

Abstract
Introduction:
Positron emission tomography (PET) using radiolabeled amyloid-binding compounds has advanced the field of Alzheimer’s disease (AD) by enabling detection and longitudinal tracking of fibrillar amyloid-β (Aβ) deposits in living people. However, this technique cannot distinguish between Aβ deposits in brain parenchyma (amyloid plaques) from those in blood vessels (cerebral amyloid angiopathy, CAA). Development of a PET radioligand capable of selectively detecting CAA would help clarify its contribution to global brain amyloidosis and clinical symptoms in AD and would help to characterize side-effects of anti-Aβ immunotherapies in AD patients, such as CAA.
Methods:
A candidate CAA-selective compound (1) from a panel of analogues of the amyloid-binding dye Congo red was synthesized. The binding affinity to Aβ fibrils and lipophilicity of compound 1 were determined and selectivity for CAA versus parenchymal plaque deposits was assessed ex-vivo and in-vivo in transgenic APP/PS1 mice and in postmortem human brain affected with AD pathology.
Results:
Compound 1 displays characteristics of Aβ binding dyes, such as thioflavin-S, in that it labels both parenchymal Aβ plaques and CAA when applied to histological sections from both a transgenic APP/PS1 mouse model of Aβ amyloidosis and AD brain. Thus, compound 1 lacks molecular selectivity to distinguish Aβ deposits in CAA from those in plaques. However, when administered to living APP/PS1 mice intravenously, compound 1 preferentially labels CAA when assessed using in-vivo two-photon microscopy and ex-vivo histology and autoradiography.
Conclusion:
We hypothesize that selectivity of compound 1 for CAA is attributable to its limited penetration of the blood-brain barrier due to the highly polar nature of the carboxylate moiety, thereby limiting access to parenchymal plaques and promoting selective in-vivo labeling of Aβ deposits in the vascular wall (i.e., “delivery selectivity”).
Keywords: Alzheimer’s disease, Amyloid, Amyloid-beta, Cerebrovascular, Immunotherapy, Positron emission tomography
1. Introduction
The neuropathologic hallmarks of Alzheimer’s disease (AD) are parenchymal deposits (plaques) of aggregated amyloid-β (Aβ) peptides and intracellular aggregates of phosphorylated tau (p-tau) protein (neurofibrillary tangles) [1]. Common to both Aβ and p-tau aggregates is a β-pleated sheet secondary structure (amyloid) that renders them insoluble in physiological fluids. In postmortem brain tissue sections, amyloid is detected histologically using amyloid-binding dyes such as Congo red (CR [2]) and its derivatives [3,4], as well as thioflavin-S [5]. One or more of these histology techniques in combination with Aβ and p-tau immunohistochemistry are used routinely in neuropathology studies. While p-tau aggregates are a common feature of many degenerative conditions collectively referred to as tauopathies, which include AD, frontotemporal dementia (FTD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Pick disease [6,7], Aβ plaques are more specific for AD and are considered a sign of subclinical AD when present in cognitively normal aged individuals [8].
Aβ deposition in the brain is not limited to parenchymal plaques. When present in the basement membrane and smooth muscle cell layers of leptomeningeal and parenchymal arteries and also in capillary basement membranes [9], it is referred to as cerebral amyloid angiopathy (CAA) [10,11]. Post-mortem histopathology studies report that the incidence of CAA increases with age, with some degree of CAA in up to 36% of brains from cognitively normal individuals older than 60 years [11]. The incidence and severity of CAA is even greater in AD [11]: autopsy studies report up to 83% of AD cases having at least a mild degree of CAA and about 25% cases with moderate to severe CAA in single or multiple cortical areas [12]. CAA is considered a pathological lesion associated with higher frequencies of hemorrhagic or ischemic lesions detected as white matter hyperintensities (WMH) using magnetic resonance imaging (MRI) (see [13,14] for reviews). CAA is also linked to reduced brain network efficiency [15] and cognitive impairment [16–18], indicating it is a clinically significant lesion. The presence of CAA may be related to the occurrence of amyloid-related imaging abnormalities (ARIA) characterized by edema or microhemorrhages frequently reported in trials of anti-Aβ immunotherapies, including recent reports of aducanumab trials which indicated a positive and disease-modifying outcome [19]. This is of particular concern as postmortem examination of an immunotherapy-treated AD subject revealed a strikingly higher extent of CAA compared to non-treated AD patients [20]. Whether CAA was a preexisting and more advanced pathology in this patient or if it developed or progressed following immunotherapy could not be determined. Development of a non-invasive and quantitative method for assessing CAA selectively in the brains of living people would help answer such questions and is thus an urgent, unmet need. Such task is challenging due to the presence of mixed Aβ pathology (CAA and plaques co-distributed in the same brain volume) in both non-demented elderly subjects and those with AD [10]. Unlike postmortem histopathological microscopy analyses which can easily distinguish CAA from Aβ plaques, current in-vivo imaging techniques are indirect as they lack the resolution required to distinguish these two lesions and/or lack specificity and sensitivity. For example, posterior predominance of WMH detected by MRI was reported as an independent predictor of autopsy-confirmed CAA [21], but the interpretation of this finding is further complicated in people with cerebrovascular disease (i.e., non-amyloid-related hemorrhage). Aβ-binding positron emission tomography (PET) radioligands such as [11C]Pittsburgh compound-B (PiB) and 18F-labeled radioligands (florbetapir/Amyvid, florbetaben/Neuraceq, and flutemetamol/Vizamyl) are capable of detecting subclinical Aβ deposits in the brains of living subjects (see [22] for review). [11C]PiB PET imaging of people with clinically suspected CAA based on the Boston criteria [23] reported that the occipital-to-global [11C]PiB retention ratio was greater in subjects with CAA compared to AD [24]. However, the contribution of CAA versus parenchymal plaques to the combined signal throughout the brain is not presently discernable with any Aβ radioligand, due to the lack of differential pharmacologic properties and also the limited spatial resolution (~5 mm full width at half maximum) of PET. Thus, the field is in need of an imaging biomarker to detect CAA selectively and monitor its progression in living people.
Recent studies reported CAA-selectivity of several compounds such as analogues of resorufin [25], llama-derived heavy chain antibody fragments [26], nanoparticle-IgG4.1 complexes [27], or [99mTc] hydroxamamide complexes with bivalent amyloid ligands [28]. However, these techniques have many limitations, such as poor solubility in physiological solutions (resorufin), low brain uptake (antibodies and nanovehicles), and poor in-vivo performance ([99mTc] hydroxamamide) and therefore require further refinement and validation. Despite the reported preponderance of Aβ1–40 over Aβ1–42 in CAA [29–31], both Aβ isoforms are detected in CAA and plaques [32–34] and therefore it is unlikely that these amyloid pathologies can be readily distinguished based on the “molecular selectivity” of candidate radioligands. We hypothesized that development of a CAA-selective imaging agent for PET would be more readily achieved using a “delivery selectivity” approach whereby a compound’s unique properties would favor its delivery to CAA with minimal or no access to Aβ plaques. To test this hypothesis, we designed a workflow of complementary in-vivo two-photon imaging and ex-vivo histopathology analyses of compounds tailored to have limited blood-brain barrier permeability. The current report presents the results obtained using our lead compound 1 (Fig. 1) in comparison to a non-selective Aβ amyloid binding compound methoxy-X04 (compound 2) that has relatively good brain entry and has been used for two-photon studies in transgenic mice [35,36].
Fig. 1.
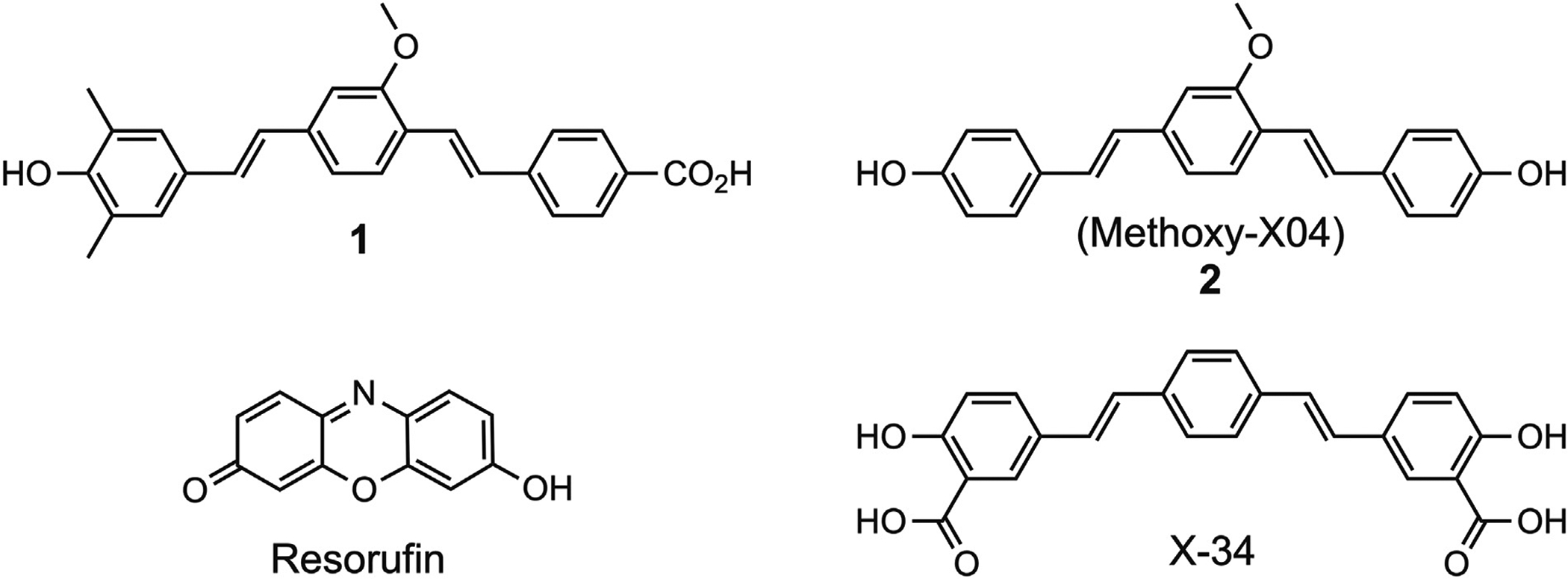
Chemical structures of 1, 2 (methoxy-X04), X-34, and resorufin.
2. Methods
2.1. Animals
Twenty female transgenic APP/PS1 mice (B6C3-Tg(APPswe, PSEN1dE9)85Dbo/Mmjax mice [37]) were purchased from Jackson Laboratory (MMRRC Stock #34829-JAX) and housed in standard cages in a 12:12 light:dark cycle with free access to food and water at an Association for Assessment and Accreditation of Laboratory Animal Care (AALAC) accredited animal facility at the University of Pittsburgh. Mice were on average 459 ± 59 (range 360–520) days old at the time of experimentation and have severe plaque and CAA pathology within this age range [38]. All studies conducted using transgenic mice or their tissues were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC).
2.2. Human brain tissue specimen
Tissue sections (thickness = 40 μm) were obtained from a 4% paraformaldehyde (4% PFA, made in 0.1 M sodium phosphate buffer, pH 7.4)-fixed tissue block of occipital cortex from a case neuropathologically diagnosed with severe AD who was enrolled in the University of Pittsburgh’s Alzheimer’s Disease Research Center (male, age = 84 years; Aβ plaque score = A3, neurofibrillary tangle stage = B3, neuritic plaque score = C3; apolipoprotein E genotype = 3/3; mini-mental score examination = 7), in compliance with the University of Pittsburgh’s Committee for Oversight of Research and Clinical Training Involving Decedents.
2.3. Chemistry general
All chemicals were used as supplied without further purification, unless noted. Characterization data were obtained at the Department of Chemistry, University of Pittsburgh. NMR spectra were obtained at room temperature on either Bruker Avance III 300 MHz or Avance III 400 MHz spectrometers, with chemical shifts reported in parts per million (ppm) relative to residual solvent signals. Mass spectra were obtained on a Micromass Ultimata Q-TOF API. Silica gel (high purity grade 9385, pore size 60 Å, 230–400 mesh particle size) was used for column chromatography purifications. Tetrahydrofuran was dried over sodium benzophenone ketyl. Radiolabeling to give [3H]1 was performed by Novandi Chemistry AB (www.novandi.se). Compound synthesis is shown in Scheme 1. Compounds 2 [36], 3 [39], 4 [40], and 5 [41] were synthesized as previously described.
Scheme 1.
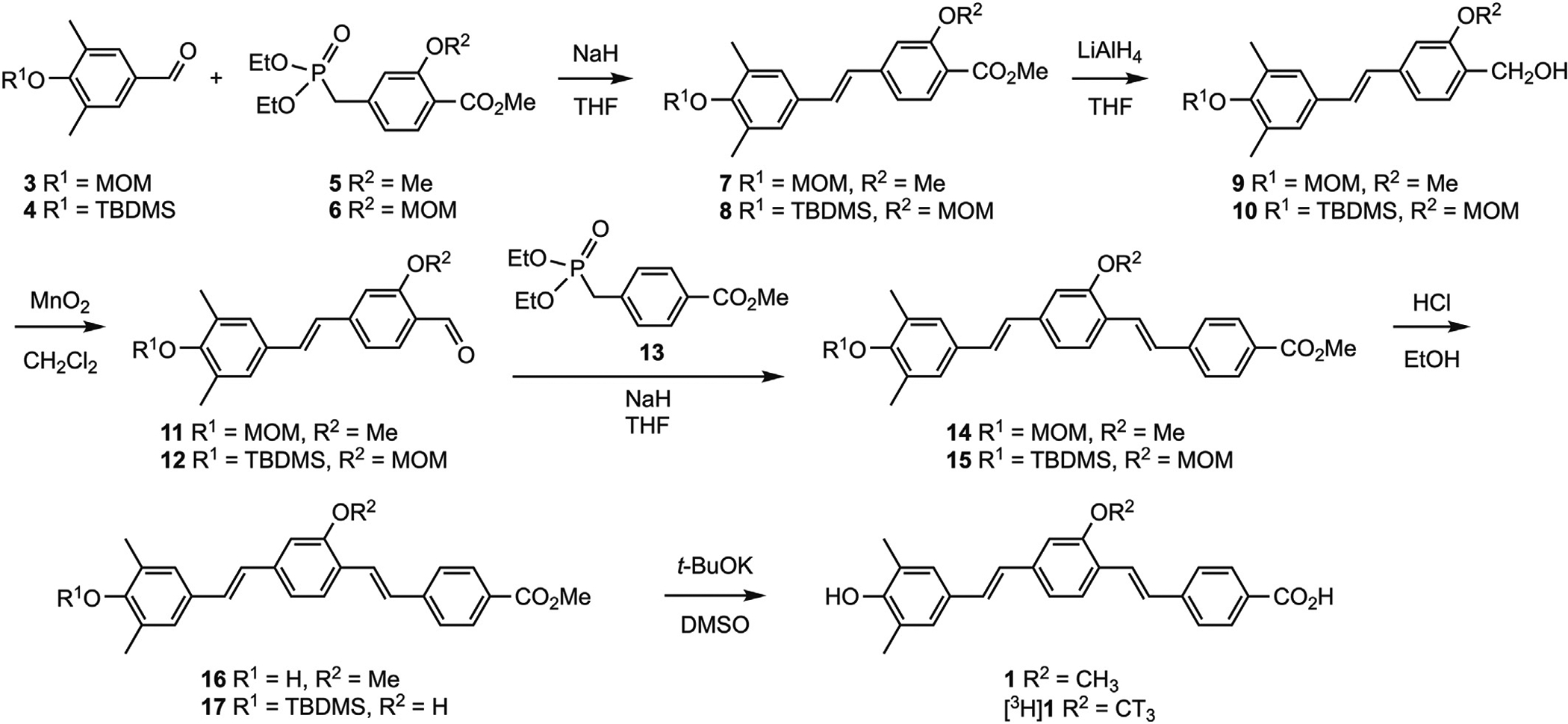
Compound synthesis.
2.4. Methyl 4-((diethoxyphosphoryl)methyl)-2-methoxymethoxybenzoate (6)
Methyl 4-bromomethyl-2-methoxymethoxybenzoate [42] (11.8 g, 40.8 mmol) was dissolved in triethylphosphite (20 ml), and the reaction mixture was heated at 100 °C for 18 h. The reaction was cooled to room temperature and the material was purified via kugelrohr distillation to afford 6 (11.4 g, 81%) as a colorless oil: 1H NMR (400 MHz, CDCl3) 1.25 (t, J = 7.2 Hz, 6H, CH3), 3.15 (d, 2JPH = 22.0 Hz, 2H, CH2), 3.50 (s, 3H, CH3), 3.87 (s, 3H, CH3), 4.01 (m, 4H, CH2), 5.24 (s, 2H, OCH2O) 6.97 (d, J = 7.6 Hz, 1H, ArH), 7.13 (s, 1H, ArH), 7.73 (d, J = 7.6 Hz, 1H, ArH).
2.5. Methyl (E)-2-methoxy-4-(4-(methoxymethoxy)-3,5-dimethylstyryl) benzoate (7)
Compound 5 [41] (1.95 g, 6.17 mmol) was dissolved in THF (50 ml) and cooled in an ice-water bath. NaH (95%, 178 mg, 7.05 mmol) was added and the mixture was stirred for 30 min. Compound 3 (1.0 g, 5.15 mmol), dissolved in THF (10 ml), was added dropwise, and the reaction mixture was allowed to stir at room temperature for 18 h. The solvent was removed at reduced pressure, the residue was dissolved in H2O, and the pH was adjusted to 7 with 5% HCl(aq). The solution was extracted with EtOAc (3 × 20 ml), the organic extracts were combined, and dried over MgSO4. The EtOAc was removed and the residue was purified by flash column chromatography (4:1 hexane/EtOAc) to afford 7 (0.83 g, 45%) as an off-white solid: 1H NMR (300 MHz, CDCl3) 2.21 (s, 6H, CH3), 3.50 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 5.25 (s, 2H, OCH2O), 6.88 (m, 2H, ArH), 7.12 (m, 3H, ArH), 7.23 (m, 2H, ArH).
2.6. Methyl (E)-4-(4-((tert-butyldimethylsilyl)oxy)-3,5-dimethylstyryl)-2-(methoxymethoxy)benzoate (8)
Compound 6 (4.2 g, 12.1 mmol) was dissolved in THF (50 ml), cooled in an ice-water bath, and NaH (95%, 330 mg, 13.1 mmol) was added. The mixture was stirred for 30 min, then compound 4 (2.7 g, 10.2 mmol), dissolved in THF (10 ml), was added dropwise. The reaction mixture was stirred at room temperature for 18 h, the solvent was removed, the residue was dissolved in water, and the pH was adjusted to 7 with 5% HCl(aq). The solution was extracted with EtOAc (3 × 20 ml), the organic extracts were combined and dried over MgSO4, and the EtOAc was removed. The residue was purified by flash column chromatography (5:1 hexane/EtOAc) to afford 8 (2.06 g, 44%) as an off-white solid: 1H NMR (300 MHz, CDCl3) 0.12 (s, 6H, CH3), 0.97 (s, 9H, CH3), 2.31 (s, 6H, CH3), 3.40 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 5.33 (s, 2H, OCH2O), 6.84 (d, J = 12.0 Hz, 1H, CH), 6.94 (d, J = 12.0 Hz, 1H, CH), 7.12 (m, 3H, ArH), 7.23 (m, 2H, ArH).
2.7. (E)-(2-methoxy-4-(4-(methoxymethoxy)-3,5-dimethylstyryl)phenyl) methanol (9)
Compound 7 (0.82 g, 2.30 mmol) was dissolved in THF (20 ml) and cooled in an ice-water bath. LiAlH4 (150 mg, 3.95 mmol) was added, the reaction mixture was allowed to warm to room temperature, and stirred overnight. The reaction mixture was quenched with Na2SO4·10 H2O, and filtrated through a pad of Celite. The filtrate was evaporated to dryness and the residue was purified by flash column chromatography (1:1 hexanes/EtOAc) to afford 9 (0.32 g, 42%) as an off-white solid: 1H NMR (300 MHz, CDCl3) 2.21 (s, 6H, CH3), 3.50 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 4.68 (s, 2H, CH2O), 5.26 (s, 2H, OCH2O), 6.91 (d, J = 12.3 Hz, 1H, CH), 6.97 (d, J = 12.3 Hz, 1H, CH), 7.11 (m, 3H, ArH), 7.23 (m, 2H, ArH).
2.8. (E)-(4-(4-((tert-butyldimethylsilyl)oxy)-3,5-dimethylstyryl)-2-(methoxymethoxy)phenyl)methanol (10)
Compound 8 (2.1 g, 4.6 mmol) was dissolved in THF (40 ml) and cooled in an ice-water bath. LiAlH4 (210 mg, 5.5 mmol) was added, the reaction mixture was allowed to warm to room temperature, and stirred overnight. The reaction mixture was quenched with Na2SO4·10 H2O, and filtrated through a pad of Celite. The filtrate was evaporated to dryness and the residue was purified by flash column chromatography (2:1 hexanes/EtOAc) to afford 10 (1.4 g, 71%) as an off-white solid: 1H NMR (300 MHz, CDCl3) 0.15 (s, 6H, CH3), 0.99 (s, 9H, CH3), 2.18 (s, 6H, CH3), 3.47 (s, 3H, OCH3), 4.64 (s, 2H, CH2O), 5.23 (s, 2H, OCH2O), 6.87 (d, J = 12.3 Hz, 1H, CH), 6.94 (d, J = 12.3 Hz, 1H, CH), 7.11 (m, 3H, ArH), 7.23 (m, 2H, ArH).
2.9. (E)-2-methoxy-4-(4-(methoxymethoxy)-3,5-dimethylstyryl)benzaldehyde (11)
Compound 9 (320 mg, 0.97 mmol) was dissolved in anhydrous CH2Cl2 (20 ml), MnO2 (3.0 g, 34.5 mmol) was added, and the reaction mixture was stirred at room temperature overnight (TLC analysis (3:1 hexane/EtOAc) showed no remaining starting material). The reaction mixture was filtered, the solvent was removed from the filtrate, and the residue was purified by flash column chromatography (4:1 hexane/EtOAc) to afford 11 (182 mg, 60%) as a yellow solid: 1H NMR (300 MHz, CDCl3) 2.22 (s, 6H, CH3), 3.54 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 5.33 (s, 2H, OCH2O), 7.08 (m, 6H, ArH), 7.80 (d, J = 8.4 Hz, 1H, ArH), 10.42 (s, 1H, CHO).
2.10. (E)-4-(4-((tert-butyldimethylsilyl)oxy)-3,5-dimethylstyryl)-2-(methoxymethoxy)benzaldehyde (12)
Compound 10 (1.4 g, 3.3 mmol) was dissolved in anhydrous CH2Cl2 (40 ml), MnO2 (7.0 g, 80.5 mmol) was added, and the reaction mixture was stirred at room temperature overnight (TLC analysis (4:1 hexane/EtOAc) showed no remaining starting material). The reaction mixture was filtered, the solvent was removed from the filtrate, and the residue was purified by flash column chromatography (5:1 hexane/EtOAc) to afford 12 (1.2 g, 86%) as a yellow solid: 1H NMR (400 MHz, CDCl3) 0.19 (s, 6H, CH3), 1.01 (s, 9H, CH3), 2.22 (s, 6H, CH3), 3.54 (s, 3H, OCH3), 5.33 (s, 2H, OCH2O), 6.93 (d, J = 16.0 Hz, 1H, CH), 7.10 (d, J = 16.0 Hz, 1H, CH), 7.30 (m, 4H, ArH), 7.79 (d, J = 8.4 Hz, 1H, ArH), 10.45 (s, 1H, CHO).
2.11. Methyl 4-((diethoxyphosphoryl)methyl)benzoate (13) [43]
Methyl 4-chloromethylbenzoate (2.0 g, 10.8 mmol) was dissolved in triethylphosphite (4 ml), and the reaction mixture was heated at 100 °C for 12 h. The reaction was cooled to room temperature and the material was purified via kugelrohr distillation to afford 13 (3.1 g, quantitative) as a colorless oil: 1H NMR (400 MHz, CDCl3) 1.22 (t, J = 7.2 Hz, 6H, CH3), 3.18 (d, 2JPH = 22.4 Hz, 2H, CH2), 3.89 (s, 3H, CH3), 3.99 (m, 4H, CH2), 7.35 (dd, J = 8.0 Hz, 4JPH = 2.0 Hz, 2H, ArH), 7.96 (d, J = 8.0 Hz, 2H, ArH).
2.12. Methyl 4-((E)-2-methoxy-4-((E)-4-(methoxymethoxy)-3,5-dimethylstyryl)styryl)benzoate (14)
Compound 13 (404 mg, 1.41 mmol) was dissolved in THF (10 ml), cooled in an ice-water bath, and NaH (95%, 38 mg, 1.50 mmol) was added. The reaction mixture was stirred for 30 min, then compound 11 (182 mg, 0.58 mmol) in THF (3 ml) was added dropwise, and the reaction mixture was stirred at room temperature for 18 h. The solvent was removed, the residue was dissolved in water, neutralized with 5% HCl(aq), and extracted with EtOAc (3 × 10 ml). The organic extracts were combined, dried over MgSO4, the solvent was removed, and the residue was purified by preparative-TLC (4:1 hexane/EtOAc) to afford 14 (112 mg, 42%) as a yellow solid: 1H NMR (400 MHz, CDCl3) 2.33 (s, 6H, CH3), 3.63 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 4.98 (s, 2H, OCH2O), 6.99 (d, J = 16.4 Hz, 1H, CH), 7.01 (m, 1H, ArH), 7.05 (d, J = 16.4 Hz, 1H, CH), 7.12 (d, J = 8.0 Hz, 1H, ArH), 7.15 (d, J = 16.4 Hz, 1H, CH), 7.21 (s, 2H, ArH), 7.58 (d, J = 8.0 Hz, 2H, ArH), 7.58 (1H – obscured), 7.59 (d, J = 16.4 Hz, 1H, CH), 8.01 (d, J = 8.0 Hz, 2H, ArH).
2.13. Methyl 4-((E)-4-((E)-4-((tert-butyldimethylsilyl)oxy)-3,5-dimethylstyryl)-2-(methoxymethoxy)styryl)benzoate (15)
Compound 13 (970 mg, 3.39 mmol) was dissolved in THF (30 ml), cooled in an ice-water bath, NaH (95%, 100 mg, 3.96 mmol) was added, and the reaction mixture was stirred for 30 min. Compound 12 (1.2 g, 2.81 mmol), dissolved in THF (5 ml), was added dropwise, and the reaction mixture was allowed to stir at room temperature for 18 h. The solvent was removed, the residue was dissolved in water, neutralized with 5% HCl(aq), and extracted with EtOAc (3 × 10 ml). The organic extracts were combined, dried over MgSO4, the solvent was removed, and the residue was purified by flash column chromatography (5:1 hexane/EtOAc) to afford 15 (1.1 g, 70%) as a yellow solid: 1H NMR (300 MHz, CDCl3) 0.21 (s, 6H, CH3), 1.04 (s, 9H, CH3), 2.24 (s, 6H, CH3), 3.56 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 5.32 (s, 2H, OCH2O), 6.93 (d, J = 16.2 Hz, 1H, CH), 7.01 (d, J = 16.2 Hz, 1H, CH), 7.14 (d, J = 16.2 Hz, 1H, CH), 7.16 (s, 2H, ArH), 7.18 (d, J = 8.1 Hz, 1H, ArH), 7.26 (1 H – obscured by CHCl3 resonance), 7.59 (d, J = 8.4 Hz, 2H, ArH), 7.60 (d, J = 8.1 Hz, 1H, ArH), 7.60 (d, J = 16.2 Hz, 1 H, CH), 8.02 (d, J = 8.4 Hz, 2H, ArH).
2.14. Methyl 4-((E)-4-((E)-4-hydroxy-3,5-dimethylstyryl)-2-methoxystyryl)benzoate (16)
Compound 14 (112 mg, 0.24 mmol) was dissolved in EtOH (2 ml), concentrated HCl(aq) (2 drops) was added, and the reaction mixture was heated at 90 °C for 5 min (TLC analysis (3:1 hexanes/EtOAc) showed no remaining starting material). The solvent was removed at reduced pressure and the residue was purified by flash column chromatography (4:1 hexanes/EtOAc) to afford 16 (96 mg, 95%) as a yellow solid: 1H NMR (400 MHz, acetone-d6) 2.26 (s, 6H, CH3), 3.89 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 7.06 (d, J = 16.4 Hz, 1H, CH), 7.21 (m, 5H, ArH + CH), 7.33 (d, J = 16.4 Hz, 1H, CH), 7.47 (br s, 1H, OH), 7.66 (d, J = 16.4 Hz, 1H, CH), 7.70 (m, 3H, ArH), 8.00 (d, J = 8.0 Hz, 2H, ArH).
2.15. Methyl 4-((E)-4-((E)-4-((tert-butyldimethylsilyl)oxy)-3,5-dimethylstyryl)-2-hydroxystyryl)benzoate (17)
Compound 15 (200 mg, 0.36 mmol) was dissolved in THF (1.1 ml), concentrated HCl(aq) (110 μl) was added, and the reaction mixture was stirred at room temperature for 9 h. H2O was added, the mixture was extracted with EtOAc (3 × 10 ml), the organic extracts were combined, and dried over MgSO4. The EtOAc was removed and the residue was purified by flash column chromatography (4:1 hexane/EtOAc) to afford 17 (104 mg, 56%) as a yellow solid: 1H NMR (400 MHz, CDCl3) 0.21 (s, 6H, CH3), 1.04 (s, 9H, CH3), 2.24 (s, 6H, CH3), 3.92 (s, 3H, OCH3), 5.08 (s, 1H, OH), 6.88 (d, J = 16.4 Hz, 1H, CH), 6.91 (unresolved d, 1H, ArH), 6.99 (d, J = 16.4 Hz, 1H, CH), 7.09 (d, J = 8.0 Hz, 1H, ArH), 7.14 (s, 2H, ArH), 7.17 (d, J = 16.4 Hz, 1H, CH), 7.50 (d, J = 16.4 Hz, 1H, CH), 7.53 (d, J = 8.0 Hz, 1H, ArH), 7.58 (d, J = 8.0 Hz, 2H, ArH), 8.02 (d, J = 8.0 Hz, 1H, ArH).
2.16. 4-((E)-4-((E)-4-hydroxy-3,5-dimethylstyryl)-2-methoxystyryl) benzoic acid (1)
Compound 16 (96 mg, 0.23 mmol) was dissolved in anhydrous DMSO (2 ml), t-BuOK (301 mg, 2.68 mmol) was added, and the reaction mixture was stirred at room temperature for 10 min. H2O was added, the solution was neutralized with 5 N HCl(aq), and extracted with EtOAc (6 × 5 ml). The combined organic extracts were washed with brine, H2O, and dried over MgSO4. The solvent was removed and the residue was purified flash column chromatography (1:1 hexanes/EtOAc) to afford 1 (84 mg, 91%) as a yellow solid: 1H NMR (400 MHz, DMSO-d6) 2.19 (s, 6H, CH3), 3.93 (s, 3H, OCH3), 7.03 (d, J = 16.4 Hz, 1H, CH), 7.20 (m, 5H, ArH), 7.32 (d, J = 16.4 Hz, 1H, CH), 7.55 (d, J = 16.4 Hz, 1H, CH), 7.67 (m, 3H, ArH) 7.88 (d, J = 8.4 Hz, 1H, ArH), 7.93 (d, J = 8.4 Hz, 1H, ArH) 8.44 (s, 1H, OH), 12.87 (br s, 1H, CO2H); HRMS (ESI) [M+H]+ calcd for C26H25O4: 401.1747; found: 401.1734.
2.17. Resorufin
A working solution of resorufin (Sigma, #424455) was prepared by diluting 1 mM stock solution to 1 μM in potassium phosphate buffer (KPB, 0.1 M, pH 7.4) and adding 1 M NaOH. Tissue sections were obtained from a 12-month old naïve APP/PS1 mouse and stained with resorufin following the same procedure used for overstaining (see Section 2.23.3 “Image acquisition” below). The same staining procedure was used on tissue sections from an AD case. Resorufin histofluorescence was imaged, slides/sections were de-coverslipped and overstained with compound 2, and subsequently re-imaged.
2.18. In vitro binding assay
The binding affinities (Ki) of compounds 1 and 2 were determined to aggregated fibrils of synthetic Aβ1–40 against [3H]chrysamine-G using published methods [36].
2.18.1. Preparation of Aβ fibrils
Aβ1–40 (Bachem Bioscience, Inc.) was dissolved in phosphate buffered saline (PBS) to a final concentration of 0.433 mg/ml (100 μM). This solution was stirred magnetically at 760 rpm for 3 days at room temperature and centrifuged at 28,000 ×g for 15 min. The supernatant was diluted 1:20 with 150 mM Tris-HCl, pH 7.0 (to 5 μM), and stored at −80 °C before it was diluted 1:50 with binding buffer (150 mM Tris-HCl, pH 7.0, with 20% ethanol) to 100 nM. Aggregated Aβ stock suspensions were stirred to maintain a homogenous suspension during removal of aliquots for the binding assays.
2.18.2. Test compound preparation
The concentration of the non-radioactive test compound was determined by quantitative NMR [44] where the sample was dissolved in a solution of 5 mM dimethyl sulfone in DMSO-d6 and the 1H NMR spectrum was obtained. The integration of the 6-proton resonance of dimethyl sulfone was set to 30 mM (5 mM × 6 protons), and then comparison to the 1H resonance integrals of the test compound enabled a determination of the concentration of the test compound. Test compound DMSO-d6 solutions were diluted first to 400 μM with DMSO, and then to 2 μM with binding buffer (final DMSO concentration 0.5% v/v). Appropriate dilutions of test compound were made with binding buffer containing 0.5% DMSO. 500 μl of each dilution of the test compound solution was diluted to a final volume of 1.0 ml with binding buffer making the final DMSO concentration 0.25% for all concentrations tested.
2.18.3. Fibril binding assays
500 μl of appropriate concentrations of non-radioactive test compound were combined with 450 μl of [3H]chrysamine-G in a volume of 950 μl of binding buffer. The assay was initiated by adding 50 μl of Aβ1–40 fibril stock suspension to a final concentration of 5 nM fibril (based on the monomer). The final concentration [3H]chrysamine-G was typically 0.1–0.3 nM. After incubation at room temperature for 60 min, the binding mixture was filtered through a Whatman GF/B glass filter and rapidly washed twice with 3 ml binding buffer. The filters were counted in Cytoscint-ES after sitting in the cocktail overnight. Complete (100%) inhibition of binding was defined as the number of counts displaced by 10 μM non-radioactive chrysamine-G. All assays were done in triplicate.
2.19. LogD determination
The determination of logD (logPoct at pH 7.4) was performed three times by a standard shake-flask method [45] using 25 ml of 1-octanol solution (~500 μM) extracted with an equal volume of 0.1 M PBS (pH 7.4). Quantification of the compound in each layer was performed in triplicate by HPLC using fluorescence emission to enhance the detection sensitivity of the compounds in the aqueous phase. A representative aliquot of the 1-octanol phase was diluted 10-fold with 1-octanol prior to HPLC separation and compound quantitation. To make each layer equivalent before HPLC, 10 μl of the 1-octanol layer was first mixed with 10 μl PBS and 80 μl mixture of acetonitrile and PBS (1:1) and 10 μl of the PBS layer was mixed with 10 μl 1-octanol and 80 μl mixture of acetonitrile and PBS (1:1).
2.20. CLogP, pKa, and tPSA estimates
The calculated logP (CLogP) and topographical polar surface area (tPSA) of compounds 1 and 2 were estimated using Chemdraw (Professional version 18) software. The log of the equilibrium acid dissociation constants (pKa) of the compounds were estimated using Marvin software (https://chemaxon.com/products/marvin/download).
2.21. Fluorescence studies
Compounds were dissolved in DMSO at 400 μM and then diluted to 3 μM final concentration. Measurements were performed on a Perkin-Elmer Model LS55 fluorometer (Perkin Elmer, Shelton, CT) using 10 nm slit widths for both excitation and emission measurements.
2.22. In-vivo two-photon microscopy
The tail of an APP/PS1 mouse was catheterized using a tail vein catheter (MTV-1, Braintree Scientific, Inc.) under isoflurane anesthesia (1.0–1.5%). Mice were then transitioned to ketamine-xylazine anesthesia (75 mg/kg and 10 mg/kg, respectively) and placed in a stereotaxic frame for surgery. A craniotomy was performed over the right parietal bone and re-sealed with a 5-mm thick glass coverslip (Warner Instruments Inc.) and dental cement to gain optical access to the brain. Two-photon imaging was then performed using an Ultima IV system, 16× water immersion lens (Nikon, Inc.) and Prairie View software (Bruker Nano Inc.). The microscope is coupled to an ultrafast laser (Insight X3, Newport Spectra-Physics Inc.) tuned to 740 nm for all two-photon imaging experiments. The emission of the fluorescent probes was filtered over 420–480 nm and captured by one of the microscope’s detectors (photo-multiplier tubes). The imaging region covered an 800 μm × 800 μm × 400 μm volume of interest. This area was imaged prior to compound administration and for 45 min post-administration. Compounds were injected into the catheter by a pump infuser at a dose of 1 mg/kg. In most experiments, compound 2 was injected afterwards and imaged for 45 min post-administration for comparison. Summary images consisted of maximum intensity projection (MIP). All animals were perfused transcardially after the imaging session concluded as described for ex-vivo experiments below. Due to its poor solubility in physiological fluids, resorufin studies were performed by infusing 1 μM directly onto the dura through a slit in the cranial window and imaged as described above.
2.23. Ex-vivo histofluorescence analyses
2.23.1. Compound preparation and injection
Compounds 1 and 2 were prepared as 6.7 mg/ml stock solutions in DMSO and diluted to a final working concentration of 0.2 mg/ml by combining 30 μl stock solution with 30 μl Cremaphor (Millipore Sigma # 238470) and 940 μl sodium phosphate buffer (NaPB, 0.1 M, pH 7.4) 5 min before injection. Prior to tail vein catheter insertion and compound injection, mice were anesthetized with 4% isoflurane in 2:1 O2/N2O and maintained on 2% isoflurane via a nose cone. Compounds (1 mg/kg body weight; injected volume = ~0.15 ml) were injected over a period of 15 s through the catheter into the tail vein and allowed to circulate for 15 min while the mice were under anesthesia.
2.23.2. Brain extraction and processing
After compound circulation, mice were perfused transcardially with 50 ml ice cold NaPB, brains were removed intact, and extracted brains were immersed in ice cold 4% PFA for 48 h at 4 °C for tissue preservation and histology. Following fixation, brains were sequentially immersed overnight in 15% and 30% sucrose made in NaPB at 4 °C. Brains were then sectioned (section thickness = 40 μm) exhaustively in the coronal plane using a freezing sliding microtome (American Optical Corp., model 860, Buffalo, NY). Sections equally spaced through the rostrocaudal extent of the forebrain were rinsed in NaPB, dipped in dH2O, mounted onto charged, silanized slides (VWR International #16004-406), coverslipped with Fluoromount-G (Thermo # 00-4958-02) and air-dried overnight in light-proof boxes.
2.23.3. Image acquisition
Sections were examined ex-vivo for fluorescence of injected/bound compound (i.e., no further processing/staining) using an Olympus BX3 microscope with fluorescent attachment (X-cite 120Q) and equipped with an Olympus DP72 digital camera connected to a Dell Precision T5500 computer running Olympus cellSens Standard 1.6 imaging software. Compound fluorescence was detected using a hydroxycoumarin filter (excites 405 nm, T425lpxr beamsplitter, 460 nm emission; Chroma, Bellows Falls, VT) and images were obtained using a 10× microscope objective (infinity-corrected, numerical aperture = 0.4; Olympus). A standard image set including fields containing pial arteries and parenchymal arterioles and capillaries was collected from coronal sections at approximately 2–2.5 mm caudal to Bregma [46]. Imaged sections/slides were then immersed in tap water to remove the coverslip and rinsed in ice cold NaPB for 5 min to remove any remaining Fluoromount G. Decoverslipped sections were then incubated in ice cold 0.1 M potassium phosphate buffer (KPB, pH 7.4) twice for 2 min each, followed by a 45-minute incubation in compound (diluted to 1 μM in KPB) at 4 °C (referred to as “Compound overstain”). Stained sections were rinsed twice (5 s each) and then differentiated for 15 min in KPB. Sections were then coverslipped with Fluoromount G and images were obtained from the same microscopic fields imaged ex-vivo. Sections/slides were then decoverslipped again, processed using X-34 following standard protocols (referred to as “X-34 overstain”; [4]), coverslipped with Fluoromount G, and images were obtained from the same microscopic fields imaged ex-vivo and following compound overstain. Fluorescence imaging and image acquisition parameters were held constant throughout the experiment. Consistency in these parameters across imaging session was confirmed by imaging a slide containing fluorescence microspheres (SPHERO Rainbow, Spherotech, Inc., Lake Forest, IL).
2.23.4. Image analysis
Fluorescence intensity of retained compounds was assessed ex-vivo in 1) parenchymal CAA (arterioles and capillaries), 2) pial CAA (large leptomeningeal arteries along the cerebral surface and penetrating cortical arteries), and 3) parenchymal plaques in images obtained from brain tissue sections that had no histological stain (“unstained”), sections with compound overstain, and sections with X-34 overstain, using a semi-quantitative rating scale: 0 = no signal detected above background; 1 = weak fluorescence; 2 = moderate fluorescence; 3 = bright fluorescence.
2.24. Ex-vivo autoradiography
Mice were anesthetized and a catheter was inserted into the tail vein as in the ex-vivo experiment. [3H]1 (5.402 × 106 Bq in ~0.15 ml NaPB) was injected through the catheter into the tail vein and allowed to circulate for 15 min while the mice were under anesthesia. Mice were then perfused transcardially with 50 ml ice cold NaPB, brains were removed intact, and extracted brains were frozen intact on dry ice and stored at −80 °C. Frozen sections (thickness = 10 μm) were obtained from approximately 2–2.5 mm caudal to Bregma using a cryostat (Reichert-Jung 2800 Frigocut E) and mounted to charged, silanized slides and stored at −80 °C. To detect radioactivity, sections were air-dried and placed in an exposure cassette (#29-1755-23, GE Healthcare Bio-Sciences AB, Uppsala, Sweden) in close apposition to a storage phosphor screen (BAS-IP TR 2025 tritium screen, Fujifilm Corp, Tokyo, Japan) for sixty days. The phosphor screen was scanned using a Typhoon FLA7000 imager (GE Healthcare) equipped with a 390 nm band-pass IP filter and images were converted to .tiff format for analysis. Sections were then over-stained using X-34 and fluorescence labeling of Aβ deposits in CAA and plaques was compared to autoradiograms.
3. Results
3.1. Resorufin
First, replication of the results of Han and co-workers [25] was attempted by examining resorufin fluorescence in CAA and plaques in brains of transgenic APP/PS1 mice. When applied directly to tissue sections using the same concentration and conditions reported by [25], resorufin labeled pial and parenchymal CAA but not parenchymal plaques. In contrast, X-34 labeled both CAA and plaques in the same microscopic field (Fig. 2A, B). For in-vivo 2-photon analysis, we were unable to inject resorufin intravenously due to the poor solubility of the compound in physiological solutions. Thus, we applied resorufin (1 μM) topically to the dura of an APP/PS1 mouse by infusing the dye through the edge of a cranial window. This experiment showed that resorufin labeled CAA in pial and parenchymal vessels (pink arrows) as well as parenchymal amyloid plaques (green arrows), in a pattern indistinguishable from compound 2 (Fig. 2C, D). In tissue sections from occipital cortex (BA17/18) of an AD autopsy case, resorufin labeled plaques (Fig. 2E) and CAA (Fig. 2G) in a pattern indistinguishable from X-34 (Fig. 2F, H).
Fig. 2.
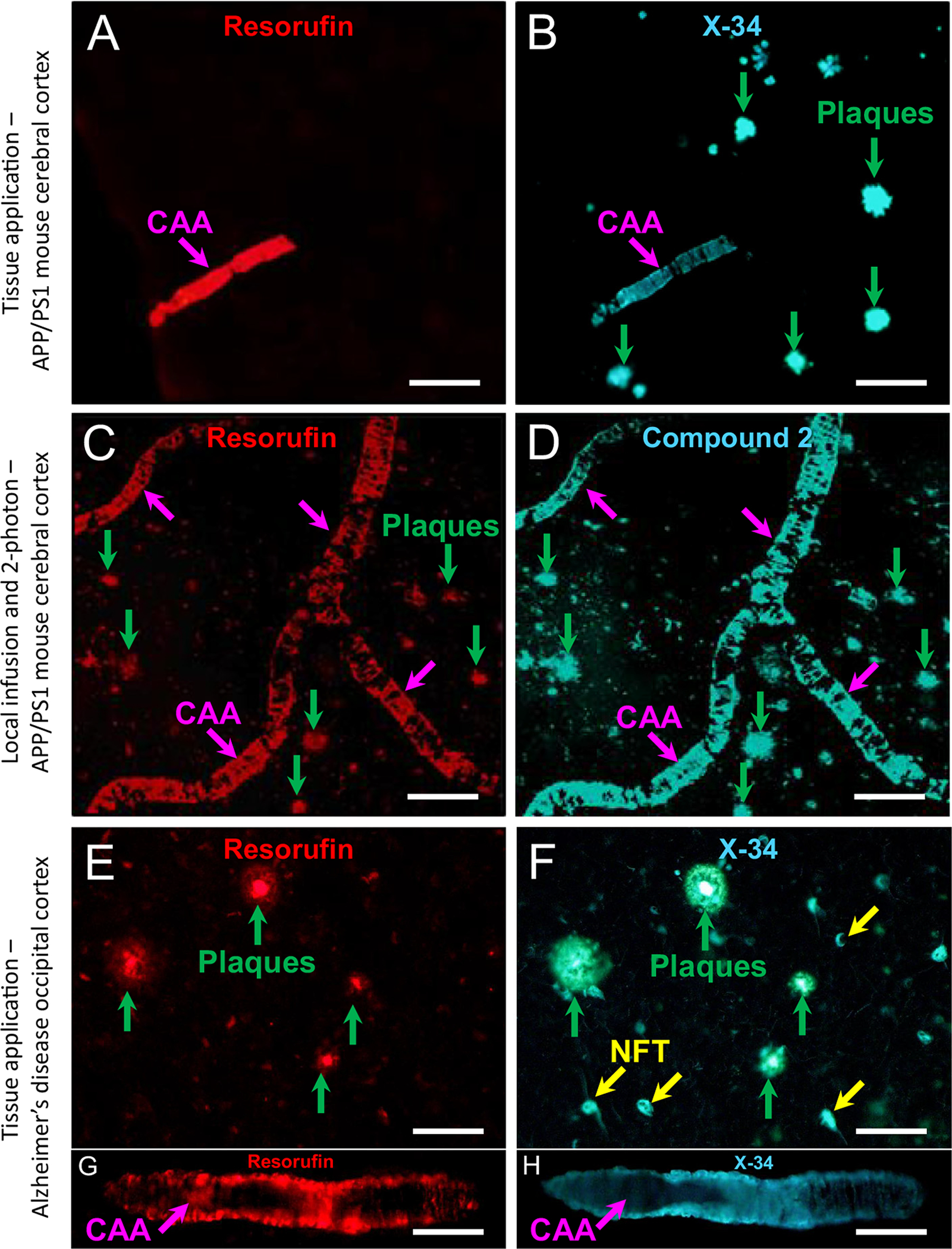
A, B. Fluorescence of 7-hydroxy-3H-phenoxazin-3-one (resorufin) to 1,4-bis(3-carboxy-4-hydroxyphenylethenyl)-benzene (X-34) or compound 2 applied directly to tissue sections from a 12-month old APP/PS1 mouse (A–D) and an AD case (E–H). Resorufin fluorescence is seen in CAA (A, pink arrow) but not plaques (green arrows in B); labeling of both CAA and plaques with X-34 overstaining of the same tissue section is shown in B. C, D. Two-photon microscopy images of resorufin after infusion into the brain through a cranial window of a 12-month old APP/PS1 mouse. Resorufin labeled both CAA (C, pink arrows) and plaques (C, green arrows). Subsequent infusion of compound 2 resulted in overstaining of the same CAA and plaques labeled with resorufin (D). E–H. In tissue sections of occipital cortex from an AD case, resorufin fluorescence is seen in both plaques (E, green arrows) and CAA (G, pink arrow). Overstaining of the same tissue section with X-34 resulted in comparable fluorescent signal in the same structures (F, H). X-34 also labeled neurofibrillary tangles (NFT, yellow arrows, F). Scale bar = 75 μm (A–F); 40 μm (G, H).
3.2. Chemical synthesis
Compound 3 [39] (Scheme 1) was reacted with 5 [41] under Horner-Wadsworth-Emmons conditions to give the (E)-alkene 7. Likewise, compound 4 [40] was reacted with 6 to give the (E)-alkene 8. Methyl esters 7 and 8 were reduced to give benzylic alcohols 9 and 10, respectively, followed by oxidation to give aldehydes 11 and 12, respectively. A second Horner-Wadsworth-Emmons reaction with 13 [43] converted 11 to 14, and 12 to 15. Acid-catalyzed removal of the MOM protecting groups of 14 and 15 then afforded compounds 16 and 17, respectively. Hydrolysis of 16 with t-BuOK in DMSO [47] then afforded 1. Compound 17 was tritiomethylated followed by deprotection to give [3H]1. Compound [3H]1, when received, was 92% radiochemically pure with molar activity 3.2 TBq/mmol (85 Ci/mmol).
3.3. Characteristics of compounds 1 and 2
Compounds 1 and 2 had high binding affinity for Aβ1–40 fibrils, with Ki’s of 8.6 nM for compound 1 and 23.6 nM for compound 2 (Table 1). The experimental logD of compound 1 was 2.40 ± 0.07 and 3.66 ± 0.25 for compound 2 (Table 1). The CLogP of the carboxylate form of compound 1 (−COO−, Table 1) was about 1 log unit higher than the experimental logD value, while the CLogP of the neutral carboxylic acid form of compound 1 (−COOH, Table 1) was about 5 log units higher than the logD value. The estimated pKa of the carboxylic acid is 4.12 (Table 1), indicating that compound 1 is highly ionized at pH 7.4 while compound 2 is not significantly ionized (pKa 8.69). The CLogP of compound 2 was about 2.5 log units higher than the experimental logD value. The calculated topographical polar surface area (tPSA) of the carboxylate form of compound 1 was 69.59, while the tPSA of compound 2 was considerably lower at 49.69. UV emission spectra for both compounds were within the desired range for effective in-vivo 2-photon microscopy studies (Table 2).
Table 1.
Aβ binding affinities, logD, ClogP, pKa, and tPSA values of compounds 1 and 2.
| Compound | Aβ binding affinity (Ki; nM) | LogD ± SD | CLogP | pKa | tPSA |
|---|---|---|---|---|---|
| 1 | 8.6 | 2.40 ± 0.07 | 7.42 (−COOH) 3.42 (−COO−) |
4.12 (−COOH) | 66.76 (−COOH) 69.59 (−COO−) |
| 2 | 23.6 | 3.66 ± 0.25 | 6.11 | 8.69 (−OH) | 49.69 |
Abbreviations: CLogP, calculated logPoct; Ki, binding affinity to synthetic Aβ1–40 fibrils; logD, logPoct at pH 7.4; pKa, calculated logKa (log of the equilibrium acid dissociation constant); SD, standard deviation; tPSA, calculated topographical polar surface area.
Table 2.
Fluorescence properties of compounds 1 and 2.
| Compound | Excitation/emission maxima (DMSO; nm) | Maximum fluorescence (3 μM in DMSO) | |
|---|---|---|---|
| Excitation | Emission | ||
| 1 | 390 | 506 | 257.7 |
| 2 | 387 | 452 | 841.3 |
Abbreviations: DMSO, dimethyl sulfoxide.
3.4. Ex-vivo fluorescence
3.4.1. Compound 2
Intravenous injection followed by 15 minute circulation of compound 2 resulted in equivalently bright fluorescent labeling of CAA in pial and parenchymal vessels and in parenchymal plaques in ex-vivo sections (Fig. 3A). Compound 2 overstaining resulted in similar fluorescence in CAA and plaques (Fig. 3B). X-34 overstaining resulted in a pattern of labeling similar to that observed following compound 2 overstain (Fig. 3C).
Fig. 3.
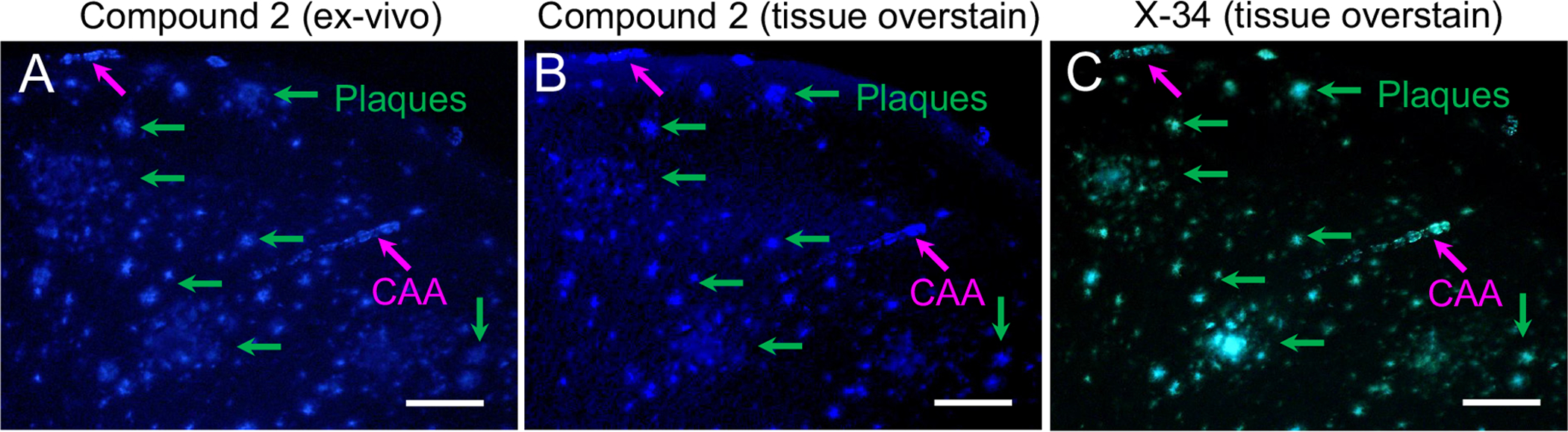
Fluorescence microscopy analyses in a tissue section from a 14-month old APP/PS1 mouse killed 15 min after injection of compound 2 (1 mg/kg in ~0.15 ml) into the tail vein. Ex-vivo fluorescence of compound 2 (A) is seen in both CAA (pink arrows) and plaques (green arrows), similar to the fluorescence signal seen in the same sections processed sequentially with compound 2 (B, after photobleaching and overstain) and X-34 (C, after photobleaching and overstain). Scale bar = 75 μm.
3.4.2. Compound 1
Intravenous injection of compound 1 resulted in bright labeling of CAA in pial vessels and penetrating arterioles (Fig. 4A, D) and was detected rarely in plaques. In contrast, compound 1 overstaining resulted in bright labeling of pial CAA (equivalent to the ex-vivo sections), CAA in parenchymal vessels, and parenchymal plaques (Fig. 4B, E). X-34 overstaining resulted in a pattern of labeling similar to that observed following compound 1 overstain, except for more extensive labeling of larger plaques (Fig. 4C, F). When applied to sections of occipital cortex from an AD case with severe CAA, compound 1 labeled both CAA and amyloid plaques (Supplement Fig. S1).
Fig. 4.
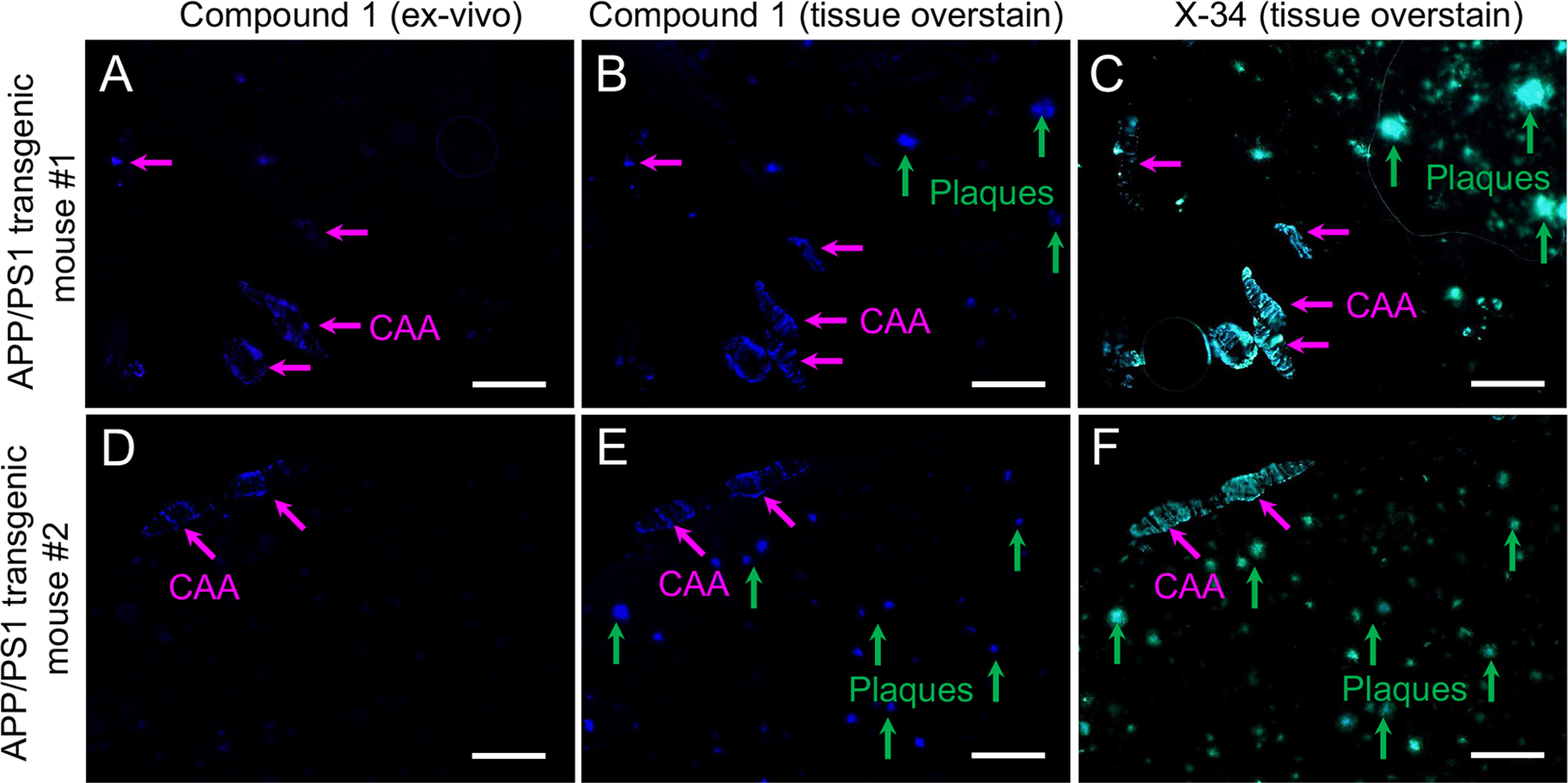
Fluorescence microscopy analyses in tissue sections from two 14-month old APP/PS1 mice (A–C and D–F) killed 15 min after injection of compound 1 (1 mg/kg in ~0.15 ml) into the tail vein. Ex-vivo fluorescence of compound 1 (A, D) is seen in CAA (pink arrows) but not in parenchymal plaques (green arrows in B–C and E–F). This contrasts the labeling of both CAA and plaques seen in the same tissue sections after overstaining sequentially with compound 1 (B and E) and X-34 (C and F). Scale bar = 75 μm.
3.5. Two-photon microscopy
Optical access to the cerebral cortex of a 13-month old APP/PS1 mouse was obtained by creating cranial window over the right cerebral hemisphere. Prior to intravenous injection of test compounds, two-photon images of the fluorescence emission between 420 and 480 nm, spanning a volume of 800 × 800 × 300 μm (rostrocaudal × mediolateral × ventral to the cortical surface, respectively), were acquired around a prominent pial artery (Fig. 5A). Dim punctate fluorescence signal corresponding to lipofuscin autofluorescence and dark outlines of pial arteries were visible within the imaged volume of interest (Fig. 5A). Following intravenous injection of compound 2 (Fig. 5B), bright fluorescence was observed in pial arteries (Fig. 5B1) and in parenchymal plaques (Fig. 5B2). Fluorescence in parenchymal arteriole CAA was also observed but less prominently (Supplemental Fig. S2). Animations of the two-photon microscopy image stack from which images illustrated in Fig. 5 were extracted is provided in Supplement Fig. S2. The same two-photon imaging parameters were used to assess compound 1. Prior to intravenous injection, dim punctate fluorescence signal corresponding to lipofuscin autofluorescence and dark outlines of pial arteries were visible within the imaged volume of interest (Fig. 6A). Following intravenous injection of compound 1 (Fig. 6B), bright fluorescence was observed in pial arteries (Fig. 6B1) and only rarely in parenchymal plaques (Fig. 6B2, B3). Dim fluorescence in parenchymal arterial CAA was also observed but less prominently (Supplemental Fig. S3A, B). After compound 1 washout, compound 2 was injected intravenously. Following compound 2 injection (Fig. 6C), bright fluorescence was detected in pial arteries (Fig. 6C1) and in numerous plaques (Fig. 6C2, C3). Animations of the two-photon microscopy image stack from which images illustrated in Fig. 6B and C were extracted is provided in Supplement Fig. S3.
Fig. 5.
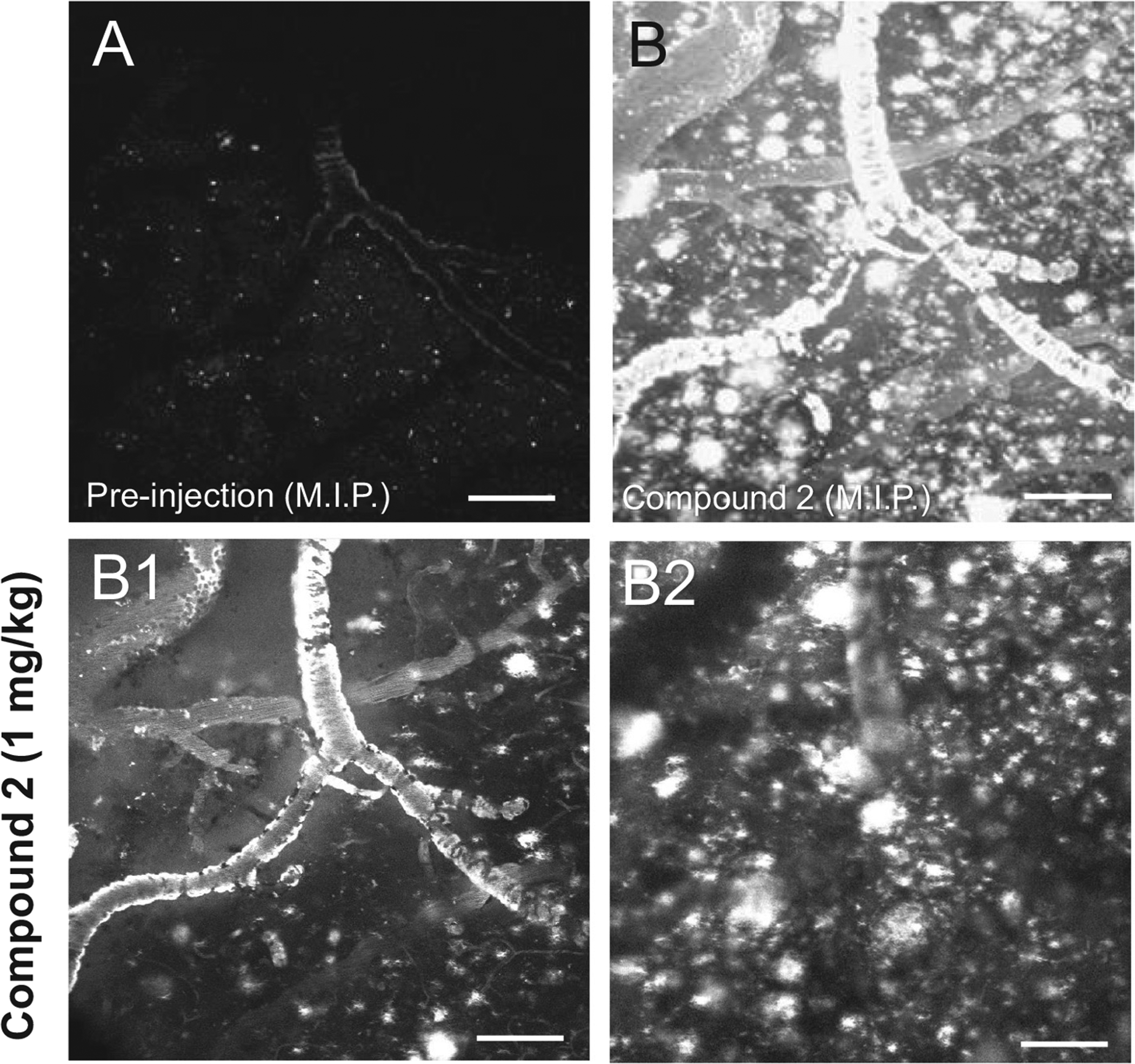
Two-photon microscopy through a cranial window in a 13-month old APP/PS1 mouse. Images were acquired prior to (A) and 30 min after (B) injection of 1 mg/kg compound 2 into the tail vein. Panels A and B are maximum intensity projections (M.I.P.) of image stacks through an 800 × 800 × 300 μm (rostrocaudal × mediolateral × ventral from the cortical surface, respectively) volume of cerebral cortex. A: Prior to injection, weak autofluorescence of lipofuscin (white specks in the brain parenchyma) and amyloid (the dim outline of a large pial artery with severe CAA) can be seen. B: After injection, compound 2 labels both CAA and plaques (bright white signal in B). B1–B2: Two frames from stack acquired during the imaging session and shown as a M.I.P. in (B) are illustrated in panels B1 and B2. Compound 2-labeled CAA and plaques are visible at the cortical surface (B1) as well as ~100 μm (B2) deeper into the cerebral cortex perpendicular to the pial surface. Scale bar = 75 μm.
Fig. 6.
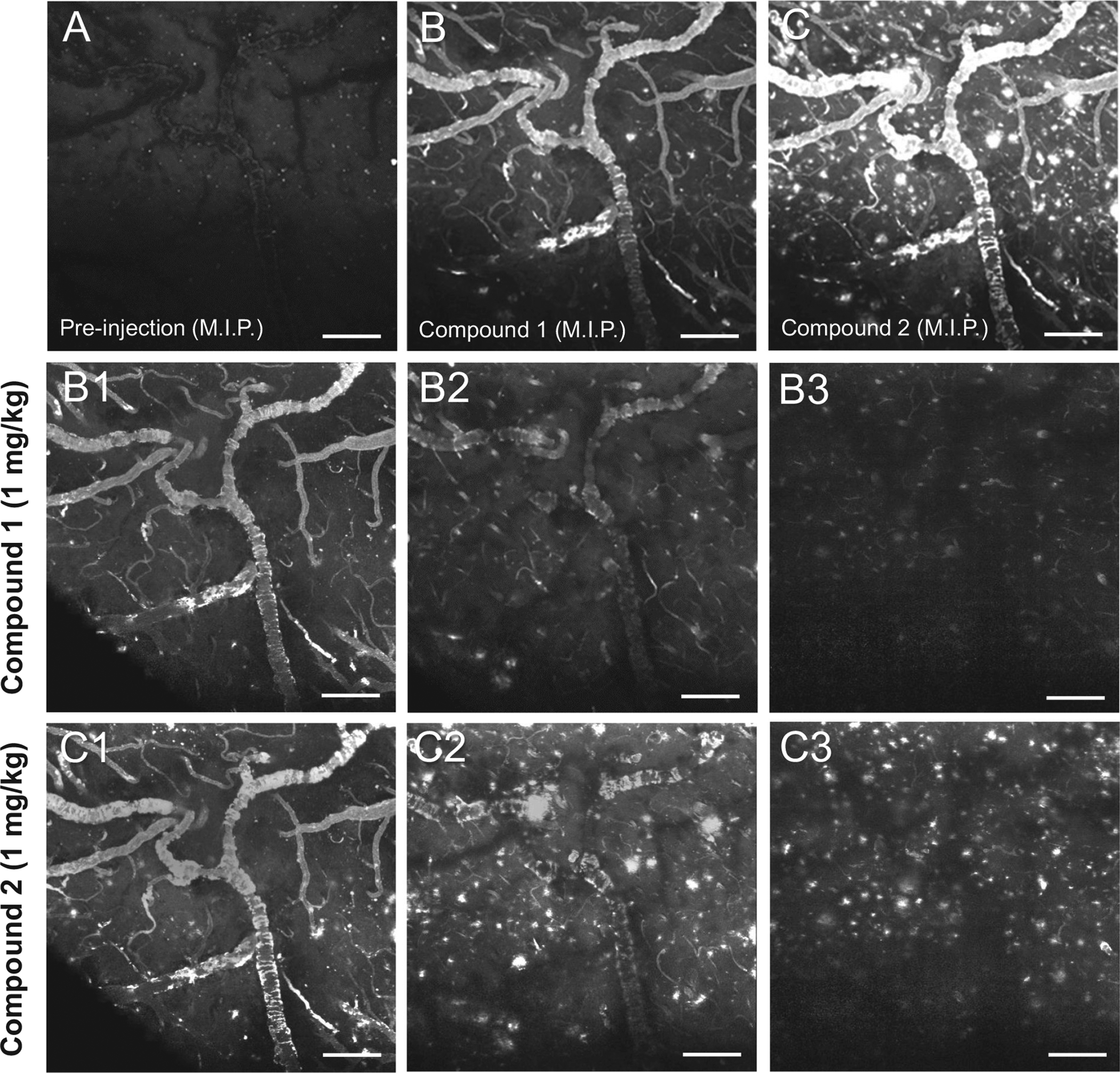
Two-photon microscopy images acquired prior to (A) and 30 min after (B) injection of 1 mg/kg compound 1, and subsequent injection of compound 2 (C) into the tail vein of a 13-month old APP/PS1 mouse. Panels A–C are maximum intensity projections (M.I.P.) of image stacks through an 800 × 800 × 300 μm (rostrocaudal × mediolateral × ventral from the cortical surface, respectively) volume of cerebral cortex. A: Prior to injection, weak autofluorescence of lipofuscin (white specks in the brain parenchyma) and amyloid (the dim outline of a large pial artery with severe CAA) can be seen. B: After injection, compound 1 labels CAA and is detected weakly in some of the plaques. C: In contrast, injection of compound 2 resulted in prominent labeling of both CAA and plaques. B1–B3: Three frames from the stack acquired during imaging of compound 1 and shown as a M.I.P. in (B) are illustrated beginning at the cortical surface (B1), and extending ~100 μm (B2) and 200 μm (B3) deeper into the cerebral cortex perpendicular to the pial surface. C1–3: Three frames acquired during imaging of compound 2 and shown as a M.I.P. in (C) are illustrated at the same cortical depths as in B1–B3. Scale bar = 100 μm.
3.6. Ex-vivo autoradiography ([3H]1)
In mice injected intravenously with [3H]1, autoradiography showed areas of focal retention of radioligand in the cerebral cortex, hippocampus, thalamus/hypothalamus. Autoradiography signal corresponded to the areas with prominent X-34-labeled CAA (Fig. 7). Areas with high densities of parenchymal plaques did not show similar evidence of compound radioligand retention (Fig. 7).
Fig. 7.
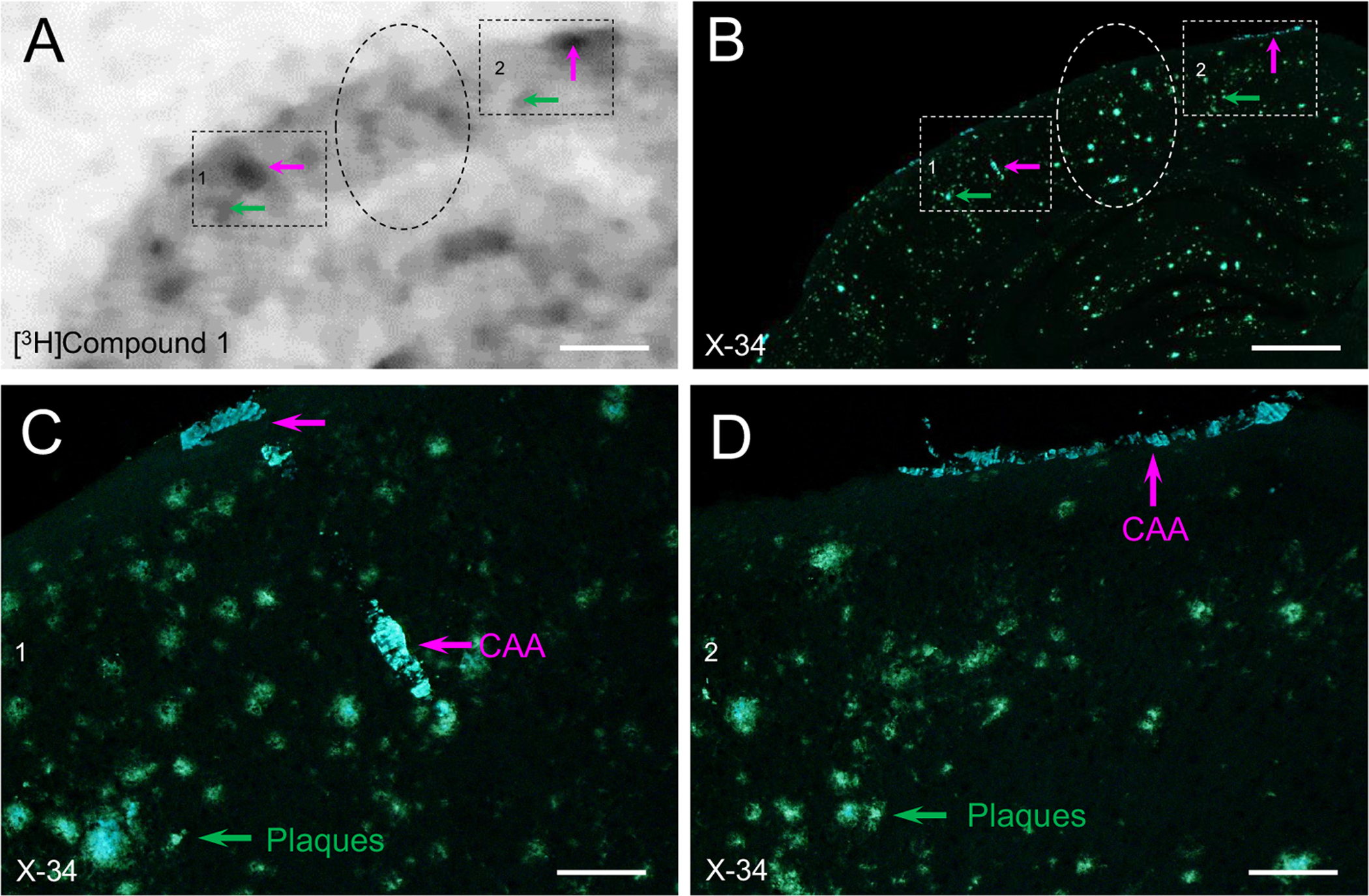
Autoradiogram showing retention of [3H]1 in cortical tissue section from a 12-month old APP/PS1 mouse injected with 5.402 × 106 Bq (in ~0.15 ml 0.1 M sodium phosphate buffer, pH 7.4) via the tail vein (A). High retention is seen in areas most affected with CAA marked by X-34 overstain of the same section (B). Panels C and D are higher magnification images corresponding to the boxed areas in B, showing that areas with heavy autoradiography signal preferentially contain CAA (pink arrows) while little or no signal is detected in areas with predominant plaques (green arrows and dashed ovals). Scale bar = 400 μm (A, B); 75 μm (C, D).
4. Discussion
CAA-selective ligands suitable for PET are critically needed for in-vivo detection and longitudinal tracking of CAA pathology in AD where CAA and Aβ plaques are often intermixed. Such a development could have a high impact in the field, as currently available Aβ PET amyloid imaging agents are unable to distinguish the relative contributions of the two amyloid pathologies to specific signal [48]. This is exemplified in a recent case report of a subject with a clinical diagnosis of probable AD dementia and a positive PiB PET scan, who at autopsy exhibited extensive CAA, but no plaques and tangles [49]. White matter hyperintensities detected by MRI and atypical regional PiB retention have value in predicting sites of potential CAA but are not ideal. For example, both autopsy [50–52] and neuroimaging [53] studies report that CAA and associated lobar hemorrhages are not uniformly distributed in the brain, but rather exhibit preferential posterior (occipital) distribution. Accordingly, the occipital to global retention ratio of PiB is more pronounced in cases with extensive CAA [24,54]. Additionally, a more posterior distribution of white matter hyperintensities detected by MRI was an independent predictor of CAA [21], and because brain regions with significant Aβ deposition are also likely to be affected with CAA-related hemorrhages, PiB-PET could predict incident hemorrhages in subjects with CAA [55]. However, these methods are either indirect or difficult to interpret due to the lack of CAA-selectivity using MRI or currently available Aβ PET ligands.
Despite several encouraging reports, a CAA-selective PET ligand remains to be demonstrated. Based on histological analysis of tissue sections from 16-month old Tg2576 mice (a mouse model of brain amyloidosis [56]) stained with analogues of resorufin, a phenoxazine derivative, Han and colleagues [25] reported CAA selectivity of these compounds. However, binding was also evident in plaque cores (see Fig. 3 from [25]). Repeating experiments described by [25], we observed in aged APP/PS1 mice that resorufin showed evidence of CAA selectivity only when applied directly to fixed tissue sections, whereas both CAA and plaques were labeled when infused into the brain and evaluated through a cranial window using two-photon microscopy. Moreover, unlike the results in tissue sections from APP/PS1 mouse brain, we did not observe CAA selectivity of resorufin in tissue sections from fixed human AD brain. The reasons for these discrepancies are unclear. Other studies have reported selective labeling of CAA using the N-terminal domain of antibodies devoid of light chains to discriminate vascular from parenchymal Aβ deposits [26], but these might not be suitable for modification for PET. Potential binding selectivity for CAA over parenchymal plaques of thioflavin-T (ThT), the charged parent analog of PiB, was suggested in an in-vivo multiphoton microscopy study using a transgenic mouse model of cerebral amyloidosis [57]. While ThT bound with equal avidity to both CAA and plaques when directly applied to the cortical surface, in-vivo access of this tracer to parenchymal plaques was reduced, likely due to its charged nature limiting effective blood brain barrier penetration. However, ThT has low affinity for fibrillar Aβ (890 nM [45]) making it unsuitable as a PET agent. The more highly charged and less lipophilic fluorescent amyloid binding dye thioflavin-S did not enter brain well and did not bind to either CAA or parenchymal plaques when injected intravenously and evaluated by 2-photon microscopy [57]. Thus, there appears to be ranges of ligand charge states and lipophilicity that govern the in-vivo binding selectivity of the tracer. The current study extends these findings and, by leveraging our experience with charged derivatives of CR dyes [58], we were able to successfully identify a compound (compound 1) that binds readily to vascular CAA after injection in-vivo but appears to have limited access to parenchymal plaques. Compound 1 is a hybrid structure derived from structural elements of both X-34 and compound 2 (Fig. 1). Dicarboxylate-containing X-34 enters brain very poorly, while compound 2 enters brain well and subsequently binds to both CAA and parenchymal plaques. The single carboxylate group of compound 1 permits some brain penetration but diminishes binding to parenchymal plaques relative to CAA.
Our delivery-selectivity approach led us to define several criteria for choosing an optimal compound, that include high binding affinity (Ki < 10 nM), tPSA > 65, and moderate lipophilicity (logD < 2.5). The tPSA and logD values of compound 1 are outside the suggested optimal ranges for effective CNS PET imaging agents, while the Ki value is in the acceptable range [59]. Compound 1, exhibited preferential labeling of CAA when injected intravenously in aged APP/PS1 mice, but it labeled both CAA and plaques when applied directly to tissue sections from aged APP/PS1 mice as well as sections of occipital cortex from an AD case. Although the mechanism is not clear and does not appear to be simply related to the logD value, we hypothesize that the selectivity of compound 1 for CAA in-vivo is due to its limited penetration of the blood-brain barrier due to the highly polar carboxylate moiety which limits access to parenchymal plaques resulting in preferential labeling of Aβ deposits in the vascular wall (i.e., “delivery selectivity”).
5. Conclusion and future directions
CAA is a lesion of significant pathological and clinical importance, with high prevalence in advanced aging and AD. The properties of an optimal CAA-selective PET radioligand still remain to be determined. Such an imaging biomarker could facilitate diagnosis of CAA as a clinical entity, support subject selection for clinical trials, and aid in identifying those individuals at high risk for CAA-related hemorrhagic stroke. Thus, identification of CAA deposits in the context of clinical AD may have implications for classification of AD subtypes as well as for safety screening to identify individuals at highest risk for anti-amyloid immunotherapy-induced edema and microhemorrhages.
Supplementary Material
Acknowledgements
This work was supported by a grant from the United States National Insititutes of Health (R01 AG052528 to MDI) and a grant from the Pittsburgh Foundation, Pittsburgh, PA, USA (MR2013-67455to CAM). We gratefully acknowledge the contributions of Ms. Lan Shao, Mr. William Paljug, Mr. Manik Debnath, and Mr. Ping Wang. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Department of Veterans Affairs, or the United States Government.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nucmedbio.2020.05.001.
Declaration of competing interest
There are no disclosures to report.
References
- [1].Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Puchtler H, Sweat F. Congo red as a stain for fluorescence microscopy of amyloid. J Histochem Cytochem. 1965;13:693–4. [DOI] [PubMed] [Google Scholar]
- [3].Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem. 2000;48:1223–32. [DOI] [PubMed] [Google Scholar]
- [4].Ikonomovic MD, Abrahamson EE, Isanski BA, Debnath ML, Mathis CA, Dekosky ST, et al. X-34 labeling of abnormal protein aggregates during the progression of Alzheimer’s disease. Methods Enzymol. 2006;412:123–44. [DOI] [PubMed] [Google Scholar]
- [5].Wisniewski HM, Wen GY, Kim KS. Comparison of four staining methods on the detection of neuritic plaques. Acta Neuropathol. 1989;78:22–7. [DOI] [PubMed] [Google Scholar]
- [6].Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22. [DOI] [PubMed] [Google Scholar]
- [7].Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59. [DOI] [PubMed] [Google Scholar]
- [8].Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Del Tredici K, Braak H. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2002;61:282–93. [DOI] [PubMed] [Google Scholar]
- [10].Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020;16:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke. 1983;14: 924–8. [DOI] [PubMed] [Google Scholar]
- [12].Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, part XV. Neurology. 1996;46:1592–6. [DOI] [PubMed] [Google Scholar]
- [13].Yamada M Cerebral amyloid angiopathy: emerging concepts. J Stroke. 2015;17: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol. 2011;70:871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reijmer YD, Fotiadis P, Martinez-Ramirez S, Salat DH, Schultz A, Shoamanesh A, et al. Structural network alterations and neurological dysfunction in cerebral amyloid angiopathy. Brain. 2015;138:179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Malek-Ahmadi M, Chen K, Perez SE, Mufson EJ. Cerebral amyloid angiopathy and neuritic plaque pathology correlate with cognitive decline in elderly non-demented individuals. J Alzheimers Dis. 2019;67:411–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35:2616–9. [DOI] [PubMed] [Google Scholar]
- [18].Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–24. [DOI] [PubMed] [Google Scholar]
- [19].Howard R, Liu KY. Questions EMERGE as Biogen claims aducanumab turnaround. Nat Rev Neurol. 2019. 10.1038/s41582-019-0295-9 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- [20].Roher AE, Maarouf CL, Kokjohn TA, Belden C, Serrano G, Sabbagh MS, et al. Chemical and neuropathological analyses of an Alzheimer’s disease patient treated with solanezumab. Am J Neurodegener Dis. 2016;5:158–70. [PMC free article] [PubMed] [Google Scholar]
- [21].Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, Ni J, Ayres A, Reed A, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology. 2014;83:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mathis CA, Lopresti BJ, Ikonomovic MD, Klunk WE. Small-molecule PET tracers for imaging proteinopathies. Semin Nucl Med. 2017;47:553–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56:537–9. [DOI] [PubMed] [Google Scholar]
- [24].Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007; 62:229–34. [DOI] [PubMed] [Google Scholar]
- [25].Han BH, Zhou ML, Vellimana AK, Milner E, Kim DH, Greenberg JK, et al. Resorufin analogs preferentially bind cerebrovascular amyloid: potential use as imaging ligands for cerebral amyloid angiopathy. Mol Neurodegener. 2011;6:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rutgers KS, van Remoortere A, van Buchem MA, Verrips CT, Greenberg SM, Bacskai BJ, et al. Differential recognition of vascular and parenchymal beta amyloid deposition. Neurobiol Aging. 2011;32:1774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jaruszewski KM, Curran GL, Swaminathan SK, Rosenberg JT, Grant SC, Ramakrishnan S, et al. Multimodal nanoprobes to target cerebrovascular amyloid in Alzheimer’s disease brain. Biomaterials. 2014;35:1967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Iikuni S, Ono M, Watanabe H, Matsumura K, Yoshimura M, Kimura H, et al. Imaging of cerebral amyloid angiopathy with bivalent (99m)Tc-hydroxamamide complexes. Sci Rep. 2016;6:25990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bourassa P, Tremblay C, Schneider JA, Bennett DA, Calon F. Beta-amyloid pathology in human brain microvessel extracts from the parietal cortex: relation with cerebral amyloid angiopathy and Alzheimer’s disease. Acta Neuropathol. 2019;137:801–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gravina SA, Ho L, Eckman CB, Long KE, Otvos L Jr, Younkin LH, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J Biol Chem. 1995;270:7013–6. [DOI] [PubMed] [Google Scholar]
- [31].Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol. 1998;57:353–9. [DOI] [PubMed] [Google Scholar]
- [32].Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, et al. Beta-amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:10836–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 1994;13: 45–53. [DOI] [PubMed] [Google Scholar]
- [34].Fukumoto H, Asami-Odaka A, Suzuki N, Shimada H, Ihara Y, Iwatsubo T. Amyloid beta protein deposition in normal aging has the same characteristics as that in Alzheimer’s disease. Predominance of A beta 42(43) and association of A beta 40 with cored plaques. Am J Pathol. 1996;148:259–65. [PMC free article] [PubMed] [Google Scholar]
- [35].Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, et al. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115:428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, et al. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. [DOI] [PubMed] [Google Scholar]
- [37].Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–70. [DOI] [PubMed] [Google Scholar]
- [38].Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–24. [DOI] [PubMed] [Google Scholar]
- [39].Ieda N, Nakagawa H, Horinouchi T, Peng T, Yang D, Tsumoto H, et al. Peroxynitrite generation from a NO-releasing nitrobenzene derivative in response to photoirradiation. Chem Commun (Camb). 2011;47:6449–51. [DOI] [PubMed] [Google Scholar]
- [40].Zhao F, Li N, Zhu YF, Han ZY. Enantioselective construction of functionalized tetrahydrocarbazoles enabled by asymmetric relay catalysis of gold complex and chiral Bronsted acid. Org Lett. 2016;18:1506–9. [DOI] [PubMed] [Google Scholar]
- [41].Boyle RG, Walker DW, Boyce RJ, Peterson S, Farouz F, Vo CH. Pharmaceutical compounds. WO 2015/120390 A1, 2015/February/09; 2015.
- [42].Nakano S, Takahashi K, Takada J, Iwamoto T, Nagae K, Maruyama Y, Shintani Y, Okada T, Ito Y, Kadowaki T, Yamauchi T, Iwabu M, Iwabu M. Spiro compound and drug for activating adiponectin receptor. WO2011142359. JP: Nissan Chemical Industries; 2013. [Google Scholar]
- [43].Moon HI, Chung IM, Jung JC, Lim E, Lee Y, Oh S, et al. The convenient synthesis and evaluation of the anticancer activities of new resveratrol derivatives. J Enzyme Inhib Med Chem. 2009;24:328–36. [DOI] [PubMed] [Google Scholar]
- [44].Bharti SK, Roy R. Quantitative 1H NMR spectroscopy. TrAC Trends Anal Chem. 2012; 35:5–26. [Google Scholar]
- [45].Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Mathis CA. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci. 2001;69:1471–84. [DOI] [PubMed] [Google Scholar]
- [46].Xiong B, Li A, Lou Y, Chen S, Long B, Peng J, et al. Precise cerebral vascular atlas in stereotaxic coordinates of whole mouse brain. Front Neuroanat. 2017;11:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chang FC, Wood NF. Reactions of potassium tert.-butoxide in dimethylsulfoxide. III. Hydrolysis of hindered carboxylic esters. Tetrahedron Lett. 1964;5:2969–73. [Google Scholar]
- [48].Farid K, Charidimou A, Baron JC. Amyloid positron emission tomography in sporadic cerebral amyloid angiopathy: a systematic critical update. Neuroimage Clin. 2017; 15:247–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ducharme S, Guiot MC, Nikelski J, Chertkow H. Does a positive Pittsburgh Compound B scan in a patient with dementia equal Alzheimer disease? JAMA Neurol. 2013;70: 912–4. [DOI] [PubMed] [Google Scholar]
- [50].Masuda J, Tanaka K, Ueda K, Omae T. Autopsy study of incidence and distribution of cerebral amyloid angiopathy in Hisayama, Japan. Stroke. 1988;19:205–10. [DOI] [PubMed] [Google Scholar]
- [51].Tomonaga M Cerebral amyloid angiopathy in the elderly. J Am Geriatr Soc. 1981;29: 151–7. [DOI] [PubMed] [Google Scholar]
- [52].Gilbert JJ, Vinters HV. Cerebral amyloid angiopathy: incidence and complications in the aging brain. I. Cerebral hemorrhage. Stroke. 1983;14:915–23. [DOI] [PubMed] [Google Scholar]
- [53].Rosand J, Muzikansky A, Kumar A, Wisco JJ, Smith EE, Betensky RA, et al. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol. 2005; 58:459–62. [DOI] [PubMed] [Google Scholar]
- [54].Ly JV, Donnan GA, Villemagne VL, Zavala JA, Ma H, O’Keefe G, et al. 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology. 2010;74:487–93. [DOI] [PubMed] [Google Scholar]
- [55].Gurol ME, Dierksen G, Betensky R, Gidicsin C, Halpin A, Becker A, et al. Predicting sites of new hemorrhage with amyloid imaging in cerebral amyloid angiopathy. Neurology. 2012;79:320–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. [DOI] [PubMed] [Google Scholar]
- [57].Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF, et al. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc Natl Acad Sci U S A. 2003;100: 12462–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mathis CA, Wang Y, Klunk WE. Imaging beta-amyloid plaques and neurofibrillary tangles in the aging human brain. Curr Pharm Des. 2004;10:1469–92. [DOI] [PubMed] [Google Scholar]
- [59].Zhang L, Villalobos A, Beck EM, Bocan T, Chappie TA, Chen L, et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J Med Chem. 2013;56:4568–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.