

ABSTRACT
The gut microbiota has emerged as a critical player in host health. Bacteroides fragilis is a prominent member of the gut microbiota within the phyla Bacteroidetes. This commensal bacterium produces unique capsular polysaccharides processed by antigen-presenting cells and activates CD4+ T cells to secrete inflammatory cytokines. Indeed, due to their immunomodulatory functions, B. fragilis and its capsular polysaccharide-A (PSA) are arguably the most explored single commensal microbiota/symbiotic factor. B. fragilis/PSA has been shown to protect against colitis, encephalomyelitis, colorectal cancer, pulmonary inflammation, and asthma. Here, we review recent data on the immunomodulatory role of B. fragilis/PSA during viral infections and therapy, B. fragilis PSA’s dual ability to mediate pro-and anti-inflammatory processes, and the potential for exploring this unique characteristic during intracellular bacterial infections such as with Mycobacterium tuberculosis. We also discuss the protective roles of single commensal-derived probiotic species, including B. fragilis in lung inflammation and respiratory infections that may provide essential cues for possible exploration of microbiota based/augmented therapies in tuberculosis (TB). Available data on the relationship between B. fragilis/PSA, the immune system, and disease suggest clinical relevance for developing B. fragilis into a next-generation probiotic or, possibly, the engineering of PSA into a potent carbohydrate-based vaccine.
KEYWORDS: Gut microbiota, Bacteroides fragilis, polysaccharide A, probiotics, viral infection, tuberculosis, Mycobacterium tuberculosis
INTRODUCTION
The gastrointestinal tract of mammals habours a complex and diverse microbial community collectively referred to as the gut microbiota. These commensal microbes play essential roles in maintaining intestinal barrier integrity, nutrient absorption, immune regulation, and energy metabolism (1–4). Perturbations in the normal constituent of the gut microbiota have been linked to an increased risk of developing disease conditions such as intestinal bowel disease (5), colon cancer (6, 7), and diabetes (8, 9). The major gut microbiota phyla include Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia; however, the most abundant are the phyla Firmicutes and Bacteroidetes (10, 11).
Bacteroides fragilis is a prominent member of the genus Bacteroides within the phylum Bacteroidetes. Indeed, B. fragilis is the most explored commensal microbe for its immunomodulatory function within this genus. It is a Gram-negative, nonspore-forming, rod-shaped obligate anaerobe (12). A large proportion of the genome of B. fragilis is committed to capsular polysaccharide synthesis. B. fragilis is reported to produce eight capsular polysaccharides (designated A-H), an astonishing number considering that most bacteria can only synthesize one capsular polysaccharide if they make it at all (13–16). B. fragilis capsular polysaccharide-A (PSA) has been extensively described (17–19).
PSA from B. fragilis is processed by antigen-presenting cells (APCs) and activates T cells in a protein-like manner (20–24), unlike many naturally occurring polysaccharide molecules, which do not activate T cells. It possesses zwitterionic properties, carrying both positive and negative charges on the same repeating sugar molecule, a characteristic possessed by only a few known bacterial polysaccharides (25, 26). B. fragilis and PSA display potent immune response-inducing capabilities. Introduction of B. fragilis or PSA to germfree (GF) mice corrected a Th1/Th2 cytokine imbalance by inducing the Th1 cytokines IFN-γ and IL-2 (27). Furthermore, B. fragilis or PSA isolated from B. fragilis have been demonstrated to protect against colitis (28), encephalomyelitis (29, 30), colorectal cancer (31, 32), pulmonary inflammation (33), and asthma (34). Nevertheless, the bacteria can cause clinically significant infections when displaced outside the gut or escapes into the bloodstream following disruption of the mucosal surface during surgery, trauma, or inflammation (12, 35). This review presents recent data detailing the protective role of B. fragilis and PSA during viral infections and therapy. We also discuss the potential for exploration of the duality of B. fragilis and PSA mediated inflammatory response during Mycobacterium tuberculosis (Mtb) infection, and the protective roles of single commensal-derived probiotic species, including B. fragilis, in lung inflammation and respiratory infection that provide cues for possible exploration of microbiota based/augmented therapies in tuberculosis (TB).
BACTEROIDES FRAGILIS AND ITS CAPSULAR POLYSACCHARIDE-A REGULATES CONSTITUTIVE LEVELS OF TYPE I INTERFERONS TO PROTECT AGAINST VIRAL INFECTIONS
Type I interferons (IFN-Is) are a family of specialized cytokines that coordinate the host immune response to viruses. IFN-Is regulate host mechanisms, which eventually kill infected cells to restrict viral spread (36). However, they are also involved in the proliferation and activation of immune cells critical for controlling other intracellular infections, anti-tumor, and anti-inflammatory responses (36, 37). The most well-defined IFN-Is in mammals are the interferon-α and β. Consistent with previous literature suggesting that indigenous microbiota regulates basal production of IFN-Is (37–39), a recent study by Stefan and colleagues (40) showed that depletion of the gut microbiota lowered the expression of both intestinal and splenic interferon-stimulated genes (ISG). Analysis of ISG levels in conventional specific-pathogen-free versus GF mice also showed reduced expression of ISG in GF mice (40). They showed that the gut microbiota induced basal levels of IFN-I response via the regulation of interferon (IFN)-β levels and was predominantly produced by colonic lamina propria dendritic cells (LP DCs) (40). Of note, in this study, the gavage of GF mice with B. fragilis alone increased IFN-β gene expression. As expected, polysaccharide-A from B. fragilis also significantly increased both in vitro and in vivo expression of ISGs and secretion of IFN-β (40).
Interestingly, the study further showed that steady-state IFN-β generated by the gut microbiota was essential in priming antiviral immune response and protection. Mice lacking IFN-β genes that were infected with vesicular stomatitis virus (VSV) were more susceptible than mice that expressed genes for IFN-β (40). Furthermore, neutralization of IFN-β before infection abolished the antiviral protection. Similarly, wild-type (WT) mice treated with antibiotics before VSV infection were more susceptible, whereas antibiotics treatment in mice lacking IFN-β gene did not affect the animals’ susceptibility (40). The authors concluded that depletion of the gut microbiota does not alter the immune response to VSV infection in the absence of IFN-β, suggesting that gut microbiota-mediated protection against VSV was explicitly through IFN-β (40). Because PSA from B. fragilis also induced IFN-β, Stefan and colleagues investigated whether treatment with PSA alone was enough to protect against VSV and influenza A virus infections. Treatment of bone marrow-derived dendritic cells (BMDCs) with PSA before infection by the viruses reduced the percentage of infected cells and increased cell viability (40). Where PSA protected WT BMDCs, protection was abrogated in the absence of IFN-β. Also, PSA treatment significantly reduced disease burden and increased survival in WT antibiotic-treated mice infected with VSV. In contrast, PSA treatment did not affect the antibiotics-treated IFN-β deficient mice (40).
Many viral infections trigger the transcription of ISGs (41); however, Stefan and colleagues’ study showed that gut microbiota-mediated regulation of basally expressed ISGs prepared the immune system to respond efficiently upon viral challenge. Depletion of the gut microbiota with antibiotics has been shown in other studies to impair viral clearance in mucosal influenza and systemic lymphocytic choriomeningitis virus mouse models (42). However, Stefan and colleagues’ work is unique not only because it showed that depletion of commensal gut microbiota tilts constitutive levels of IFN-Is in favor of the establishment of VSV infection, but also that PSA from B. fragilis alone induced homeostatic IFN-β and was enough to reestablish the antiviral protection of the entire gut microbiota (40). IFN-Is signaling pathways during Mycobacterium tuberculosis (Mtb) infection are not yet thoroughly studied. Both types I and II IFNs are produced during the early stage of TB and act synergistically to induce an optimal immune response in the lung (43). Whereas IFN-γ enhances immune cells’ ability to restrict bacterial growth, IFN-Is limit the number of potential target cells that Mtb can infect in the lungs (43). In other studies, an IFN-Is-inducible gene signature dominated blood transcriptional profiles of active TB patients (44, 45). Expression of the gene signature correlated with the extent of lung disease and reduced with successful treatment (44). Similarly, early overexpression of IFN-response genes was reported in TB contacts who progressed to active TB (46, 47), suggesting that the development of the active disease may be preceded by circulatory IFN-Is response (46). Considering the impact of ISG on Mtb infection control and the establishment of TB disease, the gut microbiota and indeed B. fragilis or PSA may play a role in defining IFN-Is responses during TB.
POLYSACCHARIDE-A FROM BACTEROIDES FRAGILIS PROTECTS AGAINST HERPES SIMPLEX ENCEPHALITIS IN A DELAYED ACYCLOVIR TREATMENT MODEL
Herpes simplex encephalitis (HSE) is an uncommon neurological disorder characterized by inflammation of the brain (48). Complications arising from infection with Herpes simplex virus (HSV) type-1 are the foremost cause of HSE (48, 49). HSV infection causes severe neurological consequences and results in a high death rate, notwithstanding antiviral treatment improvements (50, 51). Acyclovir (ACV) is the standard antiviral drug for treating HSV infection (50, 51). In any case, early initiation of ACV therapy in HSV patients is crucial in forestalling the escalation of the disease. Early drug intervention is necessary, primarily because although ACV suppresses viral replication in the brain when administered in the late stage, brainstem inflammation may continue, culminating in fatal HSE (52–54).
Ramakrishna and colleagues (55) investigated the immunomodulatory role of PSA isolated from B. fragilis in HSE in a delayed ACV administration model in mice. Their findings showed that pretreatment with six doses of PSA via oral gavage prevented the development of HSE in HSV-infected mice following delayed HCV therapy. PSA stifled the invasion of CD45high leukocytes, CD11b+ monocytes/macrophages, and Ly6Chigh inflammatory monocytes into the brainstem (BS) of WT compared to the control animals (55) (Fig. 1). The authors further showed that PSA interacted with intestinal plasmacytoid dendritic (pDC) and B cell plasmablasts (PB) and induced the secretion of interleukin (IL)-10 and interferon gamma (IFN-γ) from T and B cells (Fig. 1). Both cytokines were elevated in the BS of the PSA-treated-HSV infected mice compared to the controls (55). However, PSA failed to protect mice lacking mature B and T cells and IL-10 or IFN-γ knockout mice against HSE. The authors concluded that the protective mechanism(s) of PSA against HSE might be associated with their ability to activate lymphocytes and induce the secretion of both IL-10 and IFN-γ (55).
FIG 1.
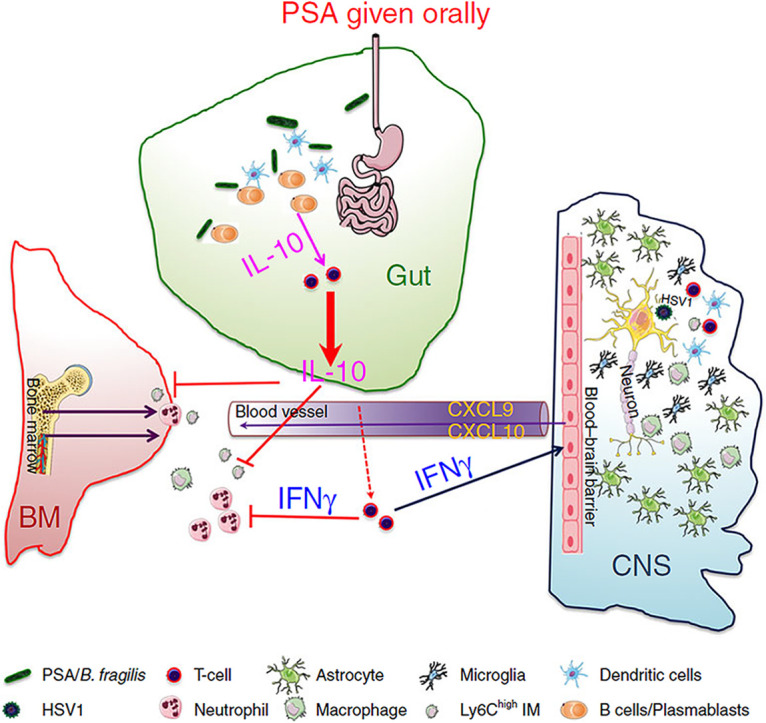
Mechanism of immune protection of PSA/B. fragilis against HSE. Adapted from Ramakrishna et al. 2019. Nature communications. http://creativecommons.org/licenses/by/4.0/. IMs and neutrophils are produced in excessive numbers in the bone marrow following infection of susceptible mice with HSV. Notwithstanding ACV intervention 4 days post HSV challenge, IMs and neutrophils continue to invade the BS culminating in fatal HSE. Binding of PSA/B. fragilis to B cell plasmablast/dendritic cells following oral gavage induced IL-10 and IFN-γ secretion from T lymphocytes together with IFN-γ inducible chemokines in the BS that dampens IMs and neutrophils infiltration resulting in prevention of fatal HSE. IM = inflammatory monocytes; HSV = Herpes simplex virus; PSA = Polysaccharide-A; BS = Brainstem; ACV = Acyclovir; HSE = Herpes simplex encephalitis; IFN-γ = Interferon gamma; IL = interleukin.
IFN-γ and IL-10 are essential pro-and anti-inflammatory cytokines, respectively. In a previous study, IFN-γ prevented fatal HSE through the regulation of innate immune responses against HSV-1 infection (56). IFN-γ mediated the suppression of granulocyte colony-stimulating factor (G-CSF) signaling, thereby increasing neutrophil apoptosis and reestablished T cell’s secretion of IL-10 in HSE-protected animals (56). G-CSF levels were elevated in IFN-γ knockout mice and led to an unrestrained proliferation of neutrophils and their infiltration into the BS (56). Indeed, immune protection in many inflammatory conditions requires a homeostatic inflammatory cytokine pool. Along these lines, one could argue that PSA’s protective effect against HSE lies in its ability to induce pro-and anti-inflammatory cytokine production. Such responses are also likely to have a significant impact on other pathogenic infections; for example, the model could be explored to investigate the effect of PSA gavage in Mtb-infected animals during treatment with anti-TB antibiotics.
THE DUALITY OF B. FRAGILIS POLYSACCHARIDE-A INFLAMMATORY PROPERTIES
It is known that the adaptive immune system can recognize extracellular protein antigens when taken up by antigen-presenting cells (APC), processed through major histocompatibility complex (MHC) class II endocytic pathway, and later presented to CD4+ T cells (57). This interaction between CD4+ T cell/MHC II is characterized by the release of Th1/Th2 cytokines. MHC II-mediated activation of CD4+ T cells has been viewed as stringently restricted to protein antigens. In contrast, stimulation of adaptive immune responses by carbohydrate antigens is thought to be independent of T cells (58). Earlier studies have shown that zwitterionic polysaccharides such as PSA produced by B. fragilis, are also recognized and engulfed by APC and adopt a similar endocytic pathway as proteins to activate CD4+ T cells (59). Processing the polysaccharides into smaller fragments occurs within endosomes and is facilitated by the release of nitric oxide through an increase in the oxidative burst (59). Further processing of PSA occurs following the fusion of lysosomes with the polysaccharide-containing endosomes (59). PSA’s processed fragments can then generate a specific pro- or anti-inflammatory immune response following CD4+ T cell/MHC II interaction (Fig. 2).
FIG 2.

B. fragilis/PSA activation of CD4+ T cell. PSA = Polysaccharide-A; MHC II = Major histocompatibility complex; CD = Cluster of differentiation; TCR = Toll-like receptor; IL = Interleukin; TNF-α = Tumor necrosis factor-alpha; CXCL = Chemokine ligand; IFN-γ = Interferon gamma; Th = T helper; Treg = Regulatory T cell.
Indeed, PSA from B. fragilis has been widely investigated for its cytokine-inducing properties and shown to orchestrate the inhibition of inflammatory processes to protect against conditions such as experimental autoimmune disease (EAE) (29, 30) inflammatory bowel disease (60), and asthma (34). Conversely, PSA from B. fragilis can also facilitate inflammatory action to induce, for example, an abscess (61, 62). In a recent study, Alvarez and colleagues characterized this duality of PSA responses (63). Here, PSA from B. fragilis upregulated the expression of many interferon-linked genes and induced the secretion of tumor necrosis factor-alpha (TNF-α), IL-6, IFN-γ, and chemokine ligand (CXCL)-10 (63) (Fig. 2). PSA treatment upregulated the expression of signal transducer and activator of transcription (STAT) 1 and 4, and T-bet (63). PSA has been shown previously to interact with DCs via toll-like receptor (TLR2) to induce IL-12 secretion. IL-12 mediates the activation of STAT 4, leading to CD4+ T cell differentiation into IFN-γ producing Th1 cells (16) (Fig. 2). PSA can also stimulate IL-4 and IL-10 from Th2 cells and Treg, respectively (15) (Fig. 2). Consistent with this pro- and anti-inflammatory paradigm of PSA, a previous study also demonstrated that antisera from PSA1 immunized mice showed a significant amount of IL-6, IL-2, IL17A, and IL-10 (64). Furthermore, engineering of PSA1 with a tumor-associated carbohydrate hapten induced predominantly IL-17A, which displayed anti-tumor effects (64). This data set showed that PSA could be engineered to generate specific and selective immune responses (64).
Alvarez and colleagues (63) further showed that T cell expression of the immune checkpoints markers, T cell immunoglobulin and mucin domain (Tim)-3, programmed cell death protein (PD)-1, and lymphocyte activation gene (Lag)-3 were upregulated by PSA from B. fragilis (63). The role of immune checkpoints has been recognized in cancers; however, they are also involved in immune regulation in infectious diseases (65). Immune checkpoints regulate host cell responses by various mechanisms. PD1 prevents early T cell activation upon encounter with PD-1 ligand in the periphery (66). Lag-3 stifles CD4+ T cell activation by binding to MHC II molecule with a higher affinity than CD4+ T cell (67). At the same time, the interaction of Tim-3 with the cell death marker, high mobility group box (HMGB)-1 protein prevents innate cell activation (68). Summarily, Alvarez and colleagues’ study showed that the immune response induced by PSA is principally both anti- and pro-inflammatory and mediated by interferon signaling (63). Their findings, therefore, suggest that the inflammatory pathway induced by PSA from B. fragilis may be dependent on the disease scenario.
DOES B. FRAGILIS OR POLYSACCHARIDE-A PLAY A ROLE DURING MYCOBACTERIUM TUBERCULOSIS INFECTION?
Host clearance of Mtb infection or immune control of the pathogen within granulomas requires a coordinated balance of innate and adaptive immune components of both pro-and anti-inflammatory immunity (69). While IFN-γ mediated inflammatory responses are crucial during Mtb infection, uncontrolled pro-inflammatory processes may promote bacterial proliferation in the lungs. On the other hand, a lung milieu dominated by anti-inflammatory cytokines may also favor bacterial survival. B. fragilis and PSA are widely reported to induce both pro-and anti-inflammatory processes and cytokines in normal and disease states, some of which are involved in the TB immune response, such as IL-10 and IFN-γ. Given this unique ability, one may speculate that B. fragilis or PSA may possibly play a role during the host response to Mtb infection. Besides, the reported effect of PSA on immune checkpoint expression in Alvarez and colleagues’ study strengthens the case for speculating that B. fragilis may contribute to the immune response against Mtb. Although controversial, studies have shown that PD-1 regulates pro-inflammatory responses during Mtb infection (70, 71). The survival rate of PD-1 deficient mice was much lower than their wild-type (WT) counterparts. These animals experienced a heightened pro-inflammatory lung environment leading to exaggerated inflammation with concomitant uninhibited Mtb proliferation (70). A similar study showed that control of Mtb was facilitated by PD-1 mediated inhibition of the lethal overproduction of IFN-γ by CD4+ T cells (71). Furthermore, in humans, patients receiving immunotherapy for cancers with immune checkpoint inhibitors were reported to develop active TB disease (72, 73). Conversely, other human studies have reported increased PD-1 levels on CD4+ T cells in active TB patients compared to healthy individuals (74) and decreased PD-1 expression on effector and responder T cells in patients receiving active TB treatment (75). Despite these findings, any suggestion that B fragilis or PSA may play a role in the control of Mtb remains a hypothesis that will require investigation in well-designed studies. If shown to be relevant in such studies, B. fragilis or PSA responses may be beneficial in the prevention of TB disease progression or assist during TB treatment.
EVIDENCE FOR THE PROTECTIVE ROLE OF THE GUT MICROBIOTA IN RESPIRATORY INFECTIONS AND LUNG INFLAMMATION
Fecal microbiota transplant ameliorates pneumonia infections.
Numerous studies have shown that alterations in intestinal microbiota composition impact the immune response to respiratory tract infections caused by bacteria and viruses, supporting the gut-lung immune cross talk hypothesis (76–78). For example, a study showed that antibiotics depletion of intestinal commensal bacteria in mice followed by intranasal infection with Streptococcus pneumoniae led to increased lung inflammation, bacterial dissemination, and higher mortality than control animals with intact microbiome (79). The levels of IL-10 and TNF-α were decreased in the microbiota-depleted mice, whereas an increase in IL-6, IL-1β, and CXCL1 was seen (79). fecal microbiome transplantation to the gut microbiome depleted mice reduced the number of bacteria in the lungs and restored IL-10 and TNF-α 6 h after infection to levels comparable to the antibiotics untreated control mice. Furthermore, the depletion of the intestinal microbiota significantly impacted the metabolism of alveolar macrophages (79). The lung resident macrophages from the microbiota-depleted animals demonstrated reduced responsiveness to lipopolysaccharide, or lipoteichoic acid, and was accompanied by diminished ability to phagocytose S. pneumoniae (79). A similar bacterial pneumonia mouse model showed that microbiota improved the ability of alveolar macrophages to kill Klebsiella and Streptococcus pneumoniae by activating the signaling of granulocyte-macrophage colony-stimulating factor (GM-CSF) (80). Microbiota regulated lung GM-CSF by increasing IL-17A levels (80). In the same study, using combined models of germfree and antibiotics treated mice colonized with commensal bacteria from four different phyla, the authors reported that nucleotide-binding oligomerization domain (Nod)-like receptor-stimulating bacteria enhanced immune defense against lung infection through GM-CSF and Nod2 (80). These bacteria include Lactobacillus crispatus and reuteri, Clostridium orbiscindens, Enterococcus faecalis, in the gut and Staphylococcus epidermidis and aureus, in the upper respiratory tract (80).
Lactobacillus and Bifidobacterium probiotics protect against respiratory infections caused by viruses.
The use of probiotics to promote host health has attracted much attention in recent years with health benefits such as improvement of inflammatory bowel disease, modulation of host immune response, enhancement of lactose metabolism, cholesterol reduction, and treatment of gastrointestinal infection attributed to the use of several probiotics (81). Bacteria within the genera, Enterococcus, Lactobacillus, Leuconostoc, Bifidobacterium, and Pediococcus, have been utilized as probiotics (82), with species within the genus Lactobacillus and Bifidobacterium being the most popular probiotics species (82, 83). Indeed, single probiotics within the Lactobacillus and Bifidobacterium genus have been investigated for their role in viral respiratory infections. For example, oral treatment of mice with Lactobacillus rhamnosus M21 induced both cell and antibody-mediated immune responses that increased host resistance against influenza virus infection (84). The mice that received L. rhamnosus probiotic developed less severe pneumonia and had an increased survival rate (84). The levels of IFN-γ and IL-2 in lung homogenates, and secretory immunoglobulin A (sIgA) in bronchoalveolar lavage fluid (BAL fluid), were elevated in the L. rhamnosus-treated animals (84). A similar study showed that Lactobacillus gasseri SBT2055, also protected against influenza virus infection. Again, the L. gasseri-treated mice had a significantly increased survival rate with reduced viral numbers in the lung (85). The expression of the antiviral genes 2′-5′ oligoadenylate synthetase 1a (Oas1a) and the Interferon-induced GTP-binding protein (Mx1) was increased, whereas IL-6 levels were reduced in the lung (85). Both Oas1a and Mx1 are induced by type I and type III interferons and are part of the host antiviral response (86, 87). Oas1a synthesizes 2′-5′-oligoadenylates (2-5A) from ATP, which binds and activates RNase L (RNase L), resulting in inhibition of viral protein synthesis and replication (88) whereas, Mx1 inhibits influenza virus replication by blocking the assembly of viral ribonucleoprotein complex (86).
In another study, L. gasseri SBT2055 was reported to protect against respiratory syncytial virus (RSV) infection (89). RSV, a common respiratory virus, is the main viral aetiologic agent of pneumonia and bronchiolitis in children (90, 91). This study by Eguchi and colleagues showed that pretreatment of mouse lung and human laryngeal epithelial cells with L. gasseri inhibited viral replication upon RSV infection (89). Furthermore, an in vivo experimental model showed that oral treatment of mice with L. gasseri significantly reduced viral lung titer in the RSV-infected mice (89). The levels of the proinflammatory cytokines and chemokines IL-1β, CCL2, TNF-α, CCL2, and IL-6 in the lung were dampened by L. gasseri treatment, whereas upregulation of IFN-β and IFN-γ genes in the lung were observed following L. gasseri administration (89). In the same vein, Bifidobacterium bifidum’s ability to enhance anti-influenza immunity was investigated by intranasally infecting mice with a lethal dose of influenza A virus (92). Like other reports, B. bifidum treatment induced both cellular and humoral immunity, reduced lung IL-6 secretion, and increased survival rate compared to the control animals (92). Another human study showed that older adults who were administered food containing Bifidobacterium longum BB536 before influenza vaccination had significantly fewer influenza infection cases than those who did not receive the probiotic (93). Notably, neutrophils’ bactericidal and NK cell activity were significantly higher at week five during B. longum administration (93).
B. fragilis capsular polysaccharide-A protects against lung inflammation.
PSA from B. fragilis has also been shown to play a role during lung inflammatory conditions. Oral treatment of mice with PSA was reported to inhibit the induction of asthma (34). PSA treatment significantly reduced the number of monocytes, neutrophils, and lymphocytes that infiltrated the lungs in treated mice compared to asthmatic controls (34). The increase in IFN-γ seen in the control animals was abolished in the PSA-treated mice. Notably, PSA from B. fragilis was able to block asthma development in both Th1 and Th2 skewed disease models of asthma and relied on the secretion of IL-10 from activated CD4+ T cells. Furthermore, the adoptive transfer of splenic CD4+ T cells from PSA-treated mice protected naive mice from developing asthma (34). A similar study also showed that protection of pulmonary inflammation was orchestrated by PSA-activated effector T cells, which induced the production of IL-10 by regulatory T cells (Tregs) (33). These studies are significant because they show that a single commensal bacterium could induce host protective immunity against respiratory infections and inflammation and could possibly be valuable as a probiotic.
Microbiota dysbiosis in tuberculosis and potential role of single commensal-derived probiotics in immune response/therapy.
Indeed, there have also been studies reporting on the potential contribution of the gut microbiota on host responses to Mtb infection. A study by Khan and colleagues showed that alterations in the composition of commensal bacteria in mice caused by treatment with broad-spectrum antibiotics increased the susceptibility of treated animals to Mtb infection (94). The antibiotics treated mice developed more severe disease with the dissemination of the bacteria to the spleen and liver. A decline in IFN-γ and TNF-α and an increase in regulatory T cells (Treg) was reported in the antibiotics-treated animals (94). Faecal microbiota transplant to the gut microbiome altered mice, restored host protective immunity and reduced lung bacterial loads (94). A similar study showed that gut microbiota dysbiosis induced by treatment of mice with broad-spectrum antibiotics reduced the number of mucosa-associated invariant T (MAIT) cells, which led to the increased early colonization of the lungs by Mtb (95). In a similar vein, treatment of mice with the narrow-spectrum anti-TB antibiotics, isoniazid/pyrazinamide, induced gut microbiota alterations, which compromised host immunity to Mtb challenge (96). There was an increase in the abundance of family Clostridiaceae in the isoniazid/pyrazinamide treated mice, with the most significant genus-level change occurring in the clostridia IV and XIV clusters. The Isoniazid/pyrazinamide instigated microbiota changes did not affect T cell-specific immune response. The number of neutrophils, alveolar and interstitial macrophages, and eosinophils were also unchanged in the lungs (96). However, the increased susceptibility of the isoniazid/pyrazinamide treated mice was linked to an impairment of alveolar macrophage metabolism, with a significant decrease in ATP generation, maximal, spare, and basal respiration, resulting in defective mycobactericidal activity (96).
Furthermore, gut microbiota dysbiosis induced by broad-spectrum antibiotics was shown to reduce the efficacy of isoniazid in killing Mtb in infected mice (97). Microbiota disruption impaired both CD4+ T cell and innate immune responses (97). However, whether whole fecal microbiota transplant from untreated mice reverses the effect of microbiota dysbiosis on the effectiveness of isoniazid in the Mtb-infected mice was not investigated in the study. A more recent report also demonstrated that gut microbiota dysbiosis favored Mtb proliferation in the lung by elevating the levels miR-21 which impaired IFN-γ secretion (98).
Although it is now well acknowledged that gut microbiota perturbations negatively impact Mtb resistance, the reported inconsistencies in commensal bacteria descriptions between studies complicate definitive conclusions. However, it is not unexpected that the resultant differential outcomes vary from study to study, observed with various choices of antibiotics, spectrum, combinations, and treatment duration. The gut microbiota is composed of a complex and dense bacterial population. Nevertheless, in as much as whole microbiota fecal transplant has been shown to restore host immune response in available TB models, it will also be fascinating to ascertain the contribution of specific microbiota phyla or commensal bacteria, especially considering the role of probiotics in lung infections and inflammation, and also considering that various beneficial commensal bacteria have been shown to induce different immune phenotypes which may perform unique functions during inflammatory diseases (99). However, no study has explored the direct effect of a single/cocktail of probiotics species in TB immunity or response to TB treatment drugs. Given the impact of alterations in the gut microbiome composition on TB and considering that dysbiosis induced by anti-TB antibiotics therapy have been hypothesized to possibly contribute to increased susceptibility to reinfection with Mtb after cure (100), probiotics or probiotics augmented therapy may contribute to an improved response to Mtb infection or TB drug treatment and, in reducing the cases of reinfection. These claims are however speculative and hypothesis-based, given available literature on the relationship between the gut microbiota and TB, and will require investigation in future hypothesis-based studies.
CONCLUSION AND POSSIBLE IMPLICATIONS FOR THE TB FIELD
Like a virus, Mtb is an intracellular parasite. The reviewed literature may well have established an essential model from which future studies could take a cue, especially in exploring B. fragilis or PSA’s possible role during Mtb infection. Whether B. fragilis or its capsular PSA (1) regulates constitutive types I and II IFN levels required to prime host responses to Mtb infection, (2) induces multiple inflammatory cytokines/upregulates immune checkpoint expression during Mtb infection that is possibly protective, requires urgent investigation. In addition, the available data on the relationship between B. fragilis or PSA isolated from B. fragilis, the immune system, and disease strengthen the potential for the development of B. fragilis into a next-generation probiotic or possibly even the engineering of PSA into a potent carbohydrate-based vaccine. However, further investigations are required to verify these claims. Furthermore, evidence for a microbiota-mediated gut-lung cross talk and the reported regulatory role of single commensal-derived probiotics (such as Lactobacillus and Bifidobacterium species) on the immune response during respiratory infections and inflammation provide an essential basis for exploring probiotics-based/augmented therapies in TB.
FUTURE DIRECTIONS
Future studies relevant to the TB field may include
-
1.
Investigating the effect of single commensal derived probiotics such as B. fragilis on the immune response and lung bacteria burdens in gut microbiota depleted Mtb-infected mice
-
2.
Characterization of the cytokine and transcriptomic profiles in Mtb-infected animals following purified polysaccharide-A (PSA) treatment
-
3.
Exploring whether B. fragilis, PSA from B. fragilis, or other single probiotics supplementation during anti-TB therapy will improve treatment response.
ACKNOWLEDGMENTS
Fig. 2 was graphically enhanced by Patrick Lane of ScEYEnce Studios.
O.A.E. and N.N.C. conceptualized the study; O.A.E. wrote the original draft; N.d.P. and N.N.C. wrote, reviewed, and edited the paper; all authors accepted the final version. All authors have read and agreed to the published version of the manuscript.
No funding was required to write this review. We declare no conflict of interest.
Contributor Information
Novel N. Chegou, Email: novel@sun.ac.za.
Anthony R. Richardson, University of Pittsburgh
REFERENCES
- 1.Takiishi T, Fenero CIM, Câmara NOS. 2017. Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers 5:e1373208. 10.1080/21688370.2017.1373208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belkaid Y, Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157:121–141. 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang G, Huang S, Wang Y, Cai S, Yu H, Liu H, Zeng X, Zhang G, Qiao S. 2019. Bridging intestinal immunity and gut microbiota by metabolites. Cell Mol Life Sci: 76:3917–3921. 10.1007/s00018-019-03190-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heiss CN, Olofsson LE. 2018. Gut microbiota-dependent modulation of energy metabolism. J Innate Immun 10:163–171. 10.1159/000481519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, Morgan XC, Kostic AD, Luo C, González A, McDonald D, Haberman Y, Walters T, Baker S, Rosh J, Stephens M, Heyman M, Markowitz J, Baldassano R, Griffiths A, Sylvester F, Mack D, Kim S, Crandall W, Hyams J, Huttenhower C, Knight R, Xavier RJ. 2014. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15:382–392. 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhan Y, Chen P-J, Sadler WD, Wang F, Poe S, Núñez G, Eaton KA, Chen GY. 2013. Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res 73:7199–7210. 10.1158/0008-5472.CAN-13-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobhani I, Bergsten E, Couffin S, Amiot A, Nebbad B, Barau C, de’Angelis N, Rabot S, Canoui-Poitrine F, Mestivier D, Pédron T, Khazaie K, Sansonetti PJ. 2019. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proc Natl Acad Sci USA 116:24285–24295. 10.1073/pnas.1912129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. 2008. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455:1109–1113. 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurung M, Li Z, You H, Rodrigues R, Jump DB, Morgun A, Shulzhenko N. 2020. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 51:102590. 10.1016/j.ebiom.2019.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, Mele MC. 2019. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 7:14. 10.3390/microorganisms7010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Doré J, Antolín M, Artiguenave F, Blottiere HM, Almeida M, Brechot C, Cara C, Chervaux C, Cultrone A, Delorme C, Denariaz G, Dervyn R, MetaHIT Consortium, et al. 2011. Enterotypes of the human gut microbiome. Nature 473:174–180. 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elsaghir H, Reddivari AKR. 2020. Bacteroides Fragilis, StatPearls [Internet]. StatPearls Publishing. [PubMed] [Google Scholar]
- 13.Pantosti A, Tzianabos A, Reinap B, Onderdonk A, Kasper D. 1993. Bacteroides fragilis strains express multiple capsular polysaccharides. J Clin Microbiol 31:1850–1855. 10.1128/jcm.31.7.1850-1855.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krinos CM, Coyne MJ, Weinacht KG, Tzianabos AO, Kasper DL, Comstock LE. 2001. Extensive surface diversity of a commensal microorganism by multiple DNA inversions. Nature 414:555–558. 10.1038/35107092. [DOI] [PubMed] [Google Scholar]
- 15.Troy EB, Kasper DL. 2010. Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Front Biosci (Landmark Ed) 15:25–34. 10.2741/3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, McLoughlin RM, Cobb BA, Charrel-Dennis M, Zaleski KJ, Golenbock D, Tzianabos AO, Kasper DL. 2006. A bacterial carbohydrate links innate and adaptive responses through Toll-like receptor 2. J Exp Med 203:2853–2863. 10.1084/jem.20062008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pantosti A, Tzianabos A, Onderdonk A, Kasper D. 1991. Immunochemical characterization of two surface polysaccharides of Bacteroides fragilis. Infect Immun 59:2075–2082. 10.1128/iai.59.6.2075-2082.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzianabos A, Pantosti A, Baumann H, Michon F, Brisson J, Jennings H, Kasper D. 1991. Structural characterization of two surface polysaccharides of Bacteroides fragilis. Trans Assoc Am Physicians 104:285–295. [PubMed] [Google Scholar]
- 19.Tzianabos A, Pantosti A, Baumann H, Brisson J, Jennings H, Kasper D. 1992. The capsular polysaccharide of Bacteroides fragilis comprises two ionically linked polysaccharides. J Biol Chem 267:18230–18235. 10.1016/S0021-9258(19)37177-7. [DOI] [PubMed] [Google Scholar]
- 20.Johnson JL, Jones MB, Cobb BA. 2015. Polysaccharide A from the capsule of Bacteroides fragilis induces clonal CD4+ T cell expansion. J Biol Chem 290:5007–5014. 10.1074/jbc.M114.621771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalka-Moll WM, Tzianabos AO, Bryant PW, Niemeyer M, Ploegh HL, Kasper DL. 2002. Zwitterionic polysaccharides stimulate T cells by MHC class II-dependent interactions. J Immunol 169:6149–6153. 10.4049/jimmunol.169.11.6149. [DOI] [PubMed] [Google Scholar]
- 22.Duan J, Kasper DL. 2011. Oxidative depolymerization of polysaccharides by reactive oxygen/nitrogen species. Glycobiology 21:401–409. 10.1093/glycob/cwq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan J, Avci FY, Kasper DL. 2008. Microbial carbohydrate depolymerization by antigen-presenting cells: deamination prior to presentation by the MHCII pathway. Proc Natl Acad Sci USA 105:5183–5188. 10.1073/pnas.0800974105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erturk‐Hasdemir D, Kasper DL. 2018. Finding a needle in a haystack: Bacteroides fragilis polysaccharide A as the archetypical symbiosis factor. Ann N Y Acad Sci 1417:116–129. 10.1111/nyas.13660. [DOI] [PubMed] [Google Scholar]
- 25.Young NM, Kreisman LS, Stupak J, MacLean LL, Cobb BA, Richards JC. 2011. Structural characterization and MHCII-dependent immunological properties of the zwitterionic O-chain antigen of Morganella morganii. Glycobiology 21:1266–1276. 10.1093/glycob/cwr018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Velez CD, Lewis CJ, Kasper DL, Cobb BA. 2009. Type I Streptococcus pneumoniae carbohydrate utilizes a nitric oxide and MHC II‐dependent pathway for antigen presentation. Immunology 127:73–82. 10.1111/j.1365-2567.2008.02924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DLJC. 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122:107–118. 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 28.Mazmanian SK, Round JL, Kasper DL. 2008. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453:620–625. 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 29.Ochoa-Reparaz J, Mielcarz D, Wang Y, Begum-Haque S, Dasgupta S, Kasper D, Kasper L. 2010. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol 3:487–495. 10.1038/mi.2010.29. [DOI] [PubMed] [Google Scholar]
- 30.Ochoa-Repáraz J, Mielcarz DW, Ditrio LE, Burroughs AR, Begum-Haque S, Dasgupta S, Kasper DL, Kasper LH. 2010. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J Immunol 185:4101–4108. 10.4049/jimmunol.1001443. [DOI] [PubMed] [Google Scholar]
- 31.Lee YK, Mehrabian P, Boyajian S, Wu W-L, Selicha J, Vonderfecht S, Mazmanian SK. 2018. The protective role of Bacteroides fragilis in a murine model of colitis-associated colorectal cancer. MSphere 3. 10.1128/mSphere.00587-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee Y-P, Chiu C-C, Lin T-J, Hung S-W, Huang W-C, Chiu C-F, Huang Y-T, Chen Y-H, Chen T-H, Chuang H-L. 2019. The germ-free mice monocolonization with Bacteroides fragilis improves azoxymethane/dextran sulfate sodium induced colitis-associated colorectal cancer. Immunopharmacol Immunotoxicol 41:207–213. 10.1080/08923973.2019.1569047. [DOI] [PubMed] [Google Scholar]
- 33.Johnson JL, Jones MB, Cobb BA. 2018. Polysaccharide-experienced effector T cells induce IL-10 in FoxP3+ regulatory T cells to prevent pulmonary inflammation. Glycobiology 28:50–58. 10.1093/glycob/cwx093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson JL, Jones MB, Cobb BA. 2015. Bacterial capsular polysaccharide prevents the onset of asthma through T-cell activation. Glycobiology 25:368–375. 10.1093/glycob/cwu117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wexler HM. 2007. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 20:593–621. 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaupp L, Muth S, Rogell L, Kofoed-Branzk M, Melchior F, Lienenklaus S, Ganal-Vonarburg SC, Klein M, Guendel F, Hain T, Schütze K, Grundmann U, Schmitt V, Dorsch M, Spanier J, Larsen P-K, Schwanz T, Jäckel S, Reinhardt C, Bopp T, Danckwardt S, Mahnke K, Heinz GA, Mashreghi M-F, Durek P, Kalinke U, Kretz O, Huber TB, Weiss S, Wilhelm C, Macpherson AJ, Schild H, Diefenbach A, Probst HC. 2020. Microbiota-Induced Type I Interferons Instruct a Poised Basal State of Dendritic Cells. Cell 181:1080–1096.e19. 10.1016/j.cell.2020.04.022. [DOI] [PubMed] [Google Scholar]
- 39.Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, Diefenbach A. 2012. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37:171–186. 10.1016/j.immuni.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 40.Stefan KL, Kim MV, Iwasaki A, Kasper DL. 2020. Commensal microbiota modulation of natural resistance to virus infection. Cell 183:1312–1324.e10. 10.1016/j.cell.2020.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, Wherry EJ, Artis D. 2012. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37:158–170. 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desvignes L, Wolf AJ, Ernst JD. 2012. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J Immunol 188:6205–6215. 10.4049/jimmunol.1200255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977. 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ottenhoff THM, Dass RH, Yang N, Zhang MM, Wong HEE, Sahiratmadja E, Khor CC, Alisjahbana B, van Crevel R, Marzuki S, Seielstad M, van de Vosse E, Hibberd ML. 2012. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS One 7:e45839. 10.1371/journal.pone.0045839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scriba TJ, Penn-Nicholson A, Shankar S, Hraha T, Thompson EG, Sterling D, Nemes E, Darboe F, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Johnson JL, Boom WH, Hatherill M, Valvo J, De Groote MA, Ochsner UA, Aderem A, Hanekom WA, Zak DE, other members of the ACS cohort study team. 2017. ACScst other members of the. Sequential inflammatory processes define human progression from M tuberculosis infection to tuberculosis disease. PLoS Pathog 13:e1006687. 10.1371/journal.ppat.1006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Hussey GD, Abrahams D, Kafaar F, Hawkridge T, Verver S, Hughes EJ, Ota M, Sutherland J, Howe R, Dockrell HM, Boom WH, Thiel B, Ottenhoff THM, Mayanja-Kizza H, Crampin AC, Downing K, Hatherill M, Valvo J, Shankar S, Parida SK, Kaufmann SHE, Walzl G, Aderem A, Hanekom WA. 2016. A prospective blood RNA signature for tuberculosis disease risk. Lancet (London, England) 387:2312–2322. 10.1016/S0140-6736(15)01316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gnann JW, Whitley RJ. 2017. Herpes simplex encephalitis: an update. Curr Infect Dis Rep 19:13. 10.1007/s11908-017-0568-7. [DOI] [PubMed] [Google Scholar]
- 49.Skelly MJ, Burger AA, Adekola O. 2012. Herpes simplex virus-1 encephalitis: a review of current disease management with three case reports. Antivir Chem Chemother 23:13–18. 10.3851/IMP2129. [DOI] [PubMed] [Google Scholar]
- 50.Gnann JW, Sköldenberg B, Hart J, Aurelius E, Schliamser S, Studahl M, Eriksson B-M, Hanley D, Aoki F, Jackson AC, Griffiths P, Miedzinski L, Hanfelt-Goade D, Hinthorn D, Ahlm C, Aksamit A, Cruz-Flores S, Dale I, Cloud G, Jester P, Whitley RJ, National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. 2015. Herpes simplex encephalitis: lack of clinical benefit of long-term valacyclovir therapy. Clin Infect Dis 61:683–691. 10.1093/cid/civ369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGrath N, Anderson N, Croxson M, Powell K. 1997. Herpes simplex encephalitis treated with acyclovir: diagnosis and long term outcome. J Neurol Neurosurg Psychiatry 63:321–326. 10.1136/jnnp.63.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lundberg P, Openshaw H, Wang M, Yang H-J, Cantin E. 2007. Effects of CXCR3 signaling on development of fatal encephalitis and corneal and periocular skin disease in HSV-infected mice are mouse-strain dependent. Invest Ophthalmol Vis Sci 48:4162–4170. 10.1167/iovs.07-0261. [DOI] [PubMed] [Google Scholar]
- 53.Lundberg P, Ramakrishna C, Brown J, Tyszka JM, Hamamura M, Hinton DR, Kovats S, Nalcioglu O, Weinberg K, Openshaw H, Cantin EM. 2008. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol 82:7078–7088. 10.1128/JVI.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramakrishna C, Openshaw H, Cantin EM. 2013. The case for immunomodulatory approaches in treating HSV encephalitis. Future Virol 8:259–272. 10.2217/fvl.12.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramakrishna C, Kujawski M, Chu H, Li L, Mazmanian SK, Cantin EM. 2019. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat Commun 10:1–13. 10.1038/s41467-019-09884-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramakrishna C, Cantin EM. 2018. IFNγ inhibits G-CSF induced neutrophil expansion and invasion of the CNS to prevent viral encephalitis. PLoS Pathog 14:e1006822. 10.1371/journal.ppat.1006822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watts C, Powis S. 1999. Pathways of antigen processing and presentation. Rev Immunogenet 1:60–74. [PubMed] [Google Scholar]
- 58.Sun L, Middleton DR, Wantuch PL, Ozdilek A, Avci FY. 2016. Carbohydrates as T-cell antigens with implications in health and disease. Glycobiology 26:1029–1040. 10.1093/glycob/cww062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cobb BA, Wang Q, Tzianabos AO, Kasper DL. 2004. Polysaccharide processing and presentation by the MHCII pathway. Cell 117:677–687. 10.1016/j.cell.2004.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazmanian SK, Round JL, Kasper DL. 2008. A microbial symbiosis factor prevents inflammatory disease. Nature 453:620–625. 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 61.Thadepalli H, Chuah SK, Qazi S, Thadepalli F, Gollapudi SV. 2001. Bacteroides fragilis-induced intra-abdominal abscess in an experimental model treated with telithromycin (HMR 3647). Chemotherapy 47:43–49. 10.1159/000048500. [DOI] [PubMed] [Google Scholar]
- 62.Gibson FC, Onderdonk AB, Kasper DL, Tzianabos AO. 1998. Cellular mechanism of intraabdominal abscess formation by Bacteroides fragilis. J Immunol 160:5000–5006. [PubMed] [Google Scholar]
- 63.Alvarez CA, Jones MB, Hambor J, Cobb BA. 2020. Characterization of Polysaccharide A Response Reveals Interferon Responsive Gene Signature and Immunomodulatory Marker Expression. Front Immunol 11:556813. 10.3389/fimmu.2020.556813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Silva RA, Appulage DK, Pietraszkiewicz H, Bobbitt KR, Media J, Shaw JJiu, Valeriote FA, Andreana PR. 2012. The entirely carbohydrate immunogen Tn-PS A1 induces a cancer cell selective immune response and cytokine IL-17. Cancer Immunol Immunother 61:581–585. 10.1007/s00262-012-1205-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dyck L, Mills KH. 2017. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol 47:765–779. 10.1002/eji.201646875. [DOI] [PubMed] [Google Scholar]
- 66.Topalian SL, Drake CG, Pardoll DM. 2012. Targeting the PD-1/B7-H1 (PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol 24:207–212. 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. 1995. CD4/major histocompatibility complex class II interaction analyzed with CD4‐and lymphocyte activation gene‐3 (LAG‐3)‐Ig fusion proteins. Eur J Immunol 25:2718–2721. 10.1002/eji.1830250949. [DOI] [PubMed] [Google Scholar]
- 68.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M. 2012. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 13:832–842. 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walzl G, Ronacher K, Hanekom W, Scriba TJ, Zumla A. 2011. Immunological biomarkers of tuberculosis. Nat Rev Immunol 11:343–354. 10.1038/nri2960. [DOI] [PubMed] [Google Scholar]
- 70.Lázár-Molnár E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, Porcelli SA, Almo SC, Nathenson SG, Jacobs WR. 2010. Programmed death-1 (PD-1)–deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci USA 107:13402–13407. 10.1073/pnas.1007394107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, Barber DL. 2016. CD4 T cell-derived IFN-γ plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog 12:e1005667. 10.1371/journal.ppat.1005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujita K, Yamamoto Y, Kanai O, Okamura M, Hashimoto M, Nakatani K, Sawai S, Mio T. Incidence of active tuberculosis in lung cancer patients receiving immune checkpoint inhibitors, p ofaa126. In (ed), Oxford University Press US; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zaemes J, Kim C. 2020. Immune checkpoint inhibitor use and tuberculosis: a systematic review of the literature. Eur J Cancer 132:168–175. 10.1016/j.ejca.2020.03.015. [DOI] [PubMed] [Google Scholar]
- 74.Shen L, Gao Y, Liu Y, Zhang B, Liu Q, Wu J, Fan L, Ou Q, Zhang W, Shao L. 2016. PD-1/PD-L pathway inhibits M. tb-specific CD4+ T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci Rep 6:38362–38369. 10.1038/srep38362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen L, Shi H, Gao Y, Ou Q, Liu Q, Liu Y, Wu J, Zhang W, Fan L, Shao L. 2016. The characteristic profiles of PD-1 and PD-L1 expressions and dynamic changes during treatment in active tuberculosis. Tuberculosis (Edinb) 101:146–150. 10.1016/j.tube.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Samuelson DR, Welsh DA, Shellito JE. 2015. Regulation of lung immunity and host defense by the intestinal microbiota. Front Microbiol 6:1085. 10.3389/fmicb.2015.01085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sencio V, Machado MG, Trottein F. 2021. The lung–gut axis during viral respiratory infections: the impact of gut dysbiosis on secondary disease outcomes. Mucosal Immunol: 14:296–299. 10.1038/s41385-020-00361-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. 2011. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci USA 108:5354–5359. 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJTH, de Boer JD, Hoogendijk AJ, de Beer R, de Vos A, Belzer C, de Vos WM, van der Poll T, Wiersinga WJ. 2016. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 65:575–583. 10.1136/gutjnl-2015-309728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brown RL, Sequeira RP, Clarke TB. 2017. The microbiota protects against respiratory infection via GM-CSF signaling. Nat Commun 8:1–11. 10.1038/s41467-017-01803-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aureli P, Capurso L, Castellazzi AM, Clerici M, Giovannini M, Morelli L, Poli A, Pregliasco F, Salvini F, Zuccotti GV. 2011. Probiotics and health: an evidence-based review. Pharmacol Res 63:366–376. 10.1016/j.phrs.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 82.Fijan S. 2014. Microorganisms with claimed probiotic properties: an overview of recent literature. Int J Environ Res Public Health 11:4745–4767. 10.3390/ijerph110504745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Song D, Ibrahim S, Hayek S. 2012. Recent application of probiotics in food and agricultural science. Probiotics 10:1–34. [Google Scholar]
- 84.Song JA, Kim HJ, Hong SK, Lee DH, Lee SW, Song CS, Kim KT, Choi IS, Lee JB, Park SY. 2016. Oral intake of Lactobacillus rhamnosus M21 enhances the survival rate of mice lethally infected with influenza virus. J Microbiol Immunol Infect 49:16–23. 10.1016/j.jmii.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 85.Nakayama Y, Moriya T, Sakai F, Ikeda N, Shiozaki T, Hosoya T, Nakagawa H, Miyazaki T. 2014. Oral administration of Lactobacillus gasseri SBT2055 is effective for preventing influenza in mice. Sci Rep 4:4638–4635. 10.1038/srep04638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Verhelst J, Parthoens E, Schepens B, Fiers W, Saelens X. 2012. Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J Virol 86:13445–13455. 10.1128/JVI.01682-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pulit-Penaloza JA, Scherbik SV, Brinton MA. 2012. Activation of Oas1a gene expression by type I IFN requires both STAT1 and STAT2 while only STAT2 is required for Oas1b activation. Virology 425:71–81. 10.1016/j.virol.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ibsen MS, Gad HH, Thavachelvam K, Boesen T, Desprès P, Hartmann R. 2014. The 2′-5′-oligoadenylate synthetase 3 enzyme potently synthesizes the 2′-5′-oligoadenylates required for RNase L activation. J Virol 88:14222–14231. 10.1128/JVI.01763-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eguchi K, Fujitani N, Nakagawa H, Miyazaki T. 2019. Prevention of respiratory syncytial virus infection with probiotic lactic acid bacterium Lactobacillus gasseri SBT2055. Sci Rep 9:1–11. 10.1038/s41598-019-39602-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pickles RJ, DeVincenzo JP. 2015. Respiratory syncytial virus (RSV) and its propensity for causing bronchiolitis. J Pathol 235:266–276. 10.1002/path.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jha DA, Jarvis H, Fraser C, Openshaw PJ. 2016. Respiratory syncytial virus. [PubMed]
- 92.Mahooti M, Abdolalipour E, Salehzadeh A, Mohebbi SR, Gorji A, Ghaemi A. 2019. Immunomodulatory and prophylactic effects of Bifidobacterium bifidum probiotic strain on influenza infection in mice. World J Microbiol Biotechnol 35:1–9. 10.1007/s11274-019-2667-0. [DOI] [PubMed] [Google Scholar]
- 93.Namba K, Hatano M, Yaeshima T, Takase M, Suzuki K. 2010. Effects of Bifidobacterium longum BB536 administration on influenza infection, influenza vaccine antibody titer, and cell-mediated immunity in the elderly. Biosci Biotechnol Biochem 74:939–945. 10.1271/bbb.90749. [DOI] [PubMed] [Google Scholar]
- 94.Khan N, Vidyarthi A, Nadeem S, Negi S, Nair G, Agrewala JN. 2016. Alteration in the gut microbiota provokes susceptibility to tuberculosis. Front Immunol 7:529. 10.3389/fimmu.2016.00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dumas A, Corral D, Colom A, Levillain F, Peixoto A, Hudrisier D, Poquet Y, Neyrolles O. 2018. The host microbiota contributes to early protection against lung colonization by Mycobacterium tuberculosis. Front Immunol 9:2656. 10.3389/fimmu.2018.02656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Khan N, Mendonca L, Dhariwal A, Fontes G, Menzies D, Xia J, Divangahi M, King IL. 2019. Intestinal dysbiosis compromises alveolar macrophage immunity to Mycobacterium tuberculosis. Mucosal Immunol 12:772–783. 10.1038/s41385-019-0147-3. [DOI] [PubMed] [Google Scholar]
- 97.Negi S, Pahari S, Bashir H, Agrewala JN. 2020. Intestinal microbiota disruption limits the isoniazid mediated clearance of Mycobacterium tuberculosis in mice. Eur J Immunol 50:1976–1987. 10.1002/eji.202048556. [DOI] [PubMed] [Google Scholar]
- 98.Yang F, Yang Y, Chen Y, Li G, Zhang G, Chen L, Zhang Z, Mai Q, Zeng G. 2020. MiR-21 Is Remotely Governed by the Commensal Bacteria and Impairs Anti-TB Immunity by Down-Regulating IFN-γ. Front Microbiol 11:512581. 10.3389/fmicb.2020.512581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zheng D, Liwinski T, Elinav E. 2020. Interaction between microbiota and immunity in health and disease. Cell Res 30:492–506. 10.1038/s41422-020-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eribo OA, Du Plessis N, Ozturk M, Guler R, Walzl G, Chegou NN. 2020. The gut microbiome in tuberculosis susceptibility and treatment response: guilty or not guilty? Cell Mol Life Sci 77:1497–1509. 10.1007/s00018-019-03370-4. [DOI] [PMC free article] [PubMed] [Google Scholar]