid conjugates obtained via click chemistry. Chem Biol Drug Design 2015;86:911–7. [DOI] [PubMed] [Google Scholar]
- 33.Krajčovičová S, Gucký T, Hendrychová D, et al. A stepwise approach for the synthesis of folic acid conjugates with protein kinase inhibitors. J Org Chem 2017;82:13530–41. [DOI] [PubMed] [Google Scholar]
- 34.Domalaon R, Idowu T, Zhanel GG, Schweizer F.. Antibiotic hybrids: the next generation of agents and adjuvants against gram-negative pathogens? Clin Microbiol Rev 2018;31:e00077–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji C, Miller PA, Miller MJ.. Iron transport-mediated drug delivery: practical syntheses and in vitro antibacterial studies of tris-catecholate siderophore-aminopenicillin conjugates reveals selectively potent antipseudomonal activity. J Am Chem Soc 2012;134:9898–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Feng Z, Wu D, et al. Enzyme-regulated supramolecular assemblies of cholesterol conjugates against drug-resistant ovarian cancer cells. J Am Chem Soc 2016;138:10758–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghawanmeh AA, Chong KF, Sarkar SM, et al. Colchicine prodrugs and codrugs: chemistry and bioactivities. Eur J Med Chem 2018;144:229–42. [DOI] [PubMed] [Google Scholar]
- 38.Bagnato JD, Eilers AL, Horton RA, Grissom CB.. Synthesis and characterization of a cobalamin-colchicine conjugate as a novel tumor-targeted cytotoxin . J Org Chem 2004;69:8987–96. [DOI] [PubMed] [Google Scholar]
- 39.Danieli B, Giardini A, Lesma G, et al. Thiocolchicine-podophyllotoxin conjugates: dynamic libraries based on disulfide exchange reaction. J Org Chem 2006;71:2848–53. [DOI] [PubMed] [Google Scholar]
- 40.Bonandi E, Foschi F, Marucci C, et al. Synthesis of thicolchicine-based conjugates: investigation towards bivalent tubulin/microtubules binders. ChemPlusChem 2019;84:98–102. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Sun H, Liu J, et al. Palladium-promoted DNA-compatible heck reaction. Org Lett 2019;21:719–23. [DOI] [PubMed] [Google Scholar]
- 42.Sharma N, Mohanakrishnan D, Shard A, et al. Stilbene–chalcone hybrids: design, synthesis, and evaluation as a new class of antimalarial scaffolds that trigger cell death through stage specific apoptosis. J Med Chem 2012;55:297–311. [DOI] [PubMed] [Google Scholar]
- 43.Malysheva YB, Combes S, Allegro D, et al. Synthesis and biological evaluation of novel anticancer bivalent colchicine-tubulizine hybrids. Bioorg Med Chem 2012;20:4271–8. [DOI] [PubMed] [Google Scholar]
- 44.Thomopoulou P, Sachs J, Teusch N, et al. New colchicine-derived triazoles and their influence on cytotoxicity and microtubule morphology. ACS Med Chem Lett 2016;7:188–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicolaus N, Zapke J, Riesterer P, et al. Azides derived from colchicine and their use in library synthesis: a practical entry to new bioactive derivatives of an old natural drug. ChemMedChem 2010;5:661–5. [DOI] [PubMed] [Google Scholar]
- 46.Kowalczyk K, Błauż A, Ciszewski WM, et al. Colchicine metallocenyl bioconjugates showing high antiproliferative activities against cancer cell lines. Dalton Trans 2017;46:17041–52. [DOI] [PubMed] [Google Scholar]
- 47.Shchegravina ES, Tretiakova DS, Alekseeva AS, et al. Phospholipidic colchicinoids as promising prodrugs incorporated into enzyme-responsive liposomes: chemical, biophysical, and enzymological aspects. Bioconjug Chem 2019;30:1098–113. [DOI] [PubMed] [Google Scholar]
- 48.Hagras M, Deeb MAE, Elzahabi HSA, et al. Discovery of new quinolines as potent colchicine binding site inhibitors: design, synthesis, docking studies, and anti-proliferative evaluation. J Enzyme Inhib Med Chem 2021;36:640–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurek J, Myszkowski K, Okulicz-Kozaryn I, et al. Cytotoxic, analgesic and anti-inflammatory activity of colchicine and its C-10 sulfur containing derivatives. Sci Rep 2021;11:9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharyya B, Panda D, Gupta S, Banerjee M.. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev 2008;28:155–83. [DOI] [PubMed] [Google Scholar]
- 51.SwissADME http://www.Swissadme.Ch/Index.Php.
- 52.Skrzypczak N, Pyta K, Ruszkowski P, et al. Anticancer activity and toxicity of new quaternary ammonium geldanamycin derivative salts and their mixtures with potentiators. J Enzyme Inhib Med Chem 2021;36:1898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ravelli RBG, Gigant B, Curmi PA, et al. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004;428:198–202. [DOI] [PubMed] [Google Scholar]
- 54.Scigress Package FJ 2.6/EU 3.1.9 ./2019; Fujitsu, Japan. [Google Scholar]
- 55.Molinspiration V2018.10 https://www.Molinspiration.Com/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.