

Abstract
While roses are today among the most popular ornamental plants, the petals and fruits of some cultivars have flavored foods for millennia. The genetic origins of these edible cultivars remain poorly investigated. We collected the major varieties of edible roses available in China, assembled their plastome sequences, and phased the haplotypes for internal transcribed spacers (ITS1/ITS2) of the 18S-5.8S-26S nuclear ribosomal cistron. Our phylogenetic reconstruction using 88 plastid genomes, of primarily maternal origin, uncovered well-supported genetic relationships within Rosa, including all sections and all subgenera. We phased the ITS sequences to identify potential donor species ancestral to the development of known edible cultivars. The tri-parental Middle-Eastern origin of R. × damascena, the species most widely used in perfume products and food additives, was confirmed as a descendent of past hybridizations among R. moschata, R. gallica, and R. majalis/R. fedtschenkoana/R. davurica. In contrast, R. chinensis, R. rugosa, and R. gallica, in association with six other wild species, were the main donors for fifteen varieties of edible roses. The domesticated R. rugosa ‘Plena’ was shown to be a hybrid between R. rugosa and R. davurica, sharing a common origin with R. ‘Fenghua’. Only R. ‘Jinbian’ and R. ‘Crimson Glory’ featured continuous flowering. All remaining cultivars of edible roses bloomed only once a year. Our study provides important resources for clarifying the origin of edible roses and suggests a future for breeding new cultivars with unique traits, such as continuous flowering.
Introduction
Domesticated roses (Rosa L.) are regarded as the queen of flowers. They represent one of the most economically important lineages of woody plants in ornamental horticulture, with more than 37 000 cultivars and ~ 200 wild species [1–3] (https://modernroses.rose.org/). However, the petals and/or accessory fruits (hips) of some rose cultivars have been bred for millennia, providing civilizations with food additives, fumigants, cosmetics, and traditional medical and/or hygienic products. As food resources, rose petals and hips offer vitamins, fibers, and secondary metabolites [4–6]. A recipe for patina de rosis is attributed to Apicius (80 BCE – 40 CE) in the first century CE [7]. The popularity of rose petal flavors derives from different concentrations of aromatic molecules, which are also found in some commercial spices, citrus fruits, and fortified wines. Domestication of roses in China probably began prior to the Eastern Han Dynasty (Shennong Bencaojing, approximately 20–220 CE), and many of these Chinese cultivars were incorporated into European horticulture from the late 18th through the 19th centuries [8]. Very likely due to frequent crossing and backcrossing with Asian genotypes that harbor continuous flowering and many other important traits, modern roses seem to feature a genetic shift from European to Asian backgrounds [9]. With petals as the most essential ingredients of aroma and flavor, Yunnan Province in southwestern China remains well known for its rose flower cakes (“Xianhuabing” in Chinese; Yanjing Suishiji, by Fucha Dunchong; Qing Dynasty). Modern Yunnan agriculture continues to maintain the largest growing area of edible roses, with more than 4 000 hectares under cultivation and an annual production of more than two billion CNY in 2017 [10].
Approximately 20 edible cultivars, most of which are called “Meigui”, have been used in China [11–14]. “Meigui” is an umbrella word referring to a combination of cultivars related to R. rugosa Thunb., the essential oil cultivars (Damask roses and R. ‘Kushui’), and other fragrant lines including R. ‘Tuwei’, YN01, and R. ‘Crimson Glory’ [12]. The only cultivar authorized as a new food resource in China is R. rugosa ‘Plena’ (‘Chongban Hongmeigui’), a variety selected presumably from R. rugosa in Pingyin County, Shandong Province [15] (https://www.nhc.gov.cn).
The phylogenetic origin of most edible roses remains elusive. Rosa ‘Kushui’, a line cultivated mainly in Yongdeng County, Gansu Province, is recorded as a natural hybrid of R. sertata Rolfe and R. rugosa Yu et Ku but without any direct genetic evidence [15]. Rosa ‘Zizhi’ may be a hybrid between R. multiflora Thunb. and R. rugosa ‘Plena’ [16, 17] or between R. davurica Pall. and R. rugosa ‘Plena’ [18]. As one of the most widely used cultivars in Yunnan, YN01 may be derived from YN02, but no book records are available. The origins of both R. ‘Tuwei’ and R. ‘Jinbian’ are in similarly ambiguous. There is also the problem that one R. rugosa line of cultivars may have several different names, although their phenotypic traits appear almost identical [15, 19].
In the last two decades, several studies have used different types of molecular markers to cluster and identify the potential progenitors of edible roses [19–21]. Currently, three major clusters – the native Rugosa group, the introduced Damask group, and the hybrid tea group – have been proposed [17, 22–25]. Rosa × damascena Herrm., the preferred culinary rose from India through Asia Minor and throughout the Middle East, was treated as a hybrid between R. gallica and R. phoenicia Bioss in the Mediterranean basin [26] but with further molecular inspection identified its triparental origin, with contributions from R. moschata Herrm. as an ovule donor and R. gallica L. and R. fedtschenkoana Regel donating pollen [20]. Due to limited markers and restricted sampling, tracing the origin of edible roses remains a challenge. However, this might change with access to high-quality chromosome-level genome and plastome sequences [27–31].
In this study, we identified the origins of cultivars of edible roses in China by reconstructing a well-supported plastome phylogeny for 88 Rosa lines in combination with haplotype phasing of ITSs. We wanted to test the hypothesis that edible cultivars used in China were derived, at least in part, from Rosa species with natural distributions in China. We also wanted to further discover the potential species contributing to the origin and domestication of R. × damascena. Identifying species and cultivars that are continuous bloomers is of immediate economic importance considering the demand for petals and hips.
Results and discussion
A well-resolved plastome phylogeny for Rosa
We collected cuttings of 23 lines representing 16 edible cultivars from eight main edible rose cultivation areas in China, 26 related wild species and/or their cultivars, and 39 genotypes with sequence information published previously (Table S1). Out of the 88 Rosa lines representing 34 wild species and 16 edible cultivars (see Materials and methods), 52 plastome sequences were newly assembled. These new plastomes were typical quadripartite and circular with 156 340–157 394 bp in length. They included a large single-copy region (LSC; 85 459–86 411 bp) and a small single-copy region (SSC; 18 715–18 875 bp) separated by two inverted repeat regions (IR; 25 973–26 100 bp). The final plastome alignment following removal of one IR was 141 366 bp in length with 10 321 variable sites (7.3%) and 4 946 parsimony informative sites (3.5%). A maximum likelihood (ML) analysis revealed a well-resolved phylogeny for the genus Rosa, with most nodes having bootstrap support above 90% (Fig. 1).
Figure 1.
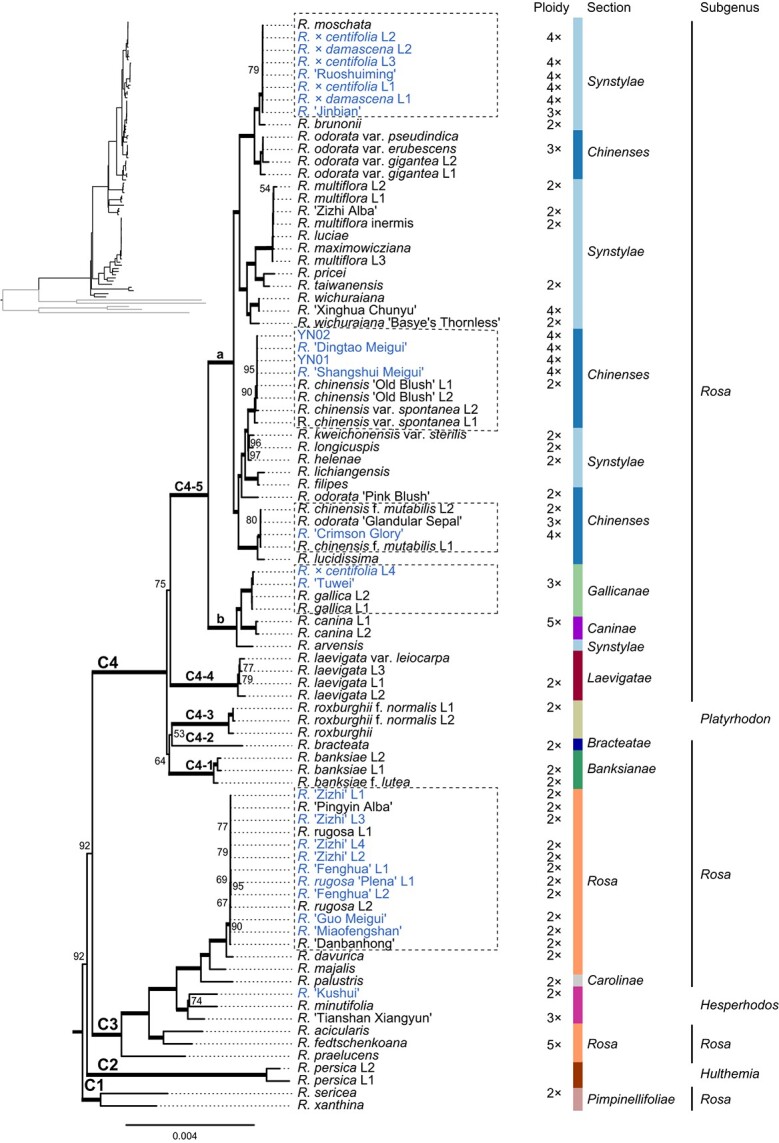
A well-supported plastome phylogeny for roses. A maximum-likelihood (ML) tree was constructed to show the phylogenetic relationships of Rosa species and cultivars. The upper left panel shows the overall phylogenetic frame with branches for outgroups marked by gray lines. Edible roses were marked in blue, while branches with thickened lines showed bootstrap support at 100%. Numbers on branches represent supports between 50% – 99%. Clades, subclades, and lineages were marked as C1 to C4, C4–1 to C4–5, and C4–5a/b, respectively. Ploidy levels, taxonomic sections and subgenera information for species [1–3, 33] are included.
Roses were grouped into four major clades: C1 – C4. C1 was sister to the other three clades, and it contained species associated within section Pimpinellifoliae DC. in subgenus Rosa. Subgenus Hulthemia (Dumort.) Focke comprised the C2 clade, which was the sister of the C3 and C4 clades. The C3 clade included genotypes from the subgenus Hesperhodos Cockerell and the sections Rosa (the former section Cinnamomeae DC.) and Carolinae DC. within subgenus Rosa. The C4 clade featured a unique 506–522 bp deletion in the psbM-trnD-GUC intergenic spacer (Fig. S1). The C4 clade clustered further into five subclades (C4–1 to C4–5). Subclades C4–1, C4–2, and C4–4 represent species in sections Banksianae Lindl., Bracteatae Thory, and Laevigatae Thory, respectively. C4–3 contained plants from subgenus Platyrhodon (Hurst) Rehder. All remaining genotypes were in subclade C4–5 with two well-supported lineages (C4–5a and C4–5b). Lineage C4–5a included only samples from sections Synstylae DC. and Chinenses DC., while C4–5b were composed of species from sections Gallicanae DC. and Caninae DC. as well as R. arvensis Huds. from section Synstylae. Therefore, our sampling covered all four subgenera and all sections in the subgenus Rosa [2, 3, 32].
Despite their centuries of ornamental and economic value to civilizations, Rosa represents one of the most difficult genera for phylogenetic reconstruction and taxonomic classification [1–3, 34–39]. Our plastome phylogenetic framework showed that most of the sections and subgenera defined previously [2, 32] were paraphyletic in origin, consistent with previous molecular phylogenetic studies [34, 37–40]. The three previous subgenera (Hesperhodos, Hulthemia, and Platyrhodon) appeared best sunk into subgenus Rosa. Early diversification within the genus Rosa occurred in two stages, with the first generating clades C1 – C4 and the second giving rise to subclades C4–1 to C4–5.
Potential maternal origin of edible roses
A well-supported plastome phylogeny allowed us to identify the potential maternal donor species involved in the natural and/or artificial evolution of edible roses. In this phylogeny, all cultivars of edible roses were embedded within the C3 clade and C4–5 subclade (Fig. 1). In the C3 clade, five edible cultivars (R. ‘Zizhi’, R. ‘Fenghua’, R. rugosa ‘Plena’, R. ‘Guo Meigui’, and R. ‘Miaofengshan’), R. ‘Pingyin Alba’, and R. ‘Danbanhong’ were all diploids (Table S1) and clustered with the diploid species R. rugosa (section Rosa). The plastomes of these genotypes differed by only a few SNPs. On the other hand, though R. ‘Kushui’ from Gansu served phylogenetically as the sister of R. minutifolia Engelm. in our analyses, these two lines varied rather significantly with more than 980 SNPs.
The remaining ten edible cultivars were grouped into the C4–5a (nine cultivars) and C4–5b (R. ‘Tuwei’ and L4 of R. × centifolia L.) subclades. Rosa gallica (section Gallicanae) appeared to be the species most closely linked to R. ‘Tuwei’ and R. × centifolia L4. It should be noted that R. ‘Tuwei’ was a triploid (Table S1). Rosa ‘Crimson Glory’, a tetraploid genotype (Table S1) widely used in Yunnan, shared a lineage with the diploid and native Chinese species R. chinensis f. mutabilis (Correvon) Rehder and R. odorata ‘Glandular Sepal’, sister to R. lucidissima H. Lév. in section Chinenses.
Rosa ‘Dingtao Meigui’ from Shandong, R. ‘Shangshui Meigui’ from Henan, and the two most widely used genotypes in Yunnan (YN01 and YN02) expressed almost no sequence divergence from R. chinensis var. spontenea (Rehder & E.H. Wilson) T.T. Yu & T.C. Ku and R. chinensis ‘Old Blush’, two of the major founding genotypes of the so-called “China roses” [8, 41–45]. These four edible roses were tetraploids (Table S1), in sharp contrast to the diploid ‘Old Blush’ [27, 28, 31].
The two R. × damascena lines, the three R. × centifolia genotypes (L1 – L3), the R. ‘Ruoshuiming’ from Henan, and the R. ‘Jinbian’ (used mainly for flavored teas in Yunnan) harbored almost no sequence variation with R. moschata in section Synstylae. Within this group, R. ‘Jinbian’ was the only cultivar of presumably triploid origin; all other lines were tetraploid (Table S1). Together, they formed the sister clade to the diploid R. brunonii Lindl.
Therefore, the five most likely maternal ancestors of commercial and edible roses were R. moschata (section Synstylae), R. chinensis var. spontanea (section Chinenses), R. chinensis f. mutabilis/R. odorata ‘Glandular Sepal’ (section Chinenses), R. rugosa (section Rosa), and R. gallica (section Gallicanae). As these edible roses were at different ploidy levels, intergenotypic hybridization may underlie their origin. Next, we used ITS haplotype phasing to identify the potential paternal genotypes for edible roses.
Incomplete concerted evolution allows haplotype phasing of rose ITSs
The genus Rosa showed incomplete concerted evolution of ITSs in the samples used in this study, a pattern also described in previous reports [37, 39]. For the 60 lines used in this study, we detected 135 and 174 haplotypes for ITS1 and ITS2 segments, respectively, with Illumina reads (Table S1). The final alignments for ITS1/ITS2 were 264/242 bp in length with 28.41% (75)/25.62% (62) variable sites, of which 78.7% (59)/83.9% (52) were parsimony informative. Only R. moschata and R. sericea Wall. ex Lindl. showed ITSs sequence homozygosity.
Concerted evolution, a process homogenizing sequence variations within repeated DNA arrays via gene conversion or unequal crossing-over [46, 47] may have been impeded by the following factors. All Rosa species and cultivars are woody and often propagated vegetatively. Roses have long generation times, as in other long-lived angiosperms [48, 49]. Interestingly, the nucleolar organizer regions (NORs) of roses, where nrDNA repeats are located, are always present on the shorter arm of submetacentric chromosomes [50–56]. Roses normally have only one NOR per genome; thus, genetic inheritance of NORs should follow the principles of Mendelian segregation. This means that the number of ITS haplotypes should be equal to or less than the number of NORs or ploidy levels. However, multiple ITS1 and ITS2 haplotypes were present in almost all of our edible roses, excluding the four lines of R. ‘Zizhi’, which had only one ITS1 haplotype but four ITS2 haplotypes (Table S1). The number of ITS haplotypes exceeded the ploidy levels in at least 17 lines, indicating that recombination might have occurred within two homologous NORs. This allows us to use ITS phasing (Fig. 2), in combination with plastome clustering, to trace the progenitor genotypes producing rose cultivars over time.
Figure 2.

Phasing strategy for ITS haplotypes in R. × damascena L1. a. Variations in ITS1 with numbers in brackets indicating the clean read numbers per variable site. The frequency of each base per site is shown as a percentage. b. A schematic strategy for assembling the ITS haplotypes using Illumina reads. The variable sites corresponded to the positions in a. Gray lines represent raw reads mapped to the reference. Sequence variation at each variable site was given as A/T/C/G, while a “-” represented a deletion. c. The assembled ITS1 haplotypes with the proportion of coverage given as a percentage.
ITS phasing reliably and accurately predicts the haplotypes of R. × damascena
We phased the haplotypes of ITS1 and ITS2 for the two lines of R. × damascena (Fig. 2). As the read coverage for both segments was ~500 times, all possible haplotypes could be recovered. For both R. × damascena roses, three and four haplotypes were identified for ITS1 and ITS2, respectively. These assembled haplotypes were nearly identical to the ITS sequences of three Damask lines reported by Iwata et al. [20] (Data S1 and S2). Sanger sequencing confirmed that all ITS haplotypes based on the Illumina reads were reliable and accurate (Figs. S2–S13). Phylogenetic clustering of these ITS haplotypes revealed that they were grouped with the sequences of three known species: R. moschata, R. gallica, and R. fedtschenkoana (Figs. S14, S15). We identified one more haplotype of ITS2 shared with R. gallica (Fig. S15). Therefore, we recovered all three expected donor species identified historically for this ancient hybrid (see also below). Together, haplotype phasing of ITSs provided a reliable and reproducible method to identify potential founder genotypes.
A phylogenetic analysis showed a similar pattern for both ITS1 and ITS2 segments (Figs. 3 and 4), although they varied in their numbers of haplotypes. However, the bootstrap supports for most branches were weak. Given that there was extensive reticulation and incomplete-concerted evolution of ribosomal repeats in Rosa [36], we focused only on the sequences most closely related to plants with edible flowers. Many sequences from different genotypes or different species showed no sequence variation. This indicated that they might share the same progenitors. We also observed that different haplotypes for ITS1 and ITS2 segments from one cultivar could be clustered into highly dispersed phylogenetic branches. This showed a complex nuclear composition for these edible roses. Hybridization between species from different branches might have occurred.
Figure 3.
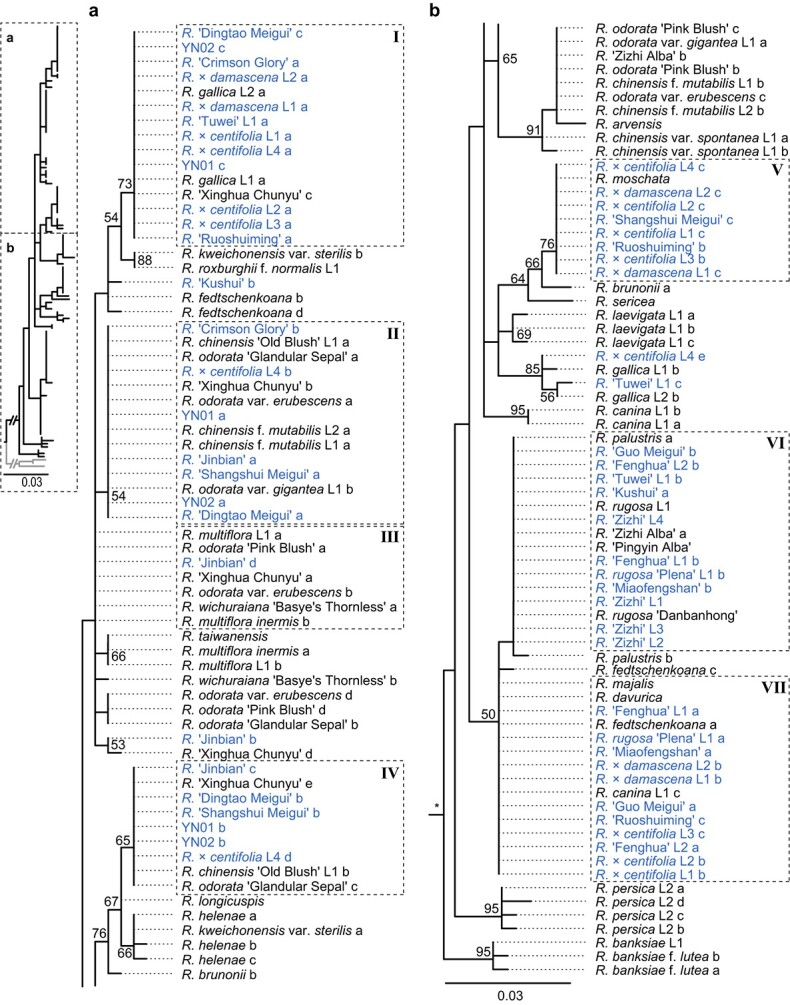
Phylogenetic clustering of ITS1 haplotypes. Bootstrap supports >50% are marked with numbers along each branch. Edible rose cultivars are labeled in blue. Alleles with identical sequences for edible roses in the same clades are marked with dashed rectangles. Their group numbers are given here as Roman numerals. a and b show the topologies for parts a and b in the left-hand panel, respectively.
Figure 4.

Phylogenetic clustering for ITS2 haplotypes. Rectangle numbers are ordered according to cooccurrence with ITS1 haplotypes (Fig. 3).
Predicting the maternal and paternal progenitors of edible roses
We next predicted the progenitors of edible roses by combining the sequence and phylogenetic information from both plastomes (Fig. 1) and ITSs (Figs. 3 and 4). While plastomes were supposed to be of maternal origin, the possibility of incomplete lineage sorting or capture cannot be excluded completely [57, 58]. ITS sequences were of both parental origins. Consequently, those haplotypes that were indistinguishable phylogenetically or close to each other were treated as belonging to the same progenitor. Wild species containing the same or similar plastome sequences with edible roses were considered potential ovule donors, especially when they shared identical or highly similar ITS haplotypes. Species sharing other alleles of ITS sequences were proposed as pollen donors. With this information, we defined the potential maternal and paternal ancestors of all edible roses (see Table 1 and Fig. 5). Based on the maternal ancestors, we classified the edible roses into six groups: Moschata, Gallica, Spontanea, Mutabilis, Rugosa, and Kushui. All cultivars of edible roses grown in China were ultimately derived from approximately nine wild species (Fig. 5) distributed naturally from the Mediterranean basin eastward through temperate and/or montane Eurasia.
Table 1.
Hypothetical progenitors for edible roses. ITS haplotype names in bold show the postulated maternal donors. Haplotypes marked with asterisks are most closely related to but not the same as those of the edible roses. The Roman numerals refer to the dashed boxes in Figs. 3 and 4. N.D., not determined
| Edible rose | Most closely related wild species | Predicted ancestors | ||
|---|---|---|---|---|
| Plastome | ITS1 | ITS2 | ||
|
Rosa × centifolia L1–L3 R. × damascena L1–L2 R. ‘Ruoshuiming’ |
R. moschata |
V (R. moschata)
I (R. gallica) VII (R. majalis/R. davurica/R. fedtschenkoana) |
V (R. moschata)
I-1 (R. gallica) I-2 (R. gallica) VII (R. majalis/R. davurica/R. fedtschenkoana) |
R. moschata × (R. gallica + R. majalis/R. fedtschenkoana/R. davurica) |
| R. ‘Jinbian’ | R. moschata | II + III + IV (R. ‘Xinghua Chunyu’) R. ‘Xinghua Chunyu’ d |
II + II + IV (R. ‘Xinghua Chunyu’) | R. moschata × R. ‘Xinghua Chunyu’ |
| R. × centifolia L4 | R. gallica* |
I (R. gallica)
R. gallica L1 b* V (R. moschata) II (R. chinensis ‘Old Blush’/R. odorata ‘Glandular Sepal’) IV (R. chinensis ‘Old Blush’/R. odorata ‘Glandular Sepal’) |
I-1 (R. gallica)
R. gallica L1 c R. gallica L1 b V (R. moschata) II (R. chinensis ‘Old Blush’/R. odorata ‘Glandular Sepal’) IV (R. chinensis ‘Old Blush’/R. odorata ‘Glandular Sepal’) |
R. gallica × (R. moschata + R. chinensis ‘Old Blush’/R. odorata ‘Glandular Sepal’) |
| R. ‘Tuwei’ L1 | R. gallica* |
I (R. gallica)
R. gallica L2 b* VI (R. rugosa) |
I-1 (R. gallica)
I-2 (R. gallica) VI-1 (R. rugosa) R. rugosa L1 c R. majalis a/R. canina b |
R. gallica × R. rugosa |
| R. ‘Crimson Glory’ |
R. chinensis f. mutabilis/ R. odorata ‘Glandular Sepal’ |
II (R. chinensis f. mutabilis/R. odorata ‘Glandular Sepal’)
I (R. gallica) |
II (R. chinensis f. mutabilis/R. odorata ‘Glandular Sepal’)
I-1 (R. gallica) |
R. chinensis f. mutabilis/R. odorata ‘Glandular Sepal’ × R. gallica |
| YN01 YN02 R. ‘Dingtao Meigui’ |
R. chinensis ‘Old Blush’ |
II + IV (R. chinensis ‘Old Blush’)
I (R. gallica) |
II + IV (R. chinensis ‘Old Blush’)
I-1 (R. gallica) |
R. chinensis ‘Old Blush’ × R. gallica |
| R. ‘Shangshui Meigui’ | R. chinensis ‘Old Blush’ |
II + IV (R. chinensis ‘Old Blush’)
V (R. moschata) |
IV + IV (R. chinensis ‘Old Blush’)
V (R. moschata) |
R. chinensis ‘Old Blush’ × R. moschata |
|
R. ‘Fenghua’ L1–L2 R. rugosa ‘Plena’ L1 R. ‘Guo Meigui’ R. ‘Miaofengshan’ |
R. rugosa |
VI (R. rugosa)
VII (R. davurica) |
VI-1 (R. rugosa)
VII-1 (R. davurica) |
R. rugosa × R. davurica |
| R. ‘Zizhi’ L1–L4 | R. rugosa/R. ‘Pingyin Alba’ | VI (R. rugosa/R. ‘Pingyin Alba’) |
VI-1 (R. rugosa/R. ‘Pingyin Alba’)
VI-2 (R. ‘Pingyin Alba’) VI-3 (R. ‘Pingyin Alba’) VII-2 (R. davurica/R. ‘Pingyin Alba’) |
R. rugosa/R. ‘Pingyin Alba’ |
| R. ‘Kushui’ | N.D. |
R. fedtschenkoana b/d* VI (R. rugosa) |
R. majalis a/R. canina b VI-1 (R. rugosa) |
? × R. rugosa |
Figure 5.
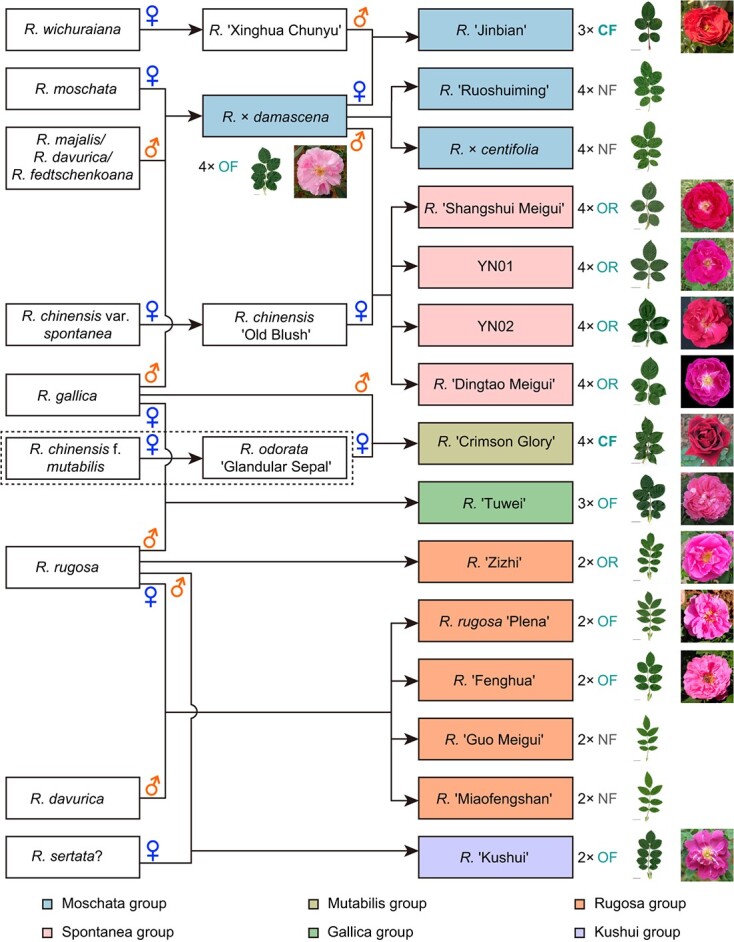
Hypothetical origins of edible roses. Potential maternal or paternal progenitors are marked here with blue ♀ or orange ♂, respectively. Ploidy levels (2x to 4x), flowering patterns (OF, once flowering; CF, continuous flowering; OR, occasional reblooming; NF marks the four lines that did not flower at the KIB.), and leaf and flower morphology are on the right (the black bar on each leaf photo represents 2 cm).
The Moschata group and new insights for the origin of R. × damascena
The Moschata group contained seven lines representing four cultivars, R. × damascena (two lines), R. × centifolia (three lines), R. ‘Ruoshuiming’ (one line), and R. ‘Jinbian’ (one line). The R. × centifolia L4 was not in this group (see below). All of these tetraploid lines featured identical plastome and ITS sequences and were most likely to have the same origins. Under our growth conditions, the only morphological variation observed was that the three lines of R. × centifolia and the one line of R. ‘Ruoshuiming’ developed fewer prickles on their stems than R. × damascena.
Our analyses confirmed that the three wild progenitors reported previously for R. × damascena included R. moschata as the potential ovule donor and R. gallica and R. fedtschenkoana as potential pollen donors (Table 1) [20]. We also noted that R. majalis and R. davurica could have also served as potential pollen donors, as both species shared a copy of ITS1 (VII) and ITS2 (VII-2) with R. fedtschenkoana (Figs. 3 and 4). Natural distributions of R. majalis and R. gallica overlapped in modern Iran (Persia), where R. × damascena originated (http://www.plantsoftheworldonline.org) [59]. It is possible that, in addition to the three species proposed by Iwata et al. [20], two additional species, R. majalis and R. davurica, may have contributed to the many lineages of Damask roses.
Rosa ‘Jinbian’ was the only triploid edible rose in the Moschata group. All ITS haplotypes of R. ‘Jinbian’ were identical and/or closely related to the tetraploid China rose, R. ‘Xinghua Chunyu’, a sister to R. × damascena (Table 1) [60]. Hence, R. × damascena and R. ‘Xinghua Chunyu’ were likely the parents of R. ‘Jinbian’. Surprisingly, the plastome and one haplotype of ITSs of R. ‘Xinghua Chunyu’ showed an almost identical sequence to R. wichuraiana ‘Basye’s Thornless’. ‘Basye’s Thornless’ is regarded as a founder genotype in the domestication of modern roses [61]. Since R. ‘Xinghua Chunyu’ and R. ‘Jinbian’ shared two ITS haplotypes with a second China rose (R. chinensis ‘Old Blush’), we propose that R. ‘Jinbian’ might be a hybrid between R. × damascena and R. ‘Xinghua Chunyu’ with additional genetic resources from R. wichuraiana ‘Basye’s Thornless’ and R. chinensis ‘Old Blush’.
The Gallica group and R. ‘Tuwei’
Maternally, the edible Rosa ‘Tuwei’ and R. × centifolia L4 clustered with R. gallica (Fig. 1). Flowers of R. ‘Tuwei’ were consumed primarily in Guangdong for more than 800 years, but its origin until now has remained obscure [62]. The leaves and petals of this variety have morphological similarities with those of R. rugosa, but the inner petals of the former curl inward, a pattern not seen in flowers of R. rugosa (Fig. 5, Fig. S16). As a China rose, R. ‘Tuwei’ was proposed to contain additional genetic resources in addition to R. rugosa due to its scent variation [62]. Haplotype phasing revealed that all its ITSs came from haplotypes of R. gallica and R. rugosa. Rosa ‘Tuwei’ then appeared to be of hybrid origin between these two wild species, with R. rugosa more likely as the pollen donor.
Rosa × centifolia L4 came from our original collection in the Kunming Botanical Garden. The initial identification of this cultivar was based on its typical deep-cup-type flowers and large number of petals amassed in each flower (Fig. S17). Congruent with a previous report [63], the plastome sequence of L4 was closer to that of R. gallica (Fig. 1). L4 had two ITS haplotypes identical to those of the other three R. × centifolia lines (Table 1), which featured two other haplotypes identical to R. chinensis ‘Old Blush’ and R. odorata ‘Glandular Sepal’. Our results suggested that our KIB L4 could likely be ascribed to R. × centifolia, while the other three lines, described above, were more likely to be R. × damascena. In corroboration with the sequence variations, morphological traits in our L4 differed significantly from those of the other three lines (Figs. S16, S17). As R. × centifolia was not widely used for food historically, our L4 was excluded from our origin model (Fig. 5).
The Spontanea group
Four tetraploid edible roses, YN01, YN02, R. ‘Dingtao Meigui’, and R. ‘Shangshui Meigui’ were clustered in the Spontanea group. All four showed similar flower and leaf morphology and identical plastome sequences (Fig. S16). In fact, all four cultivars shared the same plastome sequences as R. chinensis ‘Old Blush’ L1 but featured eight constant SNPs with R. chinensis var. spontanea lines. Two ITS haplotypes were identical to R. chinensis ‘Old Blush’. The third haplotype of R. ‘Dingtao Meigui’, YN01 and YN02, was the same as the R. gallica type (ITS1 I & ITS2 I-1). In contrast, the third haplotype of R. ‘Shangshui Meigui’ was an R. moschata type (ITS1 V & ITS2 V). Given the morphological similarity, we suggest that the ancestral pollen donor was R. × damascena, which had both the R. gallica and R. moschata types of ITS haplotypes. These four tetraploids may be derived from the same crossing combination of R. chinensis ‘Old Blush’ × R. × damascena. Our results indicate that these four edible tetraploids in the Spontanea group had the same origin.
The Mutabilis group and R. ‘Crimson Glory’
Rosa ‘Crimson Glory’ had good records, with parents identified as R. ‘Catharine Kordes’ × R. ‘W. E. Chaplin’ (by Wilhelm J.H. Kordes II, 1935; https://www. helpmefind.com/roses). Although information on the wild species contributing to the origins of its two progenitors was missing, our plastome and ITS phasing data suggested that the maternal ancestor of R. ‘Crimson Glory’ was R. chinensis f. mutabilis or R. odorata ‘Glandular Sepal’. Rosa gallica was the most likely pollen donor (Table 1).
The Rugosa group
All the edible cultivars in the Rugosa group were diploids with typical rugose leaves (Fig. 5). They clustered with the same maternal clade as R. rugosa sensu strictu (Fig. 1). All of them shared the same unique ITS1 haplotype and one identical ITS2 sequence (VI-1) with the parent species R. rugosa (Figs. 3 and 4). As R. ‘Zizhi’ and R. ‘Pingyin Alba’ (with white flowers; Fig. S18) featured the same ITS1 haplotype and the same three ITS2 haplotypes (Table 1), we propose that R. ‘Zizhi’ and R. ‘Pingyin Alba’ may have originated directly from R. rugosa, instead of the earlier hypothesis that this cultivar is of hybrid origin between R. davurica × R. rugosa ‘Plena’ or R. multiflora × R. rugosa ‘Plena’ [16–18]. Although they have been given very different names, the remaining edible cultivars in the Rugosa group (R. ‘Guo Meigui’, R. ‘Fenghua’, R. ‘Miaofengshan’, R. rugosa ‘Plena’) were more likely hybrids between R. rugosa × R. davurica (Table 1). The natural distributions of the parent species R. rugosa and R. davurica overlapped broadly in China, Japan, Korea, Mongolia, and Russia [1]. Introgression was detected previously between these two species [64]. In our study, edible cultivars in the Rugosa group seemed to feature a simpler genetic background than other groups. This may also reflect a shortcoming of having only used ITS sequences in the analysis.
The origin of R. ‘Kushui’
As the primary edible rose grown and used in Gansu, R. ‘Kushui’ had one haplotype of ITSs identical to that of R. rugosa (ITS1 VI and ITS2 VI-1). The second allele of ITS1 (b) was highly similar to the b and d copies of ITS1 from R. fedtschenkoana (Fig. 3). The second ITS2 (b) was identical to that of R. majalis (a) and R. canina L. (L1 b; Fig. 4). This indicated another complex origin. We could not identify the closest maternal contributor, as the genetic variation between R. ‘Kushui’ and R. minutifolia was extremely large, with more than 980 SNPs (Fig. 1). Rosa sertata, proposed previously as a progenitor of R. ‘Kushui’, was missing in our data matrix. A comparison using known chloroplast markers (matK, rbcL, atpB-rbcL and trnH-psbA intergenic spacers) and ITS1 sequences showed that R. ‘Kushui’ differed significantly from R. sertata (Data S3 – S6) [36, 37, 65, 66]. Further analyses using more samples should help clarify its origin.
Variation in the floral phenology of edible roses
While continuous flowering (CF) is a preferred trait in commercial roses 67,68, only two cultivars, R. ‘Jinbian’ and R. ‘Crimson Glory’, showed this flowering mode under growing conditions at the KIB campus (Fig. 5). Unfortunately, the cuttings of R. ‘Guo Meigui’, R. ‘Miaofengshan’, R. × centifolia (L1 to L3), and R. ‘Ruoshuiming’ transplanted to KIB has not yet bloomed. Rosa ‘Zizhi’, R. ‘Dingtao Meigui’, R. ‘Shangshui Meigui’, YN01 and YN02 were occasional rebloomers in autumn. All remaining cultivars flowered in mid-March for approximately four to six weeks only once a year.
In summary, via plastome phylogeny reconstruction and ITS phasing, we showed that at least nine wild species were involved in the origins of the major Chinese edible roses and that the majority of cultivars bloomed only once a year (Fig. 5). To generate new varieties featuring continuous flowering, a preferred trait in the food and flavor industry, our study offers an opportunity to select likely combinations in future crosses. With the availability of high-quality genome sequences [27, 28, 31, 69], high-density genetic maps [70, 71], single-copy nuclear markers [72], and other genetic tools, such as targeted gene editing, bulk segregation analysis, and whole-genome prediction [67, 68], our work should facilitate future genome-breeding programs in commercial roses.
Materials and methods
Plant materials and flowering behavior scoring
Edible roses were collected from eight main edible rose cultivation areas in China, and for some cultivars, such as Rosa ‘Zizhi’, R. ‘Fenghua’, R. rugosa ‘Plena’, R. × damascena and R. × centifolia, materials were collected from more than one area. Related wild species and/or their cultivars were mainly collected from Kunming Botanical Garden (KBG), with some materials obtained from the Germplasm Bank of Wild Species, Kunming Institute of Botany (CAS), and Yunnan Irose company. Rose cuttings were propagated and planted in the KBG within the campus of the Kunming Institute of Botany (KIB, Yunnan), CAS. These plants, together with an additional 39 genotypes with sequences downloaded from GenBank, covered all subgenera and all sections in the subgenus Rosa (Table S1) [2, 32]. Together, there were 88 rose lines including all potential wild progenitors, but R. foetidaHerrm. contributed to modern rose domestication [9, 42]. The identification of these species was determined primarily following the Flora of China [1]. Some species names were revised according to updated information in the plantlist (http://www.theplantlist.org). We kept the names of R. wichuraiana Crep and R. brunonii Lindl. For edible cultivars, morphological characters defining and segregating leaves, flowers, and stem prickles were photographed using specimens grown in the same garden. “China roses” in this study refer to the cultivars and wild species that originated in China and were brought to Europe before 1867 [8, 41, 42]. These materials are known to contribute important genetic traits and resources to modern rose domestication.
Flowering periods of these edible cultivars were recorded over two consecutive years (from March 2019 to April 2021) [73]. Briefly, most of the KIB collection began flowering between the middle of March and early May. Cultivars producing flowers continuously from mid-March until mid-November were categorized as continuous flowering (CF). Plants that ceased flowering after four to six weeks between mid-March and mid-May were categorized as once-flowering (OF). Cultivars observed to stop flowering after the initial mid-March/mid-May blooming season but that produced a few flowers in early autumn were categorized as occasional rebloomers (ORs).
DNA extraction, sequencing, and plastome assembly
Young fresh and healthy leaves were collected and frozen immediately in liquid nitrogen and used for total DNA extraction with a modified cetyltrimethylammonium bromide (CTAB) method [74]. DNA samples were sequenced following protocols provided by Illumina with a paired-end method at Biomarker Technologies Co., Ltd. (Beijing, China). For each line, ~3 Gb of randomly selected data was used for plastome assembly and ITS phasing. The plastome sequences were assembled de novo with SPAdes 3.12.0 [75] using the GetOrganelle pipeline [76]. Using the plastome of R. wichuraiana ‘Basye’s Thornless’ as a reference [30], the final assembly was constructed and annotated with Bandage 0.8.1 [77] and Geneious 9.1.4 (Biomatters Ltd., Auckland, New Zealand), respectively. The plastomes of five additional species in subfamily Rosoideae (Potentilla purpurea (Royle) Hook. f., Fragaria vesca L., Rubus fockeanus Kurz, Rubus niveus Thunb., and Fallugia paradoxa (D. Don) Endl. ex Torr.) were downloaded from GenBank and employed as outgroups (Table S1).
Phasing the ITS haplotypes
We used the same pipelines and software to assemble and annotate the nuclear ribosomal DNA (nrDNA) sequences by referring to the sequence for Pyrus communis L. (MN577903) in the Rosaceae. We used a rigorous mapping process to justify the status of ambiguous sites, which could be ‘corrected’ in the SPAdes within the GetOrganelle pipeline. We mapped the clean reads to the initially assembled ITS sequences in the nrDNA using bowtie2 [78] in Geneious.
Consistent with previous studies that reported certain levels of heterozygous ITS markers in several rose varieties [36, 37, 39, 79, 80], we detected obvious heterozygosity in the generated assemblies and focused on the variable sites within ITS1 and ITS2 regions of a specific line. The heterozygous sites could be caused either by allelic sequence variation or by sequencing errors, which should be random and at a relatively lower level. By post manual counting of each ambiguous position, only the nucleotide composition per ambiguous site with >5% of total mapped reads was considered allelic variation and kept for further analysis. The haplotypes were then distinguished by sorting variable reads within the overlapping read group. As ITS1 and ITS2 were separated by a highly conserved 5.8S rRNA (159 bp), which caused the assembly of a continuous ITS contig difficult with the ~150 nt Illumina reads, we assembled and analyzed the ITS1 and ITS2 sequences separately. Each inferred haplotype was labeled using a lowercase letter in a decreasing order with its proportion. A schematic diagram for haplotype phasing is shown in Fig. 2.
We next used PCR followed by Sanger sequencing to verify the accuracy of assembled haplotypes. With universal primers ITS-4 and ITS-A, we obtained the ITS fragments for ten edible roses selected at random [39, 81]. The heterozygous states for sites within ITS1 and ITS2 were checked manually and compared to those generated with Illumina reads. Additionally, PCR products for R. × damascena L1 were cloned into the pClone007 Versatile Simple Vector (Tsingke Biotechnology Co., Ltd.). Sixteen clones were sequenced and examined for their sequence identity to the phased haplotypes generated with Illumina sequencing.
Sequence alignment and phylogeny analyses
The plastome and phased ITS sequences were aligned with MAFFT in Geneious [82]. The alignment was used for a maximum-likelihood (ML) phylogenetic analysis in RAxML 8.2.11 with the best-fit model (GTR + I + G) generated with jModelTest [83, 84]. Bootstrap analyses were done with 1000 replicates.
Genome size estimation and ploidy level prediction
Flow cytometry (FCM) was used to assess the holoploid genome sizes of rose lines [85]. Young leaves were sampled for nuclear suspension with the option A (one-step protocol) according to Dolezel et al. [86]. The nuclear samples were measured with a BD FACScalibur Flow Cytometer (USA) with maize B73 (2.3 Gb) as the internal reference [87]. The relative genome size was estimated following the equations described [86]. Ploidy levels were predicted following the method described [88] using known genome sizes and ploidy levels for the same species [88–92]. We predicted diploids with genome sizes between 0.37–0.6 Gb. In comparison, triploids were predicted between 0.64–0.7 Gb, tetraploids between 0.83–1.14 Gb, and with pentaploids larger than 1.2 Gb (Table S1).
Supplementary Material
Acknowledgments
We thank Yan-Xia Jia for measuring the C-values for all samples. We appreciate Shubin Li, Shulan Chen, Hongyuan Yu, Yuanlin Lv, Xianshui Meng, Jie Cai, and Wang Xi for their help in providing and cultivating the rose plants. We are grateful to Jun-Yun Wang for providing rose information. We acknowledge De-Zhu Li and Lian-Ming Gao for useful discussions. Prof. Peter Bernhardt served as the ‘word doctor’. This work was partially facilitated by the Germplasm Bank of Wild Species and the Kunming Botanical Garden, Kunming Institute of Botany, Chinese Academy of Sciences.
Research was supported by the following grants: the Science and Technology Leading Talent Project of Yunnan, China (2017HA014), the Strategic Priority Research Program of the Chinese Academy of Sciences (CAS; XDB31000000), the CAS Pioneer Hundred Talents Program (292015312D11035), CAS Key Laboratory for Plant Diversity and Biogeography of East Asia, and Yunnan Recruitment Program of Experts in Science.
Author contributions
J-Y H conceived the project; J-Y H, X-D J, X D, and W F coordinated the research; W-H C, X-D J, W F, and Z-Q S collected the samples, extracted the genomic DNA, and performed the sequencing; W-H C, X-Y D, M-C Z, X D, and X-D J analyzed and visualized the data. X-D J, D W, and W-H C monitored the flowering time behavior and other traits. J-Y H and W-H C wrote the paper. All authors have read and approved the final manuscript.
Data availability
All sequences had been submitted to GenBank (Table S1).
Declaration of Competing Interest
The authors declare no conflict interests. Patent related to this manuscript is under protection of CN202110640825.X.
Supplementary data
Supplementary data is available at Horticulture Research Journal online.
References
- 1. Gu C, Robertson KR. Rosa Linnaeus. In: Wu CY, Raven PH, eds. Flora of China Vol. 9, Beijing/St, Louis: Science Press/Missouri Botanical Garden Press, 2003,339–81. [Google Scholar]
- 2. Wissemann V. Conventional Taxonomy (Wild Roses). In: Roberts AV, ed. Encyclopedia of Rose Science. London: Elsevier, 2003,111–7. [Google Scholar]
- 3. Wissemann V. Conventional Taxonomy (Wild Roses). In: Reference Module in Life Sciences. London: Elsevier, 2017, 1–7. [Google Scholar]
- 4. Cutler RR. Culinary Uses and Nutritional Value. In: Roberts AV, ed. Encyclopedia of Rose Science. London: Elsevier, 2003,707–16. [Google Scholar]
- 5. Friedman H, Rot I, Agami Oet al. Edible flowers: new crops with potential health benefits. Acta Hortic. 2007;755:283–90. [Google Scholar]
- 6. Cai YZ, Xing J, Sun Met al. Phenolic antioxidants (hydrolyzable tannins, flavonols, and anthocyanins) identified by LC-ESI-MS and MALDI-QIT-TOF MS from Rosa chinensis flowers. J Agric Food Chem. 2005;53:9940–8. [DOI] [PubMed] [Google Scholar]
- 7. Hollingsworth BE. Flower Chronicles. Chicago: University of Chicago Press; 1988, 302. [Google Scholar]
- 8. Le Rougetel H. A heritage of roses. Keene: Stemmer House Pub. 1988, 176. [Google Scholar]
- 9. Liorzou M, Pernet A, Li Set al. Nineteenth century French rose (Rosa sp.) germplasm shows a shift over time from a European to an Asian genetic background. J Exp Bot. 2016;67:4711–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Z. The development status and countermeasures of edible rose industry. Shaanxi Journal of Agricultural Sciences. 2017;63:84–8. [Google Scholar]
- 11. Chen Y. Comparison of aroma components and influence factors of edible rose cultivars in Yongdeng County. Master 70. Gansu Agricultural University, 2016. [Google Scholar]
- 12. Zhang W, Wang C, Zhang Jet al. Research progress of edible rose. Chinese Wild Plant Resources. 2016;35:24–30. [Google Scholar]
- 13. Zong N, Zhang Z. Research Progress in the progressing and exploitation edible roses. Farm Products Processing. 2017;440:44–52. [Google Scholar]
- 14. Fu Y, Zhao F, Fu Xet al. Countermeasure research on present situation and development of edible rose industry in Bajie Anning City of Yunnan. Journal of Kunming University. 2016;38:85–9. [Google Scholar]
- 15. Li W, Wang W. Primary research on the Rosa rugosa resources in China. Acta Horticulturae Sinica. 1983;10:211–5. [Google Scholar]
- 16. Wang W. Studies on the Pollen Morphology and Viability of Rosa rugosa. Master 76. Shandong Agricultural University, 2004. [Google Scholar]
- 17. Wu L. Phylogenetic Relationship Analyzed Using RAPD Markers Among the Germplasm of Rosa rugosa Thunb in Pingyin. Master 58. Shandong Agricultural University, 2005. [Google Scholar]
- 18. Zhao H, Wang J, Ding Xet al. Compatibility of interspecific crossing between several Rosa species and modern rose cultivars. Acta Botan Boreali-Occiden Sin. 2015;35:0743–53. [Google Scholar]
- 19. Wang H, Yao L, Cai R. Germplasm resources classification and evaluation of oil-bearing rose in China based on genetic diversity. Doctor 209. Shanghai Jiao Tong University, 2013. [Google Scholar]
- 20. Iwata H, Kato T, Ohno S. Triparental origin of damask roses. Gene. 2000;259:53–9. [DOI] [PubMed] [Google Scholar]
- 21. Deng H, Gao X, Li Xet al. Molecular evidence for hybridization origin of Rosa × sterilis (Rosaceae). J Plant Resour Environ. 2015;24:10–7. [Google Scholar]
- 22. Feng L. Study on evolution of wild germplasm resources and their genetic relationship with cultivars of Rosa rugosa. Doctor 129. Shangdong Agricultural University, 2007. [Google Scholar]
- 23. Cai F. Rosa rugosa germplasm and their medical material identification. Master 95. Peking Union Medical Colledge, Chinese Academy of Medical Sciences, 2008. [Google Scholar]
- 24. Xu Z. Analysis of Genetic Diversity and Construction of Fingerprint of Rosa rugosa by SRAP. Master 60. Shandong Agricultural University, 2011. [Google Scholar]
- 25. Wang H, Yao L, Cai Ret al. Genetic relationship analyses of oil-bearing roses in China using matK sequences. Sci Hortic. 2012;137:121–4. [Google Scholar]
- 26. Widrlechner MP. History and utilization of Rosa damascena. Econ Bot. 1981;35:42–58. [Google Scholar]
- 27. Raymond O, Gouzy J, Just Jet al. The Rosa genome provides new insights into the domestication of modern roses. Nat Genet. 2018;50:772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saint-Oyant LH, Ruttink T, Hamama Let al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nature Plants. 2018;4:473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li SB, Qu XJ, Zhong MCet al. Characterization of the complete chloroplast genome of Rosa chinensis 'Old Blush' (Rosaceae), an important cultivated Chinese rose. Acta Hortic. 2019;1232:119–24. [Google Scholar]
- 30. Cui W-H, Zhong M-C, Du X-Yet al. The complete chloroplast genome sequence of a rambler rose, Rosa wichuraiana (Rosaceae). Mitochondrial DNA Part B. 2020;5:252–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhong M-C, Jiang X-D, Yang G-Qet al. A genomic link in China roses: and they all lived prickly but water deficient ever after? Natl Sci Rev. 2021. 10.1101/2020.07.16.207795. [DOI] [Google Scholar]
- 32. Rehder A. Manual of Cultivated Trees and Shrubs. New York: MacMillan; 1940 [Google Scholar]
- 33. Wang K, Zhang W, Wang Qet al. The phylogenetic position and hybrid origination of Rosa praelucens Byhouwer. Journal of Plant Genetic Resources. 2018;19:1006–15. [Google Scholar]
- 34. Fougere-Danezan M, Joly S, Bruneau Aet al. Phylogeny and biogeography of wild roses with specific attention to polyploids. Ann Bot. 2015;115:275–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matsumoto S, Nishio H, Ueda Yet al. Phylogenetic analyses of genus Rosa: Polyphyly of section Pimpinellifoliae and origin of Rosa x fortuniana Lindl. Acta Hortic. 2001;357–63. [Google Scholar]
- 36. Ritz CM, Schmuths H, Wissemann V. Evolution by reticulation: European dogroses originated by multiple hybridization across the genus Rosa. J Hered. 2005;96:4–14. [DOI] [PubMed] [Google Scholar]
- 37. Wissemann V, Ritz CM. The genus Rosa (Rosoideae, Rosaceae) revisited: molecular analysis of nrITS-1 and atpB-rbcL intergenic spacer (IGS) versus conventional taxonomy. Bot J Linn Soc. 2005;147:275–90. [Google Scholar]
- 38. Bruneau A, Starr JR, Joly S. Phylogenetic relationships in the genus Rosa: new evidence from chloroplast DNA sequences and an appraisal of current knowledge. Syst Bot. 2007;32:366–78. [Google Scholar]
- 39. Zhu Z-M, Gao X-F, Fougere-Danezan M. Phylogeny of Rosa sections Chinenses and Synstylae (Rosaceae) based on chloroplast and nuclear markers. Mol Phylogenet Evol. 2015;87:50–64. [DOI] [PubMed] [Google Scholar]
- 40. Liu C, Wang G, Wang Het al. Phylogenetic relationships in the genus Rosa revisited based on rpl16, trnL-F, and atpB-rbcL sequences. HortScience. 2015;50:1618–24. [Google Scholar]
- 41. Hurst CC. Notes on the origin and evolution of our garden roses. J R Hortic Soc. 1941;73–82, 242,–50, 282–9. [Google Scholar]
- 42. Wylie A. The history of garden roses, part 1. J R Hortic Soc. 1955;555–69. [Google Scholar]
- 43. Soules VA. Analysis of Genetic Diversity and Relationships in the China Rose Group. Master 79. Texas A&M University, 2009. [Google Scholar]
- 44. Meng J, Fougère-Danezan M, Zhang LBet al. Untangling the hybrid origin of the Chinese tea roses: evidence from DNA sequences of single-copy nuclear and chloroplast genes. Österreichische botanische Zeitschrift. 2011;297:157–70. [Google Scholar]
- 45. Tan J, Wang J, Luo Let al. Genetic relationships and evolution of old Chinese garden roses based on SSRs and chromosome diversity. Sci Rep. 2017;7:15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dover G. Molecular drive-a cohesive mode of species evolution. Nature. 1982;299:111–7. [DOI] [PubMed] [Google Scholar]
- 47. Hillis DM, Moritz C, Porter CAet al. Evidence for biased gene conversion in concerted evolution of ribosomal DNA. Science. 1991;251:308–10. [DOI] [PubMed] [Google Scholar]
- 48. Sang T, Crawford DJ, Stuessy TF. Documentation of reticulate evolution in peonies (Peonia) using internal transcribed spacer sequences of nuclear ribosomal DNA-implications for biogeography and concerted evolution. Proc Natl Acad Sci U S A. 1995;92:6813–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alvarez I, Wendel JF. Ribosomal ITS sequences and plant phylogenetic inference. Mol Phylogenet Evol. 2003;29:417–34. [DOI] [PubMed] [Google Scholar]
- 50. Ma Y, Islam-Faridi MN, Crane CFet al. In situ hybridization of ribosomal DNA to rose chromosomes. J Hered. 1997;88:158–61. [Google Scholar]
- 51. Kirov IV, Van Laere K, Van Roy Net al. Towards a FISH-based karyotype of Rosa L. (Rosaceae). Comparative Cytogenetics. 2016;10:543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang T, Jian H, Tian Met al. Physical location of 45S rDNA and 5S rDNA in the genomes of three wild rose species. Acta Horticulturae Sinica. 2014;41:994–1000. [Google Scholar]
- 53. Zhang T, Jian H, Mo Xet al. Karyotype of Rosa longicuspis Bertol. Based on rDNA FISH. Southwest China Journal of Agricultural Sciences. 2018;31:2036–40. [Google Scholar]
- 54. Fang Q, Tian M, Zhang Tet al. Karyotype analysis of Rosa praelucens and its closely related congeneric species based on FISH. Acta Horticulturae Sinica. 2020;47:503–16. [Google Scholar]
- 55. Tan J, Wang J, Gao Het al. Distribution of 45S rDNA in 17 Rosa species of China. Acta Botan Boreali-Occiden Sin. 2019;39:1333–43. [Google Scholar]
- 56. Herklotz V, Kovarik A, Lunerova Jet al. The fate of ribosomal RNA genes in spontaneous polyploid dogrose hybrids Rosa L. sect. Caninae (DC.) Ser. exhibiting non-symmetrical meiosis. Plant J. 2018;94:77–90. [DOI] [PubMed] [Google Scholar]
- 57. Rieseberg LH, Soltis DE. Phylogenetic consequences of cytoplasmic gene flow in plants. Evolutionary Trends in Plants. 1991;5:65–84. [Google Scholar]
- 58. Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 2006;55:21–30. [DOI] [PubMed] [Google Scholar]
- 59. Kiani M, Zamani Z, Khalighi Aet al. Microsatellite analysis of Iranian Damask rose (Rosa damascena Mill.) germplasm. Plant Breed. 2010;129:551–7. [Google Scholar]
- 60. Wu G. Investigation of wild parent of Chinese old garden roses based on SSR molecular markers. Master 67. Yunnan University, 2019. [Google Scholar]
- 61. Vries DPD, Dubois LAM. Rose breeding: past, present, prospects. Acta Hortic. 1996;424:241–8. [Google Scholar]
- 62. Wang G. Old Roses in China. Beijing: Science Press; 2015, 310. [Google Scholar]
- 63. Takeuchi S, Nomura K, Uchiyama Het al. Phylogenetic relationship in the genus Rosa based on the restriction enzyme analysis of the chloroplast DNA. Journal of the Japanese Society for Horticultural Science. 2000;69:598–604. [Google Scholar]
- 64. Nagamitsu T. Genetic structure in chloroplast and nuclear microsatellites in Rosa rugosa around sea straits in northern Japan. Plant Species Biology. 2017;32:359–67. [Google Scholar]
- 65. Qiu X, Zhang H, Jian Het al. Genetic relationships of wild roses, old garden roses, and modern roses based on internal transcribed spacers and matK sequences. HortScience. 2013;48:1445–51. [Google Scholar]
- 66. Tan S-L, Luo YH, Hollingsworth PMet al. DNA barcoding herbaceous and woody plant species at a subalpine forest dynamics plot in Southwest China. Ecology and Evolution. 2018;8:7195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bendahmane M, Dubois A, Raymond Oet al. Genetics and genomics of flower initiation and development in roses. J Exp Bot. 2013;64:847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dong X, Jiang X, Kuang Get al. Genetic control of flowering time in woody plants: roses as an emerging model. Plant diversity. 2017;39:104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen F, Su L, Hu Set al. A chromosome-level genome assembly of rugged rose (Rosa rugosa) provides insights into its evolution, ecology, and floral characteristics. Horticulture Research. 2021;8:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yu C, HuiHua W, Bourke PMet al. High density genetic map and quantitative trait loci (QTLs) associated with petal number and flower diameter identified in tetraploid rose. J Integr Agric 2021;20:1287–301. [Google Scholar]
- 71. Li S, Yang G, Yang Set al. The development of a high-density genetic map significantly improves the quality of reference genome assemblies for rose. Sci Rep. 2019;9:5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Debray K, Le Paslier MC, Berard Aet al. Unveiling the patterns of reticulated evolutionary processes with Phylogenomics: hybridization and polyploidy in the genus Rosa. Syst Biol. 2021. [DOI] [PubMed] [Google Scholar]
- 73. Iwata H, Gaston A, Remay Aet al. The TFL1 homologue KSN is a regulator of continuous flowering in rose and strawberry. Plant J. 2012;69:116–25. [DOI] [PubMed] [Google Scholar]
- 74. Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin. 1987;19:11–5. [Google Scholar]
- 75. Bankevich A, Nurk S, Antipov Det al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jin J-J, Yu WB, Yang JBet al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020;21:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wick RR, Schultz MB, Zobel Jet al. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31:3350–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wissemann V. Genetic constitution of Rosa sect. Caninae (R-canina, R-jundzillii) and sect. Gallicanae (R-gallica). Journal of Applied Botany-Angewandte Botanik. 1999;73:191–6. [Google Scholar]
- 80. Wissemann V. Molecular evidence for allopolyploid origin of the Rosa canina-complex (Rosaceae, Rosoideae). Journal of Applied Botany-Angewandte Botanik. 2002;76:176–8. [Google Scholar]
- 81. White TJ, Bruns T, Lee Set al. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, eds. PCR Protocols: A Guide to Methods and Applications. New York: Academic Press, 1990,315–22. [Google Scholar]
- 82. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–6. [DOI] [PubMed] [Google Scholar]
- 84. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Greilhuber J, Doležel J, Lysák MAet al. The origin, evolution and proposed stabilization of the terms ‘genome size’ and ‘C-value’ to describe nuclear DNA contents. Ann Bot. 2005;95:255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dolezel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc. 2007;2:2233–44. [DOI] [PubMed] [Google Scholar]
- 87. Schnable PS, Ware D, Fulton RSet al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5. [DOI] [PubMed] [Google Scholar]
- 88. Roberts AV, Gladis T, Brumme H. DNA amounts of roses (Rosa L.) and their use in attributing ploidy levels. Plant Cell Rep. 2009;28:61–71. [DOI] [PubMed] [Google Scholar]
- 89. Jacob Y, Teyssier C, Reynders-Aloisi Set al. Use of flow Cytometry for the rapid determination of Ploidy level in the genus Rosa. Acta Hortic. 1996;273–8. [Google Scholar]
- 90. Yokoya K, Roberts AV, Mottley Jet al. Nuclear DNA amounts in roses. Ann Bot. 2000;85:557–61. [Google Scholar]
- 91. Jian H, Zhang T, Wang Qet al. Nuclear DNA content and 1Cx-value variations in genus Rosa L. Caryologia. 2014;67:273–80. [Google Scholar]
- 92. Li S, Zhang C, Gao X. Estimation of nuclear DNA content of 17 Chinese wild rose species by flow cytometry. Plant Science Journal. 2017;35:558–65. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequences had been submitted to GenBank (Table S1).