

Abstract
Acephalic spermatozoa syndrome is a rare type of teratozoospermia that severely impairs the reproductive ability of male patients, and genetic defects have been recognized as the main cause of acephalic spermatozoa syndrome. Spermatogenesis and centriole-associated 1 like (SPATC1L) is indispensable for maintaining the integrity of sperm head-to-tail connections in mice, but its roles in human sperm and early embryonic development remain largely unknown. Herein, we conducted whole-exome sequencing (WES) of 22 infertile men with acephalic spermatozoa syndrome. An in silico analysis of the candidate variants was conducted, and WES data analysis was performed using another cohort consisting of 34 patients with acephalic spermatozoa syndrome and 25 control subjects with proven fertility. We identified biallelic mutations in SPATC1L (c.910C>T:p.Arg304Cys and c.994G>T:p.Glu332X) from a patient whose sperm displayed complete acephalia. Both SPATC1L variants are rare and deleterious. SPATC1L is mainly expressed at the head–tail junction of elongating spermatids. Plasmids containing pathogenic variants decreased the level of SPATC1L in vitro. Moreover, none of the patient's four attempts at intracytoplasmic sperm injection (ICSI) resulted in a transplantable embryo, which suggests that SPATC1L defects might affect early embryonic development. In conclusion, this study provides the first identification of SPATC1L as a novel gene for human acephalic spermatozoa syndrome. Furthermore, WES might be applied for patients with acephalic spermatozoa syndrome who exhibit reiterative ICSI failures.
Keywords: acephalic spermatozoa syndrome, biallelic mutations, spermatogenesis and centriole-associated 1 like, whole-exome sequencing
INTRODUCTION
Acephalic spermatozoa syndrome (OMIM 617187) is an infrequent but severe type of teratozoospermia that has a genetic etiology and severely affects male fertility.1,2 Sperm integrity requires the head–tail coupling apparatus (HTCA), which is a complex component containing proximal centrioles, distal centrioles, and associated dense material.3 HTCA is made up of a variety of proteins, including coiled-coil domain containing 42 (Ccdc42), centrosomal protein 131 (Cep131), centrobin, centriole duplication and spindle assembly protein (Cntrob), family with sequence similarity 46, member C (Fam46c), intraflagellar transport 88 (Ift88), ornithine decarboxylase antizyme 3 (Oaz3), outer dense fiber of sperm tails 1 (Odf1), serine protease 21 (Prss21), spermatogenesis-associated 6 (Spata6), hook microtubule tethering protein 1 (HOOK1), polyamine modulated factor 1 binding protein 1 (PMFBP1), and Sad1 and UNC84 domain containing 5 (SUN5), and defects in its composition often result in acephalic spermatozoa syndrome and male infertility.4
Mature human sperm contains two centrioles, a typical proximal centriole and a remodeled distal centriole, which play critical roles in the development of the flagellum, the connection between the head and tail of the sperm, spindle pole formation, and early embryonic development.5 Defects in sperm centriole proteins are potential causes of acephalic spermatozoa syndrome,6 and biallelic mutations in centrosomal protein 112 (CEP112) have been identified as the pathogenesis for infertility due to acephalic spermatozoa syndrome in two men.7
Spermatogenesis and centriole-associated 1 like (Spatc1l) protein localizes to the neck region of mouse testicular sperm. The knockout of Spatc1l in the mice alters the head–tail integrity and produces acephalic spermatozoa.8 These results show that Spatc1l plays an essential role in maintaining the integrity of the sperm in mice, but whether SPATC1L plays the same role in human sperm remains unknown.
Biallelic mutations of SPATC1L were identified in an infertile man with acephalic spermatozoa syndrome in this study. The biallelic mutations were deleterious, and plasmids containing the pathogenic variants decreased the level of SPATC1L in vitro. Moreover, SPATC1L defects might affect early embryonic development. Therefore, the results showed that SPATC1L was essential for the sperm head–tail junction and defect in SPATC1L is a novel pathogeny of human acephalic spermatozoa syndrome.
PARTICIPANTS AND METHODS
Patients and control participants
A total of 22 infertile patients with acephalic spermatozoa syndrome were recruited from the Han Chinese population for genetic analysis, and 25 men with proven fertility served as control participants from January 2012 to December 2019 at The First Affiliated Hospital of Xiamen University (Xiamen, China). All the participants underwent physical examinations and routine semen analysis. Their somatic karyotypes were normal, and no microdeletions were detected on the Y chromosome. Approximately 5-ml samples of peripheral blood was obtained, and written informed consent was obtained from each participant. This study was conducted following the 1964 Helsinki Declaration and its later amendments or comparable ethical standards and approved by the ethics committee of The First Affiliated Hospital of Xiamen University (No. XMYY-2020KYSB001).
Whole-exome sequencing (WES) and Sanger sequencing validation
WES and data analysis were performed following the manufacturer's protocol and a previously reported procedure.9 Briefly, genomic DNA was extracted from peripheral blood samples and subsequently sequenced on a NovaSeq6000 platform using AIExomeV1-CNV from iGeneTech Co., Ltd. (Beijing, China). The sequencing covered an area of approximately 62 Mb, including exons, noncoding sequences, ClinVars, and copy number variation (CNVs). Whole-exome reads were aligned against University of California, Santa Cruz (UCSC) h19 using the Burrows–Wheeler Aligner tool (Wellcome Trust Sanger Institute, Cambridge, UK). Genes highly expressed or specifically expressed in the testes or sperm were screened. Homozygous or complex heterozygous mutations of rare pathogenic diseases were screened according to the American College of Medical Genetics (ACMG) Classification Standards and Guidelines for Genetic Variations.10 Sanger sequencing was further performed to verify the variants in the SPATC1L-defective patient and his parents. The primers used for the Sanger sequencing of SPATC1L (NM_001142854.2) c.910C>T and c.994G>T were as follows: F, GCGTTTACTGCAGGCAAGG; and R, GCGTTCAGCGAGTTCCTCA.
Papanicolaou staining and immunofluorescence
Papanicolaou staining and immunofluorescence analysis of the spermatozoa were performed as previously described.9 Primary antibodies against SPATC1L (HPA018979; Sigma-Aldrich, St. Louis, MO, USA), DEAD-box helicase 4 (DDX4; ab27591; Abcam, Cambridge, UK), protein kinase cAMP-dependent type I regulatory subunit alpha (PRKAR1A; PA5-79867; Thermo Fisher, Waltham, MA, USA), and acetylated tubulin (66200-1-Ig; Proteintech Group, Rosemont, IL, USA) were used for immunofluorescence analysis.
Construction of plasmids and western blot analysis
The normal cDNA sequence of SPATC1L (NM_001142854.2) and SPATC1L mutation sequences (c.910C>T or c.994G>T) were amplified by polymerase chain reaction (PCR) and inserted into the pCMV-FH-3xFlag plasmid to express Flag-tagged fusion proteins. The full-length or mutant cDNAs of SPATC1L were then transfected into HEK293 cells.
Total proteins were extracted, separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and incubated with primary antibodies at 4°C overnight. The specific primary antibodies were as follows: Flag primary antibodies (#8146; Cell Signaling Technology, Danvers, MA, USA) and beta-tubulin antibody (10068-1-AP; Proteintech Group).
Intracytoplasmic sperm injection (ICSI) and embryo culture
ICSI was performed as described previously.11 The metaphase II oocyte was injected with the sperm head and detached tail from the patient. The fertilization rate was assessed, and the embryos were cultured with Vitrolife G-Series media (Vitrolife, Goteborg, Sweden). The embryo quality was evaluated as described previously.12
RESULTS
Identification of biallelic mutations of SPATC1L from a patient with acephalic spermatozoa syndrome
The genomic DNA of 22 patients with acephalic spermatozoa syndrome was subjected to WES. The data were analyzed and filtered to exclude irrelevant or meaningless variants. SPATC1L biallelic mutations were found in one patient. This patient showed normal physical development, normal development of external genitalia, and bilateral spermatic veins. The testes of the proband were normal in size, and his hormone levels were in the normal range. However, the sperm concentration in his semen was far below the reference value, and no sperm with progressive motility could be observed. The clinical data and semen parameters of the patient are shown in Table 1. It should be noted that the patient's sperm concentration was approximately 3.5 × 106 ml−1, which indicates that he was a patient with oligospermia (Table 1). A sperm morphology analysis of the SPATC1L-deficient patient was performed using Papanicolaou staining. The spermatozoa from the control participants exhibited a normal morphology with the head and tail closely linked. The spermatozoa of the patient were all headless, and a single head without a tail could still be observed (Figure 1).
Table 1.
Clinical data of the patient with acephalic spermatozoa syndrome
| Clinical parameters | II: 1 | Reference |
|---|---|---|
| Age (year) | 35 | NA |
| Height (cm) | 167 | NA |
| Body weight (kg) | 66 | NA |
| Infertility (year) | 5 | NA |
| Semen volume (ml) | 2.4 | ≥1.5 |
| Semen pH | 7.4 | ≥7.2 |
| Sperm concentration (106 ml−1) | 3.5 | ≥15 |
| Sperm progressive motility (%) | 0 | ≥32 |
| Sperm total motility (%) | 5 | ≥40 |
| Testicular volume (ml), left/right | 12/12 | 10–15 |
| FSH (mIU ml−1) | 14.65 | 1.27–18.96 |
| LH (mIU ml−1) | 6.52 | 1.24–8.62 |
| Testosterone (ng ml−1) | 5.68 | 4.14–7.26 |
| PRL (ng ml−1) | 7.35 | 2.64–13.13 |
| E2 (pg ml−1) | 30 | 20–75 |
NA: not available; FSH: follicle-stimulating hormone; LH: luteinizing hormone; PRL: prolactin; E2: estradiol
Figure 1.
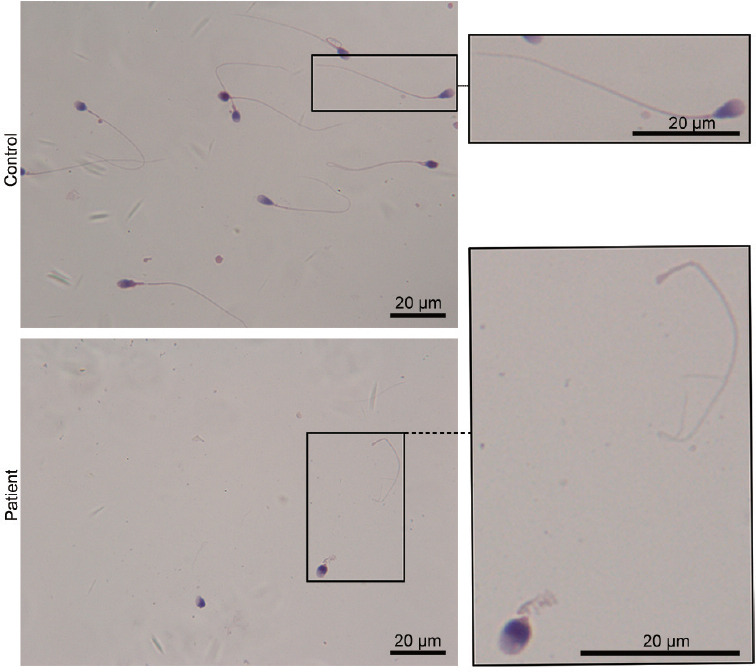
Patient with biallelic mutations in SPATC1L and acephalic spermatozoa syndrome. Morphological analysis of spermatozoa from the SPATC1L-deficient patient by Papanicolaou staining. Scale bars = 20 μm. SPATC1L: spermatogenesis and centriole-associated 1 like.
Among the list of rare and potentially pathogenic variants in this patient, only SPATC1L was closely associated with the maintenance of the integrity of the sperm head–tail junction and preferentially expressed in the testes (Supplementary Table 1). Thus, it was hypothesized that the biallelic mutations c.910C>T:p.Arg304Cys and c.994G>T:p.Glu332X of SPATC1L served as the pathogenesis of the patient's acephalic spermatozoa syndrome. The SPATC1L-mutated patient was from a nonconsanguineous family (Figure 2a). Sanger sequencing was performed to verify the biallelic mutations in the patient and his parents. As expected, the biallelic mutations c.910C>T:p.Arg304Cys and c.994G>T:p.Glu332X were verified in the patient. Furthermore, his unaffected father carried the heterozygous c.910C>T:p.Arg304Cys variant, and his mother carried the heterozygous c.994G>T:p.Glu332X variant (Figure 2b).
Supplementary Table 1.
The rare and potential pathogenic variants in the patient with acephalic spermatozoa syndrome
| Variants | Homozygous/heterozygous | rs | gnomAD_exome_ALL | SIFT | Mutation Taster | Fathmm-MKL |
|---|---|---|---|---|---|---|
| AP5Z1:NM_014855:exon17:c.G2344A:p.D782N | Heterozygous | rs751001042 | 0.00006587 | Damaging | Disease_causing | Damaging |
| AP5Z1:NM_014855:exon12:c.C1567T:p.R523C | Heterozygous | rs201067711 | 0.0001 | Damaging | Disease_causing | Damaging |
| ARSD:NM_001669:exon5:c.T719G:p.F240C | Heterozygous | rs143238998 | 0.000008385 | Tolerable | Neutral | Neutral |
| ARSD:NM_001669:exon5:c.G713T:p.C238F | Heterozygous | rs150899882 | 0.000008362 | Tolerable | Neutral | Neutral |
| BCOR:NM_001123383:exon4:c.C494T:p.A165V | Homozygous | rs538820529 | 0.00007279 | Damaging | Neutral | Damaging |
| BPTF:NM_004459:exon1:c.G149T:p.R50M | Heterozygous | . | 0 | Damaging | Neutral | Neutral |
| BPTF:NM_004459:exon1:c.G152T:p.W51L | Heterozygous | . | 0 | Tolerable | Neutral | Neutral |
| C2CD4B:NM_001007595:exon2:c.G389A:p.C130Y | Heterozygous | . | 0 | Tolerable | Neutral | Neutral |
| C2CD4B:NM_001007595:exon2:c.G386C:p.S129T | Heterozygous | . | 0 | Tolerable | Neutral | Neutral |
| CXorf67:NM_203407:exon1:c.G1504A:p.E502K | Homozygous | rs781789803 | 0.00005147 | Damaging | Neutral | Neutral |
| ESX1:NM_153448:exon4:c. 1105_1131del:p.369_377del | Homozygous | . | 0.0000341 | NA | NA | NA |
| GRIK5:NM_002088:exon19:c.C2809T:p.R937W | Heterozygous | . | 0 | Damaging | Disease_causing | Damaging |
| GRIK5:NM_002088:exon19:c.G2807T:p.C936F | Heterozygous | . | 0 | Damaging | Disease_causing | Damaging |
| HSFX3:NM_001323079:exon2:c.G988A:p.D330N | Homozygous | rs372261595 | 0 | NA | NA | NA |
| KRTAP5-7:NM_001012503:exon1:c.329_330insCTG CTGCCAGTCCAGCTGCTGTAAGCCCTGCTGCTGCCAGTCC AGCTGCTGTAAGCCCTG:p.S110delinsSCCQSSCCKPCCCQSSCCKPC | Homozygous | . | 0 | NA | NA | NA |
| MRE11:NM_001330347:exon4:c.A310T:p.S104C | Homozygous | rs748434421 | 0.00000407 | Damaging | Disease_causing | Damaging |
| MUC16:NM_024690:exon55:c.G40588A:p.G13530S | Heterozygous | . | 0 | Tolerable | Neutral | Neutral |
| MUC16:NM_024690:exon46:c.A39683C:p.K13228T | Heterozygous | . | 0 | Damaging | Neutral | Neutral |
| NOX4:NM_001291929:exon10:c.T947C:p.L316P | Homozygous | . | 0 | Damaging | Disease_causing | Damaging |
| PCSK1N:NM_013271:exon1:c.G23T:p.W8L | Heterozygous | . | 0 | Tolerable | Neutral | Neutral |
| PCSK1N:NM_013271:exon1:c.G7T:p.G3W | Heterozygous | . | 0 | Tolerable | Neutral | Neutral |
| PRDM2:NM_001007257:exon3:c.246_247del:p.D82fs | Heterozygous | rs776947041 | 0.0006 | NA | NA | NA |
| PRDM2:NM_001007257:exon3:c.249delT:p.D83fs | Heterozygous | rs770299670 | 0.0005 | NA | NA | NA |
| RPLP0:NM_001002:exon8:c.C838A:p.P280T | Heterozygous | . | 0.000004119 | Tolerable | Disease_causing | Damaging |
| RPLP0:NM_001002:exon8:c.C851A:p.A284D | Heterozygous | . | 0 | Damaging | Disease_causing | Damaging |
| SLC2A14:NM_001286236:exon5:c.C413T:p.T138M | Heterozygous | rs778496220 | 0.00008533 | Damaging | Neutral | Neutral |
| SLC2A14:NM_001286236:exon4:c.C64T:p.L22F | Heterozygous | rs751861316 | 0.00004062 | Tolerable | Disease_causing | Neutral |
| SPATC1L:NM_001142854:exon5:c.G994T:p.E332X | Heterozygous | rs553752275 | 0.00004149 | NA | Disease_causing | Neutral |
| SPATC1L:NM_001142854:exon5:c.C910T:p.R304C | Heterozygous | rs755224454 | 0.00001716 | Damaging | Disease_causing | Damaging |
| TBL1Y:NM_134258:exon7:c.G413A:p.R138Q | Homozygous | rs377026718 | 0.0003 | Tolerable | NA | Damaging |
| TPRN:NM_001128228:exon1:c.G174T:p.E58D | Heterozygous | . | 0 | Damaging | Neutral | Damaging |
| TPRN:NM_001128228:exon1:c.G161T:p.G54V | Heterozygous | . | 0 | Damaging | Disease_causing | Damaging |
NA: not available; SIFT: sorting intolerant from tolerant
Figure 2.
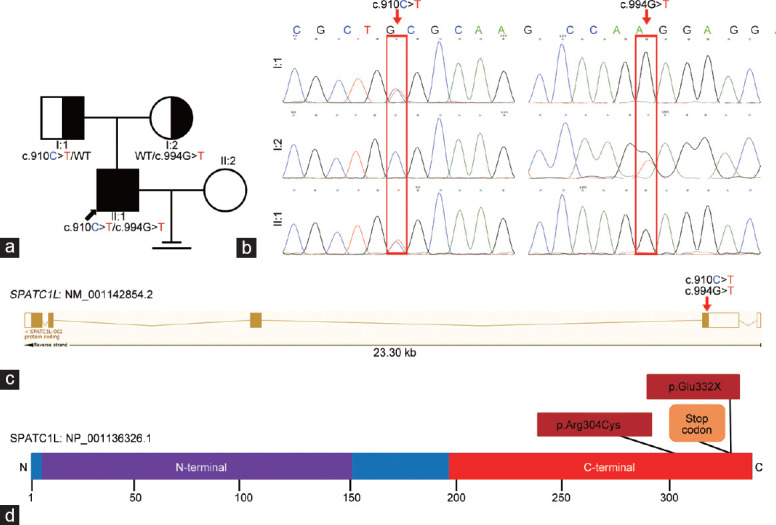
Identification of SPATC1L biallelic mutations in an infertile patient with acephalic spermatozoa syndrome. (a) Pedigree chart of the patient with SPATC1L mutations. The black arrow indicates the proband. (b) Sanger sequencing verified the biallelic mutations in the proband and his parents. The mutant sites are indicated by the red rectangles. (c) Locations of the biallelic mutations in the SPATC1L structure. (d) Affected amino acids on the protein structure of full-length SPATC1L. SPATC1L: spermatogenesis and centriole-associated 1 like.
In silico analysis of SPATC1L variants
The transcript of SPATC1L (NM_001142854.2) contains five exons, which encode full-length SPATC1L (NP_001136326.1) comprising 340 amino acids. Both the biallelic mutations c.910C>T and c.994G>T were located in the fifth exon (Figure 2c) and caused amino acid substitution at the C-terminus (Figure 2d). The c.910C>T variant resulted in a substitution of 304 amino acids from arginine to cysteine, which significantly changed the molecular weight, side chain type, charge, and polarity of the amino acid. The c.994G>T variant results in a premature stop codon and causes direct termination of amino acid translation (Figure 2d). The alignment analysis of the SPATC1L amino acid sequences in different species showed that the amino acids were affected by biallelic mutations at p.Arg304 and p.Glu332, and the subsequent amino acid sequences were highly conserved (Supplementary Figure 1 (451KB, tif) ).
The predicted results of the pathogenicity of the two variants from genetic databases, including sorting intolerant from tolerant (SIFT), likelihood ratio test (LRT), MutationTaster, and other common databases, showed that the biallelic mutations were highly deleterious (Table 2). In addition, the mutations were rare in gnomAD, ExAC, and other common databases Supplementary Table 2).
Table 2.
In silico missense prediction of biallelic mutations in spermatogenesis and centriole-associated 1 like
| Algorithm | c.910C>T:p.Arg304Cys (predicted value) | c.994G>T:p.Glu332X (predicted value) |
|---|---|---|
| SIFT | Damaging (0.001) | NA |
| LRT | Deleterious (0) | Neutral (0.004) |
| MutationTaster | Disease-causing (1) | Disease-causing (1) |
| PROVEAN | Damaging (−4.63) | NA |
| CADD | Damaging (25.2) | Damaging (41) |
| DANN | Damaging (0.999) | Damaging (0.996) |
| GenoCanyon | Damaging (1) | Damaging (1) |
| fitCons | Damaging (0.706) | Damaging (0.706) |
| ClinPred | Pathogenic (0.88554954) | NA |
NA: not available; SIFT: sorting intolerant from tolerant; LRT: likelihood ratio test; PROVEAN: protein variation effect analyzer; CADD: combined annotation dependent depletion; DANN: deleterious annotation of genetic variants using neural networks
Supplementary Table 2.
Allele frequency in a population of biallelic mutations in spermatogenesis and centriole-associated 1 like
| Dataset | c.910C>T:p.Arg304Cys | c.994G>T:p.Glu332X |
|---|---|---|
| gnomAD_All | 0.00001716 | 0.00004149 |
| ExAC_All | 0.00003071 | 0.00005343 |
| 1000 genomes_All | NA | NA |
| 1000 genomes | NA | NA |
| ESP6500_All | NA | NA |
| Kaviar_All | 0.0000194 | 0.0000259 |
| HRC_All | NA | NA |
| CG69_All | NA | NA |
NA: not available
WES data were searched for mutations in this gene to further investigate the proportion of patients with acephalic spermatozoa syndrome caused by SPATC1L pathogenic variants. Unfortunately, no SPATC1L variant could be identified in the WES data of 34 patients with acephalic spermatozoa syndrome. In addition, no SPATC1L variant appeared in 25 control participants with proven fertility.
Biallelic mutations affect SPATC1L expression and the integrity of the human sperm head–tail junction
The expression patterns of SPATC1L in the human testicular sections and ejaculated sperm were examined by immunofluorescence staining to investigate the function of SPATC1L in human spermatogenesis. SPATC1L is mainly expressed at the head–tail junction of elongated spermatids in the testis sections from a patient with obstructive azoospermia who exhibited normal spermatogenesis (Figure 3a). However, a SPATC1L-positive staining signal could not be observed in the sperm in the ejaculated semen from the control participant or the patient (Supplementary Figure 2 (200.8KB, tif) ). In the ejaculated semen of the control participant, PRKAR1A was mainly expressed in the sperm neck. A PRKAR1A-positive signal could still be detected in the sperm of the patient (Supplementary Figure 3 (887KB, tif) ). SUN5 was located in the neck of the spermatozoa, anchoring the sperm head to the tail, and a SUN5-positive signal could still be detected in the sperm tail from the patient (Supplementary Figure 4 (296KB, tif) ).
Figure 3.
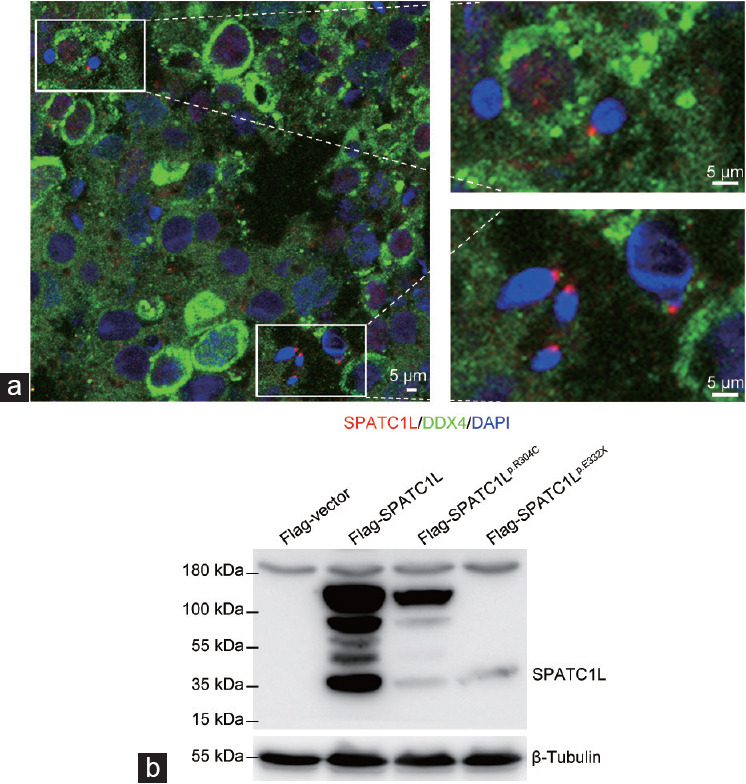
Biallelic mutations affect SPATC1L expression. (a) Immunofluorescence staining of SPATC1L (red) in the testis from a man with obstructive azoospermia. Scale bars = 5 μm. (b) Western blot analysis of the protein expression of Flag-vector, Flag-SPATC1L, and Flag-SPATC1Lp.Arg304Cys, and Flag-SPATC1Lp.Glu332X. β-Tubulin was used as the loading control. SPATC1L: spermatogenesis and centriole-associated 1 like; DDX4: DEAD-box helicase 4; DAPI: 4’,6-diamidino-2-phenylindole.
Plasmids containing full-length SPATC1L or mutant sites were constructed and transfected into the HEK293 cell line to evaluate the effects of biallelic mutations on the expression of SPATC1L. The expression of SPATC1L could be easily observed at the 37-kDa position in the lane of Flag-SPATC1L, whereas the protein expression levels of SPATC1L were significantly decreased in the lanes of Flag-SPATC1Lp.Arg304Cys and Flag-SPATC1Lp.Glu332X (Figure 3b).
Biallelic mutations in SPATC1L might affect early embryonic development
The couple (II:1 and II:2, Figure 2a) who struggled with infertility had experienced three failed assisted reproductive treatments before coming to the center. The patient's casebook showed that no transplantable embryos were formed on either occasion. At the patient's request, ICSI was performed for the last attempt. Seven metaphase II oocytes were retrieved and injected with the patient's sperm head and detached tail, and all the eggs were fertilized (Figure 4). All but one of the eggs (E3, Figure 4) could develop into blastocysts, but the number of cells formed by division was relatively small; additionally, too many cell fragments were present, and none of the blastocysts met the transplantation criteria (Figure 4). The six day-3 cleavage embryos (E1, E2, and E4–E7) were classified as 5C3, 5C3, 5C3, 2C4, 3C4, and 3C4, respectively. The outcomes of four ICSI treatments are summarized in Supplementary Table 3.
Figure 4.
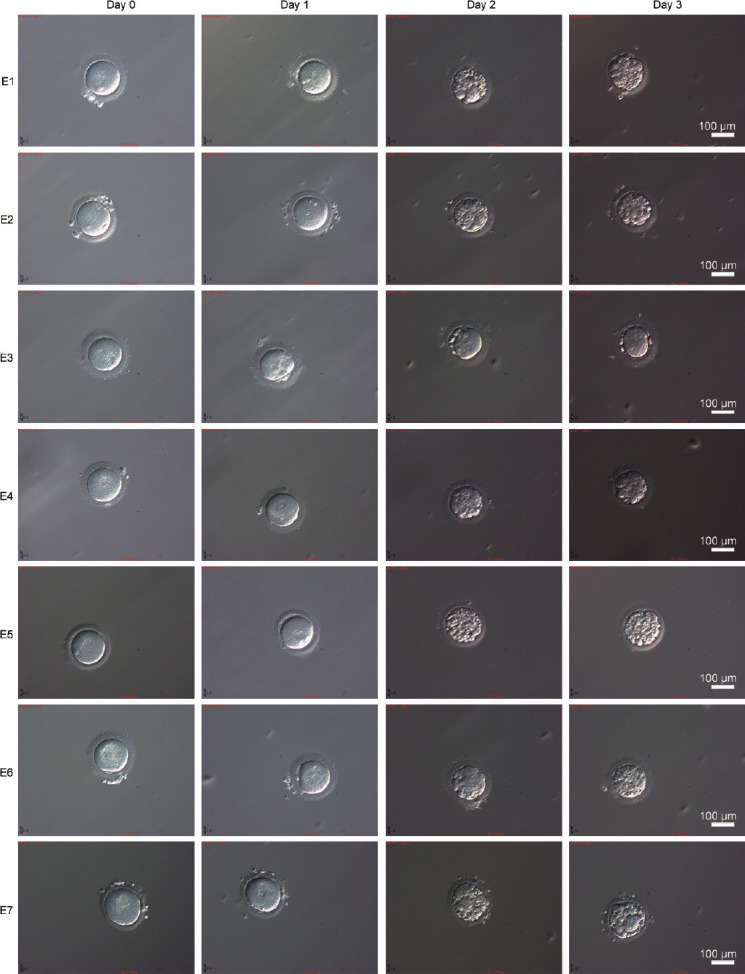
Morphology of the fertilized eggs during embryogenesis. Fertilized eggs on day 0, day 1, day 2, and day 3 of embryogenesis. Scale bars = 100 μm. E1–E7: embryos 1 to 7.
Supplementary Table 3.
Outcomes of intracytoplasmic sperm injection treatments
| Cycles | Protocol | Initiation | Number of oocytes | Insemination method | Number of MII oocytes | Number of fertilized oocytes | Number of 2PN oocytes | Number of cleaved embryos | Number of embryos on day3 | Classification of cleaved embryos |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GnRH protocol | Gonal-F 75 IU + HMG 75 IU | 12 | TESA-ICSI | 7 | 6 | 6 | 6 | 6 | 6C4*4, 5C4, and 3C4 |
| 2 | GnRH protocol | Puregon 75 IU + HMG 75 IU | 14 | TESA-ICSI | 11 | 8 | 8 | 6 | 6 | 6C3*2, 4C3*2, and 5C3*2 |
| 3 | Long-acting GnRH protocol | Puregon 100 IU + HMG 75 IU | 7 | TESA-ICSI | 4 | 4 | 4 | 4 | 4 | 4C4*3 and 3C4 |
| 4 | PPOS protocol | Puregon 100 IU + HMG 75 IU | 10 | TESA-ICSI | 7 | 7 | 6 | 6 | 6 | 5C3*3, 2C4, sand 3C4*2 |
HMG: human menopausal gonadotropin; TESA: testicular sperm aspiration; ICSI: intracytoplasmic sperm injection; PPOS: progestin-primed ovarian stimulation
DISCUSSION
This study investigated the genetic pathogenesis of patients with acephalic spermatozoa syndrome and identified biallelic mutations in SPATC1L. The SPATC1L-defective sperm was approximately 100% headless, and blastocysts derived from defective sperm with ICSI were seriously deficient. SPATC1L was specifically expressed at the head–tail junction of elongating human spermatids, and Spatc1l-knockout mice exhibit an acephalic spermatozoa phenotype.8 Taken together, the results from this study revealed that mutations in SPATC1L are a novel pathogenesis of human acephalic spermatozoa syndrome.
Acephalic spermatozoa syndrome shows a familial incidence and a suggested genetic origin, as has been well demonstrated through WES in previous studies.2,13 SUN5 localizes to the head–tail junction in normal spermatozoa, and defects in SUN5 are the most common genetic etiology of human acephalic spermatozoa syndrome because these can explain the pathogenesis of approximately one-third to one-half of the affected individuals.14,15 Consistent with this finding, Sun5 is also located in the neck of mouse spermatozoa, and the absence of functional Sun5 results in sperm HTCA detachment from the nucleus during spermatid elongation. The spermatozoa of Sun5-null mice also display an acephalic spermatozoa phenotype.16 Mutations in PMFBP1 have been identified in patients with acephalic spermatozoa syndrome and confirmed in mouse models, which demonstrated that PMFBP1 defects are another important cause of acephalic spermatozoa syndrome.17,18 Defects in SUN5 or PMFBP1 could explain 50%–70% of cases.18 Testis-specific 10 (TSGA10), bromodomain testis associated (BRDT), and CEP112 are also reportedly associated with human acephalic spermatozoa syndrome by different research groups.2,7,9,14,19,20 These results suggest that acephalic spermatozoa syndrome exhibits high genetic heterogeneity. The knockout of Spatc1l affects the head–tail integrity of sperm and causes acephalic spermatozoa in mice.8 Therefore, the results strongly indicate that a defect in SPATC1L is the pathogenesis of human acephalic spermatozoa syndrome.
In mice, Spatc1l is specifically expressed in the testis and exhibits dynamic and spatial-specific expression patterns. In detail, Spatc1l is diversely expressed in spermatocytes and then gradually localized to the neck region of testicular sperm. Strangely, the expression of Spatc1l suddenly disappears and cannot be detected in the epididymal mature sperm.8 This study found that SPATC1L could be observed in spermatocytes and was specifically highly expressed at the head–tail junction of elongated spermatids (Figure 2a). However, a SPATC1L-positive staining signal was not observed on the ejaculated sperm of the control participant or the patient (Supplementary Figure 2 (200.8KB, tif) ). The study demonstrated that SPATC1L exhibited similar temporal and spatial expression patterns in human and mouse testes, which suggested that genetic defects in SPATC1L are involved in the occurrence of human acephalic spermatozoa syndrome. The 3’,5’-cyclic adenosine monophosphate (cAMP)-dependent protein kinase (PKA) is a tetrameric holoenzyme comprising two regulatory (PKA-R) and two catalytic (PKA-C) subunits. The PKA-R subunits act as sensors of cAMP and allow PKA-C activity. One of the first signaling events observed during mammalian sperm capacitation is PKA activation. The expression of PRKAR1A was still positive in the ejaculated semen of the patient. Previous studies showed that SUN5 was located in the neck of sperm, and the head and tail of sperm were fixed together. The head and tail conjugator of sperm without SUN5 was separated from the nucleus during sperm cell elongation. However, the present study detected no obvious abnormality in SUN5, which indicated that the expression of SUN5 and PRKAR1A was not affected by the mutation of SPATC1L.
Human oocytes lack centrioles, and sperm centrioles thus play vital roles in fertilization and early embryonic development.21 Thus, abnormal centrosomes of the sperm often lead to fertilization failure or early embryonic development disorders.21 Spatc1l is a centromere-related protein. Although the absence of this protein affects the head–tail integrity of the sperm and causes an acephalic spermatozoa phenotype in mice, the effect of the absence of Spatc1l on fertilization and early embryonic development remains unknown.8 In this study, a couple who struggled with infertility had three failed assisted reproductive treatments for unknown reasons before coming to the reproductive medicine center. After various examinations and assessments, the couple was fully informed, and ICSI was performed for the couple at their request. The eggs were fertilized after ICSI and could develop into blastocysts. However, the number of cells formed by division was insufficient, and the cell fragments were too many; all the blastocysts did not meet the transplantation criteria. Considering that the examination results of the patient's wife were normal, it was highly suspected that these four similar assisted reproductive failure outcomes were due to SPATC1L defects, which affected centriole function and early embryonic development. Mouse sperm and zygotes lack centrioles.22 Therefore, other appropriate animal models are needed to further examine the role of SPATC1L during fertilization and early embryonic development.
CONCLUSIONS
In conclusion, this novel study demonstrated that biallelic mutations in SPATC1L severely impair head–tail attachment and result in human acephalic spermatozoa syndrome. This study provides further insights for clinicians and researchers regarding the genetic etiology of acephalic spermatozoa syndrome.
AUTHOR CONTRIBUTIONS
FXZ and ZXL designed and supervised this study. YZL and NL recruited the participants, collected clinical information, and analyzed the data. YWS, RFW, YLT, and YFW recruited the participants and collected clinical information. XSZ and XYZ performed the molecular experiments. WSL and XLW analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declare no competing interests.
Alignment of the amino acid sequences of SPATC1L in different species at the mutation sites of Arg304 and Glu332 and the subsequent amino acid sequences. SPATC1L: spermatogenesis and centriole-associated 1 like.
Immunofluorescence staining of SPATC1L (red) on the sperm in ejaculated semen from the control participant or the patient. SPATC1L: spermatogenesis and centriole-associated 1 like.
Expression of SUN5 (red) on the sperm in ejaculated semen from the control participant or the patient. SUN5: Sad1 and UNC84 domain containing 5.
Localization of PRKAR1A (red) on the sperm in ejaculated semen from the control participant or the patient. PRKAR1A: protein kinase cAMP-dependent type I regulatory subunit alpha.
ACKNOWLEDGMENTS
The authors thank all the participants for their support and cooperation. This study was supported by the National Natural Science Foundation of China (No. 82001616), the Natural Science Foundation of Fujian Province of China (No. 2019J01565 and 2017J01361), and the Medical and Health Guidance Project of Xiamen (No. 3502Z20209004).
Supplementary Information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Chemes HE, Carizza C, Scarinci F, Brugo S, Neuspiller N, et al. Lack of a head in human spermatozoa from sterile patients: a syndrome associated with impaired fertilization. Fertil Steril. 1987;47:310–6. doi: 10.1016/s0015-0282(16)50011-9. [DOI] [PubMed] [Google Scholar]
- 2.Liu G, Wang N, Zhang H, Yin S, Dai H, et al. Novel mutations in PMFBP1, TSGA10 and SUN5: expanding the spectrum of mutations that may cause acephalic spermatozoa. Clin Genet. 2020;97:938–9. doi: 10.1111/cge.13747. [DOI] [PubMed] [Google Scholar]
- 3.Fawcett DW, Phillips DM. The fine structure and development of the neck region of the mammalian spermatozoon. Anat Rec. 1969;165:153–64. doi: 10.1002/ar.1091650204. [DOI] [PubMed] [Google Scholar]
- 4.Wu B, Gao H, Liu C, Li W. The coupling apparatus of the sperm head and tail. Biol Reprod. 2020;102:988–98. doi: 10.1093/biolre/ioaa016. [DOI] [PubMed] [Google Scholar]
- 5.Avidor-Reiss T. Rapid evolution of sperm produces diverse centriole structures that reveal the most rudimentary structure needed for function. Cells. 2018;7:67. doi: 10.3390/cells7070067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rawe VY, Terada Y, Nakamura S, Chillik CF, Olmedo SB, et al. A pathology of the sperm centriole responsible for defective sperm aster formation, syngamy and cleavage. Hum Reprod. 2002;17:2344–9. doi: 10.1093/humrep/17.9.2344. [DOI] [PubMed] [Google Scholar]
- 7.Sha Y, Wang X, Yuan J, Zhu X, Su Z, et al. Loss-of-function mutations in centrosomal protein 112 is associated with human acephalic spermatozoa phenotype. Clin Genet. 2020;97:321–8. doi: 10.1111/cge.13662. [DOI] [PubMed] [Google Scholar]
- 8.Kim J, Kwon JT, Jeong J, Kim J, Hong SH, et al. SPATC1L maintains the integrity of the sperm head-tail junction. EMBO Rep. 2018;19:e45991. doi: 10.15252/embr.201845991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye Y, Wei X, Sha Y, Li N, Yan X, et al. Loss-of-function mutation in TSGA10 causes acephalic spermatozoa phenotype in human. Mol Genet Genomic Med. 2020;8:e1284. doi: 10.1002/mgg3.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sha YW, Sha YK, Ding L, Lin SB, Ji ZY, et al. A successful pregnancy by intracytoplasmic sperm injection using ejaculate sperm from an infertile man with 46, XX/46, XY true hermaphrodite. Asian J Androl. 2017;19:721–2. doi: 10.4103/1008-682X.190329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai NN, Goldstein J, Rowland DY, Goldfarb JM. Morphological evaluation of human embryos and derivation of an embryo quality scoring system specific for day 3 embryos: a preliminary study. Hum Reprod. 2000;15:2190–6. doi: 10.1093/humrep/15.10.2190. [DOI] [PubMed] [Google Scholar]
- 13.Chemes HE, Puigdomenech ET, Carizza C, Olmedo SB, Zanchetti F, et al. Acephalic spermatozoa and abnormal development of the head-neck attachment: a human syndrome of genetic origin. Hum Reprod. 1999;14:1811–8. doi: 10.1093/humrep/14.7.1811. [DOI] [PubMed] [Google Scholar]
- 14.Sha YW, Xu X, Ji ZY, Lin SB, Wang X, et al. Genetic contribution of SUN5 mutations to acephalic spermatozoa in Fujian China. Gene. 2018;647:221–5. doi: 10.1016/j.gene.2018.01.035. [DOI] [PubMed] [Google Scholar]
- 15.Zhu F, Wang F, Yang X, Zhang J, Wu H, et al. Biallelic SUN5 mutations cause autosomal-recessive acephalic spermatozoa syndrome. Am J Hum Genet. 2016;99:942–9. doi: 10.1016/j.ajhg.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang Y, Zhu F, Wang L, Ouyang YC, Dong MZ, et al. Essential role for SUN5 in anchoring sperm head to the tail. Elife. 2017;6:e28199. doi: 10.7554/eLife.28199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sha YW, Wang X, Xu X, Ding L, Liu WS, et al. Biallelic mutations in PMFBP1 cause acephalic spermatozoa. Clin Genet. 2019;95:277–86. doi: 10.1111/cge.13461. [DOI] [PubMed] [Google Scholar]
- 18.Zhu F, Liu C, Wang F, Yang X, Zhang J, et al. Mutations in PMFBP1 cause acephalic spermatozoa syndrome. Am J Hum Genet. 2018;103:188–99. doi: 10.1016/j.ajhg.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sha YW, Sha YK, Ji ZY, Mei LB, Ding L, et al. TSGA10 is a novel candidate gene associated with acephalic spermatozoa. Clin Genet. 2018;93:776–83. doi: 10.1111/cge.13140. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Sha Y, Wang X, Li P, Wang J, et al. Whole-exome sequencing identified a homozygous BRDT mutation in a patient with acephalic spermatozoa. Oncotarget. 2017;8:19914–22. doi: 10.18632/oncotarget.15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palermo GD, Colombero LT, Rosenwaks Z. The human sperm centrosome is responsible for normal syngamy and early embryonic development. Rev Reprod. 1997;2:19–27. doi: 10.1530/ror.0.0020019. [DOI] [PubMed] [Google Scholar]
- 22.Avidor-Reiss T, Mazur M, Fishman EL, Sindhwani P. The role of sperm centrioles in human reproduction - the known and the unknown. Front Cell Dev Biol. 2019;7:188. doi: 10.3389/fcell.2019.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alignment of the amino acid sequences of SPATC1L in different species at the mutation sites of Arg304 and Glu332 and the subsequent amino acid sequences. SPATC1L: spermatogenesis and centriole-associated 1 like.
Immunofluorescence staining of SPATC1L (red) on the sperm in ejaculated semen from the control participant or the patient. SPATC1L: spermatogenesis and centriole-associated 1 like.
Expression of SUN5 (red) on the sperm in ejaculated semen from the control participant or the patient. SUN5: Sad1 and UNC84 domain containing 5.
Localization of PRKAR1A (red) on the sperm in ejaculated semen from the control participant or the patient. PRKAR1A: protein kinase cAMP-dependent type I regulatory subunit alpha.