

Abstract
A population of neural stem cells exists in the adult mammalian central nervous system. Purification and characterization of neurospheres provide valuable tools to study the regulation and differentiation of neural stem cells both in vitro and in vivo. Successful stimulation and production of neurospheres can ultimately be used for therapeutic purposes. The currently available methods are limited by their poor yield and the large number of animals required to compensate for that. Here, we describe a procedure to purify neurospheres from adult mouse whole brain. We provide detailed steps on how to propagate, passage, and maintain the adult neurospheres, and how to differentiate the pure neurospheres into the lineage of interest. Using this method, neurospheres can be easily derived from adult mouse whole brain. The derived adult neurospheres maintain their homogenous undifferentiated status while retaining their differentiation potential. This new protocol facilitates adult neurospheres isolation, purification, maintenance, and differentiation. © 2019 by John Wiley & Sons, Inc.
Keywords: adult, brain, mouse, neurosphere, progenitor, rodent, stem cell
INTRODUCTION
The neurosphere culture system is known as a valuable technique to determine the proliferation and differentiation potential of adult neural stem cells in vitro. This system can be used to determine the effects of different exogenous and endogenous factors on proliferation and differentiation of neural precursor cells isolated from different genetic backgrounds or differentially treated animals. The potential of neural precursor cell lines can be tested over several passages (Walker & Kempermann, 2014), during which they retain a stable capacity to generate functional, mature brain cells (Gritti et al., 1996). For decades, it was believed that adult brain was not able to generate new neurons. In the 1960s, for the first time, a small group of scientists challenged this dogma. At present, we know that the adult brain produces new neurons throughout life. In the adult brain, neurogenesis primarily occurs in the subventricular zone (SVZ) of the forebrain and the subgranular zone of the dentate gyrus (DG) within the hippocampus. In rodents, the potential of SVZ progenitor cells to act as neural stem cells has been demonstrated both in vitro and in vivo (Doetsch, Caillé, Lim, García-Verdugo, & Alvarez-Buylla, 1999; Garcia, Doan, Imura, Bush, & Sofroniew, 2004; Laywell, Rakic, Kukekov, Holland, & Steindler, 2000). Neural stem cells were identified for the first time in 1992 and isolated from the SVZ of adult mammalian brain (Gritti et al., 1996; Reynolds & Weiss, 1992, Weiss et al., 1996; Weiss et al., 1996). Currently, no protocol is available on using the adult whole brain to isolate the neural cells.
Stem cells are found in different tissues and exhibit two fundamental properties: they have the ability to undergo self-renewal and they can differentiate. Due to these properties, stem cells play crucial roles in maintaining tissue homeostasis and in repairing injured tissue. Recently, stem cells have received specific attention regarding their therapeutic potential, as well as their involvement in pathological states such as tumors (Pastrana, Silva-Vargas, & Doetsch, 2011).
Two neurosphere assays (Reynolds & Weiss, 1992; Reynolds & Weiss, 1996), developed in the early 1990s, have remained the gold standards for in vitro neural stem cell culture. In these assays, the striatum of the brain and the thoracic spinal cord are used for microdissection to obtain primary tissue. Next, the tissue is dissociated into a single-cell suspension and cultured in the presence of epidermal growth factor (EGF) and fibroblast growth factor-2 (FGF2) mitogens. These growth factors induce formation of free-floating clusters called neurospheres (Reynolds & Weiss, 1992). These systems have their own disadvantages. The yield of the available isolation strategies is very poor. The use of these current protocols can become limited due to the relatively large number of animals required to acquire sufficient cells. It has been revealed that neurogenesis in adult brains contributes to brain individualization; therefore, individualized ex vivo models are needed to gain accurate data (Freund et al., 2013). The currently available protocols are mostly limited in obtaining data from the individual brain. Thus, depending on the purpose of the study, the appropriate system should be chosen. Using neurospheres is advantageous for studying intrinsic specification of cells when they are out of their external environment (Walker & Kempermann, 2014). Additionally, neurospheres are useful tools to study the extrinsic cues by assessing the number and size of neurospheres generated in response to adding the factor of interest to the growth medium (Reynolds, Tetzlaff, & Weiss, 1992). Neurospheres contain a mix of different types of cells including the stem cells, committed progenitors, and differentiated cells at the center, especially in large neurospheres. Therefore, proper passaging and maintenance of the neurospheres is required to ensure that a large portion of neurospheres are indeed true stem cells (Babu, Cheung, Kettenmann, Palmer, & Kempermann, 2007; Jessberger, Clemenson, & Gage, 2007; Reynolds et al., 1992).
Here, we present a detailed procedure for generation of adult mouse neurospheres from neural precursor cultures. We first describe isolation of adult neural stem cells from the whole brain (Basic Protocol 1). We show that adult neural stem cells can generate viable pure neurospheres (Basic Protocol 2). The neurospheres can be passaged several times while maintaining their homogenous status (Basic Protocol 3). The isolated adult neurospheres proliferate in response to bFGF and EGF. We also describe how adherent cultures can be prepared from adult neural stem cells for maintaining and/or differentiating them into multiple lineages (Basic Protocol 4). Characterization of the abovementioned neurospheres confirms their stemness properties, evidenced by expression of stem cell markers (Basic Protocol 5). Evidence of multipotency in the neurosphere-derived cells is obtained by differentiating them into neuronal and glial lineages after providing lineage-specific media and supplements (Basic Protocol 6). In addition, we outline cryopreservation of neurospheres as well as starting a new culture from the frozen stock (Basic Protocol 7). The protocols described here eliminate the need for a site-specific microdissection and use of a large number of animals to obtain enough adult neural stem cells, while at the same time achieving a higher yield from only one animal. In addition, using the whole brain significantly reduces the time required for performing the isolation and preparation of adult neural stem cells.
BASIC PROTOCOL 1
ISOLATION OF NEURAL STEM CELLS FROM ADULT MOUSE WHOLE BRAIN TISSUE
Here we provide detailed methodology on isolation of neural cells from adult mouse whole brain for use in neurosphere culture. Although isolation of neural cells from embryonic (Banker & Cowan, 1977; Brewer, Torricelli, Evege, & Price, 1993) or postnatal (Ahlemeyer & Baumgart-Vogt, 2005) brain is relatively easy, isolation of adult neural cells is problematic. The described method employs a density-gradient step to enrich for neural cells over other cell types. Additionally, using protease digestion of brain slices increases the yield of isolation from each brain, compared to other available methods (Eide & McMurray, 2005).
Materials
Adult mouse (5 to 8 months old)
70% ethanol
10% bleach
Sterile DMEM/F12 medium (ThermoFisher, Cat#12634028)
Papain dissociation system (Worthington Biochemical Corporation, Cat#LK003150)
Sterile neural stem cell (NSC) medium (see recipe)
20 μg/ml EGF (see recipe)
10 μg/ml FGF (see recipe)
Large and small sharp scissors, sharp and blunt forceps, small spatula (all surgical tools must be autoclaved prior to start of procedure)
Sterile laminar flow cell culture hood
Sterile single-use 5- and 10-ml pipettes
Petri dishes
Sterile 15- and 50-ml tubes (e.g., Corning Falcon)
37°C and 5% CO2 humidified incubator with mechanical shaker
Autoclaved glass Pasteur pipettes and bulbs
Cell strainer (Cell Star Easy Strainer, 70 μm; USA Scientific, Cat#5654–2070)
High-speed table-top centrifuge
Nonadherent cell culture flask
Additional reagents and equipment for euthanasia of rodents (Donovan & Brown, 2006) and counting viable cells by trypan blue exclusion (Takahashi, Takebe, & Taniguchi, 2018)
Experiment setup
-
1
Select mouse of appropriate age (5 to 8 months old).
Whole-brain tissue from one animal is enough to establish a neural stem/progenitor cell line; however, multiple animals may be pooled and reagent volumes adjusted according to amount of tissue.
-
2
Ensure that all surgical tools are autoclaved before use.
Tools can be autoclaved at 120°C for 30 min
-
3
Spray down laminar flow hood with 70% ethanol, then with 10% bleach, and switch on UV light for about 20 min before use.
-
4
Before dissection, ensure that the following reagents are ready: sterile DMEM/F12 at room temperature and cold EBSS (from Worthington Papain dissociation kit)
Harvesting the brain
-
5
Euthanize animal in a CO2 euthanasia chamber and perform cervical dislocation to ensure death (both procedures described in Donovan & Brown, 2006).
Harvesting of the brain should be done as soon as possible after this step to maintain cell quality and viability
-
6
Spray euthanized animal with 70% ethanol and decapitate using sharp scissors.
-
7
Cut skin along midline to expose the skull.
Make sure to exclude any skin and hair while harvesting brain tissue.
-
8
Use sharp, thin scissors to cut along the skull along the midline and expose the brain tissue. Gently coax the brain tissue out of skull and let it drop immediately into a petri dish with DMEM/F12.
It might be necessary to push the skull tissue to the side to expose and loosen the brain tissue within. Use blunt forceps to hold the head of the mouse steady while carefully cutting through the skull and moving it aside. Use a scalpel to push the brain tissue into the DMEM/F12.
-
9
From this step onward, ensure that the brain is hydrated and covered with DMEM/F12 at all times during the cell isolation. Perform all further steps inside a laminar flow hood to maintain full aseptic technique and limit any contamination issues.
Dissection of whole brain tissue
-
10
Transfer harvested whole brain tissue immediately into a new petri dish with fresh DMEM/F12 medium.
Ensure that the brain remains submerged in medium at all times, to prevent drying of the tissue.
-
11
Use forceps and small sharp scissors to mince brain tissue into small pieces to ease cell isolation in the next steps. Clean out the meninges to minimize contamination with endothelial cells.
Conduct this process in the laminar flow hood, preferably under a microscope, to make sure that the tissue is minced as finely as possible.
Make sure to not pull at the pieces, but to gently separate the cut-up portions from one another.
-
12
Pipette the medium containing minced brain tissue into a 50-ml conical tube and let the pieces settle to the bottom of the tube while preparing reagents required for the next steps.
Dissociation of whole brain tissue using the papain dissociation system
-
13
Add 32 ml EBSS to the albumin-ovomucoid inhibitor mixture (from the papain dissociation kit) and allow dissolution, while making the other mixtures as described below. Reconstitute for the first use and then store and reuse.
Ensure that no air bubbles are created in the solution when mixing.
-
14
Add 5 ml of EBSS to the papain vial provided in the papain dissociation kit. Place at 37°C for 10 min or until papain is fully dissolved and solution appears clear (the color of normal media). Solution should be used promptly, but can be stored at room temperature during dissociation.
As adult mouse brains are larger in size than embryonic/early-post embryonic mouse brains, we can adjust the volume of papain accordingly.
Prepare two full papain vials (5 ml each) for enzymatic dissociation of minced whole brain tissue from one adult brain.
-
15
Add 500 μl EBSS to the DNase vial provided with the papain dissociation kit. Mix gently, since DNase is prone to degradation. Add 250 μl of this solution to each vial containing papain (this gives you 20 U/ml papain and 0.005% DNase). Save the remaining DNase vial to use in step 22.
To keep volumes and ratios consistent, make two such DNase vials if two papain vials were made.
-
16
Carefully aspirate the medium from the 50-ml conical tube with minced tissue, making sure that no tissue is sucked up.
-
17
Immediately add papain/DNase solution (total 7.5 to 10 ml/brain) to the minced tissue. Replace the cap, to avoid too much airflow into the tube, and mix gently and avoid air bubbles.
-
18
Incubate 50-ml tube containing the tissue at 37°C with constant agitation (on shaker) for 60 to 90 min.
Gently swirl the mixture every 10 min manually to ensure that every piece of tissue is submerged in papain.
Check if the mixture becomes cloudy, indicating dissociation of cells from the minced tissue.
-
19
After 60 to 90 min, triturate the mixture with a glass Pasteur pipette 20 to 30 times until the whole solution becomes cloudy. Avoid formation of air bubbles.
Trituration for longer intervals may be necessary for adult tissue, since it is much sturdier and more fibrous than embryonic/early-post embryonic mouse brains.
-
20
Filter the cloudy mix through a cell strainer into a fresh 50-ml tube, to get rid off any bigger un-dissociated pieces (vigorous trituration = high yield of cells).
-
21
Centrifuge the freshly strained cloudy mix 5 min at 300 × g, room temperature.
-
22
During this time, prepare medium to resuspend pelleted cells. Mix 2.7 ml EBSS with 300 μl albumin-ovomucoid inhibitor solution in a sterile 15-ml tube. Add 150 μl of DNase solution saved at step 15.
Double all the volumes in this step if two vials of papain were used for dissociation in step 17.
-
23
Remove 50-ml conical tube from centrifuge. Discard supernatant and immediately resuspend cell pellet in the diluted DNase/albumin-ovomucoid inhibitor solution prepared in step 22.
Make sure to avoid air bubble formation while mixing to resuspend cells.
-
24
Prepare discontinuous density gradient. Add 5 ml of albumin-ovomucoid inhibitor solution to a 15-ml centrifuge tube, carefully layer the cell suspension on top, then centrifuge 5 min at 400 × g, room temperature.
A 50-ml tube will be required if the volumes are doubled as described in steps 22 and 24.
The interface between the two layers of the gradient should be clearly visible, but minimal mixing is fine. Dissociated cells pellet at the bottom of the tube, while membrane fragments remain at interface.
Double the volume of the albumin-ovomucoid inhibitor solution in this step if volumes were doubled in step 22.
-
25
Discard supernatant and immediately resuspend the pelleted cells in sterile neural stem cell medium.
Culturing/plating neural stem cells
-
26
Count cells (see, e.g., Takahashi et al., 2018) and plate according to requirement in neural stem cell medium, in a nonadherent cell culture flask and incubate on a shaker at 37°C. Supplement the medium with EGF (20 ng/ml) and FGF (8 to 10 ng/ml). Continue EGF/FGF supplementation every 3 to 4 days.
Incubating the cells on a shaker enhances enrichment of neural stem/progenitor cells while selecting out cells that do not grow well in suspension.
It is advisable to count cells using 1:1 dilution of the cell suspension in 4% trypan blue (Takahashi et al., 2018). This will give an estimate of the cell viability and effectiveness of the cell isolation procedure.
EGF/FGF should be used as fresh supplements each time and should not be added to the bulk medium directly as stock.
-
27
Maintain for 3 to 4 days until clumps of live cells start appearing
The initial few cultures will contain lots of debris (non-progenitor cells, dying cells, etc. (Fig. 1A).
Figure 1.
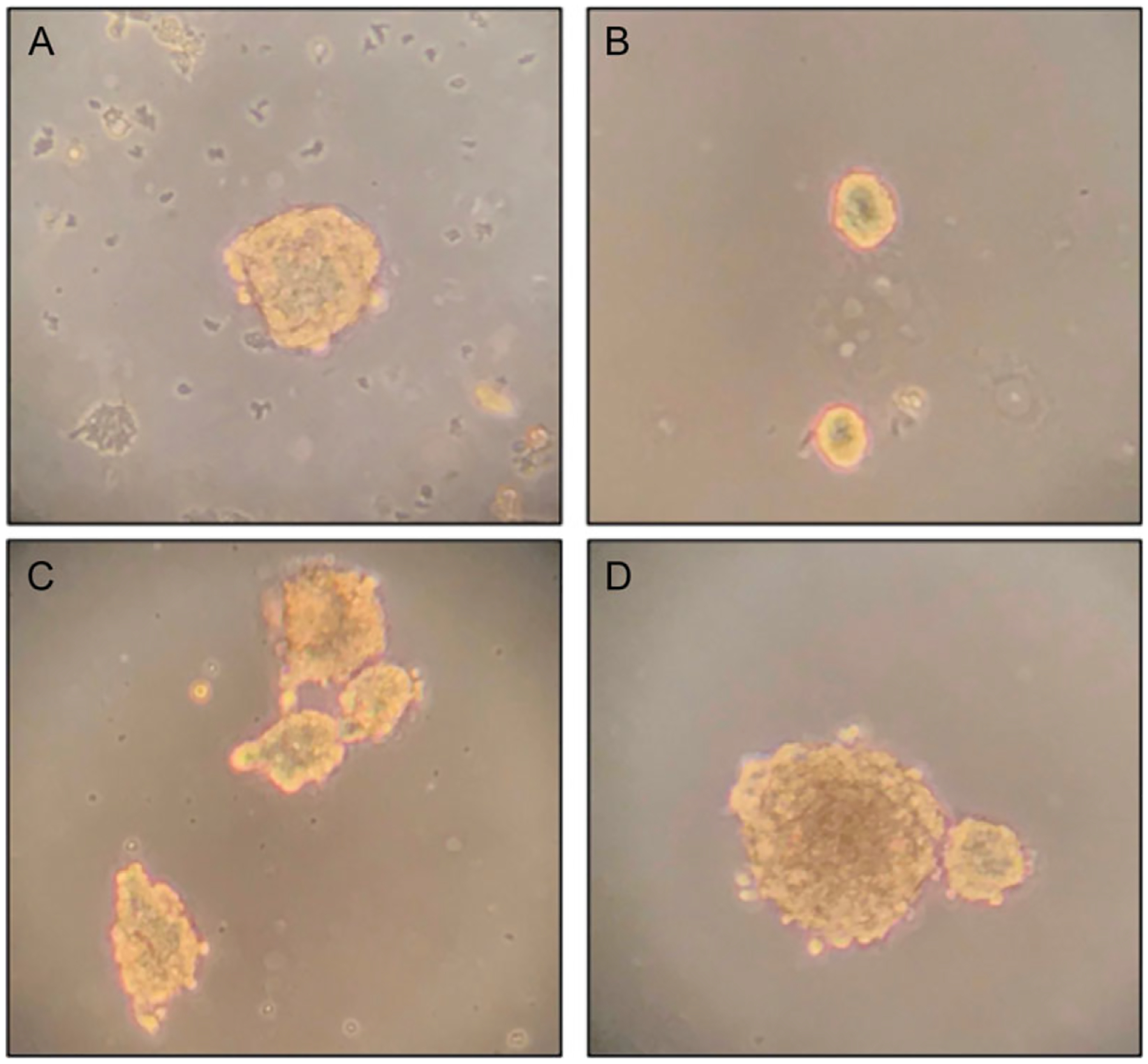
Neurosphere formation from adult murine neural progenitor/stem cells (NSCs). (A-D) Different stages of neurosphere formation. (A) NSC clump that forms around 3 to 4 days post cell isolation from whole brains. The live cell clump is seen surrounded by debris, which can be filtered out when the NSCs are allowed to settle to the bottom of a tube at first passage. This NSC clump can be isolated, triturated to form a single-cell suspension, and passaged to get pure NSC culture. (B) Small pure neurospheres generated 1 to 2 days post first passaging. (C) Larger neurospheres start forming 3 to 4 days post plating as cells proliferate and clump. These spheres are suitable for passaging even without enzymatic dissociation, and usually result in a high-viability single-cell suspension. (D) Tight spherical body, with dark areas that might indicate necrotic center. These spheres might be harder to triturate and might need enzymatic dissociation for subculturing. Images were taken at 20× magnification.
BASIC PROTOCOL 2
ISOLATION OF PURE NEURAL PROGENITOR CLUMPS/NEUROSPHERES BY TRITURATION
Formation of macroscopic spheres from stem cells isolated from different regions of central nervous system (CNS) has been previously reported (Reynolds & Weiss, 1992). Here, we describe the isolation of pure neurospheres from primary neural culture. This protocol demonstrates how to separate live cell clumps of progenitors from dead/dying debris and, next, establish neurosphere cultures.
Materials
Cell suspension (Basic Protocol 1)
Sterile neural stem cell (NSC) medium (see recipe)
20 μg/ml EGF (see recipe)
10 μg/ml FGF (see recipe)
Cell culture microscope
Sterile single-use 10-ml pipette
Sterile 50-ml conical tube
Trituration
-
Three to four days after plating, check the cell suspension under the microscope for live stem/cell clumps in suspension.
Under the microscope, pure live cell clumps of progenitors are easily distinguishable, but some live progenitor cells might be clumped together with dead/dying debris.
-
Wet a 10-ml pipette with medium so that cells do not stick to walls during the process. Triturate the mixture 5 to 10 times to dislodge debris and dead cells from live progenitor cells clumps.
Dead cells/debris are lighter and should dislodge from live cells relatively easily.
Check under microscope to confirm dislodging.
Pipette cell solution into a sterile 50-ml conical tube.
Let the tube rest until the heavier live-cell clumps settle, leaving the debris in the supernatant (10 to 15 min).
Pipette out debris and add fresh NSC medium onto the pelleted cells.
Centrifuge the debris 15 min at 2500 × g, room temperature, and use the clarified medium supernatant as conditioned medium for the neurosphere culture.
-
Pay attention to the size of the spheres. Large spheres may develop necrotic core in the center. Triturate such spheres and let them form again (Fig. 1B, C, D).
Neurospheres are multipotent and can now be differentiated into cells of different neuronal lineages on addition of relevant growth factors and medium supplements.
BASIC PROTOCOL 3
SUBCULTURING/PASSAGING NEUROSPHERES
This protocol describes passaging the neurospheres isolated in Basic Protocol 2. Using this method allows maintenance of neurosphere formation over several passages. The neurospheres continue to increase in size over time; therefore, sustaining their stem-like properties is critical. Here we show that a combination of enzymatic and physical dissociation of neurospheres into single cells or small cell clumps is effective for the maintenance of the neurospheres. The neurospheres are normally passaged once a week according to confluency and size.
Materials
Neurosphere culture (Basic Protocol 2)
Sterile neural stem cell medium (see recipe)
20 μg/ml EGF (see recipe)
10 μg/ml FGF (see recipe)
Accutase (Accumax, Innovative Cell Technologies, Cat#AM-105)
Sterile 50-ml conical tube
Sterile 5-ml pipette
Repeat pipettor and sterile 1000-μl pipette tips
Centrifuge
Microscope
Hemacytometer
Pipette the non-adherent neurosphere culture into a 50-ml conical tube. Wash the flask with 5 to 6 ml of NSC medium to collect any remaining neurospheres and add this rinse to the 50-ml tube.
-
Let the neurospheres settle to the bottom of the tube for about 10 to 15 min, depending on the size of the spheres.
Any debris or dead cells will be left in the medium supernatant.
Remove the medium, spin it down for 15 min at 3000 × g, room temperature, and save the supernatant to be used as conditioned medium for the passaged neurospheres.
Add 0.5 to 1 ml Accutase to the neurosphere pellet (depending on size of pellet). Leave the cells in the incubator (preferably on a shaker) for 4 to 5 min, until the neurospheres start to fall apart. Triturate the cells intermittently, to ensure sphere dispersion into single cells, and add NSC medium (double the volume of the Accutase added) to neutralize the Accutase once sufficient dispersion has been achieved.
-
Smaller spheres may be dissociated into single cells only with trituration. Enzymatic dissociation is not necessary. Triturate with 1000-μl micropipette tip (30 to 40 times) by pushing pipette tip down toward the bottom of the tube. Confirm dissociation under the microscope and continue trituration if required. Avoid formation of any air bubbles.
Air bubbles lead to cellular stress and lower viability.
-
Spin down the cells 1.5 min at 500 × g, room temperature, resuspend in medium. Measure cell number and viability with trypan blue (see, e.g., Takahashi et al., 2018).
Use 10 μl of cell suspension and 10 μl of 4% trypan blue. Add 10 μl of mix to hemocytometer to count cells to get cells/ml count and percentage viability (Takahashi et al., 2018).
BASIC PROTOCOL 4
PREPARING ADHERENT CULTURES OF NEURAL STEM CELLS FOR MAINTENANCE AND/OR DIFFERENTIATION INTO MULTIPLE LINEAGES
This protocol describes how to derive adherent cultures from neurospheres for their differentiation into multiple lineages. An important characteristic of neural stem cells is their multipotency to differentiate into neurons, astrocytes, and oligodendrocytes. This protocol demonstrates that the neural stem cells can form adherent cultures while maintaining their NSC status. The adherent neural stem cells can be differentiated into the specific lineage of choice by using appropriate growth factors, medium, and supplements.
Materials
Poly-ornithine hydrochloride (ThermoFisher, Cat#P2533–100MG)
Fibronectin (ThermoFisher, Cat# PHE0023)
Sterile 1× phosphate-buffered saline (PBS; Corning Cellgro, Cat#21–031-CV)
Neurospheres (Basic Protocol 3)
Sterile neural stem cell medium (see recipe)
20 μg/ml EGF (see recipe)
10 μg/ml FGF (see recipe)
Sterile cell culture growth flasks/plates of choice
Poly-ornithine (PO) and fibronectin (FN) coating of plates for neural stem cells culture
Prepare poly-ornithine: To make 200× (3 mg/ml) stock, dilute 100 mg poly-ornithine hydrochloride in 33.33 ml distilled, deionized H2O. This can be stored at 4°C for 2 to 3 months, or −20°C for long-term storage. When coating plates, dilute 1 ml in 200 ml distilled deionized H2O (final coating concentration is 15 μg/ml) for a 1× concentration.
Prepare fibronectin: To make 500 × stock, dissolve 5 mg fibronectin in 8 ml sterile 1× PBS. To coat plates, make 1× solution in sterile 1× PBS.
- On the first day, make 1× poly-ornithine hydrochloride in distilled deionized H2O as described in step 1. Add the following volumes to required culture area:
-
24-well plate: 0.5 ml/well
-
6-well plate: 2 ml/well
-
96-well plate: 100 ul/well
-
10-cm dish: 5 to 8 ml/well (If <7 ml is added, then plates need to be shaken overnight at room temperature for even coating).
-
Incubate plates at 37°C overnight. On the second day, wash plates with distilled deionized H2O twice.
Add 1× fibronectin to plates (same amount as poly-ornithine).
-
Leave plate in the cell culture hood >1.5 hr. Wash plates with distilled deionized H2O twice.
The plates can be used immediately, or stored at 4°C for about 1 month.
Plating neural stem cells
-
5
Whole neurospheres can be plated on poly-ornithine/fibronectin-coated plates, in NSC medium with regular EGF/FGF supplementation, to maintain NSC multipotent status.
Cells that grow out from the spheres will be partly adherent and partly in suspension, but will maintain NSC status if not given lineage-specific growth factors.
-
6
Single cells obtained from dissociation of neurospheres can also be plated on poly-ornithine/fibronectin-coated plates at a density of about 2–4 × 105 cells/cm2 (Fig. 2).
-
7
In addition, these cells can be differentiated into lineage of choice by addition of suitable growth medium, and supplementation with lineage-specific growth factors (see Reagents and Solutions).
Figure 2.
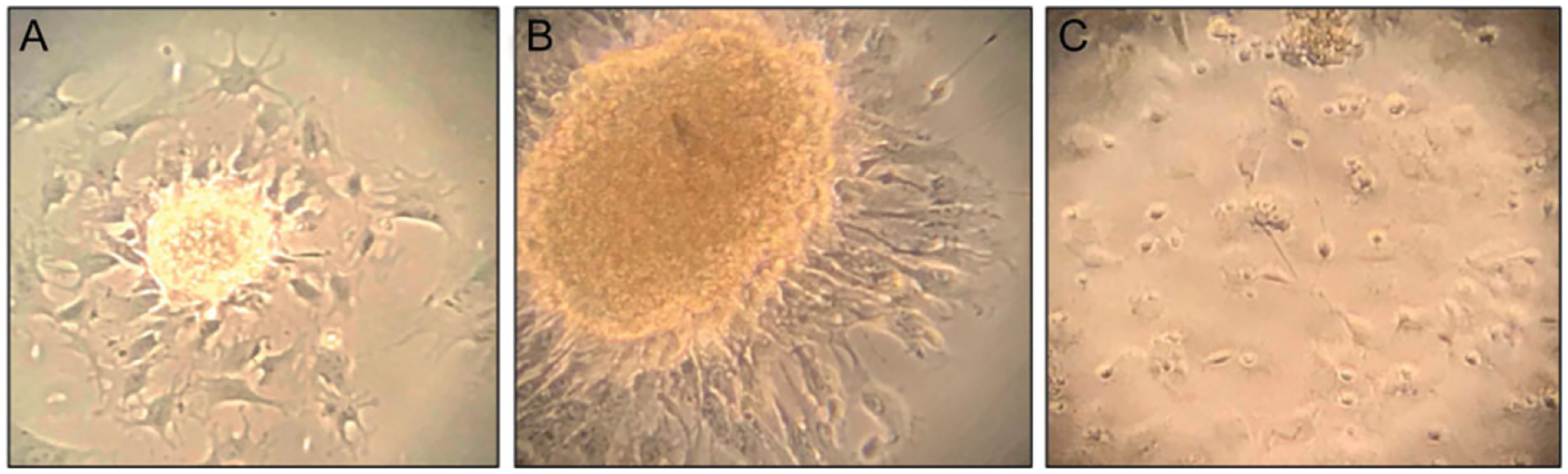
Proliferation of adherent neural stem/progenitor cells from whole neurospheres. Neurospheres were cultured on plates coated with poly-ornithine and fibronectin. (A and B) Outgrowth of cells from intact neurospheres, in a rosette formation. (C) Bipolar cells proliferating about 5 days after plating whole neurospheres on the poly-ornithine/fibronectin-coated plates. Cells may also be maintained as neural stem cells by keeping them in NSC medium with EFG/FGF supplementation. The rosette spreads out as cells from the sphere attach, and the adherent cells proliferate.
BASIC PROTOCOL 5
CONFIRMATION OF NEURAL PROGENITOR STATUS IN ISOLATED SPHERES/CELLS
NESTIN is an interstitial filament protein expressed mainly by neuronal progenitor lineage, and loses expression as cells mature (Lindahl, Zimmerman, & McKay, 1990). SOX2 is found in embryonic and adult stem cells in the brain, and helps maintain the multipotent identity of the progenitor population (Ellis et al., 2004). We compared the expression of these two markers in the isolated adult murine neurospheres to their expression levels in post-natal day 2 (PND2) murine neurons (fully differentiated) (Fig. 3).
Figure 3.
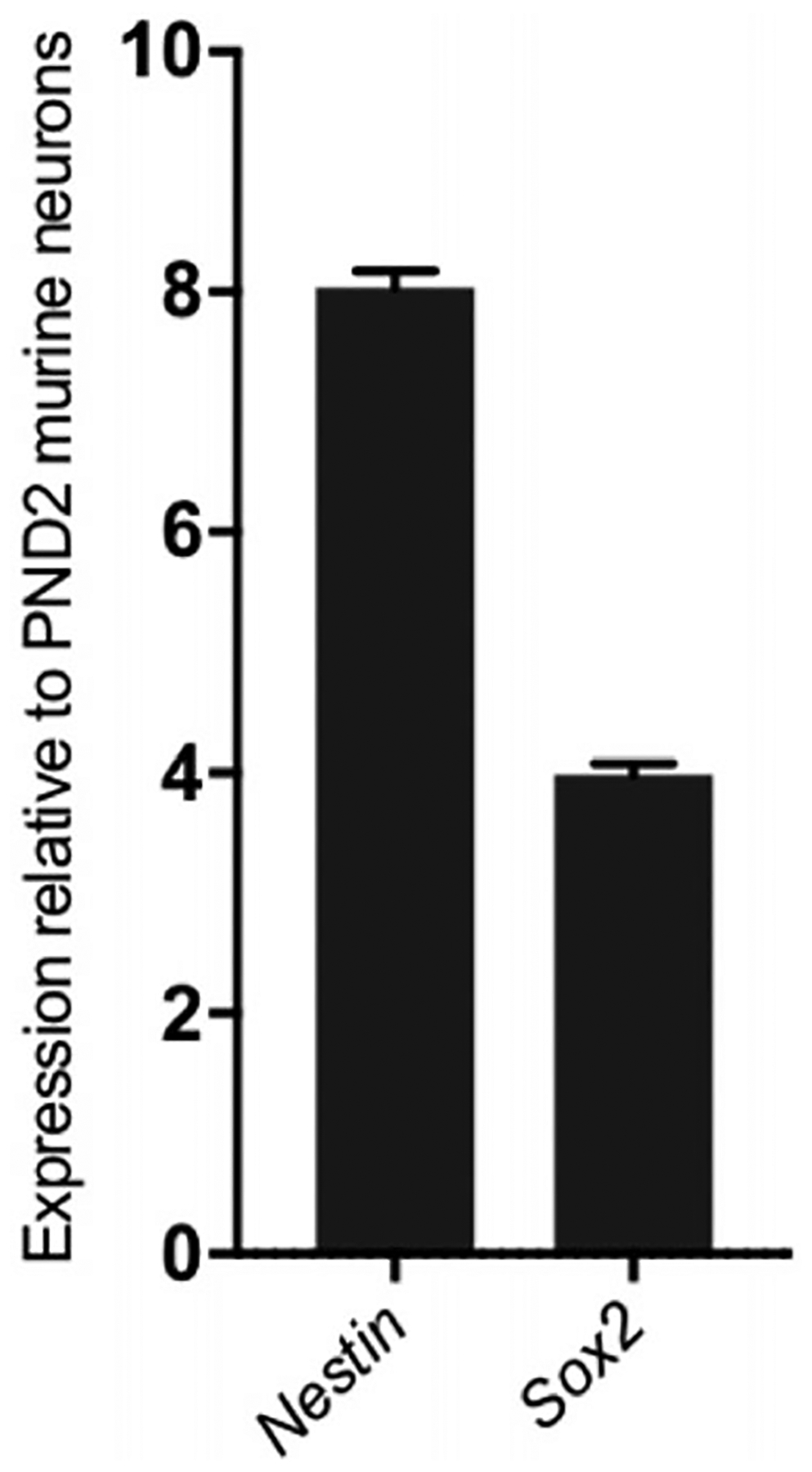
Confirmation of neurosphere multipotency. Expression of neuronal progenitor markers Nestin and Sox2 in the neurospheres. The adult murine neurospheres express significantly elevated levels of both genes compared to mature neurons isolated from post-natal day 2 mice. This is confirmatory of their stem/progenitor multipotent status.
Materials
Neurospheres, or trypsinized adherent NSC cells (see protocols above)
RNeasy RNA isolation Mini kit (Qiagen, Cat#74136)
QIAshredder (Qiagen, Cat#79656)
2-Mercaptoethanol
Sterile nuclease-free H2O
Maxima First Strand cDNA synthesis kit (ThermoFisher, Cat#K1642)
- Forward+Reverse primers for Mouse Nestin
- Primer 1: GGA AAG CCA AGA GAA GCC T
- Primer 2: CAC CTC AAG ATG TCC CTT AGT C
- Forward+Reverse primers for Mouse Sox2
- Primer 1: CTT GAC CAC AGA GCC CAT
- Primer 2: GTA CAA CTC CAT GAC CAG CTC
Preferred reagents for reagents for qPCR (e.g., PowerUp Sybr Green Master Mix; ThermoFisher, Cat#A25778)
Sterile 15-ml tube
Nanodrop microspectrophotometer
Confirmation of Nestin, Sox2, and expression
-
Collect whole neurospheres or Accutase-treated adherent NSC cells as a pellet (see Basic Protocol 3).
The cell pellet can be used fresh or can be frozen at −80 degrees to be used later for RNA isolation.
Isolate RNA from pelleted cells using the RNeasy RNA isolation Mini kit.
Measure RNA concentration and purity with Nanodrop before using it to synthesize cDNA.
Depending on the concentration of RNA eluted, use around 50 ng to 1000 μg of RNA to make cDNA using the cDNA synthesis kit (or any available and reliable method of cDNA synthesis).
Use the cDNA to confirm Nestin and Sox2 expression in the neurospheres, compared to mature PND 2 in neurons (Fig. 3).
BASIC PROTOCOL 6
LINEAGE DIFFERENTIATION FROM ADULT MURINE NEUROSPHERES
One of the most important uses of neurosphere cultures lies in the ability to be differentiated into cell populations of choice for subsequent research purposes. Neurosphere cultures contain neural precursor cells, which are a mix of stem and progenitor cell populations. Since NSC/NPCs are multipotent, they can be differentiated into neurons, astrocytes, or oligodendrocytes when provided with suitable media and supplements. This protocol describes methodology for differentiation of multipotent NPC/NSCs into neuronal and glial lineage. We confirm acquisition of lineage-specific expression in the cells via fluorescent immunocytochemistry.
Materials
Neurospheres (see protocols above)
Sterile neural stem cell medium (see recipe)
20 μg/ml EGF (see recipe)
10 μg/ml FGF (see recipe)
Sterile neuronal differentiation medium (see recipe)
Sterile glial cell differentiation medium (see recipe)
Materials for confirmation of expression of molecular markers of lineage commitment:
- Primary antibodies for:
- NSC/NPC validation:
- Nestin (Sigma Aldrich, Cat#N5413)
- Sox2 (Novus Biologicals, Cat#AF2018)
- Validation of glial lineage differentiation:
- Vimentin (Genetex, Cat#GTX100619)
- Glial fibrillary acidic protein (GFAP; Abcam, Cat#ab53554)
- CNPase (Novus Biologicals, Cat# NB110–93662)
- Myelin basic protein (MBP; Millipore Sigma, Cat#AB9348)
- Validation of neuronal lineage differentiation: Neu-N (Abcam Cat#ab104225)
Fluorescent secondary antibodies of choice, e.g.:
Anti-rabbit (Jackson ImmunoResearch, Cat#111-165-144)
Anti-goat (Jackson ImmunoResearch, Cat#705-545-147)
Anti-chicken (Abcam, Cat#ab7114)
Poly-ornithine (PO)– and fibronectin (FN)–coated (see Basic Protocol 4) glass coverslips in 24-well cell culture plates
Lineage differentiation from adult murine neurospheres
Plate neurospheres onto poly-ornithine/fibronectin-coated cell culture plates in sterile neural stem cell medium supplemented with EGF/FGF as described in Basic Protocol 1.
The next day, change medium on the neurospheres that you want to differentiate into lineage of choice to sterile neuronal differentiation medium or sterile glial differentiation medium, depending on purpose of experiment.
Maintain the neurospheres in medium of choice for 5 to 6 days, and observe for change in cellular morphology as cells commit to preferred lineage based on medium provided.
Feed the cells with preferred lineage differentiation medium every 2 to 3 days, to advance lineage commitment.
Depending on lineage, the cells will now either be non-proliferating and terminally differentiated (i.e., neurons), or become glial cells that may be passaged and re-plated for propagation.
- Confirm differentiation by analyzing expression of lineage-specific markers in cells with immunofluorescence (Fig. 4).
- Neuronal lineage: Neu-N
- Glial lineage (astrocyte): Vimentin, GFAP
- Glial lineage (oligodendrocyte): CNPase, MBP
- Maintenance of multipotency in NSCs: Nestin, Sox2.
Figure 4.
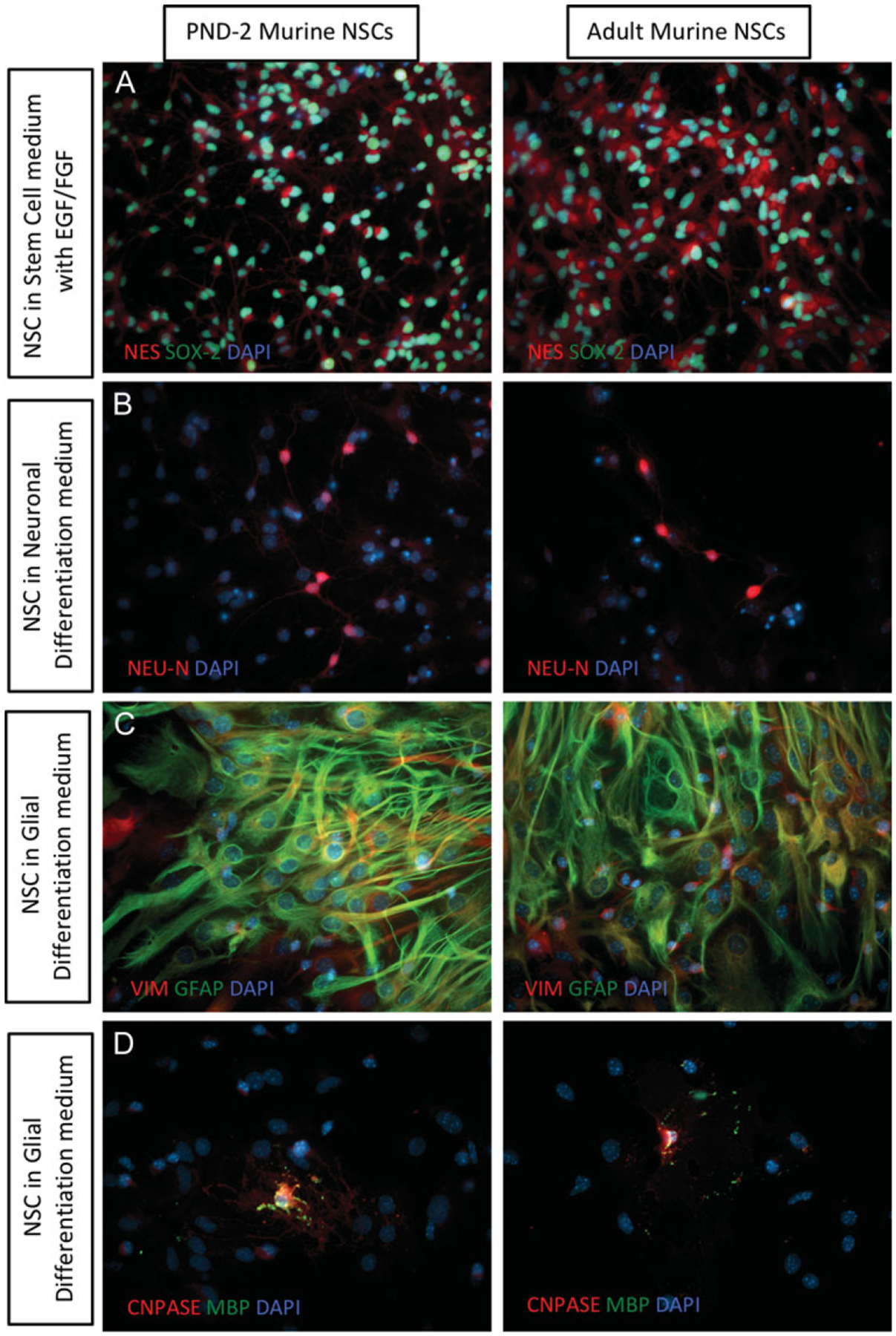
Lineage differentiation from adult murine NSCs. Lineage differentiation capacity is maintained in adult murine NSCs, when compared to post-natal day 2 (PND 2) murine NSCs. Changes in cellular and nuclear morphology, as well as protein expression, is seen as cells start differentiating into distinct lineages. (A) NESTIN (red) and SOX2 (green) expression in NSCs. (B) NSCs upregulate NEU-N (red) as they differentiate into neuronal lineage. (C) NSCs upregulate GFAP (green) and VIMENTIN (red) expression as they differentiate into glial lineage. (D) A portion of NSCs grown in glial lineage medium differentiate into oligodendrocytes, and show expression of CNPASE (red) and myelin basic protein (green), which is absent in all other cell types. All images were taken at 40× magnification.
BASIC PROTOCOL 7
CRYOPRESERVATION OF NSCs
This protocol describes cryopreservation of neurospheres, as well as starting the neurosphere culture from the frozen stock.
Materials
100% isopropanol
Neurospheres (see protocols above)
Stem cell cryopreservation medium (Wako, Cat#302–14681)
Sterile neural stem cell medium (see recipe)
Temperature controlled cryofreezing apparatus
Sterile 15-ml tubes
Cryofreeze vials
Centrifuge
Appropriate cell culture flask or plate
Freezing neurospheres
-
1
Fill a clean cryofreezing apparatus container with 100% isopropanol.
-
2
Pipette neurospheres in suspension culture into a sterile 15-ml tube and let the spheres settle for about 10 min.
-
3
Remove the medium supernatant and resuspend the neurosphere pellet in cryopreservation medium.
-
4
Transfer this cell suspension to cryovials and place vials in the cryofreezing apparatus container to freeze at −80°C.
Thawing cryopreserved neurospheres
-
5
Place frozen cryovial with neurospheres in cryopreservation medium, at 37°C until it thaws (5 to 10 min).
-
6
Take suspension and add it to sterile tube with NSC medium. Mix to resuspend neurospheres.
-
7
Spin down the tube to pellet the neurospheres 2 min at 500 × g, room temperature.
-
8
Aspirate supernatant carefully and resuspend the pellet in fresh NSC medium and add to cell culture flask/plate of choice.
REAGENTS AND SOLUTIONS
NOTE: Complete NSC medium is neural stem cell medium (see recipe below), with added supplementation of EGF and FGF. Addition of this medium to neurospheres should ideally maintain their multipotent stem/progenitor properties until differentiation is desired. EGF/FGF should be used as fresh supplements each time and should not be added to the bulk medium directly as stock.
EGF supplement, 20 μg/ml, and FGF supplement, 10 μg/ml
Reconstitute EGF (Peprotech, Cat#AF-100–15) in sterile distilled water to 20 μg/ml.
-
Reconstitute FGF (Peprotech, Cat#100–18B) in 5 mM Tris (Ph 7.6) to 10 μg/ml.
Reconstituted stocks should be aliquoted (100 μl) and stored at −80°C.
EGF and FGF should be supplemented into the NSC medium every 3 to 4 days at 20 ng/ml and 8 to 10 ng/ml, respectively. Daily supplementation is not necessary.
Glial differentiation medium, sterile
500 ml DMEM/F12 (ThermoFisher, Cat#12634028)
5 ml NG2 supplement (100×; ThermoFisher, Cat#17502–048)
5 ml antibiotic-antimycotic (100×; ThermoFisher, Cat#15240062)
-
5 ml Glutamax (100×; ThermoFisher, Cat#35050061)
This medium can be stored at 4°C and used for about a month. Do not use medium that has been made >1 month in advance.
Ensure that the bottle containing the medium is not left open for long intervals and is always tightly capped when not in use.
Complete glial differentiation medium should be used when glial differentiation from multipotent cells is desired.
Neural stem cell medium, sterile
500 ml DMEM/F12 (ThermoFisher, Cat#12634028)
10 ml B27 supplement (50×; ThermoFisher, Cat#17504044)
5 ml antibiotic-antimycotic (100×; ThermoFisher, Cat#15240062)
6.75 ml Glutamax (100×; ThermoFisher Cat#35050061)
Neuronal differentiation medium, sterile
500 ml Neurobasal-A medium (ThermoFisher, Cat#10888022)
10 ml B27 supplement (50×; ThermoFisher, Cat#17504044)
5 ml antibiotic-antimycotic (100×; ThermoFisher, Cat#15240062)
-
5 ml Glutamax (100×; ThermoFisher, Cat#35050061)
Complete neuronal differentiation medium should be used when neuronal differentiation from multipotent cells is desired.
COMMENTARY
Background Information
During the last decade, culturing neurospheres has remained as the most popular and reliable method for isolating, maintaining, and propagating NSCs. Studying NSCs from adult brain tissue has remained particularly challenging due to the low number of cells in the adult brain (0.1% to 1%) (Leong, Zhai, Kim, Yun, & Chang, 2013) and tedious cell isolation and preparation approaches (Brewer & Torricelli, 2007; Galli, Gritti, Bonfanti, & Vescovi, 2003). A drawback of most available protocols for neurosphere culture is the poor yield of isolation and requirement for a high number of animals to compensate for the low recovery. In studies with limited availability of animals, the need for six to eight animals for each round of cell isolation can be a practical obstacle. Furthermore, lack of self-renewal ability in the isolated neurospheres can be a challenge, as previously shown by two groups who used the whole hippocampal tissue for isolation (Bull & Bartlett, 2005; Seaberg & van der Kooy, 2002). The previously developed “one mouse-one culture” protocol (Guo, Patzlaff, Jobe, & Zhao, 2012; Walker & Kempermann, 2014) addresses the need for isolating NSCs from individual adult brain; however, using only the SVZ and DG regions makes microdissection laborious and results in low yield. A similar method was later developed for NSC isolation from the SCZ located between the white matter and hippocampus (Kim, Lee, & Sun, 2016). In this method, precise dissection of SCZ slices from whole brain requires obtaining brain matrix with intervals of 1 mm, and then microdissection of SCZ from other cortical regions. These latter steps negatively affect NSC preparation, isolation yield, and viability of cells. Thus, attempts have been made to develop isolation techniques for adult neural stem cells.
The procedure described above uses papain for enzymatic dissociation and a density gradient step for separation of cells from debris. Papain has several advantages over other methods of cell isolation. Being an enzyme, it is more efficient than manual mechanical tissue dissociation and results in higher yield of viable neural stem/progenitor cells. Additionally, papain is less harsh compared to trypsin, which can be too strong for the relatively fragile brain cells. The current procedure ensures the acquisition of a pure neurosphere culture by gentle trituration and allowing live cell clumps to settle. This procedure does not cause undue stress to the cells, while also getting rid of unwanted debris and inhibitory factors. Cells growing from the NSC/neurospheres could be from multiple lineages, but a portion of these are true multipotent progenitors/stem cells. If maintenance of these multipotent adult neurospheres is desired, it is advised to grow cells under EGF/FGF supplementation. This method uses a serum-free freezing medium. This ensures that the stem cells are frozen in an environment similar to their growth conditions, since the NSC medium does not contain serum either.
Critical Parameters
It is critical to make sure that the brain tissue is immersed in medium/required solution without being left dry. Exposure to oxygen/air bubbles stresses the tissue and might lead to loss of viable neural stem cells.
It is essential that the harvested brain tissue be minced into fine pieces, preferably under a microscope. Fine mincing ensures that the papain/DNase solution reaches every part of the tissue and causes efficient tissue dissociation within 60 to 90 min.
Live progenitor/stem cell clumps are heavier than debris and will settle to the bottom of the tube themselves. Centrifugation to hasten this process is not necessary and might even disturb the integrity of the developing neurospheres.
Trituration should be forceful enough to separate the debris, but formation of air bubbles should be avoided.
We suggest passaging the neurospheres before they start showing darker central cores. Larger, tighter neurospheres are harder to separate into single-cell suspension mechanically, and might require Accutase exposure for an extended period of time. Extended enzyme exposure could be harsh on NSCs and may interfere with cell viability.
Troubleshooting
In Basic Protocol 1, if the cell suspension in papain does not appear cloudy, it might help to triturate with a glass Pasteur pipette 5 to 10 times every 15 min before placing it back in the incubator. Trituration will help supplement the dissociative action of papain.
In Basic Protocol 2, steps 5 and 6 can be repeated twice. This will make sure that any residual debris remaining from the first resting period can be removed in the second iteration.
In Basic Protocol 5, if insufficient RNA concentration is achieved, it is advised that a considerable number of neurospheres be pooled together for RNA isolation.
In Basic Protocol 7, to make sure that cells are not exposed to the cryopreservation medium when in culture, the culture medium can be replaced on day 2 post culturing of the thawed neurospheres. This will help in getting any residual cryopreservation medium out of the culturing conditions and help the neurospheres acclimate faster to the culture medium.
Anticipated Results
Cells from adult murine brains are not as malleable as embryonic or early post-natal brains and respond very quickly to stress. Hence, the resulting total cell viability in Basic Protocol 1 might be lower than expected (25% to 35% live cells from one mouse brain). However, on culturing in NSC medium under shaking conditions, live, healthy neural progenitor cells survive, thrive, and proliferate to start forming neurosphere clumps within 2 to 3 days of culture.
Following Basic Protocol 2, a cell suspension of pure neurospheres/clumps is expected. These cells will continue to proliferate and form larger bodies of NSCs as long as they are supplemented with EGF/FGF.
It is expected that viable single-cell NSC suspension generated in this process will be able to self renew and form more neurospheres under floating conditions (on EGF/FGF supplementation). These cells can also be plated onto matrix coated plates and allowed to self renew in adherent conditions or differentiated into lineage of choice.
Whole neurospheres plated on to polyornithine/fibronectin-coated plates will spread onto the matrix in rosette formation and proliferate by radiating away from the sphere. These cells should maintain NSC status when supplemented with EGF/FGF every 3 to 4 days.
The neurospheres consist of a multipotent stem/progenitor cells and thus show enhanced expression of Nestin and Sox2 genes, when compared to mature neurons.
Cells thawed out of the freezing medium might take longer to adjust to culturing conditions. It is important that they are cultured in the conditions (NSC maintenance/differentiation) desired for the purpose.
Time Consideration
The entire procedure described in Basic Protocol 1 takes a total of 3 to 4 hr. Animal euthanasia, brain harvesting, and mincing take about 1 hr. The timing for all of the next steps are listed in the protocol in detail.
Basic Protocol 2 takes between 30 and 45 min depending on size of live cell clumps and the ease of dislodging live cells from debris in step 2.
Basic Protocol 3 takes between 30 and 45 min depending on size of neurospheres and ease of dissociation into single cells.
Coating the plates with poly-ornithine and fibronectin is a 2-day process as described. Therefore, it is advisable that it be planned and completed before the neurospheres/dissociated cells are ready for plating. The plating process takes 30 to 45 min.
RNA isolation takes 45 min to an hour. cDNA synthesis requires about 1 to 1.5 hr.
Acknowledgments
The following work is supported by Department of Defense (W81XWH-15-1-0426), Susan G. Komen for the Cure (CCR15332673), and American Brain Tumor Association (DG1600003, MSSF1700010).
Literature Cited
- Ahlemeyer B, & Baumgart-Vogt E (2005). Optimized protocols for the simultaneous preparation of primary neuronal cultures of the neocortex, hippocampus and cerebellum from individual newborn (P0.5) C57Bl/6J mice. Journal of Neuroscience Methods, 149(2), 110–120. doi: 10.1016/j.jneumeth.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Babu H, Cheung G, Kettenmann H, Palmer TD, & Kempermann G (2007). Enriched monolayer precursor cell cultures from micro-dissected adult mouse dentate gyrus yield functional granule cell-like neurons. PLoS One, 2(4), e388. doi: 10.1371/journal.pone.0000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker GA, & Cowan WM (1977). Rat hippocampal neurons in dispersed cell culture. Brain Research, 126(3), 397–342. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, & Torricelli JR (2007). Isolation and culture of adult neurons and neurospheres. Nature Protocols, 2(6), 1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, & Price PJ (1993). Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. Journal of Neuroscience Research, 35(5), 567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Bull ND, & Bartlett PF (2005). The adult mouse hippocampal progenitor is neurogenic but not a stem cell. The Journal of Neuroscience, 25(47), 10815–10821. doi: 10.1523/JNEUROSCI.3249-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, & Alvarez-Buylla A (1999). Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell, 97(6), 703–716. doi: 10.1016/S0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Donovan J, & Brown P (2006). Euthanasia. Current Protocols in Immunology, 73, 1.8.1–1.8.4. doi: 10.1002/0471142735.im0108s73. [DOI] [PubMed] [Google Scholar]
- Eide L, & McMurray CT (2005). Culture of adult mouse neurons. Biotechniques, 38(1), 99–104. doi: 10.2144/05381RR02. [DOI] [PubMed] [Google Scholar]
- Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S, … Pevny L (2004). SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Developmental Neuroscience, 26(2–4), 148–165. doi: 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- Freund J, Brandmaier AM, Lewejohann L, Kirste I, Kritzler M, Krüger A, … Kempermann G (2013). Emergence of individuality in genetically identical mice. Science, 340(6133), 756–759. doi: 10.1126/science.1235294. [DOI] [PubMed] [Google Scholar]
- Galli R, Gritti A, Bonfanti L, & Vescovi AL (2003). Neural stem cells: An overview. Circulation Research, 92(6), 598–608. doi: 10.1161/01.RES.0000065580.02404.F4. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, & Sofroniew MV (2004). GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nature Neuroscience, 7(11), 1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Gritti A, Parati EA, Cova L, Frolichsthal P, Galli R, Wanke E, … Vescovi AL (1996). Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. The Journal of Neuroscience, 16(3), 1091–1100. doi: 10.1523/JNEUROSCI.16-03-01091.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, & Zhao X (2012). Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nature Protocols, 7(11), 2005–2012. doi: 10.1038/nprot.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Clemenson GD Jr., & Gage FH (2007). Spontaneous fusion and nonclonal growth of adult neural stem cells. Stem Cells, 25(4), 871–874. doi: 10.1634/stemcells.2006-0620. [DOI] [PubMed] [Google Scholar]
- Kim JY, Lee JH, & Sun W (2016). Isolation and culture of adult neural stem cells from the mouse subcallosal zone. Journal of Visualized Experiments, (118). doi: 10.3791/54929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laywell ED, Rakic P, Kukekov VG, Holland EC, & Steindler DA (2000). Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proceedings of the National Academy of Sciences of the United States of America, 97(25), 13883–13888. doi: 10.1073/pnas.250471697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, & McKay RD (1990). CNS stem cells express a new class of intermediate filament protein. Cell, 60(4), 585–595. doi: 10.1016/0092-8674(90)90662-X. [DOI] [PubMed] [Google Scholar]
- Leong C, Zhai D, Kim B, Yun SW, & Chang YT (2013). Neural stem cell isolation from the whole mouse brain using the novel FABP7-binding fluorescent dye, CDr3. Stem Cell Research, 11(3), 1314–1322. doi: 10.1016/j.scr.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Pastrana E, Silva-Vargas V, & Doetsch F (2011). Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell, 8(5), 486–498. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, & Weiss S (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science, 255(5052), 1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, & Weiss S (1996). Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Developmental Biology, 175(1), 1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, & Weiss S (1992). A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. The Journal of Neuroscience, 12(11), 4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaberg RM, & van der Kooy D (2002). Adult rodent neurogenic regions: The ventricular subependyma contains neural stem cells, but the dentate gyrus contains restricted progenitors. The Journal of Neuroscience, 22(5), 1784–1793. doi: 10.1523/JNEUROSCI.22-05-01784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Takebe T, & Taniguchi H (2018). Methods for generating vascularized islet-like organoids via self-condensation. Current Protocols in Stem Cell Biology, 45, e49. 10.1002/cpsc.49. [DOI] [PubMed] [Google Scholar]
- Walker TL, & Kempermann G (2014). One mouse, two cultures: Isolation and culture of adult neural stem cells from the two neurogenic zones of individual mice. Journal of Visualized Experiments, 84, e51225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Dunne C, Hewson J, Wohl C, Wheatley M, Peterson AC, & Reynolds BA (1996). Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. The Journal of Neuroscience, 16(23), 7599–7609. doi: 10.1523/JNEUROSCI.16-23-07599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, & van der Kooy D (1996). Is there a neural stem cell in the mammalian forebrain? Trends in Neurosciences, 19(9), 387–393. doi: 10.1016/S0166-2236(96)10035-7. [DOI] [PubMed] [Google Scholar]