

Abstract
The genus Camelina (Brassicaceae) comprises 7–8 diploid, tetraploid, and hexaploid species. Of particular agricultural interest is the biofuel crop, C. sativa (gold-of-pleasure or false flax), an allohexaploid domesticated from the widespread weed, C. microcarpa. Recent cytogenetics and genomics work has uncovered the identity of the parental diploid species involved in ancient polyploidization events in Camelina. However, little is known about the maternal subgenome ancestry of contemporary polyploid species. To determine the diploid maternal contributors of polyploid Camelina lineages, we sequenced and assembled 84 Camelina chloroplast genomes for phylogenetic analysis. Divergence time estimation was used to infer the timing of polyploidization events. Chromosome counts were also determined for 82 individuals to assess ploidy and cytotypic variation. Chloroplast genomes showed minimal divergence across the genus, with no observed gene-loss or structural variation. Phylogenetic analyses revealed C. hispida as a maternal diploid parent to the allotetraploid Camelina rumelica, and C. neglecta as the closest extant diploid contributor to the allohexaploids C. microcarpa and C. sativa. The tetraploid C. rumelica appears to have evolved through multiple independent hybridization events. Divergence times for polyploid lineages closely related to C. sativa were all inferred to be very recent, at only ~65 thousand years ago. Chromosome counts confirm that there are two distinct cytotypes within C. microcarpa (2n = 38 and 2n = 40). Based on these findings and other recent research, we propose a model of Camelina subgenome relationships representing our current understanding of the hybridization and polyploidization history of this recently-diverged genus.
Introduction
Chloroplast genomes are valuable resources for phylogenetic studies and evolutionary inferences, owing to their uniparental inheritance and lack of recombination [1]. Chloroplast genomes of most flowering plants are maternally inherited, making them particularly useful for studying historical plant hybridization and inferring maternal lineages involved in polyploidization events. Previous studies have utilized chloroplast DNA to infer maternal and paternal ancestors of several polyploid crops, including in Brassica (mustards), Gossypium (cotton), Musa (banana), and Triticum (wheat) [2–6]. In the cases of Citrus (citrus fruits) and Malus (apple), chloroplast sequencing was used to date the timing of speciation and contributions of wild progenitors to domesticated varieties [7, 8]. Chloroplast haplotypes have also been proven useful for elucidating demographic history in some wild plant species, including Capsella bursa-pastoris and Palicourea padifolia [9, 10]. New techniques for preparing sequencing libraries from herbarium material [11] have further enabled the use of chloroplast genome sequencing to include taxa that are rare or extinct but still available in herbaria.
Camelina Crantz is a small genus comprising 7–8 species of herbaceous annuals in the family Brassicaceae [12, 13]. The genus includes diploid, tetraploid, and hexaploid species, including the hexaploid oilseed crop, C. sativa (L.) Crantz (gold-of-pleasure or false flax), and widespread weeds, Camelina rumelica Velen. and C. microcarpa Andrz. ex DC. In a previous study, we employed genome-wide ddRADseq markers to assess phylogenetic relationships in the genus; this work confirmed the traditional morphologically-based species delimitations and documented the domestication origin of cultivated C. sativa from the wild hexaploid species, C. microcarpa (Brock et al. 2018). However, many unanswered questions remain on phylogenetic relationships in the genus and the domestication origin of C. sativa. With respect to sampling, the previous phylogenetic study did not include a newly described diploid species, C. neglecta Brock et al., which has since been proposed as a diploid ancestor to the hexaploid crop lineage [12, 14], nor did it include other recently-collected germplasm that could potentially represent novel taxa (described below), or the rare (potentially extinct) species C. anomala Boiss. & Hausskn. Another unanswered question concerns the identity of the maternal subgenome contributor to C. sativa and maternal ancestry of the other polyploid lineages in the genus. Previous attempts at using individual chloroplast loci (trnL intron, psbA-trnH, ndh-rpL32 and trnQ-rps16) to address this and other phylogenetic questions revealed that these markers are largely invariant and thus unsuitable for phylogenetic analyses in Camelina [15]. In addition, much uncertainty remains about the domestication of C. sativa. While domestication is generally hypothesized to have occurred in central to eastern Europe [16] or western Asia, next-generation sequencing techniques have not yet been employed on a wide geographic sampling of C. microcarpa and C. sativa for the purposes of determining the geographic origins of domestication.
Camelina sativa is an ancient oilseed crop that was long cultivated in Europe and western Asia as a food and fuel source. While historically widely grown, most predominantly in Europe, C. sativa cultivation sharply declined in the 20th century in favor of other higher-yielding oilseed crops. This led to a major depletion of genetic diversity, as many landraces were abandoned. Several studies have documented this paucity of genetic diversity in modern C. sativa varieties [17–20]. In recent years, C. sativa has gained renewed interest as an oilseed source due to its short time to flowering, low input requirements, disease resistance, and high levels of seed omega-3 and long-chain fatty acids [21–23]. Of particular interest, biofuels generated from C. sativa seeds are highly suited for aviation applications and can achieve a 75% reduction in CO2 emissions over petroleum-based jet fuel [24]. However, modern breeding efforts are constrained by the limited genetic diversity of present-day germplasm.
The wild relatives of C. sativa offer a potentially valuable germplasm resource for overcoming the crop’s present-day limited genetic diversity. Crossing of wild C. microcarpa with C. sativa would provide one source of readily available diversity, as previous studies have shown evidence of interfertility between the crop and its progenitor [25–27]. However, even greater diversity enhancement could be achieved by identifying and hybridizing the extant diploid progenitors of the C. microcarpa/sativa complex to resynthesize the allohexaploid crop genome. Such approaches have been used with great success to increase genetic diversity in the allotetraploid Brassica napus crop [28, 29]. Knowledge of the maternal diploid contributor to C. sativa would facilitate this process by providing information on the direction of hybridization among the diploid and tetraploid lineages.
Recent research has established that the allohexaploid C. microcarpa/sativa lineage evolved as a product of ancient hybridization and genome duplication between two diploid progenitors, C. hispida Boiss. and C. neglecta, through a proposed intermediate, a C. neglecta-like tetraploid [14]. Other recent research suggests that two distinct cytotypes of C. microcarpa may exist [20], one 2n = 38 and the other 2n = 40. Mapping of genome-wide, genotyping-by-sequencing (GBS) data suggests that the 2n = 40 cytotype shares all three subgenomes with C. sativa (also 2n = 40); in contrast, the putative 2n = 38 C. microcarpa cytotype shares only two of its three subgenomes with the 2n = 40 cytotypes [20] and appears to be less closely related to the crop species. It remains unclear whether the two cytotypes of hexaploid C. microcarpa originated from independent polyploidization events, or if descending dysploidy in the 2n = 38 lineage after polyploidization is instead responsible for the chromosome number variation [30]. Additionally, a 2n = 26 tetraploid taxon has been discovered which, based on mapping of GBS data, appears to share its two subgenomes with both C. microcarpa cytotypes and C. sativa [20]. Our previous study had proposed the existence of a 2n = 26 tetraploid C. neglecta-like genome that hybridized with C. hispida to produce the 2n = 40 allohexaploids C. microcarpa and C. sativa [14]; however, additional evidence is required to prove that the previously identified tetraploid [20] is in fact a progenitor to the allohexaploid lineages.
In this study we used chloroplast genome sequencing to elucidate the maternal parentage of polyploid Camelina lineages, characterize phylogenetic relationships among previously unrecognized taxa, and estimate divergence time of the domesticated C. sativa in relation to its progenitor, C. microcarpa. We sequenced and assembled 84 Camelina chloroplast genomes with the goal of addressing the following specific questions: 1) Which diploid Camelina species contributed the chloroplast and maternal subgenome to the allotetraploid species C. rumelica and the allohexaploid species C. microcarpa and C. sativa? 2) Do 2n = 38 and 2n = 40 C. microcarpa cytotypes originate from different diploid/tetraploid ancestors? 3) Is the 2n = 26 C. neglecta-like taxon the previously-proposed allotetraploid intermediate with which C. hispida hybridized to give rise to the allohexaploid C. microcarpa/sativa lineage? 4) Approximately how old are the polyploidization events that produced the polyploid Camelina lineages?
Results
Karyotyping reveals cytotype variation only in C. microcarpa
A total of 82 accessions of Camelina were the subject of chromosome counting. Of these, 46 were C. microcarpa, 25 of which were found to be 2n = 38 and 21 of which were 2n = 40 (Supplemental Table 1). These two cytotype groups within C. microcarpa correspond to two previously-identified genetic subpopulations of that species [31], with all 2n = 38 accessions belonging to the “Ukraine” genetic population and all 2n = 40 accessions belonging to the “Caucasus” population. For all other species examined, we observed no variation in chromosome number. These include 8 C. laxa (2n = 12), 2 C. neglecta (2n = 12), 5 C. hispida (2n = 14), 3 C. neglecta-like (2n = 26), 10 C. rumelica (2n = 26), and 8 C. sativa (2n = 40). These results indicate that there is no chromosome number variation in most species, with the exception of C. microcarpa, which displayed two distinct cytotypes.
Chloroplast sequencing reveals low intragenic variation across Camelina
Assembled chloroplasts ranged in size from 152 986 to 154 455 bp, with a pair of inverted repeats (IR) of 26 484–27 164 bp separated by a large-single copy region (LSC) of 81 555–83 661 bp and a small-single copy region (SSC) of 17 820–17 934 bp (Table 1, Figure 1). In total, 79 protein-coding genes, 30 tRNAs, and 4 rRNAs were recovered from all samples in conserved order and orientation, with no observed gene loss or gain within the genus. A functional copy of rps16 was absent from all individuals, including a reference C. sativa chloroplast (NC_ 029337) although it is known to be present in other genera of the tribe Camelineae [32]. LSC, IR, and SSC junctions were nearly identical across all Camelina species, and no notable structural rearrangements or gene loss were observed (Supplemental Figure 1). Chloroplast genome sizes within species were relatively consistent, and variation between species was also low (Table 1). A total of 962 variable sites, including 347 singletons and 615 parsimony-informative sites were present in the final alignment of all Camelina chloroplasts studied here. Overall nucleotide diversity across all Camelina chloroplasts was π = 0.00121, with LSC π = 0.00139, IR π = 0.00014, and SSC π = 0.00199 (Supplemental Figure 2). Prominent peaks of nucleotide diversity did not occur within any coding genes or rRNAs. Conserved chloroplast genome structure and low nucleotide diversity indicate little evolutionary divergence in chloroplast genomes across the genus.
Table 1.
Description of Camelina taxa sampled here, including number of chloroplast genomes assembled, karyotypes observed, and chloroplast genome size ranges
| Species | # Plastomes | Observed Karyotypes (2n) | Chloroplast size (bp) | LSC (bp) | SSC (bp) | IR (bp) |
|---|---|---|---|---|---|---|
| Camelina anomala | 1 | N/A | 15 4007 | 8 3177 | 17860 | 2 6485 |
| Camelina hispida | 9 | 14 | 15 3351 - 15 3933 | 8 2450 - 8 3064 | 17880 - 17929 | 2 6485 - 2 6488 |
| Camelina laxa | 8 | 12 | 15 4253 - 15 4455 | 8 3456 - 8 3661 | 17820 - 17845 | 2 6486 - 2 6487 |
| Camelina microcarpa | 34 | 38/40 | 15 3002 - 15 3096 | 8 2194 - 8 2284 | 17820 - 17845 | 2 6484 - 2 6491 |
| Camelina neglecta | 3 | 12 | 15 3481 - 15 3483 | 8 2194 - 8 2284 | 17834 - 17836 | 2 6484 - 2 6485 |
| Camelina neglecta-like | 3 | 26 | 15 3056 - 15 3067 | 8 2245 - 8 2255 | 17841 - 17842 | 2 6485 |
| Camelina rumelica | 12 | 26 | 15 2986 - 15 3777 | 8 2128 - 8 2899 | 17886 - 17934 | 2 6486 - 2 6489 |
| Camelina sativa | 14 | 40 | 15 3051 - 15 3080 | 8 2246 - 8 2257 | 17825 - 17845 | 2 6485 - 2 6490 |
Figure 1.
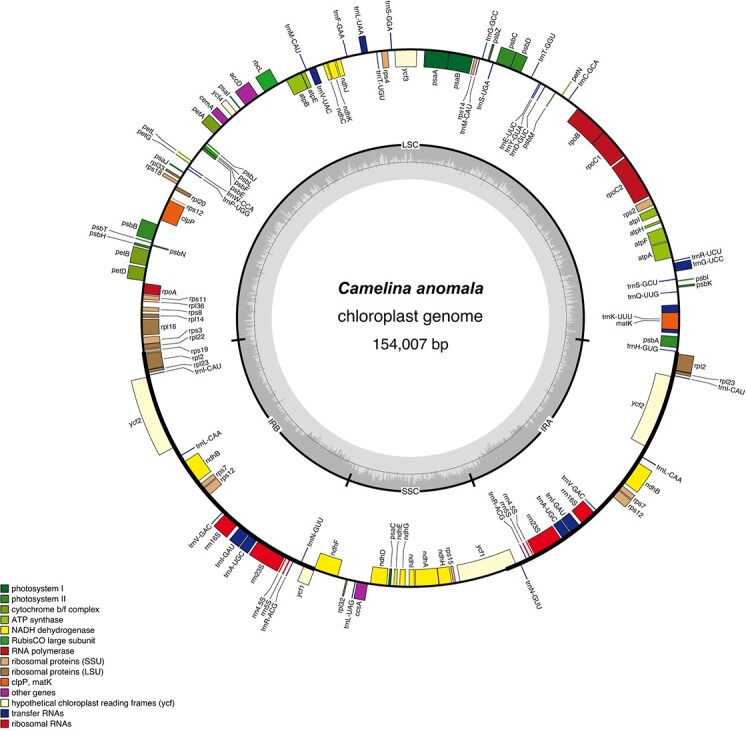
Gene map of the Camelina anomala chloroplast genome representative of gene, rRNA, and tRNA composition and orientation across the genus. Functional groups of genes are color-coded according to the key. Genes drawn on the outside of the map are transcribed clockwise and those on the inside are transcribed counterclockwise. The innermost circle represents relative GC content (in dark gray) and AT content (in light gray) of the chloroplast genome.
Phylogenomic analysis provides insights to maternal origins of several polyploid lineages
Maximum likelihood (ML) phylogenetic analyses of Camelina chloroplast genomes revealed strong support (100% bootstrap values) for monophyly of the diploid species - C. laxa, and C. neglecta as well as C. anomala (unknown ploidy). In contrast, although diploid C. hispida and tetraploid C. rumelica together form a clade with 100% bootstrap support, the two taxa do not form reciprocally monophyletic sister clades within that clade (Figure 2). This result suggests that C. hispida is the maternal parent of C. rumelica, and that there were likely multiple origins of the allotetraploid C. rumelica. The hexaploid C. microcarpa (both cytotypes) and its domesticated derivative C. sativa form a clade with short branches. Notably, the diploid C. neglecta clade is sister to the C. microcarpa/C. sativa clade; this placement indicates that C. neglecta is the closest extant diploid relative to the maternal tetraploid contributor to the allohexaploid lineages. This pattern also indicates that, by inference, the C. hispida subgenome in C. sativa must be derived through paternal ancestry. The C. anomala chloroplast genome is sister to the C. hispida/C. rumelica clade, although it is likely not a maternal parent to any polyploid lineages given its relatively high divergence and long branch length. Similarly, as the most basal species, C. laxa does not share similar chloroplast haplotypes with any other lineage and thus does not appear to have contributed as a maternal parent to any other sampled species. Herbarium specimens of C. sativa synonyms (C. linocola and C. dentata) group together with contemporary C. sativa and C. microcarpa, indicating that they do not have unique maternal origins (Figure 2). Taken together, these findings suggest that Camelina chloroplast genomes are highly useful for phylogenetic and hybridization inferences.
Figure 2.

Maximum-likelihood tree of Camelina inferred using whole chloroplast genomes. Taxon labels are colored according to taxonomy. Numbers on branches correspond to bootstrap supports, values over 50% are shown. Chromosome counts (bolded) are provided to the right of taxa where available. Scale represents evolutionary distance in number of substitutions per site. Asterisks represent previously reported chromosome numbers (see Supplemental Table 1).
Closer analysis of C. microcarpa and C. sativa chloroplasts with a haplotype network analysis provided little additional resolution beyond the ML tree and only limited grouping by cytotype or species (Supplemental Figure 3). Despite the differences in subgenome composition, the two C. microcarpa cytotypes share remarkably similar chloroplast genomes and show no clear phylogenetic divergence (Figure 2, Supplemental Figure 3). The low level of variation and lack of haplotype structure in the C. microcarpa/sativa lineage provide no clear insight on the geographic origin of domestication for the crop.
Molecular dating reveals extremely recent origin of Camelina polyploid lineages
BEAST analyses on concatenated datasets of all protein-coding genes and the LSC, IR, and SSC regions were used for molecular dating. Camelina chloroplasts were found to have diverged from the outgroup Capsella ~ 7.16 million years ago (Mya), (95% HPD: 5.30–8.72 Mya, HPD: highest posterior density); however, a low branch posterior probability value (0.4236) suggests that this relationship is uncertain (Figure 3). The most recent common ancestor (MRCA) of all Camelina chloroplast haplotypes is estimated at ~1.62 Mya (95% HPD: 0.97–2.51 Mya), whereas Capsella is ~2.05 Mya (95% HPD: 0.99–3.20 Mya) and Arabidopsis is ~5.80 Mya (95% HPD: 4.90–6.73 Mya). Within Camelina there is evidence of recent hybrid origins of several polyploid lineages. The tetraploid C. rumelica is diverged from the diploid C. hispida chloroplast haplotype by ~0.29 Mya (95% HPD: 0.11–0.50 Mya). The clade including tetraploid C. neglecta-like, hexaploid C. microcarpa cytotypes, and C. sativa has a very recent common ancestor at ~65 Kya (95% HPD: 18.7–120.7 Kya); domesticated C. sativa is estimated to have diverged from the 2n = 40 cytotype of C. microcarpa by ~17 Kya (95% HPD: 0.7–42.7 Kya), which is approximately on the time scale of the origin of agriculture ~10–12 Kya. The closest related diploid species to this group is C. neglecta at ~0.92 Mya (95% HPD: 0.45–1.44 Mya). Within Camelina, these dates suggest a likely Pleistocene divergence between all species, with much more recent diversification within the group of C. neglecta-like/C. microcarpa/C. sativa lineages. The results of molecular dating in Camelina thus indicate that while some species are of relatively older origin, three polyploid taxa (C. neglecta-like, C. microcarpa, C. sativa) appear to share a very recent common maternal ancestor.
Figure 3.
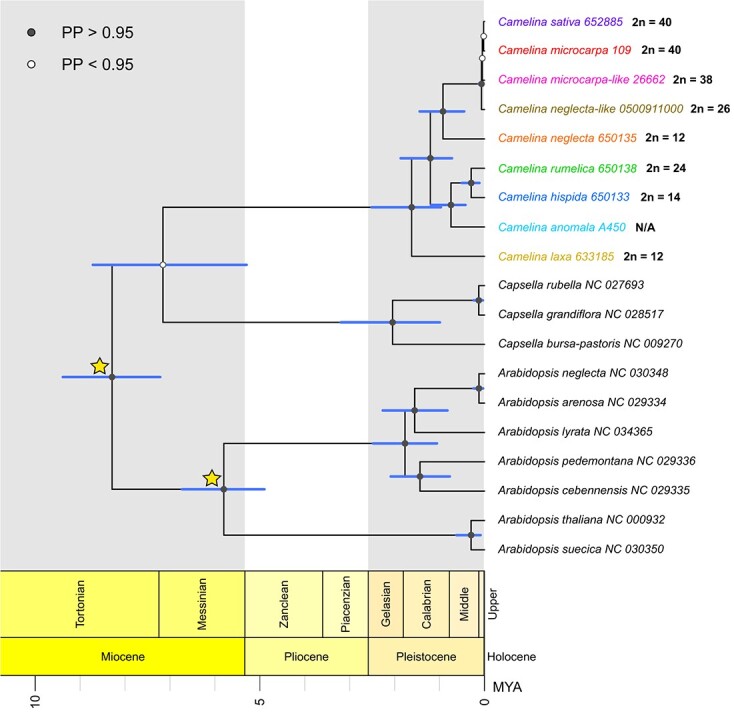
Camelineae chronogram inferred using BEAST with coding-gene and whole chloroplast sequence data partitions. Time calibration points include 8.16 Mya for the crown age and 5.97 Mya for Arabidopsis [39, 55]. Blue bars represent the 95% confidence intervals for divergence times.
Discussion
Despite the utility of chloroplast genomes for determining hybridization events and phylogenetic relationships between species, very few complete chloroplast genomes have previously existed for Camelina. The present study introduces 84 new chloroplast genomes for Camelina, including sampling from diploid, tetraploid, and hexaploid species as well as rare (potentially extinct) taxa from herbarium records. Our results indicate maternal inheritance of the C. hispida chloroplast in C. rumelica and maternal inheritance of a C. neglecta or C. neglecta-like chloroplast in C. microcarpa and C. sativa. We also uncover evidence of multiple independent maternal origins of C. rumelica from C. hispida. The high sequence similarity and monophyly of the tetraploid C. neglecta-like taxon, together with all C. microcarpa and C. sativa chloroplasts, suggests a single, shared maternal ancestor for these taxa. Molecular dating analyses indicate the MRCA of all Camelina taxa to be very recent at ~1.62 Mya, with the allotetraploid C. rumelica originating ~0.29 Mya and the allohexaploids C. microcarpa/C. sativa originating even more recently at ~65 Kya. Notably, the two cytotypes of C. microcarpa, despite lacking a common hybrid origin, display highly similar chloroplast genomes consistent with a shared maternal ancestor in the recent past. Polymorphisms present in C. microcarpa and C. sativa chloroplast genomes were not sufficient to recover a clear signature on the geographical origins of this domestication event.
Chromosome number variation
Previous work in Camelina has reported a wide range of chromosome counts, suggesting intraspecific ploidy variation (Brassibase, https://brassibase.cos.uni-heidelberg.de/). However, we find no evidence for ploidy or cytotype variation in any species other than C. microcarpa. Because some species can be difficult to distinguish from one another at various life-stages, we believe that taxonomic misidentifications are likely responsible for these historical cytotypic discrepancies.
Recently, evidence emerged of tetraploid 2n = 26 populations of C. microcarpa along the eastern Rocky Mountains of Canada [33], although it remains unclear whether these are of similar subgenome composition and origin as the 2n = 26 C. neglecta-like lineage proposed to be the intermediate tetraploid progenitor to the allohexaploid C. microcarpa [14]. Another study utilized GBS from a tetraploid (2n = 26) “C. microcarpa” and discovered that reads mapped strongly to two C. sativa subgenomes, but not to the third C. hispida subgenome [20]; this pattern indicates that the accession is not C. rumelica (also 2n = 26), as we exclusively observe C. rumelica to contain a C. hispida subgenome [14]. The geographic distribution of this tetraploid “C. microcarpa” or C. neglecta-like lineage remains unclear, and to our knowledge three records of these lineages exist in North Mongolia, South Siberia, and North Kazakhstan.
Previous work documented the genome origins in polyploid Camelina taxa [14] but included only 2n = 40 karyotypes of C. microcarpa, while the present study includes 2n = 38 and 2n = 40 cytotypes which belong to two distinct genetic populations [20, 31]. Our previous GBS sequencing study of C. microcarpa uncovered two highly distinct genetic populations in this species [31]; here we included 16 individuals from the Ukrainian genetic population and 16 individuals from the Caucasus genetic population for chromosome counting. Of these, all individuals from the Ukrainian genetic population were found to be 2n = 38, whereas all Caucasus individuals were 2n = 40. This evidence supports the high degree of genetic differentiation we previously observed between these two groups as being associated with chromosome number variation. Interestingly, while lacking any evidence of admixture, the 2n = 38 and 2n = 40 cytotypes appear to be widespread and indeed can be found in very close geographical proximity. Broadly, the 2n = 40 C. microcarpa is most common in the Caucasus region and eastern Turkey, although it is also sometimes present in the U.S. and Europe, whereas the 2n = 38 cytotype predominates in Europe and the U.S. but has not been recorded in the Caucasus or Turkey [31]. Despite the high genetic divergence and distinct chromosome numbers, the two cytotypes of C. microcarpa appear morphologically very similar, and taxonomic endeavors to identify distinct morphological characters have not yet been conducted. If morphologically diagnostic characters exist, this would further justify recognizing the two cytotypes as separate species.
The chromosomal differences in C. microcarpa cytotypes reported here also help to explain the seemingly contradictory findings of many previous studies examining interfertility of C. microcarpa and C. sativa. Whereas some studies successfully generated hybrid offspring from C. microcarpa × C. sativa crosses [25, 27], several other studies reported unsuccessful crossing attempts or offspring abnormalities and infertility with certain accessions of C. microcarpa [26, 34]. We believe this discrepancy may be explained by cytotype variation in C. microcarpa, where 2n = 38 cytotypes are much less likely to produce viable offspring with C. sativa (2n = 40). If this assumption is true, the 2n = 40 cytotype of C. microcarpa would be of higher value for breeding efforts to enhance genetic diversity in the crop.
Insights from chloroplast sequencing and phylogenetics
Our sequencing approach included five herbarium specimens to fill in potential gaps in diversity currently available, including a specimen of the rare species C. anomala (Figure 1). The placement of the C. anomala chloroplast as sister to the C. hispida/C. rumelica clade (Figure 2) provides additional evidence of its close relationship with C. hispida that was previously determined with ribotype sequencing [15]. The long branch length with high support (100% ML bootstrap support) and relatively diverged chloroplast genome suggests that C. anomala is indeed a distinct species, yet little is known about its current distribution, ploidy, or if it is extant in the wild. Our findings refute a previous hypothesis that C. anomala is basal to the Camelina genus [35], and instead we add to previous reports of C. laxa as being the most basal species in the genus [15, 36]. The feature of siliques in C. anomala should thus be considered an evolutionarily derived trait for this species, as the rest of the genus all have silicle fruits. The discovery of any extant C. anomala populations in the Levant could provide valuable material for understanding the underlying genetic basis of this morphological transition, which could yield considerable agricultural potential if integrated to the C. sativa crop.
Remarkably, the tetraploid C. neglecta-like taxon (2n = 26), the 2n = 38 C. microcarpa, and the 2n = 40 C. microcarpa/sativa chloroplasts are all monophyletic in a strongly supported clade (100% bootstrap) with shallow branches and little substructure (Figure 2). The 2n = 26 C. neglecta-like chloroplast sequences appear to be basal with weak support, along with one 2n = 40 C. microcarpa accession (JRB 118), indicating that the tetraploids may be more basal to the remainder of the clade. This basal phylogenetic position, minimal divergence from the hexaploid species, and expected chromosome number, provides support for this taxon being the 2n = 26 tetraploid that was previously proposed to be a contributor of two C. neglecta subgenomes to the allohexaploids C. microcarpa and C. sativa [14].
Our results further suggest that there may have been multiple independent origins of the tetraploid C. rumelica. Given the relatively longer branch lengths in the C. hispida/C. rumelica clade (Figure 2), and general morphological ambiguities in C. rumelica, we believe that at least two independent hybrid origins of C. rumelica from different populations of C. hispida occurred within the last ~0.29 My. Similarly, a study in Fragaria (strawberry) uncovered multiple independent origins of polyploid lineages in the genus via chloroplast genome sequencing [37]. Morphological variation within C. rumelica warrants closer scrutiny, as subtle morphological differences can in some cases distinguish polyploids of independent origin (e.g. Draba, another genus in the Brassicaceae) [38].
Our findings of two distinct cytotypes in C. microcarpa are consistent with recently published research that, based on genotyping-by-sequencing, also revealed evidence of a separate lineage of C. microcarpa with a unique karyotype (2n = 38) which is sister to the 2n = 40 C. microcarpa/sativa (2n = 40) lineage20. Our results show that the chloroplast genomes of these three groups are monophyletic with little divergence (Figure 2), indicating that they all share a common maternal ancestor and likely formed in the very recent past (Figure 4). Furthermore, these groups all share a C. neglecta subgenome, whereas 2n = 38 C. microcarpa lacks the C. hispida subgenome found in 2n = 40 C. microcarpa and C. sativa [20]. These findings suggest two origins involving distinct diploid ancestors for the two C. microcarpa “types”, despite their extraordinarily high chloroplast similarity (Figure 4). Thus, we believe that the allopolyploidization events leading to the respective C. microcarpa “types” were independent and involved different paternal subgenomes but the same maternal tetraploid, and that both evolved around the same time period.
Figure 4.
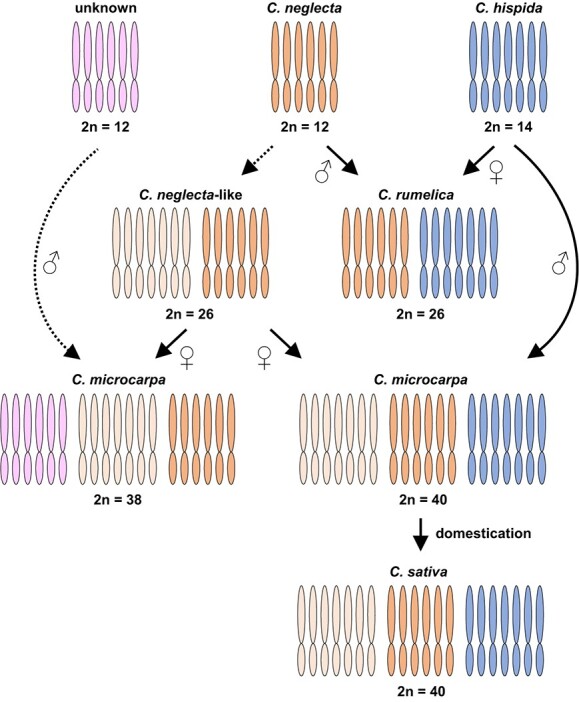
Proposed parental origins of polyploid Camelina species. Dotted arrows and outlines represent hypothetical events and subgenomes of unclear origin, respectively. Subgenome composition and origins are inferred from data presented herein along with results from previous studies [14, 20].
Between the maximum likelihood tree, BEAST analysis, and haplotype network (Supplemental Figure 3), we observe the 2n = 26 C. neglecta-like individual as basal to all allohexaploid Camelina taxa, and only divergent by a handful of markers. However, among allohexaploid taxa, the haplotype network recovers a similar pattern as the ML tree, with one clade (92% ML bootstrap support) of 2n = 38 C. microcarpa also appearing as a branch in the haplotype network and another clade with (67% ML bootstrap support), including 2n = 40 C. microcarpa and C. sativa accessions. The remaining ambiguity in the network is likely the result of the recent origin of these taxa and their very high sequence identity and minimal divergence.
Molecular dating and divergence time analyses
Molecular dating analyses conducted in BEAST provide insight to the diversification of species and the potential timing of polyploidization events within Camelina. Our results suggest that Camelina is a relatively young genus at ~1.62 Mya (Figure 3) when compared to related genera such as Arabidopsis at ~5.79 Mya [39]. Molecular dating suggests a recent common ancestor at ~0.29 Mya between the diploid C. hispida and tetraploid C. rumelica. Intriguingly, the estimated divergence date for the entire clade comprising the C. neglecta-like tetraploid, both cytotypes of C. microcarpa, and C. sativa is only ~65 Kya (Figure 3). In light of recent evidence that 2n = 38 C. microcarpa does not share the same subgenome composition as 2n = 40 C. microcarpa [20], we infer multiple origins at approximately the same time from the same or highly similar maternal contributors (Figure 4). Furthermore, the origin of the C. neglecta-like tetraploid is also unclear, leaving open the possibility of yet another polyploidization event occurring within this timeframe — in this case an auto/allopolyploidization event involving C. neglecta and/or a putative and unknown C. neglecta-like diploid. The divergence of 2n = 40 C. microcarpa and its domesticate, C. sativa, is ~17 Kya with a 95% HPD range between 0.7–42.7 Kya, consistent with the origins of agriculture ~10–12 Kya.
Due to the constraints of the uncorrelated relaxed lognormal clock implemented in BEAST, we were forced to subsample one individual per species for the time calibration analysis. Branch lengths were very short within the C. neglecta-like/C. microcarpa/C. sativa clade of the maximum likelihood tree; thus, it is unlikely that sampling would skew these dating estimates to a large extent. However, due to the mixed structure of the C. hispida/C. rumelica clade and relatively longer branch lengths, these dates would be liable to change considerably depending on sampling. Therefore, we decided to sample two widely available USDA GRIN accessions located in different highly supported clades of the maximum likelihood tree to provide a high-end dating estimate of the polyploidization event(s) producing C. rumelica. These molecular dating estimates provide insight to the relative age of the hybridization and whole-genome-duplication events in Camelina and provide strong support for a very recent origin of the allohexaploid lineages.
Implications for the understanding of Camelina subgenome evolution
Based on the topology of the maximum likelihood tree presented here and previous literature, we have constructed a diagram representing our current understanding of the evolutionary trajectory, hybridization history, and maternal subgenome inheritance of polyploid taxa in Camelina (Figure 4). Data from Mandáková et al. 2019 show that C. rumelica contains a C. neglecta and C. hispida subgenome and combined with data presented here we conclude that C. rumelica inherited a C. hispida subgenome maternally and the C. neglecta subgenome paternally. The origins of C. microcarpa and C. sativa have also been explored in Mandáková et al. 2019 regarding the 2n = 40 cytotype of C. microcarpa, which shares all subgenomes with C. sativa. The recent Chaudhary et al. 2020 study, shows that both C. microcarpa cytotypes and C. sativa share two subgenomes, one of which is highly similar to C. neglecta, and the other shares some similarity but is not identical. Based on this research and our own findings, it is clear that this tetraploid, sometimes called tetraploid C. microcarpa or C. neglecta-like, is the maternal parent to the polyploidization event leading to both C. microcarpa cytotypes and C. sativa. Thus, the C. hispida subgenome present in C. sativa and 2n = 40 C. microcarpa must be paternally inherited. Finally, at least one subgenome contributor of paternal origin in 2n = 38 C. microcarpa remains unknown (Figure 4).
We recognize that further sampling in Camelina may potentially provide definitive identification of parental lineages of polyploids [40]; additional diversity present in the native range, especially the eastern Eurasian steppe, could account for missing subgenomes and progenitors. In the case of C. neglecta, few wild individuals have been recovered, though it is possible that the species is relatively diverse and common in southern France, other regions of Europe, and perhaps even across the Eurasian steppe. It remains a distinct possibility that the missing C. neglecta-like diploid genome is extant in the wild as a cryptic species or present in areas that are historically under sampled (e.g. eastern Eurasia). If this taxon were recovered, further resolution could be brought to the understanding of the hybrid origin of the allohexaploid biofuel crop C. sativa. Additional sampling from Europe and Asia Minor may provide the pieces necessary to elucidate the evolution of the remaining unexplained subgenome detected in 2n = 38 C. microcarpa.
Given the high degree of genome-wide differentiation of these C. microcarpa cytotypes based on genotyping-by-sequencing [31], plus their differences in subgenome composition, karyotype, and their likely sexual incompatibility, we believe that it is appropriate to recognize one of the two cytotypes as a distinct species. The recently resolved designation of the proper type specimen for C. microcarpa [41] will now allow suitable application of this name in future taxonomic treatments. However, it remains difficult to ascertain the origins of the C. microcarpa type, especially because both cytotypes are likely to occur in the region where it was collected (now eastern Moldova or southwestern Ukraine). Finally, additional molecular studies and taxonomic endeavors should also be pursued on the recently characterized C. neglecta-like tetraploid, which represents an important piece of the evolutionary puzzle that produced the widespread weed, C. microcarpa, and the emerging aviation biofuel crop, C. sativa.
Methods
Collections
Species diversity within Camelina is highest in Turkey and the Caucasus, though some species (e.g. C. microcarpa and C. rumelica) are cosmopolitan with wide distributions as weeds. Seven species of Camelina were collected from the field in Turkey, Georgia, Armenia, Ukraine, United States, from the USDA GRIN and PGRC germplasm collections, and from herbarium specimens located at MO and G. Most samples in this study were previously described in Brock et al. 2018 and Brock et al. 2020. GPS coordinates and approximate localities for collections can be found in Supplemental Table 1.
Chromosome counts
A total of 82 individuals were chosen for chromosome counting. Sampled C. microcarpa included 16 accessions from a “Ukraine” genetic population and 16 from a genetically distinct “Caucasus” population; these two genetic populations were previously identified in an analysis based on GBS [31]. Root tips or anthers were used for chromosome preparation following the protocol of Mandáková et al. (2019). Root tips were harvested from germinating seeds, pre-treated with ice-cold water for 16 h, fixed in ethanol/acetic acid (3:1) fixative for 24 h at 4°C and stored at −20°C until further use. Young inflorescences were harvested from the cultivated plants and fixed in ethanol/acetic acid (3:1) fixative for 24 h at 4°C and stored at −20°C until further use. Selected root tips or flower buds were rinsed in distilled water (twice for 5 min) and citrate buffer (10 mM sodium citrate, pH 4.8; twice for 5 min), and digested in 0.3% cellulase, cytohelicase and pectolyase (all Sigma-Aldrich) in citrate buffer at 37°C for 3 h. After digestion, individual root tips or anthers were dissected on a microscope slide in 20 μl acetic acid and spread on the slide placed on a metal hot plate (50°C) for c. 30 s. Then, the preparation was fixed in freshly prepared ethanol/acetic acid (3:1) fixative by dropping the fixative around the drop of acetic acid and into it. The preparation was dried using a hair dryer and staged using a phase contrast microscope. Chromosomes were counterstained with 2 μg/ml DAPI in Vectashield. Preparations were photographed using Zeiss Z2 epifluorescence microscope and CoolCube CCD camera. Additional chromosome numbers were obtained for several accessions from previously published studies (Brock et al., 2019; Mandáková et al., 2019; Supplementary Table 1).
Chloroplast sequencing
DNA was extracted from 72 accessions, including 67 from fresh or silica-dried leaf and 5 from herbarium specimens (Supplemental Table 1). Extraction of DNA from fresh leaves was performed with a modified CTAB DNA extraction protocol [36], DNeasy Plant Mini Kit (Qiagen, Valencia, California, USA), and NucleoSpin Plant II kit (Macherey-Nagel, Düren, Germany). DNA extraction from herbarium specimens utilized an alternative extraction method to enhance yield and library quality [11]. DNA quantity was assessed using a Qubit fluorometer (Life Technologies, Carlsbad, California, USA). Depending on DNA concentration, between ~50 ng and 200 ng was used as template for whole genome sequencing library preparation using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, Massachusetts, USA) following manufacturer’s protocol with NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1, E7600S). Final libraries were quantified with Qubit fluorometry, and fragment distribution was determined with an Agilent High Sensitivity DNA Kit on an Agilent 2100 Bioanalyzer System (Agilent Technologies, Santa Clara, California, USA). Samples were pooled for sequencing, and the final pooled library contained fragment sizes between 200 bp and 1000 bp with an average length of 498 bp. The final library was sequenced on an Illumina HiSeq platform (Illumina, San Diego, California, USA) for 2x150 bp reads. DNA from 5 samples was sequenced on an Illumina HiSeq 4000 platform at the University of Illinois Roy J. Carver Biotechnology Center. DNA from an additional 7 samples was prepared from fresh or silica-dried leaf material and sequenced at the Core Facility Genomics, CEITEC, Masaryk University using the Illumina MiSeq platform generating 2x150 bp reads.
Chloroplast assembly
The GetOrganelle [42] v1.6.4 pipeline was utilized for de novo assembly of whole chloroplast genomes. This pipeline was fed raw low-coverage whole genome reads for each sample on default settings with K-mer values of 21, 45, 65, 85, 105. Bandage [43], together with manual inspection, were used to verify circularity of the chloroplast sequence and the boundaries of inverted repeats (IR), large-single copy (LSC), and small-single copy (SSC) regions. Full circular chloroplast genome sequences were annotated using CPGAVAS2 [44]. Chloroplast annotations were examined in Geneious Prime 2019.0.4 and when necessary, annotations were corrected. Chloroplast genome maps were generated with OGDRAW [45] v1.3. Junctions of the LSC, IR, and SSC regions were visualized with IRscope [46].
Chloroplast phylogenetics and evolution
Chloroplast genomes sequenced here, in addition to the reference chloroplast genome for C. sativa (NC_ 029337) [39], were aligned using MAFFT [47] v7.271, and alignments were manually checked in Geneious Prime 2019.0.4 and adjusted where necessary. All gap positions in the alignment were eliminated using Gblocks [48] v0.91b. One inverted repeat (IR) region was removed from the alignment to prevent overrepresentation of IR sequences. The GTR + I + gamma model was chosen as the best fit model implementable in RAxML based on the Akaike Information Criterion in jModelTest [49] 2 v2.1.10. Maximum likelihood trees were generated using RAxML [50] v8.2.4 with 500 bootstrap replicates and rooted on C. laxa which is the sister taxon to all other Camelina as shown in recent studies [15, 36]. Nucleotide diversity measures and the numbers of variable, singleton, and parsimony-informative sites were calculated with DnaSP [51] 6.12.03. Haplotype networks were generated in PopART [52] v1.7 using TCS [53].
Divergence time estimation
To estimate divergence times of species within Camelina, we constructed a concatenated data set of protein-coding genes from all sampled Camelina chloroplasts along with the following outgroup taxa: Arabidopsis arenosa (NC_029334), A. cebennensis (NC_029335), A. lyrata (NC_034365), A. neglecta (NC_030348), A. pedemontana (NC_029336), A. thaliana (NC_000932), C. bursa-pastoris (NC_009270), C. grandiflora (NC_028517), and C. rubella (NC_027693). A total of 79 protein-coding genes were used in the concatenated dataset, with only one gene, rps16, absent in Camelina. The following genes were included in the final concatenated data set: accD, atpA, atpB, atpE, atpF, atpH, atpI, ccsA, cemA, clpP, matK, ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhl, ndhJ, ndhK, petA, petB, petD, petG, petL, petN, psaA, psaB, psaC, psaI, psaJ, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ, rbcL, rpl2, rpl14, rpl16, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36, rpoA, rpoB, rpoC1, rpoC2, rps2, rps3, rps4, rps5, rps7, rps8, rps11, rps12, rps14, rps15, rps18, rps19, ycf1, ycf2, ycf3, ycf4. In addition to this dataset, alignments of the LSC, SSC, and IR regions were generated with MAFFT v7.271 and included in divergence time estimation.
Divergence times were estimated in BEAST [54] v2.6.2. The XML editing software BEAUti v2.6.3 was used to generate XML parameter files for the BEAST analyses. The concatenated protein-coding dataset was partitioned into the three codon positions with unlinked substitution rate, rate heterogeneity, and base frequencies. An uncorrelated relaxed lognormal clock was used along with a Yule tree prior and the GTR + I + gamma model of substitution. Divergence times for A. thaliana and other Arabidopsis species were given a prior of monophyly with a log-normal divergence date corresponding to 5.97 Mya, while the crown of Arabidopsis, Capsella, and Camelina was given a divergence date of 8.16 Mya based on previous literature [39, 55]. We ran two independent MCMC simulations of 200 million generations with a 20 million generation pre-burn-in and sampled trees every 10 000 generations. Tree outputs from both runs were combined using LogCombiner v2.6.3 with a 10% sample burn-in. Tracer v. 1.7.1 was used to inspect convergence; all effective sample sizes (ESS) were found to be over 200 before and after combining log files, indicating good mixing. A maximum clade credibility (MCC) tree with mean node heights was generated in TreeAnnotator v2.6.2 using a total of 72 000 trees. The resulting MCC tree was then visualized in R [56] using the function PHYLOCH for import [57] and the geoscalePhylo function for visualization.
Supplementary Material
Acknowledgements
We thank Ihsan Al-Shehbaz (Missouri Botanical Garden) for insightful discussions and assistance with sampling of Camelina herbarium materials. We also thank Laura Kassen (Jardin Botanique de Bordeaux) and David Dickenson for providing material of C. neglecta from southern France. We are grateful to Anže Žerdoner Čalasan, Herbert Hurka and Barbara Neuffer for providing seeds of tetraploid C. neglecta. We thank Elizabeth A. Kellogg for providing support for sequencing of an initial round of chloroplast genomes. J. Brock would like to thank Nichole M. Tiernan and Aryeh H. Miller for their helpful suggestions and insights.
This work was supported by a Washington University in St. Louis International Center for Energy, Environment and Sustainability (InCEES) grant to J.R.B. and K.M.O., by research grants from the Czech Science Foundation (21-03909S) and the CEITEC 2020 project (LQ1601) to M.A.L. The William H. Danforth Fellowship in Plant Sciences provided student funding for laboratory work, analyses, and writing to J.R.B. A National Geographic Society Committee for Research & Exploration grant (#CP-055ER-17) to J.R.B. funded field collections in Ukraine. A portion of the plastome sequencing was covered by National Science Foundation grant DEB-1457748 to E. A. Kellogg at the Donald Danforth Plant Science Center.
Contributions
The study was designed by J.R.B. Chloroplast sequencing was performed by J.R.B., M.M.K., T.M., M.A.L. Chloroplast assembly, annotation, and phylogenetic analyses were performed by J.R.B. Chromosome counting was performed by T.M. Authors J.R.B. and K.M.O. wrote the paper and all authors provided edits and approved the final manuscript.
Data availability
All annotated chloroplast genomes used here have been submitted to NCBI under GenBank accession numbers MZ343072-MZ343153, MZ351195, MZ351196.
Conflict of interests
none.
Supplementary data
Supplementary data is available at Horticulture Research online.
References
- 1. Nock CJ, Waters DLE, Edwards MAet al. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol J. 2011;9:328–33. [DOI] [PubMed] [Google Scholar]
- 2. Li P, Zhang S, Li Fet al. A phylogenetic analysis of chloroplast genomes elucidates the relationships of the six economically important brassica species comprising the triangle of U. Front Plant Sci. 2017;8:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allender CJ, King GJ. Origins of the amphiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol. 2010;10:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carreel F, Gonzalez de Leon D, Logada Pet al. Ascertaining maternal and paternal lineage within Musa by chloroplast and mitochondrial DNA RFLP analyses. Genome. 2002;45:679–92. [DOI] [PubMed] [Google Scholar]
- 5. Middleton CP, Senerchia N, Stein Net al. Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PLoS One. 2014;9:e85761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wendel JF. New World tetraploid cottons contain Old World cytoplasm. Proc Natl Acad Sci. 1989;86:4132–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carbonell-Caballero J, Alonso R, Ibañez Vet al. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus citrus. Mol Biol Evol. 2015;32:2015–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nikiforova SV, Cavalieri D, Velasco Ret al. Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Mol Biol Evol. 2013;30:1751–60. [DOI] [PubMed] [Google Scholar]
- 9. Ceplitis A, Su Y, Lascoux M. Bayesian inference of evolutionary history from chloroplast microsatellites in the cosmopolitan weed Capsella bursa-pastoris (Brassicaceae). Mol Ecol. 2005;14:4221–33. [DOI] [PubMed] [Google Scholar]
- 10. Gutiérrez-Rodríguez C, Ornelas JF, Rodríguez-Gómez F. Chloroplast DNA phylogeography of a distylous shrub (Palicourea padifolia, Rubiaceae) reveals past fragmentation and demographic expansion in Mexican cloud forests. Mol Phylogenet Evol. 2011;61:603–15. [DOI] [PubMed] [Google Scholar]
- 11. Saeidi S, McKain MR, Kellogg EA. Robust DNA isolation and high-throughput sequencing library construction for herbarium specimens. JoVE. J Vis Exp. 2018;e56837. 10.3791/56837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brock JR, Mandáková T, Lysak MAet al. Camelina neglecta (Brassicaceae, Camelineae), a new diploid species from Europe. PhytoKeys. 2019;115:51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al-Shehbaz I, Beilstein M. Camelina Flora N Am Editor Comm. 2010;7:451–3. [Google Scholar]
- 14. Mandáková T, Pouch M, Brock JRet al. Origin and evolution of diploid and allopolyploid Camelina genomes were accompanied by chromosome shattering. Plant Cell. 2019;31:2596–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Žerdoner Čalasan A, Seregin AP, Hurka Het al. The Eurasian steppe belt in time and space: phylogeny and historical biogeography of the false flax (Camelina Crantz, Camelineae, Brassicaceae). Flora. 2019;260:151477. [Google Scholar]
- 16. Ghamkhar KG, Croser J, Aryamanesh Net al. Camelina (Camelina sativa (L.) Crantz) as an alternative oilseed: molecular and ecogeographic analyses. Genome. 2010. 10.1139/G10-034. [DOI] [PubMed] [Google Scholar]
- 17. Vollmann J, Grausgruber H, Stift Get al. Genetic diversity in camelina germplasm as revealed by seed quality characteristics and RAPD polymorphism. Plant Breed. 2005;124:446–53. [Google Scholar]
- 18. Singh R, Bollina V, Higgins EEet al. Single-nucleotide polymorphism identification and genotyping in Camelina sativa. Mol Breed. 2015;35:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luo Z, Brock J, Dyer JMet al. Genetic diversity and population structure of a Camelina sativa spring panel. Front Plant Sci. 2019;10:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaudhary R, Koh CS, Kagale Set al. Assessing diversity in the Camelina genus provides insights into the genome structure of Camelina sativa. G3 (Bethesda). 2020;10:1297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berti M, Gesch R, Eynck Cet al. Camelina uses, genetics, genomics, production, and management. Industrial Crops and Products. 2016;94:690–710. [Google Scholar]
- 22. Gugel RK, Falk KC. Agronomic and seed quality evaluation of Camelina sativa in western Canada. Can J Plant Sci. 2006;86:1047–58. [Google Scholar]
- 23. Zubr J. Oil-seed crop: Camelina sativa. Ind Crops Prod. 1997;6:113–9. [Google Scholar]
- 24. Shonnard DR, Williams L, Kalnes TN. Camelina-derived jet fuel and diesel: sustainable advanced biofuels. Environ Prog Sustain Energy. 2010;29:382–92. [Google Scholar]
- 25. Martin SL, Lujan-Toro BE, Sauder CAet al. Hybridization rate and hybrid fitness for Camelina microcarpa Andrz. Ex DC (♀) and Camelina sativa (L.) Crantz(Brassicaceae) (♂). Evol Appl. 2019;12:443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Séguin-Swartz G, Nettleton JA, Sauder Cet al. Hybridization between Camelina sativa (L.) Crantz (false flax) and north American Camelina species. Plant Breed. 2013;132:390–6. [Google Scholar]
- 27. Zhang C-J, Auer C. Hybridization between Camelina sativa (L.) Crantz and common brassica weeds. Ind Crops Prod. 2020;147:112240. [Google Scholar]
- 28. Becker HC, Engqvist GM, Karlsson B. Comparison of rapeseed cultivars and resynthesized lines based on allozyme and RFLP markers. Theor Appl Genet. 1995;91:62–7. [DOI] [PubMed] [Google Scholar]
- 29. Girke A, Schierholt A, Becker HC. Extending the rapeseed genepool with resynthesized Brassica napus L. I: genetic diversity. Genet Resour Crop Evol. 2012;59:1441–7. [Google Scholar]
- 30. Mandáková T, Lysak MA. Post-polyploid diploidization and diversification through dysploid changes. Curr Opin Plant Biol. 2018;42:55–65. [DOI] [PubMed] [Google Scholar]
- 31. Brock JR, Scott T, Lee AYet al. Interactions between genetics and environment shape Camelina seed oil composition. BMC Plant Biol. 2020;20:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo X, Liu J, Hao Get al. Plastome phylogeny and early diversification of Brassicaceae. BMC Genomics. 2017;18:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin SL, Smith TW, James Tet al. An update to the Canadian range, abundance, and ploidy of Camelina spp. (Brassicaceae) east of the Rocky Mountains. Botany. 2017;95:405–17. [Google Scholar]
- 34. Tepfer M, Hurel A, Tellier Fet al. Evaluation of the progeny produced by interspecific hybridization between Camelina sativa and C. microcarpa. Ann Bot. 2020;125:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mirek Z. Monographic studies in genus Camelina Cr. 1. Camelina anomala Boiss. Et Hausskn. Acta Soc. Acta Soc Bot Pol Pol Tow Bot. 2014;53:429–32. [Google Scholar]
- 36. Brock JR, Dönmez AA, Beilstein MAet al. Phylogenetics of Camelina Crantz. (Brassicaceae) and insights on the origin of gold-of-pleasure (Camelina sativa). Mol Phylogenet Evol. 2018;127:834–42. [DOI] [PubMed] [Google Scholar]
- 37. Dillenberger MS, Wei N, Tennessen JAet al. Plastid genomes reveal recurrent formation of allopolyploid Fragaria. Am J Bot. 2018;105:862–74. [DOI] [PubMed] [Google Scholar]
- 38. Soltis DE, Soltis PS. Polyploidy: recurrent formation and genome evolution. Trends Ecol Evol. 1999;14:348–52. [DOI] [PubMed] [Google Scholar]
- 39. Hohmann N, Wolf EM, Lysak MAet al. A time-calibrated road map of Brassicaceae species radiation and evolutionary history. Plant Cell. 2015;27:2770–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edger PP, McKain MR, Bird KAet al. Subgenome assignment in allopolyploids: challenges and future directions. Curr Opin Plant Biol. 2018;42:76–80. [DOI] [PubMed] [Google Scholar]
- 41. Mosyakin SL, Brock JR. On the proper type designation for Camelina microcarpa, a wild relative and possible progenitor of the crop species C. sativa (Brassicaceae). Candollea. 2021;76:55–63. [Google Scholar]
- 42. Jin JJ, Yu WB, Yang JBet al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020;21:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wick RR, Schultz MB, Zobel Jet al. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31:3350–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi L, Chen H, Jiang Met al. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019;47:W65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52:267–74. [DOI] [PubMed] [Google Scholar]
- 46. Amiryousefi A, Hyvönen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018;34:3030–1. [DOI] [PubMed] [Google Scholar]
- 47. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52. [DOI] [PubMed] [Google Scholar]
- 49. Darriba D, Taboada GL, Doallo Ret al. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JCet al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–302. [DOI] [PubMed] [Google Scholar]
- 52. Leigh JW, Bryant D. Popart: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6:1110–6. [Google Scholar]
- 53. Clement, M., Snell, Q., Walke, P.et al. TCS: estimating gene genealogies. in Proceedings 16th International Parallel and Distributed Processing Symposium 7 pp ( IEEE, 2002). 10.1109/IPDPS.2002.1016585. [DOI] [Google Scholar]
- 54. Bouckaert R, Heled J, Kuhnert Det al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Novikova PY, Hohmann N, Nizhynska Vet al. Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating speciation and abundant trans-specific polymorphism. Nat Genet. 2016;48:1077–82. [DOI] [PubMed] [Google Scholar]
- 56. Team, R. C . othersR: A language and environment for statistical computing. In: 2013.
- 57. Heibl C. PHYLOCH: R Language Tree Plotting Tools and Interfaces to Diverse Phylogenetic Software Packages. 2008.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All annotated chloroplast genomes used here have been submitted to NCBI under GenBank accession numbers MZ343072-MZ343153, MZ351195, MZ351196.