

Abstract
Southwest China is a plant diversity hotspot. The near-cosmopolitan genus Ilex (c. 664 spp., Aquifoliaceae) reaches its maximum diversity in this region, with many narrow-range and a few widespread species. Divergent selection on widespread species leads to local adaptation, with consequences for both conservation and utilization, but is counteracted by geneflow. Many Ilex species are utilized as teas, medicines, ornamentals, honey plants, and timber, but variation below the species level is largely uninvestigated. We therefore studied the widespread Ilex polyneura, which occupies most of the elevational range available and is cultivated for its decorative leafless branches with persistent red fruits. We assembled a chromosome-scale genome using approximately 100x whole genome long-read and short-read sequencing combined with Hi-C sequencing. The genome is approximately 727.1 Mb, with a contig N50 size of 5 124 369 bp and a scaffold N50 size of 36 593 620 bp, for which the BUSCO score was 97.6%, and 98.9% of the assembly was anchored to 20 pseudochromosomes. Out of 32 838 genes predicted, 96.9% were assigned functions. Two whole genome duplication events were identified. Using this genome as a reference, we conducted a population genomics study of 112 individuals from 21 populations across the elevation range using restriction site-associated DNA sequencing (RADseq). Most populations clustered into four clades separated by distance and elevation. Selective sweep analyses identified 34 candidate genes potentially under selection at different elevations, with functions related to responses to abiotic and biotic stresses. This first high-quality genome in the Aquifoliales will facilitate the further domestication of the genus.
Introduction
Tropical and subtropical Southwest China is a global hotspot of plant species diversity due to its topographic complexity, relative climatic stability, and large continuous land area at this latitude in Asia [1, 2]. Ilex (Aquifoliaceae), the hollies, is one of the largest and most characteristic woody genera in this region, which supports almost one-third of the known species in this near-cosmopolitan genus [3]. Similar to many plant types, most Ilex species have relatively narrow distributions, with some species known to come from only a single location, while a few are widespread and occupy a range of habitats. Divergent selection in widespread woody species is expected to lead to local adaptation, with consequences for both utilization and conservation, and for predicting responses to climate change [4, 5]. However, local adaptation can be hindered by high levels of gene flow between populations, which is expected in Ilex because of its unspecialized, bee-pollinated flowers, and small, bird-dispersed fruits [3].
Variation below the species level is of more than academic interest in Ilex because many different species throughout the range of the genus have been utilized as teas and medicines, ornamentals, honey plants, timbers, and other minor uses, with a total global market value of billions of US dollars annually [3]. Subspecific variation has been utilized in horticulture for the development of new cultivars and hybrids, but not yet in the huge market for holly teas and medicines. Breeding programs to produce novel ornamental cultivars, caffeine-free teas, or improved medicines would be facilitated by the availability of a high-quality reference genome, which is currently lacking not only for the genus Ilex but also for the entire order, Aquifoliales, to which it belongs.
In this study, we therefore investigated subspecific variation and local adaptation in I. polyneura, a widespread diploid member of the deciduous East Asian clade [3] that occupies most of the elevational range available in the region. It has considerable potential as a source of cut, leafless branches with persistent red fruits, similar to the commercially important Ilex decidua (possumhaw) and I. verticillata (winterberry) in North America and, increasingly, in eastern China. Hong et al. [6] reduced this taxon to a synonym of I. micrococca, but these taxa are usually distinct in our study area, so we followed Flora of China [7] to keep them separate. First, we sequenced and assembled a high-quality chromosome-scale reference genome using PacBio long-read sequencing, Illumina HiSeq sequencing, and Hi-C techniques. Then, using this high-quality genome as a reference, we conducted a population genomics study of 112 individuals collected from 21 populations across the whole elevation range of the species using restriction site-associated DNA sequencing (RADseq). We looked for evidence of a genetic structure that is likely to be the outcome of spatially divergent selection and identified candidate genes potentially involved in adaptation to local differences in elevation.
Materials and methods
DNA extraction and genome sequencing
Young trees of Ilex polyneura were transplanted from a wild population in Xishuangbanna, Yunnan Province, China (N21.5621°, E100.3631°), into the Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences. Fresh plant tissues, including leaves, flowers, fruits, stems, and roots, were collected in the garden from a single female plant, frozen in liquid nitrogen, and then used for total DNA and RNA extractions. After extraction, sequencing libraries with 350 bp insert sizes were constructed using the library construction kit (Illumina, San Diego, CA, USA), and these libraries were then sequenced using an Illumina HiSeq X Ten platform. These short reads were used to assess the genome size and for genome survey and correction. The genome was assembled using whole genome long-read and short-read sequencing combined with Hi-C sequencing.
Genome assembly
The genome size of Ilex polyneura was estimated using flow cytometry with Carica papaya as the reference [8]. After removing the adapters, reads with more than 10% “N” nucleotides, and reads with more than 20% low-quality nucleotides, the clean reads were used for calculating K-mers (K = 17) by SOAPdenovo2 [9] to confirm the sequencing depth and corresponding K-mer count by determining the highest peak value of the frequency curve of the K-mer occurrence distribution. The genome size was then estimated by the K-mer count/sequencing depth.
NextDenovo v2.3.0 (https://github.com/Nextomics/NextDenovo) was applied to correct the single base errors of the PacBio long reads using the Illumina short reads and then to assemble the preliminary genome using default parameters except for minimap2_options_cns = −x ava-pb -t 10 -k17 -w17, seed_cutoff = 32 210, and minimap2_options_raw = −x ava-pb -t 10. The preliminary genome assembly was further polished using NextPolish v1.2.4 [10] and Purge Haplotigs v1.0.4 [11]. Again, default parameters were used except lgs_minimap2_options = −x map-pb and lgs_options = −min_read_len 1 k -max_read_len 100 k -max_depth 100 in NextPolish and -a 84 in Purge Haplotigs. Three methods were used to evaluate the quality of the assembled genome. First, we mapped the Illumina short reads onto the assembled genome using BWA-MEM [12] to check the mapping rate of reads, coverage, and average sequencing depth. Second, we mapped core eukaryotic genes onto the assembled genome using CEGMA (Core Eukaryotic Genes Mapping Approach) [13]. Third, we used the BUSCO (Benchmarking Universal Single-Copy Orthologs) test to evaluate the assembled genome’s quality and searched the assembly for gene content that was conserved among all plants with the Embryophyta_odb9 database based on default parameters [14].
Pseudochromosome construction using hi-C
For the Hi-C experiments, approximately 3 g of fresh young leaves was ground into powder in liquid nitrogen. The Hi-C library was constructed following a previously published protocol for plant tissues [15] with chromatin extraction and digestion and DNA ligation, purification, and fragmentation. We constructed chromosome-level scaffolds of the Ilex polyneura genome using ALLHiC v0.9.8 software [16] with the following parameters: --CLUSTER 20, −-MaxLinkDensity 3, −-shortest 150, −-minREs 50, −-format Sanger, −-enz DpnII, −-longest 800, and --NonInformativeRatio 0. The order and directions of the contigs on the pseudochromosomes were then adjusted by examining their interactions in the Hi-C heatmap. The completeness and quality of the final assembled genome was assessed with BUSCO tests using gene content from the Embryophyta_odb9 database [14].
Repeat annotation
A combination of homology alignment and de novo search was used for repeat annotation. RepeatModeler [17] and LTR_FINDER [18] were used for de novo repeat family identification. RepeatScout [19] was used to build the de novo repeat library, only using repeats longer than 100 bp and with a string of “N” nucleotides less than 5%. The repeat library was then combined with the Repbase library (http://www.girinst.org/repbase/) for repeat detection using RepeatMasker and RepeatProteinMask [17]. Tandem repeats were extracted using TRF [20] by ab initio prediction.
Gene prediction and functional annotation
Protein-coding genes were identified and annotated using a combination of ab initio prediction, homology-based prediction, and RNA-Seq assisted prediction. Different databases for genome annotation have different algorithms for detecting genes or motifs [21]. Additionally, some incompatible transcripts cannot be transferred to different databases [22], some databases include data from other databases (e.g. Pfam shares data with interPro), but others do not [23], and some databases include only completed genome sequences, while others also include genes or proteins without a completed genome [24], so annotations based on different databases give different results. Thus, several databases were used for ab initio prediction here, including Augustus v3.2.3 [25], Geneid v1.4.4 [26], Genescan [27], GlimmerHMM v3.0.4 [28], and SNAP [29]. Sequences of homologous proteins from Helianthus annuus, Lactuca sativa, Handroanthus impetiginosus, Nicotiana sylvestris, and Solanum lycopersicum were downloaded from Ensembl [30] and NCBI (https://www.ncbi.nlm.nih.gov/protein/) for homology-based prediction. These five species are either campanulids with completed genomes or model plant species. Protein sequences were aligned to the assembled Ilex polyneura genome using TblastN v2.2.26 (a maximal E-value of 1e-5), and the matching proteins were then aligned to the homologous genome sequences with GeneWise v2.4.1 [31], which was used to predict the gene structure in each protein region. For RNA-Seq assisted prediction, the RNA-Seq reads from different tissues, including leaves, flowers, fruits, stems, and roots, were aligned to the genome fasta file using TopHat v2.0.11 [32] to identify exon regions and splice positions with the default parameters. These results were used as inputs for Cufflinks v2.2.1 [33] for genome-based transcript assembly, again with default parameters. The nonredundant reference gene set was generated by merging the genes predicted by these three methods with EvidenceModeler (EVM, v1.1.1) using PASA (Program to Assemble Spliced Alignment) terminal exon support and including masked transposable elements as input into the gene prediction [34].
Gene functions were assigned by aligning the protein sequences to those in the Swiss-Prot database using Blastp (with a threshold of a maximal E-value of 1e-5) and choosing the best match. The motifs and domains were annotated using InterProScan (v5.31) by searching against publicly available databases, including Swiss-Prot [35], NCBI (http://www.ncbi.nlm.nih.gov/protein), Pfam [36], KEGG [37], and InterPro [38]. The Gene Ontology (GO) IDs for each gene were assigned according to the corresponding InterPro entry. We predicted the protein functions by transferring annotations from the closest BLAST hits (a maximal E-value of 1e-5) in the Swiss-Prot and NCBI databases. We also mapped the gene set to KEGG pathways and identified the best match for each gene.
Noncoding RNA annotation
The tRNAs were predicted using the program tRNAscan-SE [39]. For rRNAs, which are highly conserved, we used the rRNA sequences of related species in BLAST. Other noncoding RNAs (including miRNAs and snRNAs) were identified by searching against the Rfam database with default parameters using infernal software [40].
Whole-genome duplication (WGD) analysis
First, the collinearity of protein-coding genes within the genome of Ilex polyneura, as well as between I. polyneura genome and those of five species, Daucus carota, H. annuus, L. sativa, Mimulus guttatus, and S. lycopersicum, was identified using BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) and MCscanX with the default parameters [41]. Then, the fourfold degenerate synonymous sites were located, and the 4DTV values were calculated. The WGD events in the I. polyneura genome were determined by plotting the distribution frequency of the 4DTV values.
Positively selected genes in the genome of Ilex polyneura
The single-copy orthologous genes identified above were aligned using MUSCLE v.3.8.31 [42] and then used for calculating the ratios of the nonsynonymous (Ka) and synonymous (Ks) substitution rates using the branch-site model in the program Codeml with the default parameters in the package PAML v4.9 [43]. Ilex polyneura was set as the foreground branch, while five species (S. lycopersicum, D. carota, H. annuus, L. sativa, and M. guttatus) were set as the background branches in the analyses. The positively selected single-copy orthologous genes were identified based on the likelihood ratio test (LRT).
Restriction site-associated DNA sequencing and SNP calling
A total of 112 individuals were collected from 21 populations at the tropical-subtropical transition (21.79–26.99°N) in Yunnan Province, Southwest China, to sample the whole elevational range of I. polyneura (675–2362 m above sea level; Supplementary Table 5). RADseq data were produced from each individual following a previously reported protocol [44]. Approximately 5 g of leaves dried in silica gel per individual was used for genomic DNA extraction. Approximately 200 ng of high-quality genomic DNA per individual was used for RADseq library preparation. Genomic DNA was digested with the restriction enzyme EcoR1 and fragmented randomly. The digested DNA fragments were ligated with an adaptor for Illumina sequencing and a unique barcode. Then, polymerase chain reaction was conducted with the prepared libraries after pooling and random shearing to select the DNA within a certain length range (between 200 and 400 bp). Subsequently, paired-end sequencing (2 x 150 bp) was performed on an Illumina NovaSeq 6000 (Illumina, USA).
The raw paired-end reads for each individual were filtered with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) with default parameters to examine the read quality. Trimmomatic was used to remove the adaptors and bases with low quality (Phred score < 20) at the beginning or end of reads [45]. Filtered reads were mapped with the new genome as a reference using BWA-MEM with default parameters [12]. Sites in repetitive sequences were examined and removed using SAMtools [46]. SNP calling was run in SAMtools [46] and further filtered in GATK [47]. We only kept high-quality SNPs that were biallelic and had a sequencing depth > 5, minor allele frequency > 0.05, and missing call rate < 0.3.
Phylogenetic and genetic structure analyses
TreeBeST v1.9.2 (http://treesoft.sourceforge.net/treebest.shtml) was used for phylogenetic analysis based on the neighbor-joining (NJ) method. Bootstrap values were calculated based on 1000 replications. Principal component analysis (PCA) was conducted based on the filtered SNPs using GCTA [48]. The input file to the tool was produced using the population genotype file in PLINK [49]. The PED file produced by PLINK was also used for population genetic structure analysis in FRAPPE [50]. The best number of clusters (K value) was determined by the lowest cross-validation error rate, which was tested from 2 to 8.
Linkage disequilibrium and selective sweep analyses
Linkage-disequilibrium (LD) decay was calculated based on the squared correlation coefficient (r2) values between pairwise SNPs using PopLDdecay [51]. In the selective sweep analyses, two approaches were used for the identification of genome-wide selective sweeps: estimating the fixation index (Fst) and the nucleotide diversity (π) for 40 kb sliding windows with a 20 kb step size in high-elevation and low-elevation populations using VCFtools v.0.1.14 [52]. Sites over the top 5% threshold of Fst and π were selected for identification of their functions using GO and KEGG pathway analyses.
Results
Genome assembly and construction of pseudochromosomes
We first produced 70.32 Gb of Illumina paired-end short reads. The 17-mer frequency of the Illumina short reads with the highest peak occurred at a depth of 70 (Supplementary Fig. 1). We then estimated the heterozygosity rate and percentage of repeat content, which were 1.18% and 52.95%, respectively. The genome size was estimated to be approximately 729.31 Mb, which is close to the size estimated by flow cytometric analyses (741.48 Mb).
The SMRT sequencing technology generated 120.82 Gb reads. These were corrected and assembled to produce a 727.10 Mb genome with a contig N50 size of 5 124 369 bp, contig N90 size of 575 529 bp, scaffold N50 size of 36 593 620 bp, and scaffold N90 size of 25 719 167 bp (Table 1). The GC content of the assembled genome based on the SMRT reads was 36.08%. The total contig length was 727 102 167 bp, and the number of contigs was 483. The longest contig was 23 338 240 bp. The total scaffold length was 727 144 267 bp, and the number of scaffolds was 62. The longest scaffold was 64 863 080 bp. All three approaches to assessing the completeness of the genome supported high completeness. A total of 97.61% of the short reads could be mapped onto the assembled genome, the average sequencing depth was 90.25, and the coverage of the mapped short reads on the assembled genome was 99.18%. A total of 95.56% of the CEGMA genes were found in the assembled genome. A total of 97.6% of the universal single-copy orthologs in the BUSCO scores (1614 complete orthologs) were detected in the assembled genome (Table 1), suggesting that the I. polyneura genome is nearly complete and of high quality.
Table 1.
Assembly statistics of the Ilex polyneura genome based on PacBio and Hi-C sequencing
| Assembly feature | Value |
|---|---|
| Assembly size (bp) | 718 958 801 |
| GC content (%) | 36.08 |
| Total contig length (bp) | 727 102 167 |
| Number of contigs | 483 |
| Contig N50 size (bp) | 5 124 369 |
| Contig N90 size (bp) | 575 529 |
| Longest contig length (bp) | 23 338 240 |
| Total scaffolds length (bp) | 727 144 267 |
| Number of scaffolds | 62 |
| Scaffold N50 size (bp) | 36 593 620 |
| Scaffold N90 size (bp) | 25 719 167 |
| Longest scaffold length (bp) | 64 863 080 |
| Gap (%) | 3.10 |
| Repeat region % of assembly | 57.62 |
| BUSCO | C:97.6%[S:90.2%,D:7.4%],F:0.9%,M:1.5%,n:1614* |
*C: Complete BUSCO, S: Complete and single-copy BUSCO, D: Complete duplicated BUSCO, F: Fragmented BUSCO, M: Missing BUSCO, and n: Total BUSCO groups searched.
We then anchored all these scaffolds from the SMRT reads to 20 pseudochromosomes using the Hi-C data. The Hi-C technique generated 1.60 Gb raw data, which was filtered to 1.48 Gb clean data for reconstructing physical maps by reordering and clustering the assembled scaffolds. We anchored 98.87% of the assembly onto 20 pseudochromosomes. Chromatin interaction data were used to assess the quality of the Hi-C assembly and showed that it was high (Supplementary Fig. 2). The lengths of the pseudochromosomes ranged from 23 672 964 bp to 64 863 080 bp, with a scaffold N50 length of 36 593 620 bp (Supplementary Table S1).
Repeat and gene annotations
The method combining ab initio and homology-based approaches identified 57.62% of the assembled genome as repetitive sequences, including DNA transposons (1.6%), retrotransposons (50.04%), and other kinds. Long terminal repeat (LTR) retrotransposons accounted for 49.23% of the genome (Table 2; Supplementary Table S2). A total of 32 838 genes were annotated by combining de novo, homology-based, and transcriptome-based approaches (Table 2; Supplementary Tables S3 and S4). The average transcript length, average coding-sequence length, average exon length, and average intron length were 5461.06 bp, 1090.15 bp, 231.91 bp, and 1181.10 bp, respectively. On average, there were 4.70 exons per gene (Table 2). Overall, 31 826 (96.90%) out of the 32 838 genes were assigned functions based on the results of the six protein databases. A total of 23 160 of these genes had homologies in the Swiss-Prot database, while 29 421 and 22 611 had annotated proteins in the NCBI and Pfam databases, respectively. Further functional annotations using InterPro estimated that 30 879 of the genes contained conserved protein domains, and 17 535 of the genes were classified by Gene Ontology (GO) terms, with 23 402 genes mapped to known plant biological pathways based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Supplementary Table S3).
Table 2.
Statistics of gene prediction for the Ilex polyneura genome
| Annotation feature | Value |
|---|---|
| Number of protein-coding genes | 32 838 |
| Number of transcripts | 31 826 |
| Average transcript length (bp) | 5461.06 |
| Average CDS length (bp) | 1090.15 |
| Average exon length (bp) | 231.91 |
| Average intron length (bp) | 1181.10 |
| Average number of exons per gene | 4.70 |
Whole-genome duplication (WGD) analysis
Two peaks were identified in the distribution plot of the 4DTv values in the Ilex polyneura genome, one at 4DTv = 0.30 and the other at 4DTv = 0.66 (Fig. 1). This suggests that the ancestors of the genus Ilex underwent one ancient and one recent WGD (Fig. 1). The later WGD event occurred between the times of two WGD events in the D. carota genome, at 4DTv = 0.24 and 4DTv = 0.39 (Fig. 1). We found two peaks that were shared between I. polyneura and S. lycopersicum (4DTv = 0.42 and 0.66), while only one was shared with M. guttatus (4DTv = 0.48) (Fig. 1), suggesting that I. polyneura and S. lycopersicum shared one WGD event not present in M. guttatus. Three peaks were found in S. lycopersicum (4DTv = 0.30, 0.69, and 0.78), suggesting that S. lycopersicum experienced three WGD events.
Figure 1.
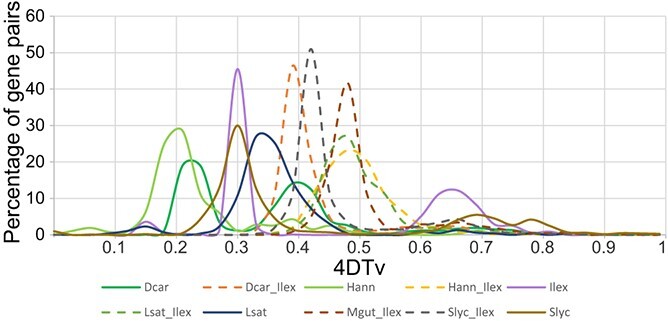
Whole-genome duplication (WGD) events detected in the genomes of Ilex polyneura (Ilex), Daucus carota (Dcar), Helianthus annuus (Hann), Lactuca sativa (Lsat), Mimulus guttatus (Mgut), and Solanum lycopersicum (Slyc), with the distribution of values for the fourfold degenerate sites (4DTV).
Positively selected genes
A total of 178 of the single-copy orthologous genes were identified as positively selected in I. polyneura compared to the other five representative species using the LRT method. Chromosome 1 had the highest number of positively selected genes (16), followed by chromosome 4 (14), chromosome 2 (12), and chromosome 6 (12) (Supplementary Fig. 3). Chromosome 17 had the smallest number (2) (Supplementary Fig. 3). KEGG pathway analysis of the positively selected genes suggested that they were enriched in pathways related to resistance to herbivores and pathogens (e.g. map00240 and map00350) and photosynthesis (e.g. map00710) (Supplementary Table S5). GO enrichment analysis of the positively selected genes suggested that they were enriched in genes related to pollination (e.g. GO:0010183), protein methylation (e.g. GO:0006479), transmembrane transport (e.g. GO:0071918), DNA structure (e.g. GO:0006310), and protein activity (e.g. GO:0004824) (Supplementary Table S6).
Population genetic diversity and structure
The population genetic analyses using RADseq produced 2 486 218 696 clean reads, with a mean of 22198381.21 clean reads, from the 21 populations (Supplementary Fig. 4). These reads produced genomes with a total length of 372932.80 Gb and a mean length of 3329.76 Gb. Additionally, 21037588.55 clean reads were mapped with the reference genome on average, with a mean length of 3061.83 Gb. The maximum mapping rate was 96.09%, the minimum was 92.24%, and the mean was 94.79%. The maximum depth was 22.07 x, the minimum was 13.41 x, and the mean was 18.09 x (Supplementary Table S7; Supplementary Fig. 4).
After filtering, 862 714 SNPs remained for population genetic analyses. In the NJ phylogeny, four clades were supported, reflecting geographic proximity and elevation. Populations in Clades A and B were from low elevations in southeastern-most Yunnan. Populations in Clade C were from medium elevation environments slightly northwest of those of Clades A and B. Populations in Clade D were from medium elevations in southeastern Yunnan and high elevations in central to northern Yunnan (Fig. 2a, b; Supplementary Table S7). Most populations were monophyletic, but six individuals (out of 112) in four populations (out of 21) were not clustered with the other individuals in their populations.
Figure 2.

Ilex polyneura population localities sampled in this study and population genetic analyses. a, Localities of sampled populations projected on an elevation map based on SMRT data with 30 arc-second resolution (https://www.usgs.gov/centers/eros/science/usgs-eros-archive-digital-elevation-shuttle-radar-topography-mission-srtm-1-arc?qt-science_center_objects=0#qt-science_center_objects) using QGIS software v.3.6.3 (http://www.qgis.org). b, Phylogenetic analysis of the 112 individuals with the neighbor-joining (NJ) method using the filtered SNPs. Green, blue, and red strips next to the individual names indicate low, medium, and high elevations. Populations are labeled by their names. Six individuals who were not clustered with other individuals in the same population are labeled with black empty circles and linked with their home populations by pink lines. c, Population genetic structure with the best K value (K = 4). d and e, PCA of genetic variation in the 21 populations. f, Cross-validation error rate of different K values from the population genetic structure analysis. g, Analysis of linkage disequilibrium (LD) decay in the 21 populations, in which the y-axis indicates that the linkage disequilibrium decays with genomic pairwise distance (x-axis) in low elevation populations (green line), medium elevation populations (blue line), high elevation populations (red line), and all populations (black line).
The population genetic structure analysis identified four clusters (K = 4) (Fig. 2c and f) as the best based on tests of K = 1–8 (Supplementary Fig. 5). There were two clusters each at low and high elevations, plus a few individuals with mixed ancestry from similar elevations. All individuals from medium elevation populations had mixed ancestry from both low and high elevations (Fig. 2c). In total, 58 out of 112 individuals had mixed ancestry. The PCA also showed that medium elevation populations were intermediate between those from low and high elevations (Fig. 2d and e).
Genomic adaptation of Ilex polyneura to different elevations
In the LD analyses, high elevation populations had the highest mean r2 value (0.230) at 0.1 kb, followed by low elevation populations (mean r2 = 0.277), medium elevation populations (mean r2 = 0.275), and all populations (mean r2 = 0.198) (Fig. 2g). Subsequently, the mean r2 value dropped rapidly to 0.151 in high elevation populations, 0.141 in low elevation populations, 0.164 in medium elevation populations, and 0.121 in all populations at 20 kb. Overall, medium elevation populations had the highest mean r2 value, followed by high elevation populations, low elevation populations, and all populations (Fig. 2g). A total of 1042 and 1332 genes were identified as candidate genes in the genome-wide scan of Fst and π values, respectively (Fig. 3a, b; Supplementary Tables S8 and S9). Only 34 candidate genes were identified by both scans (Supplementary Fig. 6; Supplementary Table S10). Subsequently, 50 terms and 15 pathways were identified in the GO and KEGG pathway analyses (Supplementary Tables S11 and S12; Supplementary Figs. 7 and 8).
Figure 3.
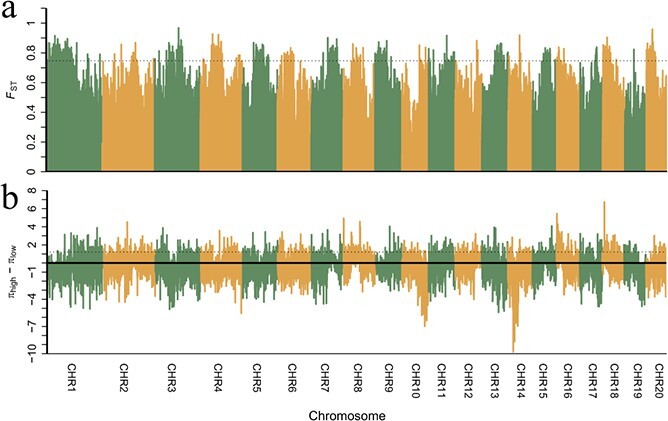
Plots of selection signature distributions on the genome, identified by Fst value (a) and nucleotide diversity (θπ) reduction (πhigh – πlow) (b). The dashed line indicates the threshold value (the top 5%) for the identification of selection signatures.
Discussion
Using a combination of Illumina short reads, PacBio single-molecule real-time sequencing technology, and Hi-C techniques, we assembled the whole genome of Ilex polyneura. The assembled genome was approximately 727.10 Mb with a contig N50 size of 5 124 369 bp and a scaffold N50 size of 36 593 620 bp. A BUSCO analysis produced 97.6% universal single-copy orthologs, indicating high contiguity and completeness of the assembly, and 98.9% of the genome assembly was anchored to 20 pseudochromosomes (Table 1). This is the first published genome at chromosome-scale in the huge genus Ilex as well as in the order Aquifoliales. Moreover, this is the only genome available for a woody member of the campanulids (asterids II), which includes six additional orders, and it is one of the highest quality genomes on the basis of contig size, BUSCO score, and completeness of the annotation for functional analysis [53–55]. In the I. polyneura genome, the average transcript length, average coding-sequence length, average exon length, and average intron length were 5461.06 bp, 1090.15 bp, 231.91 bp, and 1181.10 bp, respectively. In the Coriandrum sativum genome, these values were 3150.11 bp, 1079.77 bp, 4.66 bp, and 231.86 bp, respectively [54]. In the Erigeron breviscapus genome, the average coding sequence length and average exon length were 1136.30 bp and 214.31 bp [55], respectively. The I. polyneura genome had fewer annotated genes (32838) than the genomes of the two campanulids (40 747 in C. sativum and 43 514 in E. breviscapus) (Supplementary Table S4).
Population genetic analyses revealed evidence of divergent selection leading to genomic adaptation of Ilex polyneura to different elevations. Population genetic structure analysis showed that both low- and high-elevation populations consisted of two genetic clusters, while medium-elevation populations were genetic mixtures between low- and high-elevation populations (Fig. 2c, d, e). The two low-elevation genetic clusters had relatively longer phylogenetic branches than the high-elevation clusters in the NJ tree (Fig. 2b). Populations in Clades A and B, corresponding to the two low-elevation genetic clusters in the population genetic structure result, are separated by the Tanaka Line, which runs from northwest to southeast and separates the Sino-Japanese and Sino-Himalayan floristic subregions [56, 57]. Its significance as a biogeographical boundary seems to largely reflect divergent climatic conditions associated with the monsoons. The LD analysis provided further evidence that populations at low elevations and high elevations have experienced different environmentally selective pressures (Fig. 2g).
Selective sweep analyses identified some candidate genes that were positively selected at different elevations (Fig. 3a and b) and had functions possibly relating to elevation adaptation in the GO and KEGG pathway analyses (Supplementary Tables S11 and S12; Supplementary Figs. 7 and 8). Some of these functions were also identified in plant adaptations to the Qinghai-Tibetan Plateau (>4000 m) [58], e.g. protein phosphorylation (GO:0006468) (Supplementary Table S11). However, most of the functions were not identified in previous studies of adaptation to the QTP, but they are important in responses to environmental stresses [59, 60] and to defense responses against insects [61] and pathogens [62] (Supplementary Tables S11 and S12).
This study thus provides evidence for genetic divergence and significant local adaptation in the face of the expected high levels of gene flow between populations in Ilex. This result has consequences for conservation planning for I. polyneura and other widespread species in the genus, since it will be necessary to protect multiple populations across the range to conserve the full genetic and ecological potential of such species. None of the candidate genes identified as potentially involved in adaptation to local differences in elevation are of direct relevance to the farming of this species for decorative fruiting branches, but the amount of genetic variation suggests that it will be worthwhile to screen multiple wild populations for useful characteristics, including climatic tolerance, phenology, and fruit display. Some of the other commercially important holly species have narrow natural ranges, but others, such as the important traditional medicine Isotachis chinensis, are widespread and would probably also benefit broader searches for useful traits.
Conclusion
Using a combination of Illumina short reads, single-molecule real-time (SMRT) sequencing technology from Pacific Biosciences (PacBio), and Hi-C techniques, a chromosome-scale genome of Ilex polyneura of approximately 727.10 Mb in length is reported here. This is the first published genome at the chromosome scale in the genus Ilex as well as in the order Aquifoliales and is the only genome available for a woody member of the campanulids. The BUSCO score was 97.6%, and 98.9% of the genome assembly was anchored to 20 pseudochromosomes. Out of the 32 838 genes predicted, 31 826 (96.9%) were assigned functions in the gene annotation. The studied populations clustered into four monophyletic clades in the NJ tree in relation to geographic location and elevation. Populations at low and high elevations had simpler genetic structures, while populations at medium elevations had mixed ancestry from both low and high elevations. The selective sweep analyses identified 34 candidate genes potentially under selection at different elevations, with functions related to responses to abiotic and biotic stresses. Our results confirm the high contiguity, completeness, and annotation of the genome and demonstrate how it can provide a basis for further research into the evolution of the genus, as well as the selection of useful cultivars for horticulture and the production of holly teas and medicines.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (41901067), the CAS “Light of West China Program” (Y9XB071B01), the CAS 135 program (2017XTBG-T03) to X.Y., and Yunnan Fundamental Research Projects (grant NO.202001AT070120) to X.Y. We wish to thank Huan Fan for useful discussions and the Public Technology Service Center, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, for their help with experiments.
Author contributions
X.Y. and R.T.C. conceived and supervised the study. X.Y., Z.L. and Y.S. collected the samples. X.Y., X.H. and Z.L. carried out comparative genomics analysis and phylogenetic analysis. X.Y. and R.T.C. wrote the paper. All authors read, edited, and approved the final paper.
Data availability
The PacBio long reads and Illumina short reads for the genome were uploaded to the NCBI SRA database under BioProject PRJNA742488. The final chromosome-scale genome assembly with annotation is available at the Genome Warehouse in the National Genomics Data Center (https://ngdc.cncb.ac.cn) under accession number GWHBDNW00000000 and Figshare (10.6084/m9.figshare.14882928). The Illumina short reads of the population genomic data were uploaded to the NCBI SRA database under BioProject PRJNA742243.
Conflict of interests
The authors declare that they have no known conflicts of interest.
Supplementary data
Supplementary data is available at Horticulture Research online.
References
- 1. Xu X, Dimitrov D, Shrestha Net al. A consistent species richness-climate relationship for oaks across the northern hemisphere. Glob Ecol Biogeogr. 2019;28:1051–66. [Google Scholar]
- 2. Li R, Yue J. A phylogenetic perspective on the evolutionary processes of floristic assemblages within a biodiversity hotspot in eastern Asia. J Syst Evol. 2020;58:413–22. [Google Scholar]
- 3. Yao X, Song Y, Yang JBet al. Phylogeny and biogeography of the hollies (ilex L., Aquifoliaceae). J Syst Evol. 2021;59:73–82. [Google Scholar]
- 4. Martins K, Gugger PF, Llanderal-Mendoza Jet al. Landscape genomics provides evidence of climate-associated genetic variation in Mexican populations of Quercus rugosa. Evol Appl. 2018;11:1842–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Y, Wang H, Jiang Zet al. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature. 2021;590:600–5. [DOI] [PubMed] [Google Scholar]
- 6. Hong DY. A taxonomical revision of ilex (Aquifoliaceae) in the pan-Himalaya and unraveling its distribution patterns. Phytotaxa. 2015;230:151–71. [Google Scholar]
- 7. Chen SK, Ma H, Feng Yet al. 2008 Aquifoliaceae. In: Wu ZY, Raven PH, Hong DY, eds. Flora of China. Beijing: Science Press; St. Louis: Missouri Botanical Garden Press, 2008. [Google Scholar]
- 8. Ming R, Hou S, Feng Yet al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature. 2008;452:991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo R, Liu B, Xie Yet al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu J, Fan J, Sun Zet al. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 2020;36:2253–5. [DOI] [PubMed] [Google Scholar]
- 11. Roach MJ, Schmidt SA, Borneman AR. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 2018;19:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997. 2013.
- 13. Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7. [DOI] [PubMed] [Google Scholar]
- 14. Simão FA, Waterhouse RM, Ioannidis Pet al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2. [DOI] [PubMed] [Google Scholar]
- 15. Louwers M, Splinter E, Driel RVet al. Studying physical chromatin interactions in plants using chromosome conformation capture (3C). Nat Protoc. 2009;4:1216–29. [DOI] [PubMed] [Google Scholar]
- 16. Zhang X, Zhang S, Zhao Qet al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on hi-C data. Nat Plants. 2019;5:833–45. [DOI] [PubMed] [Google Scholar]
- 17. Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2004;5: 4.10.1-4.10.14. [DOI] [PubMed] [Google Scholar]
- 18. Zhao X, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–8. [DOI] [PubMed] [Google Scholar]
- 20. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koonin EV, Galperin MY. Sequence — Evolution — Function. Boston, MA: Springer US; 2003. [PubMed] [Google Scholar]
- 22. Yin J, McLoughlin S, Jeffery IBet al. Integrating multiple genome annotation databases improves the interpretation of microarray gene expression data. BMC Genomics. 2010;11:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mitchell AL, Attwood TK, Babbitt PCet al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019;47:D351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanehisa M, Sato Y, Kawashima Met al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stanke M, Diekhans M, Baertsch Ret al. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–44. [DOI] [PubMed] [Google Scholar]
- 26. Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr Protoc Bioinformatics. 2007;Chapter 4: 4.3.1-4.3.28. [DOI] [PubMed] [Google Scholar]
- 27. Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. [DOI] [PubMed] [Google Scholar]
- 28. Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open-source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–9. [DOI] [PubMed] [Google Scholar]
- 29. Li S, Ma L, Li Het al. Snap: an integrated SNP annotation platform. Nucleic Acids Res. 2007;35:D707–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hunt SE, Mclaren W, Gil Let al. Ensembl variation resources. Database (Oxford) bay119. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Birney E, Durbin R. Using GeneWise in the drosophila annotation experiment. Genome Res. 2000;10:547–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trapnell C, Roberts A, Goff Let al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7:562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haas BJ, Salzberg SL, Zhu Wet al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. The UniProt Consortium . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El-Gebali S, Mistry J, Bateman Aet al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kanehisa M, Furumichi M, Sato Yet al. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hunter S, Apweiler R, Attwood TKet al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lowe TM, Chan PP. tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kalvari I, Nawrocki EP, Argasinska Jet al. Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinformatics. 2018;62:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Tang H, Debarry JDet al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. [DOI] [PubMed] [Google Scholar]
- 44. Baird NA, Etter PD, Atwood TSet al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li H, Handsaker B, Wysoker Aet al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DePristo M, Banks E, Poplin Ret al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang J, Lee SH, Goddard MEet al. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Purcell S, Neale B, Todd-Brown Ket al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tang H, Peng J, Wang Pet al. Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol. 2005;28:289–301. [DOI] [PubMed] [Google Scholar]
- 51. Zhang C, Dong SS, Xu JJet al. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics. 2019;35:1786–8. [DOI] [PubMed] [Google Scholar]
- 52. Danecek P, Auton A, Abecasis Get al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pu X, Li Z, Tian Yet al. The honeysuckle genome provides insight into the molecular mechanism of carotenoid metabolism underlying dynamic flower coloration. New Phytol. 2020;227:930–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Song X, Wang J, Li Net al. Deciphering the high-quality genome sequence of coriander that causes controversial feelings. Plant Biotechnol J. 2020;18:1444–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. He S, Dong X, Zhang Get al. High quality genome of Erigeron breviscapus provides a reference for herbal plants in Asteraceae. Mol Ecol Resour. 2021;21:153–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fan DM, Yue JP, Nie ZLet al. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka-Kaiyong line’, an important phytogeographic boundary in Southwest China. Mol Ecol. 2013;22:4270–88. [DOI] [PubMed] [Google Scholar]
- 57. Qian LS, Chen JH, Deng Tet al. Plant diversity in Yunnan: current status and future directions. Plant Divers. 2020;42:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen J, Huang Y, Brachi Bet al. Genome-wide analysis of cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat Commun. 2019;10:5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hasanuzzaman M, Nahar K, Anee TIet al. Glutathione in plants: biosynthesis and physiological role in environmental stress tolerance. Physiol Mol Biol Plants. 2017;23:249–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vriese KD, Pollier J, Goossens Aet al. Dissecting cholesterol and phytosterol biosynthesis via mutants and inhibitors. J Exp Bot. 2021;72:241–53. [DOI] [PubMed] [Google Scholar]
- 61. Kang K, Yue L, Xia Xet al. Comparative metabolomics analysis of different resistant rice varieties in response to the brown planthopper Nilaparvata lugens Hemiptera: Delphacidae. Metabolomics. 2019;15:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang L, Paasch BC, Chen Jet al. An important role of l-fucose biosynthesis and protein fucosylation genes in Arabidopsis immunity. New Phytol. 2019;222:981–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PacBio long reads and Illumina short reads for the genome were uploaded to the NCBI SRA database under BioProject PRJNA742488. The final chromosome-scale genome assembly with annotation is available at the Genome Warehouse in the National Genomics Data Center (https://ngdc.cncb.ac.cn) under accession number GWHBDNW00000000 and Figshare (10.6084/m9.figshare.14882928). The Illumina short reads of the population genomic data were uploaded to the NCBI SRA database under BioProject PRJNA742243.