

Abstract
The hypothalamic peptides named the orexins, or hypocretins, were discovered in 1998. In 1999 it was established that genetic narcolepsy could be caused by mutations in the genes synthesizing these peptides or their receptors. In September of 2000 it was found that most human narcolepsy is caused by loss of hypocretin cells, most likely as a result of a degenerative process. This paper reviews these events and their implications for our understanding of brain arousal and motor control systems.
Keywords: Hypocretin, Orexin, Narcolepsy, Sleep
Advances within the last three years have transformed our understanding of narcolepsy and suggested new possibilities for diagnosis and treatment of this disease. They have also exemplified the power of basic research to offer insights into human sleep disorders. The inter-relation of basic and human work in the field of sleep and psychiatric research is an approach that Chris Gillin has pioneered. This can be seen in his work on basic brainstem mechanisms of sleep control, the cholinergic role in REM sleep expression, brain imaging and the relation of these same parameters to human depression, sleep disorders, addiction and narcolepsy (Gillin et al. 1976, 1978, 1979, 1981; Shiromani et al. 1987, 1988a,b,c; Sutin et al. 1986; Kennedy et al. 1982; Sitaram et al. 1976, 1980; Fredrick et al. 1977; Buchsbaum et al. 1982; Shiromani, Gillin, 1987;). The hypocretin/narcolepsy story is a dramatic example of the application of basic science techniques to the analysis of sleep disorders.
THE OREXIN/HYPOCRETIN STORY
The story begins not with the search for the cause of narcolepsy, but rather with the search for a possible treatment for obesity. It was reasoned that genes expressed selectively in the hypothalamus might have medically useful effects because of the role of the hypothalamus in control of appetite, thirst, pituitary hormones and many other autonomic and arousal functions. Sutcliffe’s group in San Diego used subtractive hybridization to identify such genes (De Lecea et al. 1998). Two peptides derived from a single gene were identified and named the hypocretins, for their hypothalamic location and sequence homology to secretin. The two co-lead authors on the publication were Luis de Lecea and Thomas Kilduff. A bizarre irony is that Dr. Kilduff was former director of the narcoleptic dog colony at Stanford University, but had abandoned narcolepsy research to focus on the mastery and application of molecular biological techniques.
At about the same time, Masashi Yanagisawa’s group was searching for ligands for “orphan receptors,” DNA sequences with strong homologies to known G protein receptors but with no identified endogenous ligand. By purifying brain extracts that activated these orphan receptors expressed in cell lines, they were able to identify two novel peptides, which were synthesized only in the hypothalamus. They found that when these peptides was injected into the 3rd ventricle they induced feeding. They named the peptides orexins, after the Greek word for appetite (Sakurai et al. 1998). The peptides detected by Yanagisawa’s group turned out to be identical to those detected by Sutcliffe’s group. The Sutcliffe and Yanagisawa papers appeared within a month of each other.
Yanagisawa’s group proceeded to further investigate the role of the peptide in feeding by using transgenic techniques to construct a null mutant mouse that did not produce either orexin peptide. They found that the knockout mice had reduced food intake as had been predicted. However, the reduction was not dramatic and had relatively little effect on weight. They were determined to analyze the nature of the feeding deficit in their model and employed infrared video observation to document food ingestion. Careful observation by Chemelli et al. revealed that these animals would often abruptly cease movement in what they hypothesized was a cataplectic attack (Chemelli et al. 1999). In follow up work they created mice lacking either of the two identified orexin (hypocretin) receptors. They found that mice without the hypocretin-2 receptor experienced slow cessations of movement that they concluded were sleep attacks. They found that mice lacking the hypocretin-1 receptor had disrupted sleep but showed fewer of the signs of narcolepsy than the type 2 receptor knockouts (Chemelli et al. 2000).
Immunohistochemistry demonstrated that the peptides encoded by the mRNAs selectively expressed in the hypothalamus were localized to cell bodies in the lateral and medial hypothalamic regions (Peyron et al. 1998). No hypocretin cells were found in the brain outside the hypothalamus, although the hypocretin cells had projections throughout the brain, especially to monoaminergic, cholinergic and limbic cell groups. The lateral hypothalamus has long been known to constitute a kind of feeding “center” (Shoham and Teitelbaum 1982). Lesions of this region cause a prolonged cessation of eating and weight loss, whereas lesions of more medial regions caused overeating and obesity.
Hypocretin/Orexin and Genetic Narcolepsy
It had previously been shown by Dement and collaborators that canine genetic narcolepsy was caused by an autosomal recessive gene (Baker and Dement 1985; Mitler and Dement 1977). Mignot’s group sought to identify the gene that caused canine narcolepsy by a systematic chromosomal analysis. They identified the gene responsible for canine narcolepsy as a mutated, non-functional version of the hypocretin-2 gene. In a second great coincidence, Mignot’s group and Yanagisawa’s group reported their complementary genetic findings within a month of each other (Chemelli et al. 1999; Lin et al. 1999).
Hypocretin/Orexin and Human Narcolepsy
The striking similarity of the symptoms expressed by canine and mouse genetic mutants to human narcoleptics suggested that hypocretin might be involved in human narcolepsy. However, from the outset it was clear that the cause could not be identical to the mutations seen in these animals. Human narcolepsy is typically discordant in identical twins (Hublin et al. 1994). It does not generally run in families, in contrast to the genetic dog and mouse models (Guilleminault and Anognos 2000).
We reasoned that damage to the hypocretin system, rather than a mutation of the peptide or receptor genes, might be the cause of human narcolepsy. We secured four human narcoleptic brains and attempted to immunohistochemically stain the hypocretin cells. Unfortunately, since these brains had been in fixative for a minimum of five years, overfixation had obstructed antibody access to the hypocretin antigen. Fortunately, we happened to have a number of normal human brains in the laboratory that had been stored in similar fixation conditions for as long or longer than the narcoleptic brains. These brains also did not initially stain immunohistochemically for hypocretin. We were able to adapt “antigen retrieval” techniques to the overfixed normal tissue and stain hypocretin neurons and then applied the techniques to the narcoleptic tissue. We found a massive loss of hypocretin neurons, averaging a 93% reduction compared with control tissue (Thannickal et al. 2000a). In a third major coincidence, our paper reporting these findings (Thannickal et al. 2000c) and a paper by Mignot’s group coming to a similar conclusion both appeared in September, 2000.
Although our work and Mignot’s group’s work are in agreement in implicating hypocretin in human narcolepsy, there are several significant differences in our findings. We see approximately 70,000 hypocretin neurons in our normal subjects versus 15–20,000 reported by Peyron et al. Figure 1 plots our count in comparison to the counts reported in recent animal studies. A second difference is the Peyron et al. (2000) report of a total absence of hypocretin containing cells in the two narcoleptic hypothalami they examined, implying either a loss of cells or a complete cessation of hypocretin production in these cells. This is in keeping with the total loss of hypocretin detectable by their assay in the cerebrospinal fluid of narcoleptics (Nishino et al. 2000). In contrast, we find surviving cells and axons in all of our subjects. Our work suggests that hypocretin might be detectable in the CSF of these subjects with a sufficiently sensitive assay.
Figure 1.
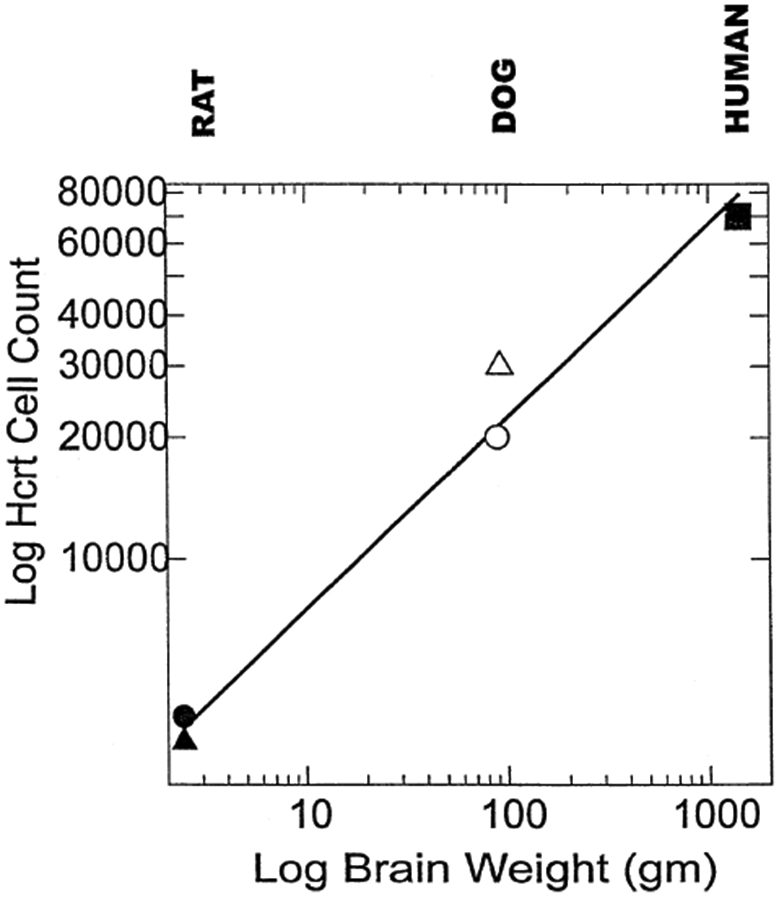
Hypocretin (Hcrt) cell number and brain weight of the three species so far quantified displayed on a log-log plot. Filled circle, rat count (3,436) from Harrison et al. (1999); filled triangle, rat count (1128) from Peyron et al. (1998); open circle, narcoleptic dog count (20,000) from Nishino (2000), open triangle, narcoleptic/normal dog count (30,000) from Thannickal et al. (2000b) and closed square, human count (70,000) from Thannickal et al. (2000b).
Hypocretin Administration as a Treatment for Narcolepsy
Since hypocretin cell loss underlies the symptoms of narcolepsy, we reasoned that it would be possible to reverse the symptoms of narcolepsy by administration of hypocretin. Because Kastin et al. had reported that hypocretin, unlike many other peptides, crossed the blood-brain barrier by diffusion (Kastin and Akerstrom 1999), we studied the effect of systemically administered hypocretin on symptoms of narcolepsy in the narcoleptic dog. We found that intravenously administered hypocretin produced an immediate increase in activity level, with amplitude and time course similar to that seen with injection into the 3rd ventricle. At the same time, hypocretin administration decreased cataplexy (John et al. 2000b). However, higher doses of hypocretin actually produced a significant increase in cataplexy. We interpret this finding in the context of our recent microinjection studies in the rat (Kiyashchenko et al. 2000). These studies have shown that microinjection of hypocretin-1 into locus coeruleus enhances muscle tone. However, microinjection just ventral to the locus coeruleus in the pontine inhibitory area suppresses muscle tone. The administration of high doses systemically could be affecting cataplexy in a similar way. According to this hypothesis low doses act on the densely hypocretin innervated locus coeruleus, which facilitates muscle tone, whereas higher doses activate not only the locus coeruleus but also the potent inhibitory circuits in the subcoeruleus region, producing a net facilitation of cataplexy. In contrast to the dose dependent short duration effects of injected hypocretin on cataplexy, repeated administration of hypocretin every other day produced a complete cessation of cataplexy for as long as six days in some of the subjects (John et al. 2000a). The mechanism for this prolonged effect is unknown but may involve changes in receptor sensitivity and interactions between hypocretin and the immune system (Ichinose et al. 1998).
UNANSWERED QUESTIONS
Symptom Variability
One important goal of future research is to explain the variability in the symptoms experienced by narcoleptics. Human narcoleptics vary in the amount of sleepiness they exhibit. Some human narcoleptics have disabling cataplexy, whereas others have no cataplexy or only rare episodes. Standard clinical practice is to label patients with unexplained sleepiness, sleep-onset REM, sleep paralysis and hypnagogic hallucinations as narcoleptic even without the presence of cataplexy. However such patients are typically not used in research because of the concern that they might have a different underlying disease process.
In our anatomic study (Thannickal et al. 2000c) we found that even a patient diagnosed as narcoleptic on the basis of the consensus clinical criteria but reporting no cataplexy had a massive loss of hypocretin cells. This finding supports the common clinical diagnostic criteria for narcolepsy, and indicates that some and perhaps most narcoleptic patients without cataplexy have the same disease process as those with cataplexy. Nevertheless, there must be a neurological correlate for the difference in symptom expression in narcoleptics. Identification of such correlates would be useful in the understanding of sleep control as well as in the care of these patients.
Even more puzzling than the variability seen in human narcoleptics is the variability seen in the symptoms shown by genetic models, specifically by the narcoleptic dog. All such animals have mutations in the hypocretin receptor 2 gene. Two breeds with this mutation have been identified, the narcoleptic Doberman and the narcoleptic Labrador retriever (Riehl et al. 1998). The hypocretin-2 receptor mutation in the Labrador is different than the hypocretin-2 mutation in the Doberman, which may partially account for the much greater symptom severity in Dobermans (Peyron et al. 2000). However, it is equally likely that genetic and environmental factors unrelated to the mutation but differing in Labradors and Dobermans breeds may contribute to the difference in symptom severity. Females of both breeds are typically more affected than males (Riehl et al. 1998).
There is also a marked developmental component to the variability (Baker and Dement 1985; Riehl et al. 1998). At birth and during the early stages of weaning, most puppies show no symptoms. At 2–4 months of age symptoms appear and intensify until about one year. At this point, symptoms diminish slowly in most dogs. By 4–5 years of age, many dogs are completely asymptomatic. However, in these dogs the underlying disease process can be exposed by administration of cholinesterase inhibitors or α1 blockers (Siegel et al. 1991; Wu et al. 1999). These drugs will precipitate symptoms in genetically narcoleptic animals, but not in normals at any dose. When given to younger narcoleptic animals symptoms will always intensify.
Adult dogs of the same breed and comparable ages show enormous symptom variability. Some “show dogs” will consistently have cataplexy regardless of the circumstances or audience, whereas others, including littermates, have no symptoms. Since the genetic component is fixed, these data suggest that the genetic defect, although causal, is not sufficient to produce symptoms. In prior work we demonstrated that neuronal degeneration was occurring at the time of symptom onset in the hypocretin-2 receptor mutated dogs (Siegel et al. 1999). It is likely that the intensity of this degenerative process may be linked to the eventual severity of symptoms developed. Factors that modulate the time course of this degeneration should modulate the development of symptoms as well.
In the human case, one may speculate that the cell loss that we have seen may similarly be just the first step in symptom development. In other words, cell loss may precipitate degenerative or remodeling processes that lead to symptoms. The nature of the remodeling process and how it contributes to symptomatology remains to be determined. The study of conditional knockouts could test this hypothesis. Understanding and reversal of such processes may be an effective means of treating the human disorder.
What Changes the Activity of Brainstem Motor Control Systems to Produce Cataplexy?
Muscle tone is controlled by brainstem effector mechanisms. Although these mechanisms are under the control of forebrain systems, they can operate in the absence of these controls. For example, the decerebrate animal (with complete removal of the forebrain, including the hypothalamus) shows REM sleep and its associated muscle atonia (Jouvet 1962). Cataplexy, because of its similarity to REM sleep in loss of muscle tone and reflex response, has long been hypothesized to result from the activation of REM sleep motor control mechanisms (Rechtschaffen et al. 1963). However, unlike REM sleep, consciousness is preserved in cataplexy. This provides an important “experiment of nature” that allows us to separate brainstem systems possibly linked to muscle tone from those linked to consciousness.
In the medial medulla, we find a group of cells in the nucleus magnocellularis that is selectively active during REM sleep (Siegel et al. 1991). Activation of these cells in the decerebrate animal by electrical and chemical stimulation produces muscle tone suppression (Lai and Siegel 1991) and lesion of these cells causes REM sleep without atonia (Schenkel and Siegel 1989). These cells are activated prior to and during cataplexy (Siegel et al. 1991). This evidence clearly indicates that these cells are linked to loss of muscle tone.
The locus coeruleus is comprised of noradrenergic cells with both ascending and descending projections. These cells reduce firing in nonREM sleep and cease discharge in REM sleep. We found that they also cease discharge, to the same or greater extent than their cessation in REM sleep, during cataplexy (Wu et al. 1999). This finding, combined with evidence that norepinephrine from locus coeruleus is released onto motoneurons, that norepinephrine depolarizes motoneurons and facilitates motor reflexes, and that inactivation or lesion of locus coeruleus reduces ipsilateral muscle tone (Pompeiano 2000), indicates that the cessation of locus coeruleus discharge is linked to the loss of muscle tone in cataplexy. In contrast to locus coeruleus, the REM-off serotonergic neurons in the raphe do not show a comparable reduction in activity in cataplexy (Wu et al. 2000). The locus coeruleus activity pattern is consistent with a link to muscle tone, but not to consciousness.
We Know That Canine Narcolepsy Is Caused by a Mutation in the Hypocretin-2 Receptor. How Does This Receptor Cause the Activity Changes in Locus Coeruleus and Nucleus Magnocellularis?
In the case of locus coeruleus the evidence is tantalizing but incomplete. The locus coeruleus receives the “largest extrahypothalamic projection” of the hypocretin system (Peyron et al. 1998). Microinjection of hypocretin into locus coeruleus facilitates muscle tone (Kiyashchenko et al. 2001). These facts are consistent with the observed correlation between cataplexy and loss of the hypocretin receptor. However, the locus coeruleus appears to be devoid of the hypocretin-2 receptor (Trivedi et al. 1998), the receptor that is mutated in genetic narcolepsy. Only hypocretin-1 receptors are present. One may speculate that dogs are unique in having hypocretin-2 receptors in the locus coeruleus. However, it was found that hypocretin-2 knockout mice, who do not have hypocretin-2 receptors in locus coeruleus, also show cataplexy, whereas hypocretin-1 knockout mice do not. This finding makes it implausible that the presence of mutated hypocretin-2 receptors in the locus coeruleus is the cause of the offset of locus coeruleus discharge in canine cataplexy (Chemelli et al. 2000). There are hypocretin projections to the nucleus magnocellularis (Peyron et al. 1998). We find that microinjection of hypocretin into this area produces loss of muscle tone. However, loss of this hypocretin response would logically be expected to increase muscle tone rather than explaining the loss of tone seen in cataplexy.
A more fundamental problem for making a simple link between the receptor mutation and the physiology of cataplexy is the nature of cataplexy itself. Narcoleptic humans and animals are not generally afflicted with a tonic hypotonia. Rather they have phasic losses of muscle tone linked to emotion. These losses occur on a background of normal muscle tone. Although our microinjection work has clearly shown major effects of hypocretin on muscle tone, the phasic nature of the losses seen in narcolepsy clearly involve more than a tonic loss of motor facilitation. There appears to be some reduction in the tonic level of discharge in the LC cell population in narcoleptics, with the mean waking discharge rate of these cells in canine narcoleptics lower than that seen in cats, rats and monkeys (Aston-Jones and Bloom 1981; Foote et al. 1983; Rasmussen et al. 1986). However, much more pronounced than this apparent reduction in tonic discharge rate, is the sudden cessation of discharge in locus coeruleus and activation of nucleus magnocellularis in narcoleptics during waking emotions, a change that does not happen in normals. The cause of cataplexy must be instability in a system responsible for the facilitation of muscle tone during times of strong emotion. We hypothesize the existence of motor inhibition or disfacilitation linked to emotion whose net effect is normally counteracted by neurons linked to the hypocretin system. Thus we hypothesize that abnormal operation of intrahypothalamic circuitry impairs the phasic release of hypocretin in critical brainstem circuitry during emotions triggering cataplexy. It is this hypothalamic circuitry rather than the brainstem receptors for this system, that requires normal hypocretin-2 receptors. The dynamics of this system remain to be elucidated. Similar considerations are likely to apply to the sleepiness that characterizes narcolepsy (Methippara et al. 2000; Siegel 1999).
In conclusion, major elements of human and genetic canine and murine narcolepsy have been unraveled in the last two years. Clearly, alterations in the hypocretin system are causal for these disorders. Nevertheless, the synaptic mechanisms responsible for the elaboration of symptoms in narcoleptics are still unclear. Clarification of these mechanisms will have important implications for our understanding of the normal operation of the hypocretin system and for our analysis of the symptom expression in human narcolepsy.
Acknowledgments
Supported by the Medical Research Service of the Dept. of Veterans Affairs, NS14610, MH64109 and HL60296.
Contributor Information
J.M. Siegel, Department of Psychiatry and Brain Research Institute, UCLA and Neurobiological Research (151A3), VA GLAHCS, North Hills, CA 91343
R. Moore, Department of Neurology, University of Pittsburgh, Pittsburgh, PA 15213
T. Thannickal, Department of Psychiatry and Brain Research Institute, UCLA and Neurobiological Research (151A3), VA GLAHCS, North Hills, CA 91343
R. Nienhuis, Department of Psychiatry and Brain Research Institute, UCLA and Neurobiological Research (151A3), VA GLAHCS, North Hills, CA 91343
REFERENCES
- Aston-Jones G, Bloom FE (1981): Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1:876–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Dement WC (1985): Canine narcolepsy-cataplexy syndrome: Evidence for an inherited monoaminergic-cholinergic imbalance. In McGinty DJ, Drucker-Colin R, Morrison A, Parmeggiani PL (eds), Brain Mechanisms of Sleep. New York, Raven Press, pp 199–234 [Google Scholar]
- Buchsbaum MS, Mendelson WB, Duncan WC, Coppola R, Kelsoe J, Gillin JC (1982): Topographic cortical mapping of EEG sleep stages during daytime naps in normal subjects. Sleep 5:248–255 [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton C, Elmquist J, Scammell T, Lee C, Richardson J, Williams S, Xiong Y, Kisanuki Y, Fitch T, Nakazato M, Hammer R, Saper C, Yanagisawa M (1999): Narcolepsy in orexin knockout mice: Molecular genetics of sleep regulation. Cell 98:437–451 [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Sinton CM, Yanagisawa M (2000): Polysomnographic characterization of orexin-2 receptor knockout mice. Sleep 23:A296–A297 [Google Scholar]
- De Lecea L, Kilduff T, Peyron C, Gao XB, Foye PE, Danielson PE, Fukahara C, Battenberg ELF, Gautvik VT, Barlett FS, Frankel WN, van den Pol AN, Bloom F, Gautvik KM, Sutcliffe JG (1998): The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95:322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote SL, Bloom FE, Aston-Jones G (1983): Nucleus locus ceruleus: New evidence of anatomical and physiological specificity. Physiol Rev 63:844–914 [DOI] [PubMed] [Google Scholar]
- Fredrick GC, Quinn CP, Gillin JG, Wyatt RJ (1977): Deprivation of rapid eye movement sleep and nesting behavior in rats. Physiology and Behavior 18:341–344 [DOI] [PubMed] [Google Scholar]
- Gillin JC, Horwitz D, Wyatt RJ (1976): Pharmacologic studies of narcolepsy involving serotonin, acetylcholine, and monoamine oxidase. In Guilleminault C, Dement WC, Passouant P (eds), Narcolepsy. New York, Spectrum Publications, Inc., pp 585–604 [Google Scholar]
- Gillin JC, Mendelson WB, Sitaram N, Wyatt RJ (1978): The neuropharmacology of sleep and wakefulness. Ann Rev Pharmacol Toxicol 18:563–579 [DOI] [PubMed] [Google Scholar]
- Gillin JC, Sitaram N, Duncan WC (1979): Muscarinic supersensitivity: A possible model for the sleep disturbance of primary depression? Psych Res 1:17–22 [DOI] [PubMed] [Google Scholar]
- Gillin JC, Van Kammen DP, Post RM, Sitaram N, Wyatt RJ, Bunney J (1981): What is the role of dopamine in the regulation of sleep-wake activity? In Corsini GU, Gessa GL (eds), Clinical Pharmacology. New York, Raven Press, pp 157–164 [Google Scholar]
- Guilleminault C, Anognos A (2000): Narcolepsy. In Kryger MH, Roth T, Dement WC (eds), Principles and Practice of Sleep Medicine. Philadelphia, W.B. Saunders, pp 676–686 [Google Scholar]
- Harrison TA, Chen CT, Dun NJ, Chang JK (1999): Hypothalamic orexin A-immunoreactive neurons project to the rat dorsal medulla. Neurosci Lett 273:17–20 [DOI] [PubMed] [Google Scholar]
- Hublin C, Kaprio J, Partinen M, Koskenvuo M, Heikkila K, Koskimies S, Guilleminault C (1994): The prevalence of narcolepsy: an epidemiological study of the Finnish twin cohort. Ann Neurol. 35:709–716 [DOI] [PubMed] [Google Scholar]
- Ichinose M, Asai M, Sawada M, Sasaki K, Oomura Y (1998): Induction of outward current by orexin-B in mouse peritoneal macrophages. FEBS Lett 440:51–54 [DOI] [PubMed] [Google Scholar]
- John J, Wu MF, Siegel JM (2000a): Hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep 23:A12. [PMC free article] [PubMed] [Google Scholar]
- John J, Wu M, Siegel J (2000b): Systemic administration of hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep Res Online 3:23–28 http://www.sro.org/2000/John/23/. [PMC free article] [PubMed] [Google Scholar]
- Jouvet M (1962): Recherches sur les structures nerveuses et les mechanismes responsables des differentes phases du sommeil physiologique. Arch Ital Biol 100:125–206. [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V (1999): Orexin A but not orexin B rapidly enters brain from blood by simple diffusion. J Pharmacol Exp Ther 289:219–223 [PubMed] [Google Scholar]
- Kennedy C, Gillin JC, Mendelson W, Suda S, Miyaoka M, Ito M, Nakamura RK, Storch FI (1982): Local cerebral glucose utilization in non-rapid eye movement sleep. Nature 297:325–327 [DOI] [PubMed] [Google Scholar]
- Kiyashchenko LI, Mileykovskiy BY, Lai YY, Siegel JM (2001): Increased and decreased muscle tone with orexin (hypocretin) microinjections in the locus coeruleus and pontine inhibitory area. J Neurophysiol. 85:2008–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Siegel JM (1991): Ponto-medullary glutamate receptors mediating locomotion and muscle tone suppression. J Neurosci 11:2931–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Kadotani H, Rogers W, Lin X, Qui X, de Jong P, Nishino S, Mignot E (1999): The REM sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor gene. Cell 98:365–376 [DOI] [PubMed] [Google Scholar]
- Methippara MM, Alam MN, Szymusiak R, McGinty D (2000): Effects of lateral preoptic area application of orexin-A on sleep-wakefulness. Neuroreport 11:3423–3426 [DOI] [PubMed] [Google Scholar]
- Mitler MM, Dement WC (1977): Sleep studies on canine narcolepsy: pattern and cycle comparisons between affected and normal dogs. Electroenceph Clin Neurophysiol 43:691–699 [DOI] [PubMed] [Google Scholar]
- Nishino S (2000): Clinical and etiological aspects of canine narcolepsy. Retreat on Narcolepsy And Hypocretins (Orexins) August 14–15, 2000 Westin Hapuna Beach Prince Hotel [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E (2000): Hypocretin (orexin) deficiency in human narcolepsy. The Lancet 355:39–41 [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, Nishino S, Mignot E (2000): A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 6:991–997 [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, De Lecea L, Heller HC, Sutcliffe JG, Kilduff TS (1998): Neurons containing hypocretin (orexin) project to multiple neuronal systems. Journal of Neuroscience 18:9996–10015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompeiano O (2000): Role of the locus coeruleus in the static and dynamic control of posture. Arch Ital de Biologie (in press) [PubMed] [Google Scholar]
- Rasmussen K, Morilak DA, Jacobs BL (1986): Single unit activity of locus coeruleus neurons in the freely moving cat. I. During naturalistic behaviors and in response to simple and complex stimuli. Brain Res 371:324–334 [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Wolpert EA, Dement WC, Mitchel SA, Fisher C (1963): Nocturnal sleep of narcoleptics. Electroenceph Clin Neurophysiol. 15:599–609 [DOI] [PubMed] [Google Scholar]
- Riehl J, Nishino S, Cederberg R, Dement WC, Mignot E (1998): Development of cataplexy in genetically narcoleptic dobermans. Exp Neruol. 152:292–302 [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M (1998): Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–585 [DOI] [PubMed] [Google Scholar]
- Schenkel E, Siegel JM (1989): REM sleep without atonia after lesions of the medial medulla. Neurosci Lett. 98:159–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiromani PJ, Armstrong DM, Berkowitz A, Jeste DV, Gillin JC (1988b): Distribution of choline acetyltransferase immunoreactive somata in the feline brainstem: implications for REM sleep generation. Sleep 11:1–16 [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Armstrong DM, Bruce G, Hersh LB, Groves PJ, Gillin C (1987): Relation of pontine choline acetyltranferase immunoreactive neurons with cells which increase discharge during REM sleep. Brain ResBull. 18:447–455 [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Armstrong DM, Gillin JC (1988a): Cholinergic neurons from the dorsolateral pons project to the medial pons: a WGA-HRP and choline acetyltransferase immunohistochemical study. Neurosci Lett. 95:19–23 [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Gillin JG (1987): Acetylcholine and the regulation of REM sleep: Basic mechanisms and clinical implications for affective illness and narcolepsy. Ann Rev Pharmacol Toxicol. 27:137–156 [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Overstreet D, Levy D, Goodrich CA, Campbell SS, Gillin JC (1988c): Increased REM sleep in rats selectively bred for cholinergic hyperactivity. Neuropscyhopharm. 1:127–133 [DOI] [PubMed] [Google Scholar]
- Shoham S, Teitelbaum P (1982): Subcortical waking and sleep during lateral hypothalamic “somnolence” in rats. Physiol Behav. 28:323–333 [DOI] [PubMed] [Google Scholar]
- Siegel JM (1999): Narcolepsy: A key role for hypocretins (orexins). Cell 98:409–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Fahringer H, Paul R, Shiromani P, Dement WC, Mignot E, Chiu C (1991): Neuronal activity in narcolepsy: identification of cataplexy related cells in the medial medulla. Science 262:1315–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Gulyani S, Ouyang S, Wu MF, Mignot E, Switzer RC, Cornford M (1999): Neuronal degeneration in canine narcolepsy. J Neurosci 19:248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitaram N, Nurnberger JI, Gershon ES, Gillin JC (1980): Faster cholinergic REM sleep induction in euthymic patients with primary affective illness. Science 208:200–202 [DOI] [PubMed] [Google Scholar]
- Sitaram N, Wyatt RJ, Dawson S, Gillin JC (1976): REM sleep induction by physostigmine infusion during sleep. Science 191:1281–1283 [DOI] [PubMed] [Google Scholar]
- Sutin EL, Shiromani PJ, Kelsoe J, Storch FI, Gillin JC (1986): Rapid-eye movement sleep and muscarinic receptor binding in rats are augmented during withdrawal from chronic scopolamine treatment. Life Sci 39:2419–2427 [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Aldrich M, Albin R, Cornford M, Siegel JM (2000a): Human narcolepsy is linked to reduced number, size and synaptic bouton density in hypocretin-2 labeled neurons. Abstr Soc Neurosci 26: 2061 [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM (2000c): Reduced number of hypocretin neurons in human narcolepsy. Neuron 27:469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Nienhuis R, Ramanathan L, Gulyani S, Turner K, Chestnut B, Siegel JM (2000b): Preservation of hypocretin neurons in genetically narcoleptic dogs. Sleep 23:A296 [Google Scholar]
- Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, Guan XM (1998): Distribution of orexin receptor mRNA in the rat brain. FEBS Lett 438:71–75 [DOI] [PubMed] [Google Scholar]
- Wu MF, Gulyani S, Yao E, Mignot E, Phan B, Siegel JM (1999): Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience 91:1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, John J, Nguyen GB, Siegel JM (2000): Serotonergic dorsal raphe rem-off cells reduce discharge but do not shut off during cataplexy. Sleep 23:A2–A3 [Google Scholar]