

 This work is licensed under a
This work is licensed under a Abstract
Classic galactosemia is an inborn error of carbohydrate metabolism associated with early-onset primary ovarian insufficiency (POI) in young women. Our understanding of the consequences of galactosemia upon fertility and fecundity of affected women is expanding, but there are important remaining gaps in our knowledge and tools for its management, and a need for continued dialog so that the special features of the condition can be better managed. Here, we review galactosemic POI and its reproductive endocrinological clinical sequelae and summarize current best clinical practices for its management. Special consideration is given to the very early-onset nature of the condition in the pediatric/adolescent patient. Afterward, we summarize our current understanding of the reproductive pathophysiology of galactosemia, including the potential action of toxic galactose metabolites upon the ovary. Our work establishing that ovarian cellular stress reminiscent of endoplasmic reticulum (ER) stress is present in a mouse model of galactosemia, as well as work by other groups, are summarized.
Lay summary
Patients with the condition of classic galactosemia need to maintain a strict lifelong diet that excludes the sugar galactose. This is due to having mutations in enzymes that process galactose, resulting in the buildup of toxic metabolic by-products of the sugar. Young women with classic galactosemia often lose the function of their ovaries very early in life (termed 'primary ovarian insufficiency'), despite adherence to a galactose-restricted diet. This means that in addition to the consequences of the disease, these women also face infertility and the potential need for hormone replacement therapy. This article summarizes current strategies for managing the care of galactosemic girls and women and also what is known of how the condition leads to early primary ovarian insufficiency.
Key Words: primary ovarian insufficiency, ovary, follicle, reproductive endocrinology, fertility preservation
Introduction
Classic galactosemia is an inborn error of carbohydrate metabolism associated with early-onset primary ovarian insufficiency (POI) in young women. Our understanding of the consequences of galactosemia upon fertility and fecundity of affected women is expanding, but there are important remaining gaps in our knowledge and tools for its management. For example, while clinical consensus is building about how best to manage patients with galactosemia in prepubertal life and the reproductive years, there is a general lack of established ‘best practice’ strategies. These gaps also include open questions about the mechanism(s) by which galactosemic metabolic perturbations impact reproductive organs, in particular, the ovary. Indeed, systemic production of ‘toxic metabolites’ of galactosemia is often referenced as a causal factor in compromised physiology generally, but direct evidence that individual metabolites can induce ovarian damage has been elusive. In this review, we attempt to fill in some of these gaps. First, we review the condition and its reproductive endocrinological clinical sequelae and summarize current best clinical practices for the management of this quite severe, early-onset POI. Afterward, we set out the state of our understanding of the reproductive pathophysiology of galactosemia, including the potential action of toxic galactosemic metabolites on the ovary. Our work establishing that ovarian cellular stress reminiscent of endoplasmic reticulum (ER) stress is present in a mouse model of galactosemia, as well as work by other groups, are summarized in this latter review section.
Classic galactosemia
Galactosemia results in the deficiency of enzymes in the Leloir pathway (Holden et al. 2003) including galactokinase (GALK1), galactose-1-phosphate uridylyltransferase (GALT), galactose mutarotase (GALM), or UDP-galactose 4′-epimerase (GALE), any of which can compromise the metabolism of galactose (Wada et al. 2019). The most common and severe form of galactosemia is GALT deficiency, also called classic galactosemia (CG), and affects 1/14,000–1/80,000 individuals worldwide (Jumbo-Lucioni et al. 2012) and 1:53,554 births in the United States of America (Therrell et al. 2014). CG is inherited in an autosomal recessive pattern. Newborns in many countries are screened through screening programs for CG after birth by measuring GALT enzyme activity or galactose in dried blood spots. Infants that screen positive have further diagnostic testing to allow a timely diagnosis early in life. Diagnosis of CG includes detection of reduced GALT enzyme activity in red blood cells, elevated erythrocyte galactose-1-phosphate (gal-1P) levels, and, in general, the identification of pathogenic variants in the GALT gene (Pyhtila et al. 2015). Initial symptoms for infants include emesis and poor feeding with failure to thrive, lethargy, jaundice, liver failure, and Gram-negative sepsis. Infants who survive the acute presentation or are treated early because of newborn screening still suffer chronic long-term complications including developmental delay, speech and language delays, motor function abnormalities, cataracts, absent or delayed puberty or premature menopause as a result of ovarian insufficiency (Fridovich-Keil et al. 2011, Berry 2020).
Treatment for CG includes a galactose-restricted diet to avoid exposure to sugar. This results in the reduction, but not in the normalization of metabolites that accumulate inappropriately due to the deficiency of the GALT enzyme. The metabolites galactitol and gal-1P are known to have toxic effects upon cells and are thought to contribute to CG pathophysiology (Thakur et al. 2017). Dietary galactose restriction resolves initial neonatal symptoms, but chronic complications persist with varying prevalence and severity despite adherence to dietary management. Although a galactose-restricted diet is started as soon as the diagnosis is confirmed, the exact timing of diet initiation does not have long-term effects on the development of chronic conditions later in life (Jumbo-Lucioni et al. 2012).
The persistent elevation of galactitol and gal-1P is likely due to the endogenous galactose production from UDP-galactose via UPD-glucose, which results in elevated levels of galactose, galactitol, and gal-1P in CG patients despite being on a galactose-restricted diet (Berry et al. 2004). Mean plasma galactose levels in CG patients on a galactose-restricted diet are on average 2.72 ± 0.70 μM (0.05 ± 0.01 mg/dL) compared to 0.52 ± 0.08 μM (0.01 ± 0.001 mg/dL) in non-galactosemic subjects on a lactose-free diet (Ning & Segal 2000); red blood cell gal-1P levels ranges from 1.1–3.5 mg/dL (0.04–0.13 mM) as compared to < 1 mg/dL (<0.038 mM) in non-galactosemic individuals (Berry et al. 1997).
Complications of classic galactosemia
Beginning in infancy, individuals with CG are assessed periodically for cognitive development, neurological and motor functions, speech and language delays, psychosocial development, bone health, and cataracts (Welling et al. 2017). Further, gal-1P levels are checked regularly to determine dietary compliance, and adjustments are made for elevated levels. An observational study that included 33 adult participants (17 men and 16 women) with CG on a galactose-restricted diet found the following complications affecting participants: cataracts (21%), low bone density (24%), tremor (46%), ataxia (15%), dysarthria (24%), and apraxia of speech (9%) (Waisbren et al. 2012).
Primary ovarian insufficiency in classic galactosemia
POI is one of the most common chronic complication associated with CG and affects 80–90% of female patients following a galactose-restricted diet (Fridovich-Keil et al. 2011). POI is defined as amenorrhea greater than 3 months in young adolescents and adult women who are less than 40 years of age with two follicle-stimulating hormone (FSH) levels in the menopausal range within a month of testing and decreased serum estradiol levels (Nelson 2009). POI can lead to infertility, hypoestrogenism, as well as mental distress (Nelson 2009).
The hypoestrogenic environment resulting from POI can result in poor bone health with an increased risk of fracture (Marino & Misra 2011, Batey et al. 2013), heightened risk of cardiovascular events (Roeters Van Lennep et al. 2016), and menopausal symptoms (Sullivan et al. 2016). Estrogen is the primary hormone that promotes secondary sexual characteristics that develop during puberty such as breasts, bone maturation, uterine growth, and the onset of menses. Many females with POI before pubertal onset will not develop these secondary sex characteristics. Hormone replacement therapy (HRT) with estrogen and progesterone is the mainstay treatment for patients with POI (see also 'Management,' subsequently).
The onset of primary ovarian insufficiency in classic galactosemia
Increasing evidence in CG POI suggests female infants have normal-appearing ovaries at birth that become damaged over time. A review of gonadal function in patients with CG by Rubio-Gozalbo and colleagues in 2010 included 23 female patients whose ovaries were evaluated either histologically, by imaging, or laparoscopy (Rubio-Gozalbo et al. 2010). Of the 23 patients, two were neonates and had morphologically normal ovaries with numerous oocytes (Levy et al. 1984, Levy 1996). The remaining patients, excluding one, were > of age and all but two had hypoplastic ovaries, and evidence of ovarian dysfunction with either fibrous ovarian tissue, significantly diminished primordial follicles, or absent growing follicles (Rubio-Gozalbo et al. 2010). There was a single patient that had normal-appearing ovaries at the age of seven, but repeat laparoscopy at age 17 revealed streak gonads (Kaufman et al. 1981). These findings are consistent with recent data from ovarian tissue cryopreservation (OTC) in prepubertal CG patients. Six prepubertal girls with CG underwent ovarian tissue cryopreservation for fertility preservation; five of the girls were less than five years of age and had normal ovarian morphology and number of follicles whereas the remaining girl was 11.7 years of age and had absent ovarian follicles (Mamsen et al. 2018). This further suggests that females with CG have normal oocyte numbers during infancy and early childhood with rapid loss over time. The trajectory of gonadotrophins to mirror menopausal levels throughout childhood into adolescence can further corroborate the biopsy/ultrasounds and suggest a progression of ovarian failure; although evidence of abnormal gonadotrophin levels have been seen as early as nine months in galactosemia (Kaufman et al. 1981, Levy et al. 1984). A retrospective chart review of 11 females with CG followed by our clinic was conducted. When available, information regarding genotype, the timing of menses/amenorrhea, the use of HRT and labs including gal-1P, FSH, anti-Müllerian hormone (AMH), luteinizing hormone (LH) and estradiol were recorded longitudinally for each patient. Our data revealed nine of 11 patients with evidence of POI including abnormal hormonal values. Of those nine patients, FSH levels before and after the age of 10 were available for six patients (summarized in Fig. 1). Four of the six had normal/borderline FSH levels prior to age 10 that sharply increased at age 10. The other two patients had elevated FSH levels prior to age 10. Six patients had data for AMH, with only two showing normal/high AMH levels before age 10 or when measured (Table 1).
Figure 1.
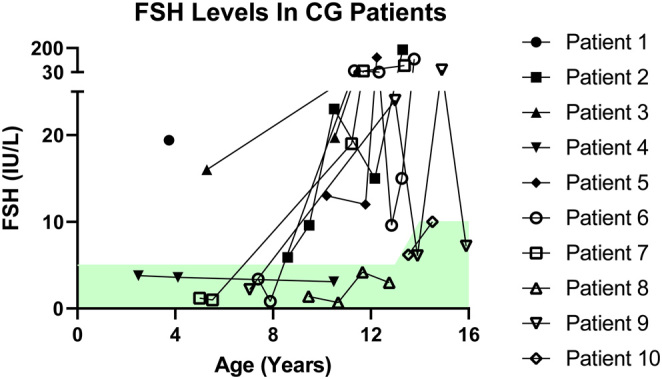
Longitudinal follicle-stimulating hormone (FSH) levels for female patients with classic galactosemia (CG) . FSH levels are provided for a cohort of child and adolescent CG patients. The green shaded region represents normal values. The timing of hormone replacement initiation was not available for this patient set; this may have influenced FSH levels in some patients.
Table 1.
Patient data for genotype and timing of POI.
| Patient no. | Genotype | Spontaneous menses | Time of amenorrhea | Hormone replacement therapy | Anti-Müllerian hormone (AMH) | Ovary images |
|---|---|---|---|---|---|---|
| 1 | Q188R/Q212X | Prepubertal | Prepubertal | Not indicated at this time | Low at 7 months and age 4 | N/A |
| 2 | Q188R/Q188R | Yes | ~16 years | Yes, periods stopped when hormones stopped | Low at age 28 | N/A |
| 3 | S135L/IVS2-2G | Prepubertal | Prepubertal | Not indicated at this time | Low at ages 5–11 | N/A |
| 4 | Q188R/R258C | Yes | None, regular periods | No | Normal range at age 16 | N/A |
| 5 | Q188R/Q188R | Yes | ~16 years | Yes, periods stopped when hormones stopped | Low at age 25 | N/A |
| 6 | Q188R/unknown | Yes | 21 years | Birth control as contraception | Low at ages 18–27 | Yes; smaller than postmenopausal volume at age 27 |
| 7 | Q188R/Q188R | Yes | None, regular periods | Yes, periods regular on birth control | Low at ages 18–22 | N/A |
| 8 | Q188R/E203K | Yes | Unknown | No | High at ages 9–13 | N/A |
| 9 | Q188R/Q188R | Yes | ~16 years | Unknown | N/A | N/A |
| 10 | R201H/M336L | Yes | Unknown | Unknown | N/A | N/A |
| 11 | Q188R/Q188R | Yes | No | Birth control as a contraceptive | N/A | Yes; scan for second pregnancy at > 30 years of age showed no visible ovaries |
While 32–55% of patients with CG do not achieve spontaneous menarche, a fraction that do can cycle regularly into their late 20s (van Erven et al. 2017, Frederick et al. 2018). Most cases of POI in CG are diagnosed in the pre-teen to teenage years due to ovarian insufficiency not being clinically evident until puberty (Fridovich-Keil et al. 2011). In a recent study that evaluated these parameters in the largest cohort of patients with CG to date, 68% of the 102 post-pubertal females with CG achieved spontaneous menarche. Spontaneous menarche occurred at an average age of 13.8 years (range 10–19), and HRT-assisted menarche was 14.2 years (range 11–18) (Frederick et al. 2018). If spontaneous menarche was achieved, only 50% of patients still cycled regularly after three years, 35% after five years, and fewer than 15% had regular menstrual cycles after 10 years. In the patients that did not receive HRT, approximately one-third of the 102 patients with CG had regular menstrual cycles in their late teens, and the number decreased to approximately 10% by their mid-20s (Frederick et al. 2018). In our small set of patients (Table 1), the majority did achieve spontaneous menarche, but only 50% were still cycling regularly after age 16 without the use of hormonal therapies (Table 1).
Due to the variability in the timing of POI onset among CG patients, and the lack of a reliable early screening tool for ovarian function/ovarian reserve, patients are often not officially diagnosed with POI until their peri-pubertal years when secondary sexual characteristics are absent (see also subsequent section). This makes counseling and prospective management of POI in CG difficult. Even so, some potential risk factors for the development of POI in CG patients have been identified. For example, there is a higher probability of POI and other long-term complications if the GALT mutation is Q188R/Q188R (Guerrero et al. 2000) or if the mean erythrocyte gal-1P is > 2.0 mg/dL on a galactose-restricted diet (Yuzyuk et al. 2018). The p.Q188R mutation is one of the most prevalent missense mutations in the GALT gene that leads to higher instability of the protein (Kumar et al. 2020). In addition to POI, homozygous Q188R mutations are also associated with cognitive impairment and verbal dyspraxia (Robertson et al. 2000, Antshel et al. 2004).
Pathophysiology of primary ovarian insufficiency
Oocytes, which are located within ovarian follicles, are progressively lost throughout the female life cycle. The maximum number of oocytes (approximately 6–7 million) is before birth at around 20 weeks gestation, which is also when primordial follicles begin to form. At birth, there are approximately 1–2 million oocytes that drop to 300,000–400,000 at puberty, and only 400–500 are ovulated during a lifespan (Strauss III & Williams 2019). Oocytes are female germ cells that are capable of fertilizing with sperm to create embryos. Follicles consist of the cell types that envelop the oocyte, such as the granulosa cells and theca cells, and the oocyte itself. Intricate cellular communication between the oocyte and the surrounding granulosa cells determines the health and longevity of the ovary. LH and FSH are gonadotropins produced by the anterior pituitary that are important in regulating follicular growth, ovulation, and ovarian hormone production. Estrogen, which is a significant steroid hormone produced by granulosa cells of preovulatory follicles, is created via a two-cell process. LH stimulates theca cells to produce androgens, and FSH stimulates granulosa cells to aromatize the androgens into estrogen, which is then released into the circulation (Strauss III & Williams 2019). In women with POI, there is an accelerated loss of follicles by either destruction or atresia resulting in diminished ovarian function. Loss of ovarian function leads to long-term complications related to hypoestrogenism in addition to infertility. Multiple factors can cause POI including genetic variants, autoimmune disorders, metabolic causes, infectious causes, iatrogenic and toxic causes (Panay et al. 2020).
Folliculogenesis and cellular signaling pathways
To delineate the potential pathophysiology of ovarian demise in CG, it is important to appreciate the normal lifespan of the ovary and follicle development. Folliculogenesis occurs in essentially four stages, beginning with the transition of a primordial follicle to the primary follicle, then to a secondary, antral, and finally, preovulatory follicle. Even in healthy ovaries, the vast majority of oocytes and follicles will not achieve ovulation but will die by atresia (Liu et al. 2006, Adhikari & Liu 2009). In utero, primordial follicles undergo meiosis and remain arrested at prophase I of the cell cycle. At around 24 weeks gestation, the first wave of primordial follicles become activated and grow into primary follicles, and after birth, the majority grow and are arrested at the pre-antral stage. At puberty, gonadotrophin production begins and selected follicles can progress to the antral stage with a select number maturing to ovulate an oocyte (Ford et al. 2020). The growth of primordial follicles to ultimately ovulatory follicles is a continuous process throughout a woman’s reproductive life, and the number of primordial follicles is considered the ovarian reserve; thus, the appropriate timing is integral in preserving ovarian function until anticipated menopause.
Various animal models of POI and cultured human ovaries have elucidated different signaling pathways involved in folliculogenesis at different stages of development; of note for this review are the phosphatidylinositol 3-kinase/AKT signaling pathway (PI3K/AKT), and the unfolded protein response (UPR), specifically EIF2A phosphorylation (Ernst et al. 2017). Many other well-known signaling pathways, including nuclear receptor signaling (Komatsu & Masubuchi 2017, Chakravarthi et al. 2020), TGFB (Yoon et al. 2013, Chang et al. 2016), Hippo (Hsueh & Kawamura 2020), and the NFKB pathway (Wright et al. 2020) have all been shown to regulate normal folliculogenesis. Here, we will limit our discussion to signaling pathways that have been investigated in the context of ovarian dysregulation in the CG mouse model.
Tightly regulated control of PI3K/AKT signaling is paramount for proper initiation of follicle growth and primordial follicle survival (Liu et al. 2006). Loss of regulators of PI3K/AKT signaling such as Pten, Foxo3a, Pdk1, and Tsc 1/2 in mouse oocytes and granulosa cells can lead to abnormal activation of primordial follicles, and thus, POI with depletion of the ovarian reserve (John et al. 2008, Jagarlamudi et al. 2009, Reddy et al. 2009, Adhikari et al. 2010). Disruption of upstream regulators of PI3K/AKT such as semaphorin 6C (SEMA6C) and cell division cycle 42 (CDC42) can also influence follicle activation through PI3K/AKT signaling (Yan et al. 2018, Zhou et al. 2018). The phosphorylation of AKT is central in PI3K/AKT signaling and controls growth in the cell. In mice with Pten deleted in their oocytes, increased pAKT was associated in a stage-specific manner with the mass activation of follicles (Jagarlamudi et al. 2009). Additionally, stimulation of AKT phosphorylation and inhibition of PTEN in cortical fragments of ovaries from human patients receiving chemotherapy were able to activate primordial follicles and produced two live human births (Kawamura et al. 2013, Suzuki et al. 2015). Changes in PI3K/AKT signaling are also associated with a high-fat diet; Nteeba and coworkers saw increased levels of mRNA for AKT and Kit ligand indicating enhanced PI3K/AKT signaling in older mice after a high-fat diet (Nteeba et al. 2013). While PI3K/AKT signaling is involved in the activation and growth of follicles, the exact mechanism and timing of activity in this pathway are poorly understood.
The UPR is a time-dependent, paradoxical survival or death response in the cell in response to ER stress that culminates in either the cell restoring homeostasis or dying via apoptosis and/or autophagy (Donnelly et al. 2013). Improper folding of proteins (a surge of protein synthesis), abnormalities in glycosylation, increased oxidative stress, and perturbations of Ca2+ homeostasis are all activators of ER stress and the UPR (Oslowski & Urano 2011). Harada and coworkers found evidence of increased UPR molecular markers in the granulosa cells of late secondary follicles in mice suggesting a normal physiological role of UPR in meeting the needs of increased protein synthesis as the follicle nears maturation (Harada et al. 2015). True to the roles of the UPR as both pro-survival and death promoter, in goat ovaries, ER stress marker protein BiP was present in both atretic and non-atretic granulosa cells but increased in the granulosa cells of follicles destined for atresia; early atretic follicles also showed increased BiP (Lin et al. 2012). In contrast, the apoptosis marker CHOP was only present in granulosa cells of atretic follicles (Lin et al. 2012). In vitro and in vivo models of obesity-induced POI exhibited increased ER stress markers and saw less viable oocytes ovulated; administration of a UPR modulator (Salubrinal) reversed these poor outcomes (Wu et al. 2010, 2012, 2015). As the UPR is a phenomenon in both follicle growth and atresia, any disruption of the UPR likely will be detrimental to the oocyte, follicles, and ovarian health (Lin et al. 2012, Babayev et al. 2016, Xiong et al. 2017, Takehara et al. 2020).
One key regulator of the UPR is the phosphorylation of EIF2A. In times of cellular stress, the phosphorylation of EIF2A serves to slow down protein translation in the cell to restore homeostasis (Donnelly et al. 2013). Beyond the UPR, EIF2A is an important component of normal protein translation, specifically to the 43S pre-initiation complex which acts with the EIF4F complex to initiate synthesis of new polypeptides during cellular growth (Colegrove-Otero et al. 2005). The phosphorylation of EIF2A by various kinases blocks the initiation of protein translation by inhibiting EIF2B, the nucleotide exchange factor involved in the 43S pre-initiation complex (Donnelly et al. 2013). In the oocyte, phosphorylated EIF2A appears to be involved in regulating meiosis and keeping follicles dormant until their predestined activation (Le Bouffant et al. 2008, Alves et al. 2009). As keeping the lifespan of follicles dormant is critical for ovarian longevity, EIF2A regulation is likely a key player in developing POI. In humans, mutations in EIF4ENIF1,an important transport protein for components crucial to the EIF4F complex, result in POI (Kasippillai et al. 2013, Zhao et al. 2019). Mutations in EIF2B are also associated with POI with concurrent neurological complications, providing more evidence that protein initiation factors are involved in maintaining ovarian health (Fogli et al. 2004).
Human and animal model examples
POI is associated with a variety of different conditions such as X chromosome abnormalities (Turner syndrome, FMR1), autosomal recessive disorders (CG, mitochondrial diseases), autoimmune diseases (Addison’s disease), specific single-gene defects (EIF4ENIF1, EIF2B, FOXL2, GDF9, FOXO3, FSHR,etc.), and iatrogenic causes (chemotherapy) (Rossetti et al. 2017). Also, metabolic dysfunction such as obesity is associated with problems in reproduction, including POI (Hohos & Skaznik-Wikiel 2017). While the etiology of POI in all causes is largely unknown, the pathophysiology of POI can be divided into two major categories: developmental problems with primordial follicles resulting in atresia before growth or by follicle depletion through accelerated growth and atresia (Panay et al. 2020).
We can look to other examples of POI caused by damaging environments such as autoimmune, iatrogenic, metabolic syndrome (obesity/high-fat diet), and models of galactose-intoxicated animals to speculate how the follicle damage is caused by abnormal metabolites in CG. Various studies exploring autoimmune causes of POI show that the oocyte, granulosa cells, zona pellucida, steroid hormone-producing cells in developing follicles, and the pituitary–hypothalamic axis can all be a target of harmful antibodies (Forges et al. 2004, Altuntas et al. 2006, Sharif et al. 2019). Induced oophoritis in animal models reveals damage mainly to growing and developing follicles (Domniz & Meirow 2019). Targeted antibody attack of one negative regulator of FSH led to increased FSH and follicle depletion due to increased follicle activation early in life (Altuntas et al. 2006). Chemotherapy agent cyclophosphamide appears to cause POI by damaging DNA, specifically in the primordial oocyte and granulosa cells of developing follicles. The damage to the primordial oocyte leads to a brief moment of growth in the oocyte but then to apoptosis without differentiation to a primary follicle at day one after administration of cyclophosphamide (Luan et al. 2019). There are conflicting interpretations of whether chemotherapy causes primordial follicle death and no increased activation of primordial follicles (Luan et al. 2019) or POI due to increased activation (Kalich-Philosoph et al. 2013); regardless, it is clear that primordial follicle damage contributes to developing POI. Researchers also saw changes in AMH levels after the administration of cyclophosphamide in mice. Three days after administration, AMH fell dramatically, similar to what is seen in humans with follicle damage, and then steadily increased to control levels seven days post-treatment, presumably due to improvement in follicle health after the initial insult (Luan et al. 2019).
Obesity, specifically a high-fat diet, can lead to reproductive dysfunction such as follicular and oocyte damage, lower responses to in vitro fertilization (IVF), and problems in the hypothalamic–pituitary–ovarian (HPO) axis likely due to high levels of insulin (Luke et al. 2011, Broughton & Moley 2017, Hohos and Skaznik-Wikiel 2017). Limited mechanistic studies of obesity/high-fat diet and the ovary are available in humans beyond association data; however, women fed a high-fat diet for two to four menstrual cycles experienced characteristics of POI such as cycle disruption, a shorter time between menses, increased FSH, and decreased estradiol (Hill et al. 1984, Reichman et al. 1992). Machtinger and colleagues assessed the quality of oocytes from obese women that failed fertilization during IVF and found disruptions in spindle and chromosome alignment (Machtinger et al. 2012). Additionally, women with POI are at a greater risk of developing type 2 diabetes, suggesting a metabolic relationship between the two disorders (Anagnostis et al. 2019). Animal studies have shed more light on the potential mechanism causing the disruptions related to obesity such as oxidative stress, mitochondrial dysfunction, HPO axis disruption, and problems in PI3K/AKT signaling (Wu et al. 2010, 2012, Nteeba et al. 2013). In mouse models of obesity, high-fat diet or genetically obese mice have ovarian aging and evidence of POI. Female mice fed high-fat diets had fewer primordial follicles, increased developing follicles, and more corpus luteum than normal caloric or restricted caloric mice (Wang et al. 2014).
Galactose-intoxicated rat models can give some clues to the toxic effects of galactose on the ovary (Rostami Dovom et al. 2019). Early prenatal administration (before 15 days post-conception) of 35% of calories from galactose to mother rats resulted in decreased primordial follicles, increased apoptosis, and disruption in germ cell formation in the pups, suggesting follicle damage in utero (Bandyopadhyay et al. 2003a,b). Other studies found that prenatal and postnatal administration of high galactose (>30%) caused increased FSH and LH and decreased estradiol, similar to human gonadotrophic presentations in POI (Chen et al. 1981). In these studies, the primary metabolite measured was gal-1P; however, the proximal mechanism that contributes to the follicle demise in these models is unknown (Rostami Dovom et al. 2019). Postnatal exposure to high galactose (40%) after weaning increased galactitol concentrations in the ovary and resulted in impaired folliculogenesis; administration of an aldose reductase inhibitor ameliorated the effects of galactitol toxicity suggesting that galactitol and the oxidative stress it creates when it is reduced are potential contributors to ovarian damage in POI in experimental hypergalactosemia. In this study, gal-1P was not measured.
Pathophysiology of primary ovarian insufficiency in galactosemia
The aforementioned models of POI and human examples give clues about what types of insults can cause POI. However, while informative to the development of POI, these models are not specific to CG, as there are presumed normal functions of the GALT enzyme. Indistinct prenatal insults are possible in CG female ovaries due to endogenous production of galactose; however, this has only been examined in galactose-intoxicated rat models of POI and not measured in human fetuses (Bandyopadhyay et al. 2003b). While the exact mechanism of ovarian failure is not known, overwhelming evidence suggests that excess gal-1P is the main culprit in CG POI (Guerrero et al. 2000, Bandyopadhyay et al. 2003a, Tang et al. 2014, Thakur et al. 2017, Vanoevelen et al. 2018, Yuzyuk et al. 2018, Balakrishnan et al. 2019). Females heterozygous for pathogenic variants in GALT and with other types of galactosemia, such as GALK1 deficiency, do not have chronically elevated levels of gal-1P, have a normal ovarian reserve, and do not undergo menopause at a premature age (Knauff et al. 2007, Badik et al. 2011, Rubio-Gozalbo et al. 2021). A summary of studies that evaluate the 'toxic' action of specific galactose metabolites is presented in Table 2.
Table 2.
Evidence for galactosemia metabolite toxic action.
| Species/Aberrant metabolites | Consequences | Mechanism | References |
|---|---|---|---|
| Humans | |||
| Gal-1P | Increased risk of long-term complications in humans | Unknown | Yuzyuk et al. (2018) |
| Gal-1P | Increased risk of POI in humans | Unknown | Guerrero et al. (2000) |
| Gal-1P | Best predictor for verbal dyspraxia | Unknown | Webb et al. (2003) |
| Gal-1P | Inhibits growth in fibroblasts derived from galactosemic patients | UDP-hexose deficiency | Lai et al. (2003) |
| Gal-1P | Increased stress in GALT negative human-derived fibroblasts | Accumulation of unfolded proteins, altered calcium homeostasis, and ER stress | Slepak et al. (2007) |
| Mouse | |||
| D-galactose Gal-1P |
Damages MII mouse oocytes and hinders embryo development | Increased ROS and disruption of spindle structure and chromosomal alignment | Thakur et al. (2017) |
| Galactose | Decreased oocyte number in offspring | Unknown | Chen et al. (1981) |
| Gal-1P | Subfertility, follicular dysfunction, and growth restriction in GALT-deficient mouse models | Tang et al. (2014) | |
| Gal-1P | Reduced growth of mutant mouse fibroblasts | Increased ER stress in mouse fibroblasts via regulation of PI3K/Akt signaling | Balakrishnan et al. (2016) |
| Gal-1P | Ovarian dysfunction in galactosemia mouse model | Increased ER stress via regulation of PI3K/Akt signaling | Balakrishnan et al. (2019) |
| Gal-1P | Increased ataxia in galactosemia mouse model | Stress-related cellular damage via regulation of PI3K/Akt signaling | Chen et al. (2017) |
| Galactose, galactitol, Gal-1P | Follicular atresia | Unknown | Bandyopadhyay et al. (2003a) |
| Galactose | Adverse germ cell migration with initial low pool of germ cells | Unknown | Bandyopadhyay et al. (2003b) |
| Galactitol | Cataracts | Unknown | Ai et al. (2000) |
| Yeast | |||
| Gal-1P | Growth arrest in yeast models | Increased environmental stress | Slepak et al. (2005) |
| Gal-1P | Growth arrest in galactosemia yeast models | Unknown | Ross et al. (2004) |
| Gal-1P | Decreased growth rate in yeast | Decrease in intracellular phosphate levels | Machado et al. (2017) |
| Gal-1P | Growth arrest of GALT negative yeast | Mumma et al. (2008) | |
| Gal-1P | Decreased growth rates in galactosemic yeast model | Inhibition of phosphoglucomutase | de Jongh et al. (2008) |
| Zebrafish | |||
| Gal-1P | Reduction in motor function and fertility potential | unknown | Vanoevelen et al. (2018) |
The GALT knockout zebrafish and mouse model have the POI phenotype similar to CG humans (Tang et al. 2014, Vanoevelen et al. 2018). Zebrafish with galt-knockout show increased gal-1P, decreased number of eggs after fertilization, and increased galk1 and gale activity in the ovary (Vanoevelen et al. 2018). This model showed aberrant glycosylation and disturbed sialyation in the ovary (Haskovic et al. 2020). Previously, our lab developed a new homozygous GalT-gene trapped mouse model to study the pathophysiology of CG on a multicellular organism (Tang et al. 2014). Galactose sensitivity, reduced fertility in females, hypogalactosylation of IgG, growth restriction, and motor impairment are seen in the GalT-deficient mice in both sexes, similar to humans with CG (Balakrishnan et al. 2016, 2017, Chen et al. 2017).
In our mouse model, we see subfertility, reduced follicles, increased corpus luteum, increased ER/UPR stress markers, and decreased PI3K/AKT signaling in two-month-old ovaries compared to WT ovaries (Balakrishnan et al. 2017, 2019). While there were significantly fewer primordial follicles at two months of age, follicle counts at postnatal day eight (unpublished data) showed no difference in primordial follicles when compared to WT mice, suggesting that follicle loss occurs progressively between birth and sexual maturity in a mouse, similar to case reports in females with CG (Kaufman et al. 1981, Balakrishnan et al. 2019). Recently, we used Salubrinal to modulate the UPR signaling in our GALT-deficient mouse model (Balakrishnan et al. 2019). Salubrinal inhibits the dephosphorylation of EIF2A, keeping protein translation reduced in the cell. The administration of Salubrinal at five weeks of life improved the fertility of GALT-deficient female mice by normalizing estrus cycles and increasing the number of pups born per litter (Balakrishnan et al. 2019). Histologic analysis of ovaries from Salubrinal-treated mice showed an increased number of primordial follicles when compared to untreated GALT-deficient mice (Balakrishnan et al. 2019).
It is reasonable to assume that the administration of Salubrinal, which allows for increased pEIF2A, may help to keep primordial follicles quiescent, and thus, preserve ovarian function. However, it is unknown if ovarian failure in CG is due to premature activation of primordial follicles or follicle atresia before activation. Preliminary data from our lab comparing one-month-old ovaries showed an increased ratio of primary to primordial follicles in GALT-deficient mice suggesting premature follicle activation and thus, depletion before sexual maturity (unpublished data). Immunofluorescent histological studies show evidence of increased ER stress marker BiP at two weeks of life in the granulosa cells of follicles in GALT-deficient ovaries (unpublished data).
Our lab found decreased markers of PI3K/AKT signaling in GALT-deficient fibroblasts and whole ovaries at two months of age compared to age-matched WT mice (Balakrishnan et al. 2016, 2017). The ovarian histology revealed a significant decrease in ovarian follicles of all stages and increased corpus luteum tissue (Balakrishnan et al. 2017). Interestingly, after administration of Salubrinal to GALT-deficient mice, molecular markers of PI3K/AKT signaling were improved in the whole ovary as well as an increased number of follicles (Balakrishnan et al. 2019). The relationship between the UPR and PI3K/AKT signaling is not well understood, but it appears that there is a communication between the phosphorylation of EIF2A and the phosphorylation of AKT (Qin et al. 2010, Tian et al. 2011). As PI3K/AKT signaling is tightly regulated in follicle activation, we speculate that by keeping eIF2ɑ phosphorylated, proper signaling is restored in the primordial follicle, allowing for follicle quiescence. Differences in PI3K/AKT signaling at earlier ages have not been explored in our mouse model of CG or the specific cell types involved. If there is evidence of early, mass primordial follicle activation in GALT-deficient mice, immunofluorescent studies may reveal increased PI3K/AKT signaling in oocytes/granulosa cells at earlier time points in development (Jagarlamudi et al. 2009). Modulators of PI3K/AKT signaling could also be used as a potential treatment if indicated, once the pathophysiology of POI in galactosemia is elucidated.
Clinical considerations
Evaluation of POI in galactosemia
The clinical presentation for CG-induced POI can manifest as delayed or absent puberty, primary or secondary amenorrhea, oligomenorrhea, or infertility (Fridovich-Keil et al. 2011, Thakur et al. 2018). In 2017, members of The Galactosemia Network (GalNet) developed evidence-based guidelines for the diagnosis, treatment, and follow-up of CG including specific recommendations related to endocrinology and fertility (Welling et al. 2017). The guidelines recommend that all females with CG be screened for POI by the age of 12 with absent secondary sexual characteristics or if they reach age 14 with irregular menses, and screening should include serum FSH and estradiol measurements. Additionally, the guidelines also recommend that females with CG that have undergone puberty with regular ongoing menstrual periods be monitored annually for signs or symptoms of POI and FSH measurement if symptoms arise (Welling et al. 2017). We provide a summary of guidelines for clinical management specific to CG and more broadly to POI in Table 3.
Table 3.
Clinical management of galactosemia and POI.
| Age | Guidelines | Unknowns | Considerations | Supporting references |
|---|---|---|---|---|
| Newborn | Screening for galactosemiaImplementing dietary management | |||
| 1–11 years | Monitor Gal-1P levels regularly to ensure dietary compliance | Utility of monitoring ovarian reserve to evaluate for imminent POIAge to consider fertility preservationThe role ER stress blockers play in maintaining fertility potential | Draw FSH and AMH annuallyConsider fertility preservation options (OTC vs oocyte cryopreservation) under research protocol as follicular numbers appear to be normal in younger patients | (Mamsen et al. 2018, Frederick et al. 2018, Yuzyuk et al. 2018, Azem et al. 2020, Poirot et al. 2019, Segers et al. 2020, Sanders et al. 2009) |
| 12–14 years | Monitor secondary sexual characteristics and menarcheScreen for POI (FSH, E2) if no secondary sexual characteristics by age 12 or irregular menses by age 14Initiate HRT if POI diagnosed | Is there a role for routine fertility preservation in this age groupThe role ER stress blockers play in maintaining fertility potential | Recommend adding AMH to screening protocol for POI as it appears to be predictive of spontaneous menarcheConsider fertility preservation in patients with evidence of adequate ovarian reserve | (Mamsen et al. 2018, Frederick et al. 2018, Cobo et al. 2016, Poirot et al. 2019, Segers et al. 2020) |
| 14+ years | Monitor annually for menstrual changes and symptoms of POI in women who underwent puberty. Screen for POI with concerns (FSH)Initiate HRT for newly diagnosed POICounsel on spontaneous conception and implement contraception if neededRefer to REI for fertility considerationsScreen for emotional well-being | Is there a role for routine fertility preservation for patients with adequate ovarian reserveThe role ER stress blockers play in maintaining fertility potential in patients who have completed puberty | Include AMH as routine screening for POIConsider fertility preservation (OTC vs oocyte cryopreservation) in patients with evidence of adequate ovarian reserve | (Mamsen et al. 2018, Frederick et al. 2018, Yuzyuk et al. 2018, Cobo et al. 2016, Poirot et al. 2019, Segers et al. 2020, Sanders et al. 2009) |
Alternative methods for screening for POI have been evaluated including measuring AMH levels. AMH is produced by granulosa cells of pre-antral and antral follicles, and its use has become more widespread in measuring ovarian reserve (Hagen et al. 2010, Hansen et al. 2011). While the guidelines currently recommend against using AMH as a screening tool for POI in this patient population (Welling et al. 2017), there have been several studies that have evaluated AMH levels in CG-induced POI (Spencer et al. 2013, Frederick et al. 2018). Studies show that patients with CG have significantly lower AMH values than controls, and AMH values can be assessed before menarche) or after initiation of HRT (Sanders et al. 2009, Spencer et al. 2013) supporting its potential utility for screening. More recently, Frederick and colleagues (Frederick et al. 2018) measured both AMH and FSH in patients with CG and tested for possible association(s) with spontaneous menarche. They found that a detectable AMH level (≥0.04 ng/mL) was highly predictive of spontaneous menarche. FSH values, unlike AMH, did not associate with spontaneous menarche. AMH values are also useful in screening for POI in patients with Turner syndrome (Hagen et al. 2010, Lunding et al. 2015). In prepubertal patients, an AMH less than 0.56 ng/mL predicted the absence of puberty in Turner syndrome girls (Lunding et al. 2015). With recent advances in AMH assay sensitivity as well as evidence of its utility in predicting menarche, we recommend routinely checking AMH levels in prepubertal patients with CG and those that have not been diagnosed with POI.
Management of POI in galactosemia
In CG patients with primary amenorrhea and absent puberty, the GalNet guidelines recommend starting with low-dose estradiol and increasing the dosage in a step-wise fashion to mimic physiological ovarian hormone function, with progesterone supplementation at a later time to induce withdrawal bleeding for endometrial protection (Welling et al. 2017). The exact timing of when to start HRT for young girls with CG and POI has not been well established but is generally started once the patient is at a peri-pubertal age, and the diagnosis of POI has been made.
According to the ESHRE guidelines on POI in the general population, low-dose estradiol should be started between ages 12–13 if no secondary sexual characteristics are present, and FSH is elevated. Estradiol should be gradually increased every 6–12 months for 2–3 years until an adult dosage is reached, and cyclic progesterone should be started after two years of estradiol or with the onset of breakthrough bleeding (Webber et al. 2016). There are many different routes of estrogen replacement including oral, transdermal, and transvaginal (Sullivan et al. 2016) with no form showing superiority in regards to improvement in symptoms (Webber et al. 2017); however, transdermal and transvaginal routes show decreased occurrence of venous-thromboembolism (VTE) in postmenopausal patients, but this has not been shown in younger populations (Webber et al. 2017, Vinogradova et al. 2019). However, given the duration of therapy in this population and the paucity of long-term longitudinal studies in pre-menopausal women with POI, mitigating VTE risk with transdermal and transvaginal administration may be prudent (Canonico et al. 2014).
Progesterone supplementation is traditionally given orally in either a micronized progesterone form or as a synthetic progestogen such as medroxyprogesterone acetate in a cyclical manner to induce regular menstrual cycles; it also can be given continuously in patients that do not desire a menstrual cycle. Alternative forms of progesterone are available such as transdermal, transvaginal, and intrauterine including the levonorgestrel intrauterine device (LNG-IUD) which supplies endometrial protection while also allowing for contraception (Webber et al. 2017). Some patients may prefer combined oral contraceptive pills (COCPs) as an alternative to traditional HRT as this is often more socially acceptable in their age group. However, studies have suggested that COCPs are inferior to HRT in maintaining bone health, but sample sizes have been small (Crofton et al. 2010, Cartwright et al. 2016). Patients who elect COCP use for HRT long-term should also be advised of the lack of improvement in associated genitourinary symptoms with this formulation compared to 17-beta estradiol as well as the lack of potential cardiovascular protection. Patients with CG should be counseled to take the COCP continuously and skip the placebo pills which would render them without estrogen or progesterone for 25% of the time (Webber et al. 2017). Finally, individuals should be counseled that HRT is not a form of contraception, and if pregnancy is not desired, a form of contraception should be recommended.
Psychosocial parameters including social well-being and social functioning are lower in patients with CG compared to the general population (Hoffmann et al. 2012). These patients are also prone to anxiety and depression with one observational study showing 67% of CG subjects reporting anxiety and 39% reporting depression (Waisbren et al. 2012). Also, decreased emotional well-being, anxiety, and depression are also significant factors associated with POI (Nelson 2009). Young women diagnosed with 'early menopause' may feel stigmatized and often devasted at the diagnosis especially when understanding the current incurable nature of the disease, related infertility, and being different than their peers (Sterling & Nelson 2011). A cross-sectional and case-control study by Davis and colleagues comparing spontaneous POI in women with 46,XX with a mean age of 32.4 years to age-matched controls found that those with POI had increased anxiety, depression, and negative affect as well as decreased overall well-being (Davis et al. 2010). Patients also perceive a lack of psychosocial support after receiving the diagnosis of POI, and few receive appropriate follow-up from social workers or a psychologist (Kanj et al. 2018). Appropriate counseling regarding the diagnosis as well as psychological support is extremely important in this patient population, and perhaps a multidisciplinary team approach is the best management method.
Fertility management
Infertility is a major long-term effect in patients with POI, although there are reports of spontaneous pregnancy in this population. The ovarian function in these women is limited, but erratic and unpredictable ovulation can still occur. In the broader POI population, 5–10% spontaneously conceive (van Kasteren & Schoemaker 1999). As mentioned previously, patients with CG continue to have ovarian compromise despite being on a galactose-restricted diet and many eventually experience POI. Yet, recent data has suggested fertility rates are higher in those patients with CG-induced POI compared to other causes of POI. In a study of 22 CG women, nine attempted to conceive spontaneously and four were successful (44% pregnancy rate) (Gubbels et al. 2008). Similar findings were detected in a recent observational study that evaluated 85 women with CG. In those patients, 21 actively attempted to conceive and nine achieved pregnancy (42.9% pregnancy rate) (van Erven et al. 2017). Even in CG patients with undetectable AMH levels and menopausal FSH levels, pregnancy can be achieved (Gubbels et al. 2009) (see as an example, Table 1). Ovarian reserve markers like FSH, AMH, and antral follicle count can help predict ovarian reserve and fertility potential but are not definitive in determining which patients with POI will and will not conceive. It is important to counsel patients appropriately on the chance of spontaneous conception and despite the diagnosis of POI, and for patients that do not desire pregnancy, contraception is recommended (Welling et al. 2017).
For those patients that do not achieve spontaneous conception and desire to conceive, referral to a reproductive endocrinologist to discuss fertility options such as oocyte and embryo donation should occur with adoption also being an alternative option (Baker 2011). Patients with POI are generally not ideal candidates to undergo IVF using autologous oocytes due to the extremely low chances of success secondary to depleted follicular reserve; while oocyte donation from a donor < 35 years of age would yield a live birth rate of approximately 44% (Hogan et al. 2019). Maintaining uterine health with estradiol and progesterone replacement therapy is thus important in helping these women achieve their family building goals.
Fertility preservation
Fertility preservation options are studied more in patients undergoing gonadotoxic therapy such as chemotherapy for cancer treatment that would render them at high risk for ovarian insufficiency and infertility, but it should also be offered to people with more benign conditions that put them at high risk for ovarian insufficiency (Donnez & Dolmans 2017). Historically, the two standard methods for fertility preservation have been oocyte cryopreservation and embryo cryopreservation which have been utilized for over 30 years. With either method, the patient receives exogenous gonadotropins for ovarian stimulation, and the resulting growing ovarian follicles are then aspirated to retrieve the oocytes. The oocytes can either be frozen to be used for embryo creation at a later date, or the fresh oocytes can be fertilized with partner or donor sperm after the retrieval to create embryos. The resulting embryos are grown to the blastocyst stage and are frozen to be thawed and transferred at a later date. In patients with a good prognosis (i.e. 35 years of age or younger), 10 oocytes cryopreserved yield a cumulative live birth rate of 60.5%, and 15 oocytes yield a cumulative live birth rate of 85.2.% (Cobo et al. 2016). Unfortunately, due to the rapid depletion of the ovarian reserve pool often before puberty in patients with CG, oocyte and embryo cryopreservation are generally not a practical option for fertility preservation in this population. To date, there are no studies evaluating the utility of oocyte cryopreservation in post-puberal patients with CG that have retained ovarian function. Due to the immaturity of the HPO axis in prepubertal girls, exogenous stimulation with gonadotropins for oocyte cryopreservation has historically not been viewed as a viable option, but a recent publication has challenged this approach. Azem and colleagues report the case of a seven-year-old female with Turner syndrome mosaicism that underwent controlled ovarian hyperstimulation with six mature oocytes cryopreserved (Azem et al. 2020). Successful oocyte cryopreservation in a prepubertal patient at high risk for developing POI is an exciting potential option for patients with CG but will require further studies. Considerations include patient maturity to undergo stimulation at this age as well as utilization of transabdominal ultrasound for follicular monitoring with transvaginal retrieval under anesthesia.
OTC, which requires surgical removal of ovarian tissue cortex that is subsequently frozen and re-implanted at a later date, is becoming a more widespread and promising option that has also resulted in > 130 live births (Donnez & Dolmans 2017, Gellert et al. 2018). Recently, ASRM lifted the experimental label for OTC that can now be offered as a standard of care (Medicine ECotASfR 2018). Traditionally in prepubertal females, OTC has been seen as the only option for fertility preservation which was generally completed under a research protocol (Medicine ECotASfR 2018, Nahata et al. 2020). Currently, there is limited evidence regarding the safety and effectiveness of OTC in prepubertal patients with only one live birth as a result of OTC from a prepubertal patient (Matthews et al. 2018). However, there have been several reports of successful induction of puberty with tissue harvested from prepubertal girls, cryopreserved, and transplanted in early adolescence supporting the efficacy of prepubertal OTC (Ernst et al. 2013, Poirot et al. 2019). Regarding CG, there are no data concerning OTC in postpubertal patients, but recent data show a potential benefit for OTC in pre-pubertal patients with CG. Six girls with galactosemia below the age of 12 underwent OTC for fertility preservation. Normal ovarian follicular density and morphology were observed in five of the girls (all ages 5 or younger) compared to controls that underwent OTC before receiving gonadotoxic therapy. The remaining girl with CG that underwent OTC was 11.7 years, and there were no ovarian follicles detected (Mamsen et al. 2018).
In vitro maturation (IVM) is the process of maturing cumulus-oocyte complexes (COCs) to the metaphase II stage in a culture which can then be used for embryo creation or oocyte cryopreservation. Improvements in lab techniques have resulted in significant improvement in IVM success. A recent retrospective study included 77 patients who underwent OTC with the maturation of COCs obtained during ovarian tissue processing. IVM rate of immature oocytes to the metaphase II stage was 39%. Of the 77 patients, 64 had oocytes cryopreserved, and the live birth rate per patient that underwent ovarian tissue oocyte IVM followed by fertilization and embryo transfer was 43% (Segers et al. 2020).
Overall, there is a lack of evidence of the utility of fertility preservation in patients with non-oncologic causes of POI specifically in the prepubertal population. In particular, in patients with CG-induced POI, there is concern that the ovarian damage might have already occurred yielding OTC futile, and this process of removing ovarian tissue will further decrease the ovarian reserve in a population that is at high risk for POI with spontaneous conception still being possible (van Erven et al. 2013). Partial oophorectomy for OTC may be considered to allow retention of tissue in the event of spontaneous ovulation and conception. Currently, the guidelines do not recommend for patients with CG to undergo routine fertility preservation techniques, and these procedures should only be offered under research studies at young prepubertal ages (van Erven et al. 2013, Welling et al. 2017). Given the recent studies, albeit limited numbers, it appears young prepubertal girls less than five years of age with CG have normal ovarian follicular numbers and morphology. These findings coupled with the advancements in OTC and IVM show this as a promising method for fertility preservation in this patient population. Further studies need to be completed, but it is reasonable to discuss this option with patients and refer them to specialized centers as needed.
Limitations and future directions
There are limitations to our review, including the lack of fertility preservation data on postpubertal patients with CG. Due to the high prevalence of early-onset POI, there are no studies evaluating fertility preservation in this age group. The considerations for fertility preservation in this population are extrapolated from the successes in similar patients with adequate ovarian reserve, which may influence clinical recommendations. The ER stress modulator Salubrinal is for research only and not safe for human use. Additionally, no data exists using ER stress modulators in human models of POI, making this target difficult to translate to a clinical setting. Finding FDA-approved drugs or nutraceuticals with similar mechanisms of action could be considered. Finally, the review of human and animal models of POI presented here are not specific to GALT deficiency, and the mechanisms that cause POI in the other models may not apply to CG. The mechanism of follicle demise may be very specific to the metabolites and cellular metabolism present in CG.
After over 50 years of research, the true pathophysiology of galactosemia remains unclear (Rubio-Gozalbo et al. 2019). No treatments for POI in galactosemia or the general population are used except for HRT, which only counters the effects of early menopause and does not prevent ovarian failure (Hadji 2008). Currently, gene/mRNA therapies and GALK1 inhibitors are being developed to treat GALT deficiency (Lai et al. 2014, Hu et al. 2019, Balakrishnan et al. 2020). However, it may be several years before this is available to human patients. Inhibitors of aldose reductase (AR), the enzyme responsible for converting excess galactose to galactitol, are also being explored as a potential treatment for galactosemia. AR inhibitors have the potential to be effective in protecting the ovary in particular from oxidative stress, which has been studied in a galactose-intoxicated rat model, but its relevance in patients with a galactose-restricted diet remains to be seen (Meyer et al. 1992). While Salubrinal showed improved fertility and increased primordial follicles in GALT-deficient mice (Kobayashi et al. 1999), its safety in humans is not well studied. At this time, compounds with a similar mechanism of action to Salubrinal and specific dietary supplements that are either FDA-approved or generally recognized as safe (GRAS) in humans are being tested as a treatment for POI in galactosemia. If a potential therapy is found, the timing of treatment for females with galactosemia remains to be determined.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding
This work was supported the National Institutes of Health (grant number R01HD089933, PI: Lai), and by University of Colorado School of Medicine, Department of Obstetrics and Gynecology, Division of Reproductive Endocrinology and Infertility, and Florence Crozier Cobb Research Funds (to J J). McPherson Family Funds are also gratefully acknowledged.
Author contribution statement
J J and K L conceived of the manuscript, contributed to its writing, and supervised revisions. S H-L and J R wrote and revised manuscript drafts, collected data, and summarized literature for tables. A A collected data and reviewed manuscript, L A and N L contributed to manuscript content and provided clinical context.
Acknowledgements
De-identified patient data are reported under the auspices of approved University of Utah protocol IRB_00085855 – Natural history of patients with metabolic disorders.
References
- Adhikari D, Liu K. 2009. Molecular mechanisms underlying the activation of mammalian primordial follicles. Endocrine Reviews 30 438–464. ( 10.1210/er.2008-0048) [DOI] [PubMed] [Google Scholar]
- Adhikari D, Zheng W, Shen Y, Gorre N, Hämäläinen T, Cooney AJ, Huhtaniemi I, Lan ZJ, Liu K. 2010. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Human Molecular Genetics 19 397–410. ( 10.1093/hmg/ddp483) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai Y, Zheng Z, O’Brien-Jenkins A, Bernard DJ, Wynshaw-Boris T, Ning C, Reynolds R, Segal S, Huang K, Stambolian D. 2000. A mouse model of galactose-induced cataracts. Human Molecular Genetics 9 1821–1827. ( 10.1093/hmg/9.12.1821) [DOI] [PubMed] [Google Scholar]
- Altuntas CZ, Johnson JM, Tuohy VK. 2006. Autoimmune targeted disruption of the pituitary-ovarian axis causes premature ovarian failure. Journal of Immunology 177 1988–1996. ( 10.4049/jimmunol.177.3.1988) [DOI] [PubMed] [Google Scholar]
- Alves VS, Motta FL, Roffe M, Delamano A, Pesquero JB, Castilho BA. 2009. GCN2 activation and eIF2alpha phosphorylation in the maturation of mouse oocytes. Biochemical and Biophysical Research Communications 378 41–44. ( 10.1016/j.bbrc.2008.10.161) [DOI] [PubMed] [Google Scholar]
- Anagnostis P, Christou K, Artzouchaltzi AM, Gkekas NK, Kosmidou N, Siolos P, Paschou SA, Potoupnis M, Kenanidis E, Tsiridis Eet al. 2019. Early menopause and premature ovarian insufficiency are associated with increased risk of type 2 diabetes: a systematic review and meta-analysis. European Journal of Endocrinology 180 41–50. ( 10.1530/EJE-18-0602) [DOI] [PubMed] [Google Scholar]
- Antshel KM, Epstein IO, Waisbren SE. 2004. Cognitive strengths and weaknesses in children and adolescents homozygous for the galactosemia Q188R mutation: a descriptive study. Neuropsychology 18 658–664. ( 10.1037/0894-4105.18.4.658) [DOI] [PubMed] [Google Scholar]
- Azem F, Brener A, Malinger G, Reches A, Many A, Yogev Y, Lebenthal Y. 2020. Bypassing physiological puberty, a novel procedure of oocyte cryopreservation at age 7: a case report and review of the literature. Fertility and Sterility 114 374–378. ( 10.1016/j.fertnstert.2020.03.009) [DOI] [PubMed] [Google Scholar]
- Babayev E, Lalioti MD, Favero F, Seli E. 2016. Cross-talk Between FSH and endoplasmic reticulum stress: a mutually suppressive relationship. Reproductive Sciences 23 352–364. ( 10.1177/1933719115602770) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badik JR, Castañeda U, Gleason TJ, Spencer JB, Epstein MP, Ficicioglu C, Fitzgerald K, Fridovich-Keil JL. 2011. Ovarian function in Duarte galactosemia. Fertility and Sterility 96 469, .e461–473.e. ( 10.1016/j.fertnstert.2011.05.088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker V.2011. Life plans and family-building options for women with primary ovarian insufficiency. Seminars in Reproductive Medicine 29 362–372. ( 10.1055/s-0031-1280921) [DOI] [PubMed] [Google Scholar]
- Balakrishnan B, Chen W, Tang M, Huang X, Cakici DD, Siddiqi A, Berry G, Lai K. 2016. Galactose-1 phosphate uridylyltransferase (GalT) gene: a novel positive regulator of the PI3K/Akt signaling pathway in mouse fibroblasts. Biochemical and Biophysical Research Communications 470 205–212. ( 10.1016/j.bbrc.2016.01.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan B, Nicholas C, Siddiqi A, Chen W, Bales E, Feng M, Johnson J, Lai K. 2017. Reversal of aberrant PI3K/Akt signaling by Salubrinal in a GalT-deficient mouse model. Biochimica et Biophysica Acta: Molecular Basis of Disease 1863 3286–3293. ( 10.1016/j.bbadis.2017.08.023) [DOI] [PubMed] [Google Scholar]
- Balakrishnan B, Siddiqi A, Mella J, Lupo A, Li E, Hollien J, Johnson J, Lai K. 2019. Salubrinal enhances eIF2α phosphorylation and improves fertility in a mouse model of classic galactosemia. Biochimica et Biophysica Acta: Molecular Basis of Disease 1865 165516. ( 10.1016/j.bbadis.2019.07.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan B, An D, Nguyen V, DeAntonis C, Martini PGV, Lai K. 2020. Novel mRNA-based therapy reduces toxic galactose metabolites and overcomes galactose sensitivity in a mouse model of classic galactosemia. Molecular Therapy 28 304–312. ( 10.1016/j.ymthe.2019.09.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Chakrabarti J, Banerjee S, Pal AK, Goswami SK, Chakravarty BN, Kabir SN. 2003a. Galactose toxicity in the rat as a model for premature ovarian failure: an experimental approach readdressed. Human Reproduction 18 2031–2038. ( 10.1093/humrep/deg414) [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Chakrabarti J, Banerjee S, Pal AK, Bhattacharyya D, Goswami SK, Chakravarty BN, Kabir SN. 2003b. Prenatal exposure to high galactose adversely affects initial gonadal pool of germ cells in rats. Human Reproduction 18 276–282. ( 10.1093/humrep/deg058) [DOI] [PubMed] [Google Scholar]
- Batey LA, Welt CK, Rohr F, Wessel A, Anastasoaie V, Feldman HA, Guo CY, Rubio-Gozalbo E, Berry G, Gordon CM. 2013. Skeletal health in adult patients with classic galactosemia. Osteoporosis International 24 501–509. ( 10.1007/s00198-012-1983-0) [DOI] [PubMed] [Google Scholar]
- Berry GT.2020. Classic galactosemia and clinical variant galactosemia. In GeneReviews® Available at https://www.ncbi.nlm.nih.gov/books/NBK1518/ (Accessed: 10 September 2020). [Google Scholar]
- Berry GT, Nissim I, Gibson JB, Mazur AT, Lin Z, Elsas LJ, Singh RH, Klein PD, Segal S. 1997. Quantitative assessment of whole body galactose metabolism in galactosemic patients. European Journal of Pediatrics 156 (Supplement 1) S43–S49. ( 10.1007/pl00014271) [DOI] [PubMed] [Google Scholar]
- Berry GT, Moate PJ, Reynolds RA, Yager CT, Ning C, Boston RC, Segal S. 2004. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Molecular Genetics and Metabolism 81 22–30. ( 10.1016/j.ymgme.2003.08.026) [DOI] [PubMed] [Google Scholar]
- Broughton DE, Moley KH. 2017. Obesity and female infertility: potential mediators of obesity’s impact. Fertility and Sterility 107 840–847. ( 10.1016/j.fertnstert.2017.01.017) [DOI] [PubMed] [Google Scholar]
- Canonico M, Plu-Bureau G, O’Sullivan MJ, Stefanick ML, Cochrane B, Scarabin PY, Manson JE. 2014. Age at menopause, reproductive history, and venous thromboembolism risk among postmenopausal women: the Women’s Health Initiative Hormone Therapy clinical trials. Menopause 21 214–220. ( 10.1097/GME.0b013e31829752e0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright B, Robinson J, Seed PT, Fogelman I, Rymer J. 2016. Hormone replacement therapy versus the combined oral contraceptive pill in premature ovarian failure: a randomized controlled trial of the effects on bone mineral density. Journal of Clinical Endocrinology and Metabolism 101 3497–3505. ( 10.1210/jc.2015-4063) [DOI] [PubMed] [Google Scholar]
- Chakravarthi VP, Ghosh S, Roby KF, Wolfe MW, Rumi MAK. 2020. A gatekeeping role of ESR2 to maintain the primordial follicle reserve. Endocrinology 161. ( 10.1210/endocr/bqaa037) [DOI] [PubMed] [Google Scholar]
- Chang HM, Qiao J, Leung PC. 2016. Oocyte-somatic cell interactions in the human ovary-novel role of bone morphogenetic proteins and growth differentiation factors. Human Reproduction Update 23 1–18. ( 10.1093/humupd/dmw039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT, Mattison DR, Feigenbaum L, Fukui H, Schulman JD. 1981. Reduction in oocyte number following prenatal exposure to a diet high in galactose. Science 214 1145–1147. ( 10.1126/science.7302587) [DOI] [PubMed] [Google Scholar]
- Chen W, Caston R, Balakrishnan B, Siddiqi A, Parmar K, Tang M, Feng M, Lai K. 2017. Assessment of ataxia phenotype in a new mouse model of galactose-1 phosphate uridylyltransferase (GALT) deficiency. Journal of Inherited Metabolic Disease 40 131–137. ( 10.1007/s10545-016-9993-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobo A, García-Velasco JA, Coello A, Domingo J, Pellicer A, Remohí J. 2016. Oocyte vitrification as an efficient option for elective fertility preservation. Fertility and Sterility 105 755, .e8–764.e8. ( 10.1016/j.fertnstert.2015.11.027) [DOI] [PubMed] [Google Scholar]
- Colegrove-Otero LJ, Minshall N, Standart N. 2005. RNA-binding proteins in early development. Critical Reviews in Biochemistry and Molecular Biology 40 21–73. ( 10.1080/10409230590918612) [DOI] [PubMed] [Google Scholar]
- Crofton PM, Evans N, Bath LE, Warner P, Whitehead TJ, Critchley HO, Kelnar CJ, Wallace WHB. 2010. Physiological versus standard sex steroid replacement in young women with premature ovarian failure: effects on bone mass acquisition and turnover. Clinical Endocrinology 73 707–714. ( 10.1111/j.1365-2265.2010.03868.x) [DOI] [PubMed] [Google Scholar]
- Davis M, Ventura JL, Wieners M, Covington SN, Vanderhoof VH, Ryan ME, Koziol DE, Popat VB, Nelson LM. 2010. The psychosocial transition associated with spontaneous 46, XX primary ovarian insufficiency: illness uncertainty, stigma, goal flexibility, and purpose in life as factors in emotional health. Fertility and Sterility 93 2321–2329. ( 10.1016/j.fertnstert.2008.12.122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jongh WA, Bro C, Ostergaard S, Regenberg B, Olsson L, Nielsen J. 2008. The roles of galactitol, galactose‐1‐phosphate, and phosphoglucomutase in galactose‐induced toxicity in Saccharomyces cerevisiae. Biotechnology and Bioengineering 101 317–326. ( 10.1002/bit.21890) [DOI] [PubMed] [Google Scholar]
- Domniz N, Meirow D. 2019. Premature ovarian insufficiency and autoimmune diseases. Best Practice and Research: Clinical Obstetrics and Gynaecology 60 42–55. ( 10.1016/j.bpobgyn.2019.07.008) [DOI] [PubMed] [Google Scholar]
- Donnelly N, Gorman AM, Gupta S, Samali A. 2013. The eIF2alpha kinases: their structures and functions. Cellular and Molecular Life Sciences 70 3493–3511. ( 10.1007/s00018-012-1252-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnez J, Dolmans M-M. 2017. Fertility preservation in women. New England Journal of Medicine 377 1657–1665. ( 10.1056/NEJMra1614676) [DOI] [PubMed] [Google Scholar]
- Ernst E, Kjærsgaard M, Birkebæk NH, Clausen N, Andersen CY. 2013. Case report: stimulation of puberty in a girl with chemo- and radiation therapy induced ovarian failure by transplantation of a small part of her frozen/thawed ovarian tissue. European Journal of Cancer 49 911–914. ( 10.1016/j.ejca.2012.09.028) [DOI] [PubMed] [Google Scholar]
- Ernst EH, Grondahl ML, Grund S, Hardy K, Heuck A, Sunde L, Franks S, Andersen CY, Villesen P, Lykke-Hartmann K. 2017. Dormancy and activation of human oocytes from primordial and primary follicles: molecular clues to oocyte regulation. Human Reproduction 32 1684–1700. ( 10.1093/humrep/dex238) [DOI] [PubMed] [Google Scholar]
- Fogli A, Gauthier-Barichard F, Schiffmann R, Vanderhoof VH, Bakalov VK, Nelson LM, Boespflug-Tanguy O. 2004. Screening for known mutations in EIF2B genes in a large panel of patients with premature ovarian failure. BMC Women’s Health 4 8. ( 10.1186/1472-6874-4-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford EA, Beckett EL, Roman SD, McLaughlin EA, Sutherland JM. 2020. Advances in human primordial follicle activation and premature ovarian insufficiency. Reproduction 159 R15–R29. ( 10.1530/REP-19-0201) [DOI] [PubMed] [Google Scholar]
- Forges T, Monnier-Barbarino P, Faure GC, Béné MC. 2004. Autoimmunity and antigenic targets in ovarian pathology. Human Reproduction Update 10 163–175. ( 10.1093/humupd/dmh014) [DOI] [PubMed] [Google Scholar]
- Frederick AB, Zinsli AM, Carlock G, Conneely K, Fridovich-Keil JL. 2018. Presentation, progression, and predictors of ovarian insufficiency in classic galactosemia. Journal of Inherited Metabolic Disease 41 785–790. ( 10.1007/s10545-018-0177-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E. 2011. Ovarian function in girls and women with GALT-deficiency galactosemia. Journal of Inherited Metabolic Disease 34 357–366. ( 10.1007/s10545-010-9221-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert SE, Pors SE, Kristensen SG, Bay-Bjørn AM, Ernst E, Andersen CY. 2018. Transplantation of frozen-thawed ovarian tissue: an update on worldwide activity published in peer-reviewed papers and on the Danish cohort. Journal of Assisted Reproduction and Genetics 35 561–570. ( 10.1007/s10815-018-1144-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbels CS, Land JA, Rubio-Gozalbo ME. 2008. Fertility and impact of pregnancies on the mother and child in classic galactosemia. Obstetrical and Gynecological Survey 63 334–343. ( 10.1097/OGX.0b013e31816ff6c5) [DOI] [PubMed] [Google Scholar]
- Gubbels CS, Kuppens SM, Bakker JA, Konings CJ, Wodzig KW, de Sain-van der Velden MG, Menheere PP, Rubio-Gozalbo ME. 2009. Pregnancy in classic galactosemia despite undetectable anti-Müllerian hormone. Fertility and Sterility 91 1293.e13–1293.e16. ( 10.1016/j.fertnstert.2008.12.031) [DOI] [PubMed] [Google Scholar]
- Guerrero NV, Singh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ. 2000. Risk factors for premature ovarian failure in females with galactosemia. Journal of Pediatrics 137 833–841. ( 10.1067/mpd.2000.109148) [DOI] [PubMed] [Google Scholar]
- Hadji P.2008. Menopausal symptoms and adjuvant therapy-associated adverse events. Endocrine-Related Cancer 15 73–90. ( 10.1677/ERC-07-0193) [DOI] [PubMed] [Google Scholar]
- Hagen CP, Aksglaede L, Sørensen K, Main KM, Boas M, Cleemann L, Holm K, Gravholt CH, Andersson AM, Pedersen ATet al. 2010. Serum levels of anti-Müllerian hormone as a marker of ovarian function in 926 healthy females from birth to adulthood and in 172 Turner syndrome patients. Journal of Clinical Endocrinology and Metabolism 95 5003–5010. ( 10.1210/jc.2010-0930) [DOI] [PubMed] [Google Scholar]
- Hansen KR, Hodnett GM, Knowlton N, Craig LB. 2011. Correlation of ovarian reserve tests with histologically determined primordial follicle number. Fertility and Sterility 95 170–175. ( 10.1016/j.fertnstert.2010.04.006) [DOI] [PubMed] [Google Scholar]
- Harada M, Nose E, Takahashi N, Hirota Y, Hirata T, Yoshino O, Koga K, Fujii T, Osuga Y. 2015. Evidence of the activation of unfolded protein response in granulosa and cumulus cells during follicular growth and maturation. Gynecological Endocrinology 31 783–787. ( 10.3109/09513590.2015.1062862) [DOI] [PubMed] [Google Scholar]
- Haskovic M, Coelho AI, Lindhout M, Zijlstra F, Veizaj R, Vos R, Vanoevelen JM, Bierau J, Lefeber DJ, Rubio-Gozalbo ME. 2020. Nucleotide sugar profiles throughout development in wildtype and galt knockout zebrafish. Journal of Inherited Metabolic Disease 43 994–1001. ( 10.1002/jimd.12265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill P, Garbaczewski L, Haley N, Wynder EL. 1984. Diet and follicular development. American Journal of Clinical Nutrition 39 771–777. ( 10.1093/ajcn/39.5.771) [DOI] [PubMed] [Google Scholar]
- Hoffmann B, Dragano N, Schweitzer-Krantz S. 2012. Living situation, occupation and health-related quality of life in adult patients with classic galactosemia. Journal of Inherited Metabolic Disease 35 1051–1058. ( 10.1007/s10545-012-9469-y) [DOI] [PubMed] [Google Scholar]
- Hogan RG, Wang AY, Li Z, Hammarberg K, Johnson L, Mol BW, Sullivan EA. 2019. Oocyte donor age has a significant impact on oocyte recipients’ cumulative live-birth rate: a population-based cohort study. Fertility and Sterility 112 724–730. ( 10.1016/j.fertnstert.2019.05.012) [DOI] [PubMed] [Google Scholar]
- Hohos NM, Skaznik-Wikiel ME. 2017. High-fat diet and female fertility. Endocrinology 158 2407–2419. ( 10.1210/en.2017-00371) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden HM, Rayment I, Thoden JB. 2003. Structure and function of enzymes of the Leloir pathway for galactose metabolism. Journal of Biological Chemistry 278 43885–43888. ( 10.1074/jbc.R300025200) [DOI] [PubMed] [Google Scholar]
- Hsueh AJW, Kawamura K. 2020. Hippo signaling disruption and ovarian follicle activation in infertile patients. Fertility and Sterility 114 458–464. ( 10.1016/j.fertnstert.2020.07.031) [DOI] [PubMed] [Google Scholar]
- Hu X, Zhang YQ, Lee OW, Liu L, Tang M, Lai K, Boxer MB, Hall MD, Shen M. 2019. Discovery of novel inhibitors of human galactokinase by virtual screening. Journal of Computer-Aided Molecular Design 33 405–417. ( 10.1007/s10822-019-00190-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagarlamudi K, Liu L, Adhikari D, Reddy P, Idahl A, Ottander U, Lundin E, Liu K. 2009. Oocyte-specific deletion of Pten in mice reveals a stage-specific function of PTEN/PI3K signaling in oocytes in controlling follicular activation. PLoS ONE 4 e6186. ( 10.1371/journal.pone.0006186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GB, Gallardo TD, Shirley LJ, Castrillon DH. 2008. Foxo3 is a PI3K-dependent molecular switch controlling the initiation of oocyte growth. Developmental Biology 321 197–204. ( 10.1016/j.ydbio.2008.06.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumbo-Lucioni PP, Garber K, Kiel J, Baric I, Berry GT, Bosch A, Burlina A, Chiesa A, Pico MLC, Estrada SCet al. 2012. Diversity of approaches to classic galactosemia around the world: a comparison of diagnosis, intervention, and outcomes. Journal of Inherited Metabolic Disease 35 1037–1049. ( 10.1007/s10545-012-9477-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalich-Philosoph L, Roness H, Carmely A, Fishel-Bartal M, Ligumsky H, Paglin S, Wolf I, Kanety H, Sredni B, Meirow D. 2013. Cyclophosphamide triggers follicle activation and ‘burnout’; AS101 prevents follicle loss and preserves fertility. Science Translational Medicine 5 185ra62. ( 10.1126/scitranslmed.3005402) [DOI] [PubMed] [Google Scholar]
- Kanj RV, Ofei-Tenkorang NA, Altaye M, Gordon CM. 2018. Evaluation and management of primary ovarian insufficiency in adolescents and young adults. Journal of Pediatric and Adolescent Gynecology 31 13–18. ( 10.1016/j.jpag.2017.07.005) [DOI] [PubMed] [Google Scholar]
- Kasippillai T, MacArthur DG, Kirby A, Thomas B, Lambalk CB, Daly MJ, Welt CK. 2013. Mutations in eIF4ENIF1 are associated with primary ovarian insufficiency. Journal of Clinical Endocrinology and Metabolism 98 E1534–E. ( 10.1210/jc.2013-1102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman FR, Kogut MD, Donnell GN, Goebelsmann U, March C, Koch R. 1981. Hypergonadotropic hypogonadism in female patients with galactosemia. New England Journal of Medicine 304 994–998. ( 10.1056/NEJM198104233041702) [DOI] [PubMed] [Google Scholar]
- Kawamura K, Cheng Y, Suzuki N, Deguchi M, Sato Y, Takae S, Ho CH, Kawamura N, Tamura M, Hashimoto Set al. 2013. Hippo signaling disruption and Akt stimulation of ovarian follicles for infertility treatment. PNAS 110 17474–17479. ( 10.1073/pnas.1312830110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauff EA, Richardus R, Eijkemans MJ, Broekmans FJ, de Jong FJ, Fauser BC, Bosch AM. 2007. Heterozygosity for the classical galactosemia mutation does not affect ovarian reserve and menopausal age. Reproductive Sciences 14 780–785. ( 10.1177/1933719107308614) [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono Het al. 1999. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nature Genetics 22 159–163. ( 10.1038/9667) [DOI] [PubMed] [Google Scholar]
- Komatsu K, Masubuchi S. 2017. The concentration-dependent effect of progesterone on follicle growth in the mouse ovary. Journal of Reproduction and Development 63 271–277. ( 10.1262/jrd.2016-154) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S U, Kumar D T, Siva , Doss C GP, Zayed H. 2020. An extensive computational approach to analyze and characterize the functional mutations in the galactose-1-phosphate uridyl transferase (GALT) protein responsible for classical galactosemia. Computers in Biology and Medicine 117 103583. ( 10.1016/j.compbiomed.2019.103583) [DOI] [PubMed] [Google Scholar]
- Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ. 2003. GALT deficiency causes UDP‐hexose deficit in human galactosemic cells. Glycobiology 13 285–294. ( 10.1093/glycob/cwg033) [DOI] [PubMed] [Google Scholar]
- Lai K, Boxer MB, Marabotti A. 2014. GALK inhibitors for classic galactosemia. Future Medicinal Chemistry 6 1003–1015. ( 10.4155/fmc.14.43) [DOI] [PubMed] [Google Scholar]
- Le Bouffant R, Boulben S, Cormier P, Mulner-Lorillon O, Bellé R, Morales J. 2008. Inhibition of translation and modification of translation factors during apoptosis induced by the DNA-damaging agent MMS in sea urchin embryos. Experimental Cell Research 314 961–968. ( 10.1016/j.yexcr.2007.12.014) [DOI] [PubMed] [Google Scholar]
- Levy HL.1996. Reproductive effects of maternal metabolic disorders: implications for pediatrics and obstetrics. Turkish Journal of Pediatrics 38 335–344. [PubMed] [Google Scholar]
- Levy HL, Driscoll SG, Porensky RS, Wender DF. 1984. Ovarian failure in galactosemia. New England Journal of Medicine 310 50. ( 10.1056/nejm198401053100114) [DOI] [PubMed] [Google Scholar]
- Lin P, Yang Y, Li X, Chen F, Cui C, Hu L, Li Q, Liu W, Jin Y. 2012. Endoplasmic reticulum stress is involved in granulosa cell apoptosis during follicular atresia in goat ovaries. Molecular Reproduction and Development 79 423–432. ( 10.1002/mrd.22045) [DOI] [PubMed] [Google Scholar]
- Liu K, Rajareddy S, Liu L, Jagarlamudi K, Boman K, Selstam G, Reddy P. 2006. Control of mammalian oocyte growth and early follicular development by the oocyte PI3 kinase pathway: new roles for an old timer. Developmental Biology 299 1–11. ( 10.1016/j.ydbio.2006.07.038) [DOI] [PubMed] [Google Scholar]
- Luan Y, Edmonds ME, Woodruff TK, Kim SY. 2019. Inhibitors of apoptosis protect the ovarian reserve from cyclophosphamide. Journal of Endocrinology 240 243–256. ( 10.1530/JOE-18-0370) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke B, Brown MB, Stern JE, Missmer SA, Fujimoto VY, Leach R. & SART Writing Group 2011. Female obesity adversely affects assisted reproductive technology (ART) pregnancy and live birth rates. Human Reproduction 26 245–252. ( 10.1093/humrep/deq306) [DOI] [PubMed] [Google Scholar]
- Lunding SA, Aksglæde L, Anderson RA, Main KM, Juul A, Hagen CP, Pedersen AT. 2015. AMH as predictor of premature ovarian insufficiency: a longitudinal study of 120 Turner syndrome patients. Journal of Clinical Endocrinology and Metabolism 100 E1030–E1038. ( 10.1210/jc.2015-1621) [DOI] [PubMed] [Google Scholar]
- Machado CM, De-Souza EA, De-Queiroz ALFV, Pimentel FSA, Silva GFS, Gomes FM, Montero-Lomelí M, Masuda CA. 2017. The galactose-induced decrease in phosphate levels leads to toxicity in yeast models of galactosemia. Biochimica et Biophysica Acta: Molecular Basis of Disease 1863 1403–1409. ( 10.1016/j.bbadis.2017.02.014) [DOI] [PubMed] [Google Scholar]
- Machtinger R, Combelles CM, Missmer SA, Correia KF, Fox JH, Racowsky C. 2012. The association between severe obesity and characteristics of failed fertilized oocytes. Human Reproduction 27 3198–3207. ( 10.1093/humrep/des308) [DOI] [PubMed] [Google Scholar]
- Mamsen LS, Kelsey TW, Ernst E, Macklon KT, Lund AM, Andersen CY. 2018. Cryopreservation of ovarian tissue may be considered in young girls with galactosemia. Journal of Assisted Reproduction and Genetics 35 1209–1217. ( 10.1007/s10815-018-1209-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino R, Misra M. 2011. Bone health in primary ovarian insufficiency. Seminars in Reproductive Medicine 29 317–327. ( 10.1055/s-0031-1280917) [DOI] [PubMed] [Google Scholar]
- Matthews SJ, Picton H, Ernst E, Andersen CY. 2018. Successful pregnancy in a woman previously suffering from β-thalassemia following transplantation of ovarian tissue cryopreserved before puberty. Minerva Ginecologica 70 432–435. ( 10.23736/S0026-4784.18.04240-5) [DOI] [PubMed] [Google Scholar]
- Medicine ECotASfR 2018. Fertility preservation and reproduction in patients facing gonadotoxic therapies: an Ethics Committee opinion. Fertility and Sterility 110 380–386. ( 10.1016/j.fertnstert.2018.05.034) [DOI] [PubMed] [Google Scholar]
- Meyer WR, Doyle MB, Grifo JA, Lipetz KJ, Oates PJ, DeCherney AH, Diamond MP. 1992. Aldose reductase inhibition prevents galactose-induced ovarian dysfunction in the Sprague-Dawley rat. American Journal of Obstetrics and Gynecology 167 1837–1843. ( 10.1016/0002-9378(9291784-8) [DOI] [PubMed] [Google Scholar]
- Mumma JO, Chhay JS, Ross KL, Eaton JS, Newell-Litwa KA, Fridovich-Keil JL. 2008. Distinct roles of galactose-1P in galactose-mediated growth arrest of yeast deficient in galactose-1P uridylyltransferase (GALT) and UDP-galactose 4′-epimerase (GALE). Molecular Genetics and Metabolism 93 160–171. ( 10.1016/j.ymgme.2007.09.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahata L, Woodruff TK, Quinn GP, Meacham LR, Chen D, Appiah LC, Finlayson C, Orwig KE, Laronda MM, Rowell EEet al. 2020. Ovarian tissue cryopreservation as standard of care: what does this mean for pediatric populations? Journal of Assisted Reproduction and Genetics 37 1323–1326. ( 10.1007/s10815-020-01794-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LM.2009. Clinical practice. Primary ovarian insufficiency. New England Journal of Medicine 360 606–614. ( 10.1056/NEJMcp0808697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning C, Segal S. 2000. Plasma galactose and galactitol concentration in patients with galactose-1-phosphate uridyltransferase deficiency galactosemia: determination by gas chromatography/mass spectrometry. Metabolism: Clinical and Experimental 49 1460–1466. ( 10.1053/meta.2000.9512) [DOI] [PubMed] [Google Scholar]
- Nteeba J, Ross JW, Perfield JW, Keating AF. 2013. High fat diet induced obesity alters ovarian phosphatidylinositol-3 kinase signaling gene expression. Reproductive Toxicology 42 68–77. ( 10.1016/j.reprotox.2013.07.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslowski CM, Urano F. 2011. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods in Enzymology 490 71–92. ( 10.1016/B978-0-12-385114-7.00004-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panay N, Anderson RA, Nappi RE, Vincent AJ, Vujovic S, Webber L, Wolfman W. 2020. Premature ovarian insufficiency: an International Menopause Society White Paper. Climacteric 23 426–446. ( 10.1080/13697137.2020.1804547) [DOI] [PubMed] [Google Scholar]
- Poirot C, Brugieres L, Yakouben K, Prades-Borio M, Marzouk F, de Lambert G, Pacquement H, Bernaudin F, Neven B, Paye-Jaouen Aet al. 2019. Ovarian tissue cryopreservation for fertility preservation in 418 girls and adolescents up to 15 years of age facing highly gonadotoxic treatment. Twenty years of experience at a single center. Acta Obstetricia et Gynecologica Scandinavica 98 630–637. ( 10.1111/aogs.13616) [DOI] [PubMed] [Google Scholar]
- Pyhtila BM, Shaw KA, Neumann SE, Fridovich-Keil JL. 2015. Newborn screening for galactosemia in the United States: looking back, looking around, and looking ahead. In JIMD Reports, Vol. 15, pp. 79–93. Springer. ( 10.1007/8904_2014_302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wang Z, Tao L, Wang Y. 2010. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 6 239–247. ( 10.4161/auto.6.2.11062) [DOI] [PubMed] [Google Scholar]
- Reddy P, Adhikari D, Zheng W, Liang S, Hamalainen T, Tohonen V, Ogawa W, Noda T, Volarevic S, Huhtaniemi Iet al. 2009. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Human Molecular Genetics 18 2813–2824. ( 10.1093/hmg/ddp217) [DOI] [PubMed] [Google Scholar]
- Reichman ME, Judd JT, Taylor PR, Nair PP, Jones DY, Campbell WS. 1992. Effect of dietary fat on length of the follicular phase of the menstrual cycle in a controlled diet setting. Journal of Clinical Endocrinology and Metabolism 74 1171–1175. ( 10.1210/jcem.74.5.1569164) [DOI] [PubMed] [Google Scholar]
- Robertson A, Singh RH, Guerrero NV, Hundley M, Elsas LJ. 2000. Outcomes analysis of verbal dyspraxia in classic galactosemia. Genetics in Medicine 2 142–148. ( 10.1097/00125817-200003000-00005) [DOI] [PubMed] [Google Scholar]
- Roeters Van Lennep JE, Heida KY, Bots ML, Hoek A. & collaborators of the Dutch Multidisciplinary Guideline Development Group on Cardiovascular Risk Management after Reproductive Disorders 2016. Cardiovascular disease risk in women with premature ovarian insufficiency: a systematic review and meta-analysis. European Journal of Preventive Cardiology 23 178–186. ( 10.1177/2047487314556004) [DOI] [PubMed] [Google Scholar]
- Ross KL, Davis CN, Fridovich‐Keil JL. 2004. Differential roles of the Leloir pathway enzymes and metabolites in defining galactose sensitivity in yeast. Molecular Genetics and Metabolism 83 103‐116. ( 10.1016/j.ymgme.2004.07.005) [DOI] [PubMed] [Google Scholar]
- Rossetti R, Ferrari I, Bonomi M, Persani L. 2017. Genetics of primary ovarian insufficiency. Clinical Genetics 91 183–198. ( 10.1111/cge.12921) [DOI] [PubMed] [Google Scholar]
- Rostami Dovom M, Noroozzadeh M, Mosaffa N, Zadeh-Vakili A, Piryaei A, Ramezani Tehrani F. 2019. Induced premature ovarian insufficiency by using D galactose and its effects on reproductive profiles in small laboratory animals: a systematic review. Journal of Ovarian Research 12 96. ( 10.1186/s13048-019-0565-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Gozalbo ME, Gubbels CS, Bakker JA, Menheere PP, Wodzig WK, Land JA. 2010. Gonadal function in male and female patients with classic galactosemia. Human Reproduction Update 16 177–188. ( 10.1093/humupd/dmp038) [DOI] [PubMed] [Google Scholar]
- Rubio-Gozalbo ME, Haskovic M, Bosch AM, Burnyte B, Coelho AI, Cassiman D, Couce ML, Dawson C, Demirbas D, Derks Tet al. 2019. The natural history of classic galactosemia: lessons from the GalNet registry. Orphanet Journal of Rare Diseases 14 86. ( 10.1186/s13023-019-1047-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Gozalbo ME, Derks B, Das AM, Meyer U, Möslinger D, Couce ML, Empain A, Ficicioglu C, Juliá Palacios N, De Los Santos De Pelegrin MMet al. 2021. Galactokinase deficiency: lessons from the GalNet registry. Genetics in Medicine 23 202–210. ( 10.1038/s41436-020-00942-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safety and pharmacokinetics of AT-007 in healthy subjects and in adult subjects with classic galactosemia. (available at: https://ClinicalTrials.gov/show/NCT04117711) [Google Scholar]
- Sanders RD, Spencer JB, Epstein MP, Pollak SV, Vardhana PA, Lustbader JW, Fridovich-Keil JL. 2009. Biomarkers of ovarian function in girls and women with classic galactosemia. Fertility and Sterility 92 344–351. ( 10.1016/j.fertnstert.2008.04.060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segers I, Bardhi E, Mateizel I, Van Moer E, Schots R, Verheyen G, Tournaye H, De Vos M. 2020. Live births following fertility preservation using in-vitro maturation of ovarian tissue oocytes. Human Reproduction 35 2026–2036. ( 10.1093/humrep/deaa175) [DOI] [PubMed] [Google Scholar]
- Sharif K, Watad A, Bridgewood C, Kanduc D, Amital H, Shoenfeld Y. 2019. Insights into the autoimmune aspect of premature ovarian insufficiency. Best Practice and Research: Clinical Endocrinology and Metabolism 33 101323. ( 10.1016/j.beem.2019.101323) [DOI] [PubMed] [Google Scholar]
- Slepak T, Tang M, Addo F, Lai K. 2005. Intracellular galactose-1-phosphate accumulation leads to environmental stress response in yeast model. Molecular Genetics and Metabolism 86 360–3. ( 10.1016/j.ymgme.2005.08.002) [DOI] [PubMed] [Google Scholar]
- Slepak TI, Tang M, Slepak VZ, Lai K. 2007. Involvement of endoplasmic reticulum stress in a novel classic galactosemia model. Molecular Genetics and Metabolism 92 78–87. ( 10.1016/j.ymgme.2007.06.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JB, Badik JR, Ryan EL, Gleason TJ, Broadaway KA, Epstein MP, Fridovich-Keil JL. 2013. Modifiers of ovarian function in girls and women with classic galactosemia. Journal of Clinical Endocrinology and Metabolism 98 E1257–E1265. ( 10.1210/jc.2013-1374) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling EW, Nelson LM. 2011. From victim to survivor to thriver: helping women with primary ovarian insufficiency integrate recovery, self-management, and wellness. In Seminars in Reproductive Medicine, p. 353. NIH Public Access. ( 10.1055/s-0031-1280920) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss III JF, Williams CJ. 2019. Ovarian life cycle. In Yen and Jaffe’s Reproductive Endocrinology, pp. 167, .e169–205. Elsevier. [Google Scholar]
- Sullivan SD, Sarrel PM, Nelson LM. 2016. Hormone replacement therapy in young women with primary ovarian insufficiency and early menopause. Fertility and Sterility 106 1588–1599. ( 10.1016/j.fertnstert.2016.09.046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Yoshioka N, Takae S, Sugishita Y, Tamura M, Hashimoto S, Morimoto Y, Kawamura K. 2015. Successful fertility preservation following ovarian tissue vitrification in patients with primary ovarian insufficiency. Human Reproduction 30 608–615. ( 10.1093/humrep/deu353) [DOI] [PubMed] [Google Scholar]
- Takehara I, Igarashi H, Kawagoe J, Matsuo K, Takahashi K, Nishi M, Nagase S. 2020. Impact of endoplasmic reticulum stress on oocyte aging mechanisms. Molecular Human Reproduction 26 567–575. ( 10.1093/molehr/gaaa040) [DOI] [PubMed] [Google Scholar]
- Tang M, Siddiqi A, Witt B, Yuzyuk T, Johnson B, Fraser N, Chen W, Rascon R, Yin X, Goli Het al. 2014. Subfertility and growth restriction in a new galactose-1 phosphate uridylyltransferase (GALT) – deficient mouse model. European Journal of Human Genetics 22 1172–1179. ( 10.1038/ejhg.2014.12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur M, Shaeib F, Khan SN, Kohan-Ghadr HR, Jeelani R, Aldhaheri SR, Gonik B, Abu-Soud HM. 2017. Galactose and its metabolites deteriorate metaphase II mouse oocyte quality and subsequent embryo development by disrupting the spindle structure. Scientific Reports 7 231. ( 10.1038/s41598-017-00159-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur M, Feldman G, Puscheck EE. 2018. Primary ovarian insufficiency in classic galactosemia: current understanding and future research opportunities. Journal of Assisted Reproduction and Genetics 35 3–16. ( 10.1007/s10815-017-1039-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therrell Jr BL, Lloyd-Puryear MA, Camp KM, Mann MY. 2014. Inborn errors of metabolism identified via newborn screening: ten-year incidence data and costs of nutritional interventions for research agenda planning. Molecular Genetics and Metabolism 113 14–26. ( 10.1016/j.ymgme.2014.07.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T, Zhao Y, Nakajima S, Huang T, Yao J, Paton AW, Paton JC, Kitamura M. 2011. Cytoprotective roles of ERK and Akt in endoplasmic reticulum stress triggered by subtilase cytotoxin. Biochemical and Biophysical Research Communications 410 852–858. ( 10.1016/j.bbrc.2011.06.078) [DOI] [PubMed] [Google Scholar]
- van Erven B, Gubbels CS, van Golde RJ, Dunselman GA, Derhaag JG, de Wert G, Geraedts JP, Bosch AM, Treacy EP, Welt CKet al. 2013. Fertility preservation in female classic galactosemia patients. Orphanet Journal of Rare Diseases 8 107. ( 10.1186/1750-1172-8-107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Erven B, Berry GT, Cassiman D, Connolly G, Forga M, Gautschi M, Gubbels CS, Hollak CEM, Janssen MC, Knerr Iet al. 2017. Fertility in adult women with classic galactosemia and primary ovarian insufficiency. Fertility and Sterility 108 168–174. ( 10.1016/j.fertnstert.2017.05.013) [DOI] [PubMed] [Google Scholar]
- van Kasteren YM, Schoemaker J. 1999. Premature ovarian failure: a systematic review on therapeutic interventions to restore ovarian function and achieve pregnancy. Human Reproduction Update 5 483–492. ( 10.1093/humupd/5.5.483) [DOI] [PubMed] [Google Scholar]
- Vanoevelen JM, van Erven B, Bierau J, Huang X, Berry GT, Vos R, Coelho AI, Rubio-Gozalbo ME. 2018. Impaired fertility and motor function in a zebrafish model for classic galactosemia. Journal of Inherited Metabolic Disease 41 117–127. ( 10.1007/s10545-017-0071-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova Y, Coupland C, Hippisley-Cox J. 2019. Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ 364 k4810. ( 10.1136/bmj.k4810) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Kikuchi A, Arai-Ichinoi N, Sakamoto O, Takezawa Y, Iwasawa S, Niihori T, Nyuzuki H, Nakajima Y, Ogawa Eet al. 2019. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genetics in Medicine 21 1286–1294. ( 10.1038/s41436-018-0340-x) [DOI] [PubMed] [Google Scholar]
- Waisbren SE, Potter NL, Gordon CM, Green RC, Greenstein P, Gubbels CS, Rubio-Gozalbo E, Schomer D, Welt C, Anastasoaie Vet al. 2012. The adult galactosemic phenotype. Journal of Inherited Metabolic Disease 35 279–286. ( 10.1007/s10545-011-9372-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Luo LL, Xu JJ, Xu MY, Zhang XM, Zhou XL, Liu WJ, Fu YC. 2014. Obesity accelerates ovarian follicle development and follicle loss in rats. Metabolism: Clinical and Experimental 63 94–103. ( 10.1016/j.metabol.2013.09.001) [DOI] [PubMed] [Google Scholar]
- Webb AL, Singh RH, Kennedy MJ, Elsas LJ. 2003. Verbal dyspraxia and galactosemia. Pediatric Research 53 396–402. ( 10.1203/01.PDR.0000049666.19532.1B) [DOI] [PubMed] [Google Scholar]
- Webber L, Davies M, Anderson R, Bartlett J, Braat D, Cartwright B, Cifkova R, de Muinck Keizer-Schrama S, Hogervorst E. 2016. ESHRE Guideline: management of women with premature ovarian insufficiency. Human Reproduction 31 926–937. ( 10.1093/humrep/dew027) [DOI] [PubMed] [Google Scholar]
- Webber L, Anderson RA, Davies M, Janse F, Vermeulen N. 2017. HRT for women with premature ovarian insufficiency: a comprehensive review. Human Reproduction Open 2017 hox007. ( 10.1093/hropen/hox007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, Grünewald S, Gubbels CS, Knerr I, Labrune Pet al. 2017. International Clinical Guideline for the management of classical galactosemia: diagnosis, treatment, and follow‐up. Journal of Inherited Metabolic Disease 40 171–176. ( 10.1007/s10545-016-9990-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CJ, Cari EL, Sandoval J, Bales E, Sam PK, Zarate MA, Polotsky AJ, Kallen AN, Johnson J. 2020. Control of murine primordial follicle growth activation by IκB/NFκB signaling. Reproductive Sciences 27 2063–2074. ( 10.1007/s43032-020-00225-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LL, Dunning KR, Yang X, Russell DL, Lane M, Norman RJ, Robker RL. 2010. High-fat diet causes lipotoxicity responses in cumulus-oocyte complexes and decreased fertilization rates. Endocrinology 151 5438–5445. ( 10.1210/en.2010-0551) [DOI] [PubMed] [Google Scholar]
- Wu LL, Russell DL, Norman RJ, Robker RL. 2012. Endoplasmic reticulum (ER) stress in cumulus-oocyte complexes impairs pentraxin-3 secretion, mitochondrial membrane potential (DeltaPsi m), and embryo development. Molecular Endocrinology 26 562–573. ( 10.1210/me.2011-1362) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LL, Russell DL, Wong SL, Chen M, Tsai TS, St John JC, Norman RJ, Febbraio MA, Carroll J, Robker RL. 2015. Mitochondrial dysfunction in oocytes of obese mothers: transmission to offspring and reversal by pharmacological endoplasmic reticulum stress inhibitors. Development 142 681–691. ( 10.1242/dev.114850) [DOI] [PubMed] [Google Scholar]
- Xiong Y, Chen H, Lin P, Wang A, Wang L, Jin Y. 2017. ATF6 knockdown decreases apoptosis, arrests the S phase of the cell cycle, and increases steroid hormone production in mouse granulosa cells. American Journal of Physiology: Cell Physiology 312 C341–C353. ( 10.1152/ajpcell.00222.2016) [DOI] [PubMed] [Google Scholar]
- Yan H, Zhang J, Wen J, Wang Y, Niu W, Teng Z, Zhao T, Dai Y, Zhang Y, Wang Cet al. 2018. CDC42 controls the activation of primordial follicles by regulating PI3K signaling in mouse oocytes. BMC Biology 16 73. ( 10.1186/s12915-018-0541-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SH, Choi YM, Hong MA, Kim JJ, Lee GH, Hwang KR, Moon SY. 2013. Association study of anti-Mullerian hormone and anti-Mullerian hormone type II receptor polymorphisms with idiopathic primary ovarian insufficiency. Human Reproduction 28 3301–3305. ( 10.1093/humrep/det384) [DOI] [PubMed] [Google Scholar]
- Yuzyuk T, Viau K, Andrews A, Pasquali M, Longo N. 2018. Biochemical changes and clinical outcomes in 34 patients with classic galactosemia. Journal of Inherited Metabolic Disease 41 197–208. ( 10.1007/s10545-018-0136-9) [DOI] [PubMed] [Google Scholar]
- Zhao M, Feng F, Chu C, Yue W, Li L. 2019. A novel EIF4ENIF1 mutation associated with a diminished ovarian reserve and premature ovarian insufficiency identified by whole-exome sequencing. Journal of Ovarian Research 12 119. ( 10.1186/s13048-019-0595-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Yan W, Shen W, Cheng J, Xi Y, Yuan S, Fu F, Ding T, Luo A, Wang S. 2018. Low expression of SEMA6C accelerates the primordial follicle activation in the neonatal mouse ovary. Journal of Cellular and Molecular Medicine 22 486–496. ( 10.1111/jcmm.13337) [DOI] [PMC free article] [PubMed] [Google Scholar]