

Abstract
Polyploid giant cancer cells (PGCC) are common in tumors and have been associated with resistance to cancer therapy, tumor relapse, malignancy, immunosuppression, metastasis, cancer stem cell production, and modulation of the tumor microenvironment. However, the molecular mechanisms that cause these cells to form are not yet known. In this study, we discover that Aurora kinases are synergistic determinants of a switch from the proliferative cell cycle to polyploid growth and multinucleation in lung cancer cell lines. When Aurora kinases were inhibited together, lung cancer cells uniformly grew into multinucleated polyploid giant cancer cells. These cells adopted an endoreplication in which the genome replicates, mitosis is omitted, and cells grow in size. Consequently, such cells continued to safely grow in the presence of antimitotic agents. These PGCC re-entered the proliferative cell cycle and grew in cell number when treatment was terminated. Thus, PGCC formation might represent a fundamental cellular response to Aurora kinase inhibitors and contribute to therapy resistance or tumor relapse.
Keywords: Aurora kinases, cell cycle, endoreplication, drug resistance, lung carcinoma
Introduction
The goal of cancer medicine is to eradicate tumors entirely. However, a major challenge in treating cancer is posed by intratumor heterogeneity (1, 2). The body of a tumor contains a diverse collection of cells harboring distinct molecular profiles with differential levels of sensitivity to therapies. The tumor population as a whole has the potential to access all the cellular mechanisms encoded in the genome and drug treatment selects for those cells that happen to have adopted an escape mechanism. To combat this, a goal of cancer medicine is to better understand the compositional and functional complexity of tumors in order to find more effective strategies to overcome the disease.
A well-known cancer cell type in tumor populations is called polyploid giant cancer cells (3–5). These cells contain multiple nuclei with significantly elevated genomic content (sometimes more than 100 fold) when compared to other cancer cells within the same tumor. The abundance of giant cells can increase markedly in high-grade malignant cancers or following anticancer treatment. They can remain viable and metabolically active for extended periods of time. They can secrete an array of growth factors, cytokines and chemokines (3, 6). Immunosuppressive proteins including programmed death-ligand 1 (PD-L1) were also found to be overexpressed in these cells (7). Therefore, they are responsible of contributing to a microenvironment advantageous for tumor growth and survival.
PGCCs can repopulate, and form macroscopic colonies and spheroids in vitro as they generate tumors when inoculated into mice (8–10). These cells gain the characteristics of cancer stem cells and generate daughter cells through asymmetric cell division patterns (11–13). In comparison to diploid cancer cells, these daughter cells lower the expression of epithelial markers and acquire a mesenchymal phenotype, which is a key transformation for cancer development, progression and metastasis (10, 14, 15).
The ratio of polyploid giant cancer cells is commonly higher in high-grade malignant tumors than in low-grade tumors and in relapsing tumors post-chemotherapy than in tumors before therapy (16). Such cells are known to escape from cytotoxicity induced by major antimitotic agents including taxanes and vinca alkaloids (17–21). Emerging evidence has demonstrated that polyploid giant cancer cells arise in lung cancer (20), cervical carcinoma (22), ovarian cancer (10, 23), prostate cancer (24, 25), gliobastoma (26), colorectal cancer (11, 16, 27), and breast cancer (15, 28, 29). The role of polyploid giant cancer cells and their daughter cells in resistance to therapy, tumor relapse, metastasis, immunosuppression, modulation of the tumor microenvironment and generation of cancer stem cells is well documented. Therefore, revealing the molecular events that cause PGCCs to form can lead to clinically relevant approaches for treating recurrent and metastatic disease, which remains a major global health issue despite decades of substantial research.
In the course of studying hypersensitivity to inhibitors of Aurora kinase A (30), we previously reported an unusual dose response relationship to Aurora kinase inhibitors (AURKi). In some non-small cell lung cancer (NSCLC) lines, more cells survived the inhibitor treatment and kept growing at higher than at lower drug concentrations. We show in the work presented below that many NSCLC cell lines grow and uniformly form multinucleated PGCCs by adopting an endoreplication when Aurora kinases are inhibited. Endoreplications omit mitosis, do not require the mitotic machinery and therefore, provide a safe path for growth by avoiding the catastrophic consequences of antimitotic perturbagens. Thus, at concentrations that induce endoreplication, we confirmed that Aurora kinase inhibitors protect NSCLC cells from the cytotoxicity induced by all antimitotic cancer agents tested; but not agents targeting mechanisms functional in interphase. These endoreplicating NSCLC cells can re-enter the proliferative cell cycle if Aurora kinase inhibitors are withdrawn. We also demonstrate that chemical programming of the cells to endoreplication with Aurora kinase inhibitors can serve as a methodology to identify antimitotic agents in a simple, high-throughput fashion.
Materials and Methods
Cell lines
NSCLC and immortalized HBEC lines were generated by the laboratories of John Minna and Adi Gazdar and their identities have been confirmed by short tandem repeat analysis of cellular DNA (Promega Corp, Madison, WI, USA). All cell lines were determined to be mycoplasma free by testing with the e-Myco Plus Mycoplasma PCR Detection Kit (Bulldog Bio, 2523448). NSCLC lines were cultured in RPMI-1640 medium (GIBCO, 11875) supplemented with 10% fetal bovine serum (v/v) (Atlanta Biological, S11550). HBEC lines were grown in Keratinocyte-SFM medium (GIBCO, 17005) supplemented with EGF and keratinocyte extract (provided by the manufacturer). HBEC3-KT, HBEC30-KT and HBEC34-KT are HBECs immortalized with stable expression of CDK4 and hTERT (31).
Reagents
MLN8237 (Alisertib) (ChemieTek, CT-M8237), VX-689 (Selleckchem, S2770), AZD1152 (Barasertib) (Selleckchem, S1147), GSK1070916 (Selleckchem, S2740), VX-680 (Tozasertib) (Chemietek, CT-VX680), PHA-739358 (Danusertib) (Selleckchem, S1107), Paclitaxel (LC Labs, P-9600), Docetaxel (Sigma-Aldrich, 01885), Vinorelbine detartrate salt hydrate (Sigma-Aldrich, V2264), Vinblastine sulfate (Sigma-Aldrich, V1377), BI 6727 (Volasertib) (Selleckchem, S2235), GSK923295 (Selleckchem, S7090), Ispinesib (Cayman Chemical Company, 18014), VS-83 (Calbiochem, 178273), Gemcitabine (ChemieTek, CT-GEM), Carboplatin (Sigma-Aldrich, C2538), Cisplatin (Sigma-Aldrich, 232120), Etoposide (LC Labs, E-4488), Topotecan (Tocris, 4562), Doxorubucin hydrochloride (Sigma-Aldrich, D1515), Pemetrexed (Tocris, 6185), Methotrexate (Tocris, 1230), 5-Fluorouracil (Sigma-Aldrich, F6627), Aphidicollin (Calbiochem, 178273), Bafilomycin A (Sigma-Aldrich, B1793), Erlotinib (LC Labs, E-4497) were purchased in powder form and stored at 10 mM or 20 mM stock concentrations in dimethyl sulfoxide (DMSO) at −20 °C. Phospho-AURKA (Thr288)/AURKB (Thr232)/AURKC (Thr198) (Cell Signaling, rabbit mAb, 2914, 1:1,000 dilution), Phospho-Histone H3 (Ser10) (Cell Signaling, rabbit mAb, 33770, 1:1,000 dilution), β-Actin (MP Biomedicals, mouse mAb, 69100, 1:10,000 dilution), p16 INK4A (Cell Signaling, rabbit mAb, 80772, 1:1,000 dilution) , p21 Waf1/Cip1 (Cell Signaling, rabbit mAb, 2947, 1:1,000 dilution) and p27 Kip1 (Cell Signaling, rabbit mAb, 3686, 1:1,000 dilution) antibodies were used for immunoblotting assays. Lamin A/C (Cell Signaling, mouse mAb, 4777, 1:100 dilution), PCM1 (Cell Signaling, rabbit pAb, 5213, 1:500 dilution), Tubulin (Abcam, rat mAb, ab6160, 1:1000 dilution) and Alexa Fluor 594™-conjugated (ThermoFisher, 1:1000 dilution) antibodies, Texas Red™-X Phalloidin (ThermoFisher Scientific, T7471, 1:100 dilution), and Hoechst 33342 reagent (ThermoFisher Scientific, H3570, 1:2000 dilution) were used for immunofluorescence assays.
Microplate drug sensitivity assays
NCI-H1693 cells (3,000 per well) were seeded in 96-well plates and incubated at 37 °C for 24 h. Treatment with vehicle (DMSO) or serial dilutions of the indicated compounds in 9 concentrations from 1 to 33,300 nM were performed in triplicate (final DMSO concentration=0.34% in all wells). After 96 h, media were removed and cell viability was measured with a CellTiter-Glo assay (Promega, G7573).
A dose–response study of a panel of NSCLC and HBEC cell lines with MLN8237 (Alisertib), VX-689 and Erlotinib was performed in 384-well plate format. Cells were dispensed at a density of 1,000–1,500 cells per well with an automated liquid dispenser (BioTek MicroFlo; Thermo-Fisher, Inc.). Each concentration was screened in triplicate. Cells were incubated for 24 h at 37 °C in the presence of 5% CO2. Drugs/chemicals were dosed in DMSO at 12 concentrations ranging from 50 pM to 50 μM (half-log dilutions) in triplicate, using an Echo 555 acoustical transfer instrument. After incubating the cells at 37 °C for 96 h, cell viability was measured with a CellTiter-Glo assay. The comparative tests of three agents were performed on the same days, in identical conditions. Dose responses of each cell line to each agent were assessed at the indicated concentrations individually. Concentrations that caused increase in the measured viability after the initial cytotoxic effect of respective agent were colored in red.
Microplate apoptosis assay
Treatments of the indicated compounds were performed in the same manner with the microplate drug sensitivity assays in 96-well plate format. 48 hours after treatments, culture media were removed by centrifuging the assay plates upside down in liquid-collecting containers at 30g for 1 min. Caspase 3 and/or 7 activity was assessed with Caspase-Glo 3/7 assay purchased from Promega (G8090).
Crystal violet staining assay
NCI-H1693 cells (1 × 105 per well) were seeded in 6-well plates and incubated at 37 °C for 24 h. Treatment with vehicle (DMSO) or serial dilutions of the indicated compounds in 10 indicated concentrations were performed (final DMSO concentration=0.34% in all wells). After seven days, media were removed and cell growth was assessed with a crystal violet staining assay solution (6% glutaraldehyde, 0.5% crystal violet (w/v) in water). Colonies formed after the treatments with the indicated compounds were analyzed with the ImageJ software bundled with ColonyArea plugin and graphed (32).
Western blotting
In all, 2 × 105 cells were treated with the indicated compounds at indicated concentrations in six-well format. Forty-eight hours after treatments, the culture medium was aspirated and cells were washed with PBS. Cells were lysed in RIPA lysis buffer (50 mM Tris buffer pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium, 0.1% SDS, 0.5% Sodium deoxycholate) supplemented with protease and phosphatase inhibitors. The protein concentrations of cell lysates were measured with the Protein DC assay (Bio-Rad, 500–0111). Phosphorylation of histone H3 and Aurora kinases A, B, C, expression of p16, p21, p27 and β-Actin (internal control) were monitored by immunoblotting with monoclonal antibodies. Protein band intensities were measured with the ImageQuant software and graphed, if indicated.
Light microscope and immunofluorescence assay
Cells in transparent bottom plates were fixed with 4% formaldehyde in PBS for 15 min at room temperature. After washing with PBS, cells were permeabilized with 0.25% Triton-X in PBS for 15 min at room temperature. After washing with PBS, cells were incubated with 10% Bovine Serum Albumin (BSA) blocking solution for 1 h. Next, cells were immunostained with Texas Red™-X Phalloidin, Lamin A/C, PCM1 or Tubulin antibodies overnight. Alexa Fluor 594™-conjugated secondary antibodies were used. Hoechst 33342 reagent was used to stain DNA. Microscopic observations were performed on a Cytation 5 or EVOS FL Cell Imaging System. Mean nuclear area of cells after the treatments with the indicated compounds were calculated with the ImageJ software and graphed. RGB images of cells were converted to CMYK, and visualized with monochromic magenta (Red and blue) or cyan (Green and blue) filters to reduce the endogenous autofluorescence and acquire a detailed view of the cellular structures of interest such as nucleus and centrosome, if indicated.
Microplate cell viability assay with RNAi-based gene silencing
Pooled siRNAs used for knockdowns of AURKA and AURKB were purchased from Dharmacon. They were dissolved in DNAse/RNase-free water overnight to make a final concentration of 5 μM. siRNA pools at a final concentration of 50 nM were mixed with RNAiMax transfection reagent (Life Technologies, 13778–150) in OptiMEM medium and dispensed into wells, which had 1,000–2,000 cells in 96-well plate format. After incubating the transfected cells for 96 hours, cell viability was measured with a CellTiter-Glo assay. Triple biological replicates were tested for each siRNA.
Results
NSCLC cells exhibit three distinct dose response phenotypes to Aurora kinase A inhibitors
Aurora kinase A is an important regulator of mitosis and functions in the assembly of the mitotic spindle (33–35). Several different Aurora A inhibitors have been developed and are currently in clinical trials as anti-cancer drug candidates (36). In a previous study, we investigated these inhibitors and observed an unusual drug response relationship with an Aurora kinase A inhibitor, MLN8237/Alisertib (MLN8237 henceforth) (30). As shown in Fig. 1A–F, in contrast to the standard sigmoidal or monophasic dose-response phenotypes (Fig. 1A, C), a subgroup of cell lines treated with MLN8237 exhibited a multiphasic “camelback” drug-response pattern (Fig. 1E). VX-689, another Aurora kinase A inhibitor, also caused a similar response pattern in the same panel of non-small cell lung cancer (NSCLC) lines (Fig. 1B, D, F).
Figure 1. There are distinctly different response phenotypes to Aurora kinase inhibition.
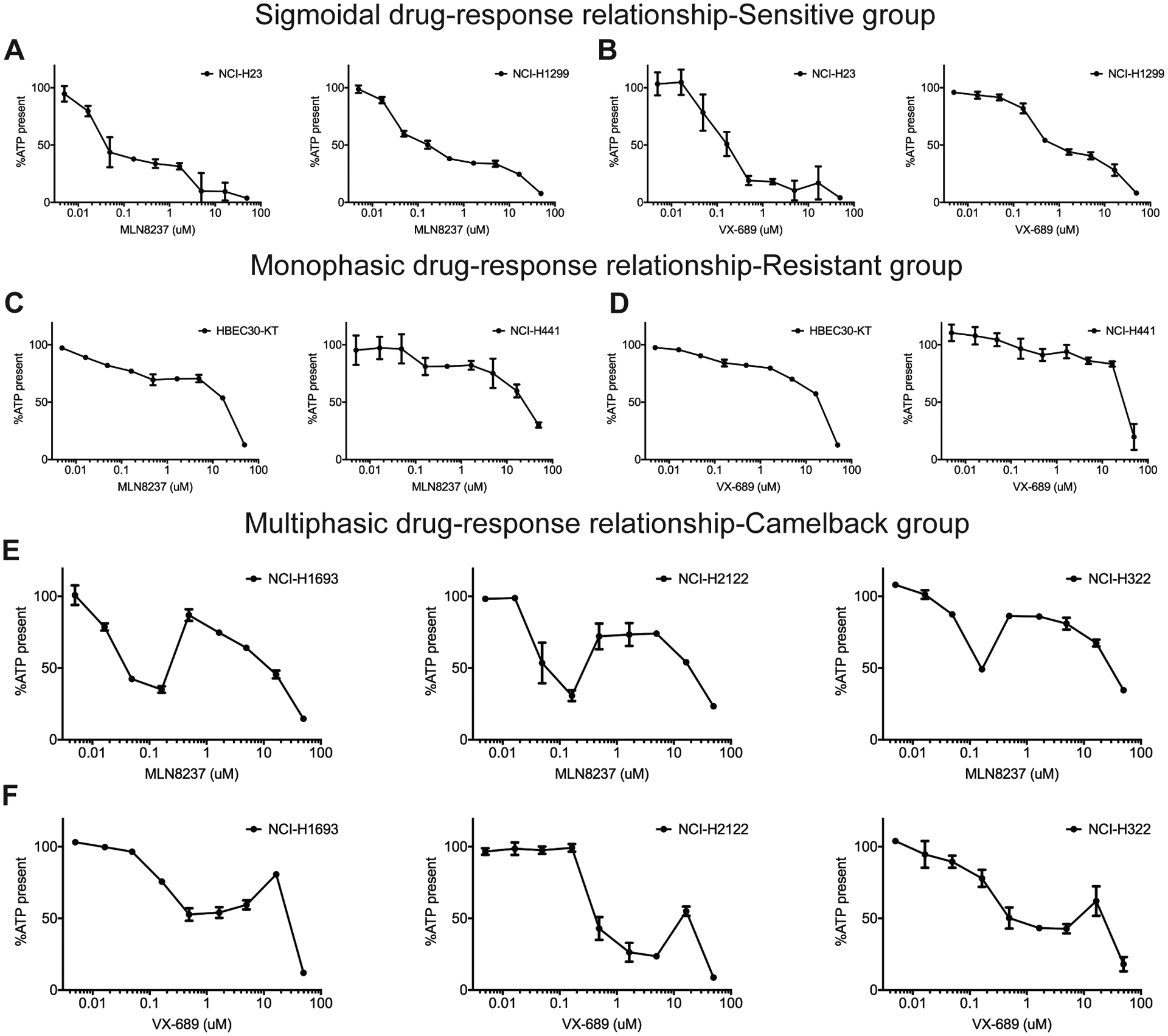
A-F, Cells were treated with Aurora A selective inhibitors MLN8237 or VX-689 at the concentrations shown and cell viability was measured with a CellTiter-Glo assay, which measures cellular ATP as an indicator of living cells. A, B, Cell lines representative of sigmoidal dose-response phenotype, C, D, monophasic dose-response phenotype and E, F, multiphasic dose-response phenotype are shown. Data are means of triplicate biological replicates and error bars are standard deviations (s.d). Some error bars are smaller than the data symbols.
This unexpected drug response pattern of increased cell survival at higher drug concentrations suggested that secondary targets of these inhibitors might control a mechanism leading to drug resistance. This would be of concern, since peak plasma concentrations of MLN8237 in humans reach 3 μM, well above the concentration at which it selectively inhibits its primary target (37). Thus, it was of interest to identify the secondary targets of Aurora A inhibitors and the respective drug resistance mechanism.
Inactivation of Aurora kinases in combination restores cell survival and growth
NSCLC line NCI-H1693 was selected as representative of cell lines capable of this unusual dose-response because it had the most dramatic phenotype with 35% of the cells growing at 87% of the rate of an untreated population in the presence of higher, potentially non-selective concentrations of MLN8237 (Fig. 2A). As demonstrated in Fig. 2A–D, NCI-H1693 cells had a first phase EC50 of 23 nM for MLN8237 and 64 nM for VX-689 (Fig. 2A, C). The EC50s of MLN8237 and VX-689 for the second, “restorative” phase in which the viability increased, was 180 nM and 26 μM, respectively. At the highest and clinically irrelevant concentrations (Phase 3), MLN8237 gains a “general toxin” characteristic, which was not investigated in this study. In parallel, we measured Caspase 3/7 activity to determine if apoptotic cell death occurred in the presence of MLN8237 and VX-689 at any of concentrations measured. Apoptosis was first significantly detected at 16 nM for MLN8237 and 47 nM for VX-689. However, caspase activation was not detected during the second phase of the response to each drug (Fig. 2B, D). In an orthogonal assay for cell growth, NCI-H1693 cells were stained with crystal violet after seven days treatment with either MLN8237 or VX-689 and cell densities were compared with the density of cells stained on day 0. As summarized in Fig. 2E–H, we found that growth of NCI-H1693 cells was inhibited at Aurora A-selective concentrations and was restored at 410 nM and 33.3 μM of MLN8237 and VX-689, respectively (Fig. 2E, G). Thus, the second phase of response to these two inhibitors not only helped NCI-H1693 cells survive, but also restored their growth.
Figure 2. Aurora A inhibitors at higher concentrations antagonize their own cytotoxic activity and restore growth.
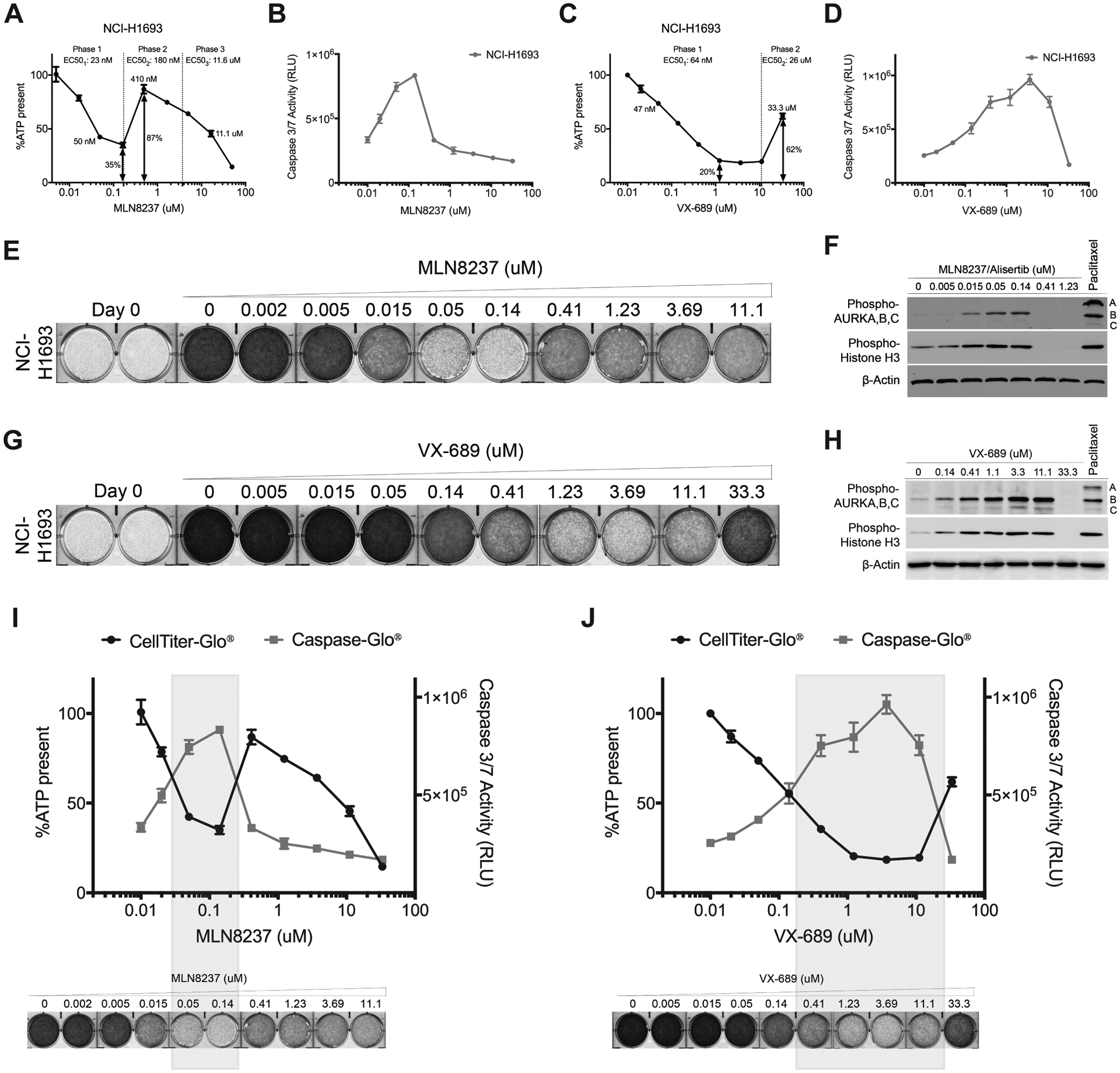
A, NCI-H1693 cells were treated with serial concentrations of MLN8237 for four days and cell viability was measured with a CellTiter-Glo assay, which measures ATP. B, NCI-H1693 cell line was treated with serial concentrations of MLN8237 for two days and active apoptosis was measured with a CaspaseGlo 3/7 assay. C, D, CellTiter-Glo and CaspaseGlo 3/7 assays were repeated with VX-689 treatments. Data are means of triplicate biological replicates with s.d. Some error bars are smaller than the data symbols. E, NCI-H1693 cells were treated with serial concentrations of MLN8237 or G, VX-689 for seven days and cell growth quantified with a crystal violet staining assay. F, Cell lysates of NCI-H1693 cell line were collected 48-hours after treating with MLN8237 or H, VX-689 and immunoblotted to measure phosphorylated histone H3 and phosphorylated Aurora A, B, and C. Paclitaxel was used in both sets of immunoblots as positive control and β-Actin was used as internal immunoblotting control. I, J, Overlay of cell viability, caspase 3/7 activity and crystal violet staining images with immunoblotting results at Aurora A kinase selective and non-selective (pan-Aurora kinase) inhibitory concentrations of MLN8237 and VX-689.
Kinase inhibitors frequently are selective for their intended target only over a narrow concentration range (38–40). Aurora kinases B and C are two orthologues of Aurora A in humans and their high degree of homology is a well-known challenge for creating inhibitors specific to Aurora kinase A. Therefore, Aurora B and Aurora C were obvious candidates for the proteins responsible for reversing cytotoxic effects at higher concentrations of Aurora A inhibitors. Aurora kinases auto-phosphorylate when bound to their activators and their phosphorylation levels directly indicate their activation status in cells. In addition, Histone H3 is a well-characterized substrate of Aurora kinases B and C, and its phosphorylation at Serine 10 is a direct reporter of Aurora B or C activity (41–44). For these reasons, the phosphorylation statuses of Aurora kinases A, B, C and Histone H3 were measured 48 hours after treating NCI-H1693 cells with MLN8237 or VX-689 at a series of concentrations covering their two distinct phases of response. In these experiments, Paclitaxel at its EC100 (20 nM) was used as a positive control to inhibit the cell cycle at the mitotic stage where Aurora kinases are active. We observed increased phosphorylation of Aurora B and C, and Histone H3 starting at concentrations 50 nM of MLN8237 and 140 nM of VX-689. However, starting at 410 nM of MLN8237 and 33.3 μM of VX-689, phosphorylation of Aurora B and C, and Histone H3 was completely undetectable (Fig. 2F, H). Thus, there was a precise overlap between cell death, apoptosis and selective inhibition of Aurora A and between lack of apoptosis, continued cell growth and inhibition of Aurora B and C (Fig. 2I, J).
To test the hypothesis that Aurora B and C are the secondary targets causing the increase in survival and growth of cancer cells, we used two selective Aurora B and C inhibitors, AZD1152/Barasertib (AZD1152 henceforth) and GSK1070916 (45, 46). We first verified their selectivity for Aurora B and C compared to Aurora A by measuring auto-phosphorylation levels of Aurora kinases A, B and C, and phosphorylation of Histone H3 in NCI-H1693 cells. Paclitaxel at its EC100 was used as a positive control. At 50 nM AZD1152 or 15 nM GSK1070916 auto-phosphorylation of Aurora B, C and Histone H3, was not detected; but Aurora A was phosphorylated (Fig. 3A, B). This indicated that at those concentrations, NCI-H1693 cells were arrested in mitosis at a point after activation of Aurora A. However, starting at 1.23 μM for AZD1152 or 140 nM for GSK1070916, phosphorylation of Aurora A was not detected, indicating that both drugs also inhibit Aurora A at higher concentrations.
Figure 3. Aurora B, C inhibitors exhibit a camelback drug response pattern similar to Aurora A inhibitors.
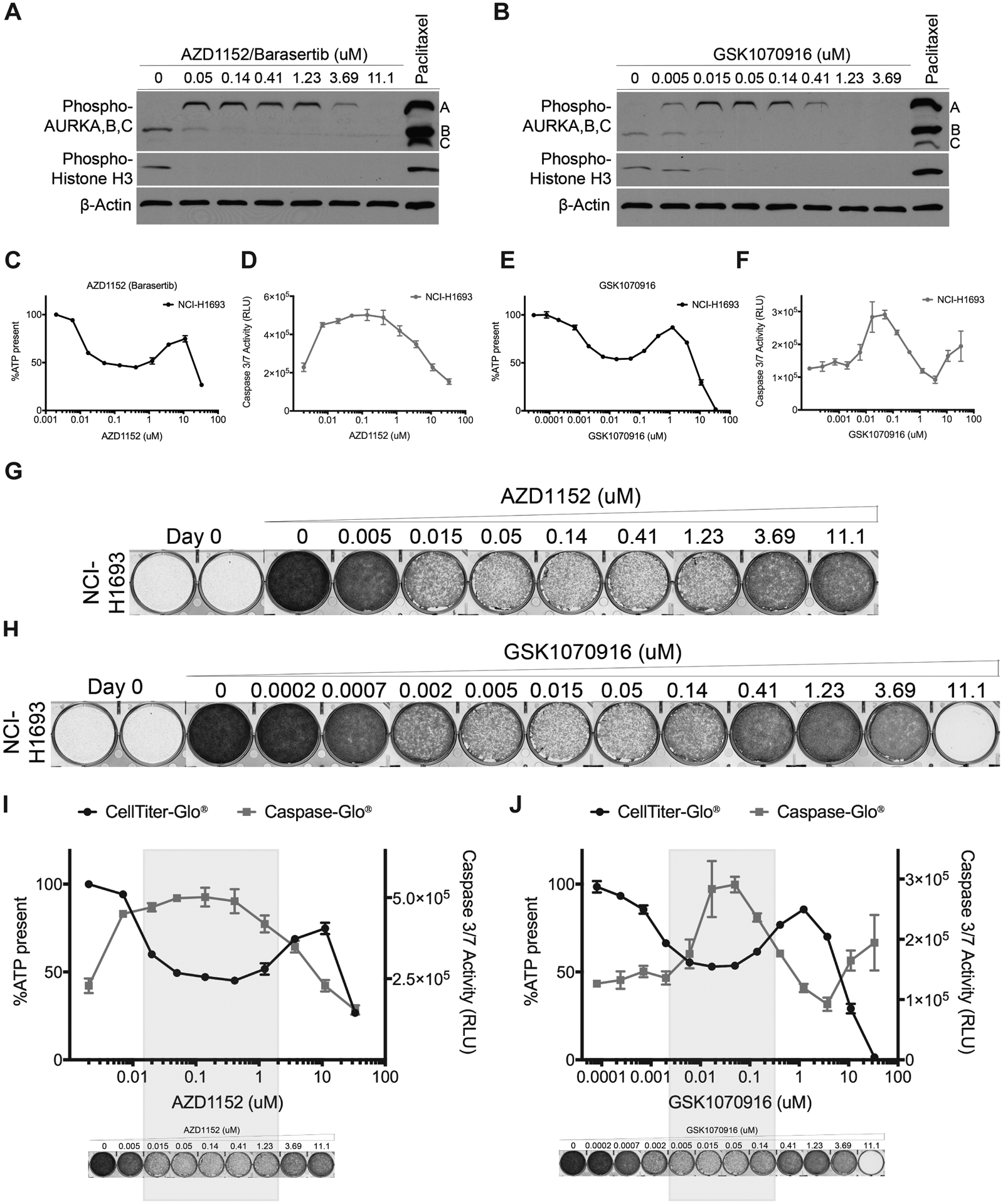
A, Cell lysates of NCI-H1693 cells were collected 48-hours after treating with AZD1152 (Barasertib) or B, GSK1070916 and immunoblotted to monitor phosphorylated histone H3 and phosphorylated Aurora A, B, C levels. Paclitaxel was used in both sets of immunoblots as positive control and β-Actin was used as internal immunoblotting control. C, NCI-H1693 cells were treated with serial concentrations of AZD1152 for four days and cell viability was measured with a CellTiter-Glo assay. D, NCI-H1693 cells were treated with serial concentrations of AZD1152 for two days and cell death was measured with a CaspaseGlo 3/7 assay. E, F, Assays were repeated with GSK1070916 treatments. Data are means of triplicate biological replicates with s.d. Some error bars are smaller than the data symbols. G, NCI-H1693 cell line was treated with serial concentrations of AZD1152 or H, GSK1070916 for seven days and cell growth was measured with a crystal violet staining assay. I, J, Overlay of cell viability, caspase 3/7 activity and crystal violet staining images with immunoblotting results at selective and non-selective concentrations of AZD1152 and GSK1070916.
The dose-response pattern of viability of NCI-H1693 cells to AZD1152 and GSK1070916 resembled the multiphasic “camelback” response of those cells to Aurora A selective inhibitors MLN8237 and VX-689 (Fig. 3C–F). AZD1152 and GSK1070916 each caused significant reduction in viability of NCI-H1693 cells at concentrations selective for Aurora B and C. However, growth was restored when NCI-H1693 cells were treated with concentrations above 1.23 μM and 140 nM of AZD1152 and GSK1070916, respectively (Fig. 3G–H), where they began to antagonize Aurora A as a secondary target. These data suggested that inhibiting Aurora A or Aurora B and C activities alone caused significant mitotic catastrophe leading to cytotoxicity; but inhibition of all three Aurora kinases together did not (Fig. 3I, J).
To ensure that Aurora kinases A, B and C, but not a previously unrecognized fourth target, are solely responsible of restoring viability induced with individual treatments of each of these four inhibitors, we treated NCI-H1693 cells with these four inhibitors as doublet combinations at concentrations where they are selective for their primary targets (Fig. 4A, B). As summarized in Fig. 4C–J, combination therapy of Aurora A and Aurora B, C inhibitors did not cause additive or synergistic cytotoxicity; but instead allowed cell survival and growth (Fig. 4C, E). We measured the activities of Caspase 3/7 (Fig. 4D, F) and used crystal violet staining (Fig. 4G, I) to assess apoptosis and cell growth. Inhibiting the Aurora kinases together allowed NCI-H1693 cells to survive and grow whereas inhibiting Aurora A or B selectively was inhibitory. Consequently, concentrations of Aurora B, C inhibitors selective for their target could prevent toxicity by Aurora A inhibitors when combined together in NCI-H1693 cells (Fig. 4H, J).
Figure 4. Combining Aurora A and B, C inhibitors at concentrations selective for their primary targets prevents cytotoxicity and allows cell growth.
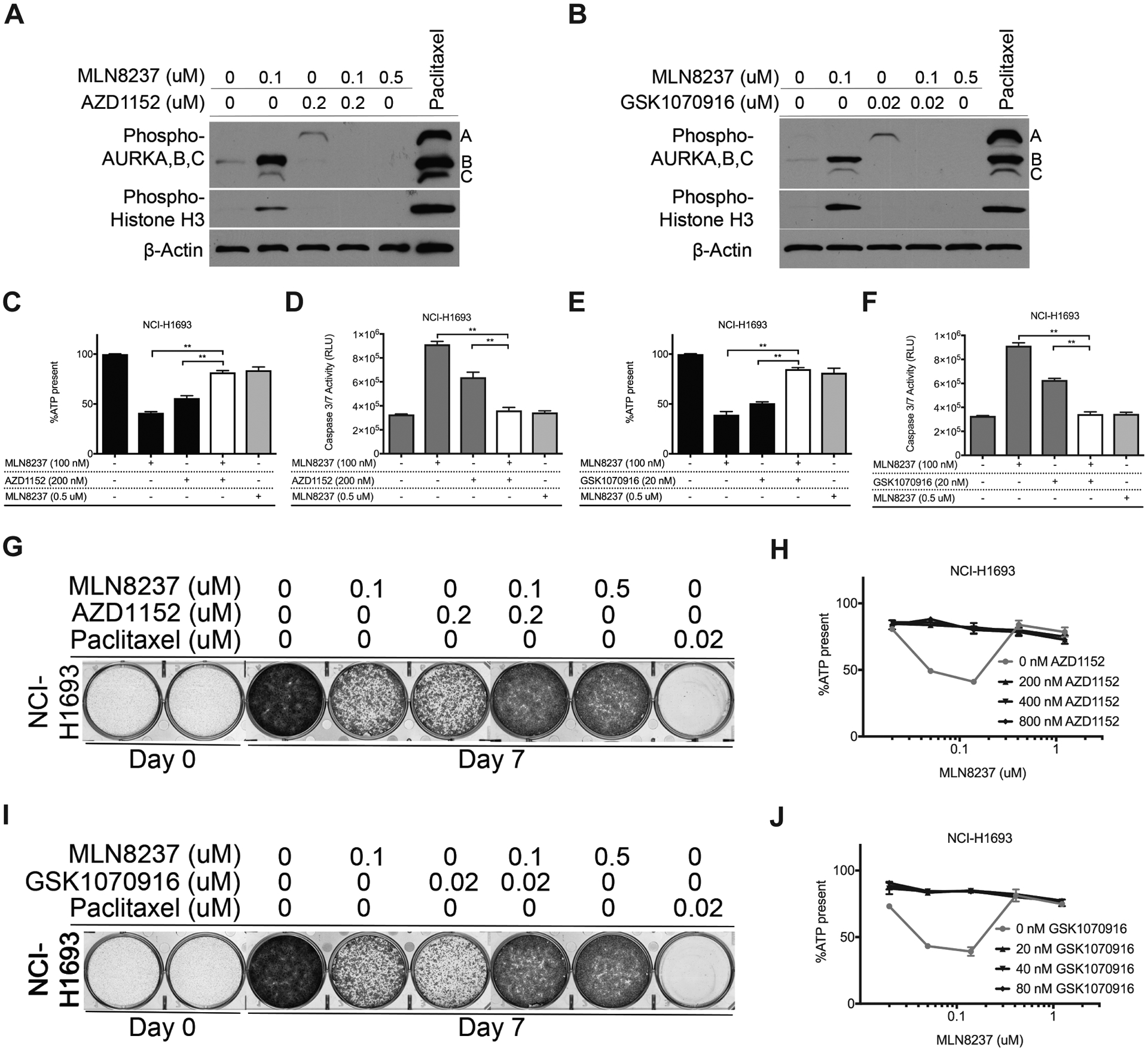
A, Cell lysates of NCI-H1693 cells were collected 48-hours after treating with combinations of MLN8237 and AZD1152 or B, MLN8237 and GSK1070916 and immunoblotted to detect phosphorylated histone H3 and phosphorylated Aurora A, B, C. Paclitaxel was used in both sets of immunoblots as a positive control and β-Actin was used as an internal immunoblotting control. C, NCI-H1693 cells were treated with combinations of MLN8237 and AZD1152 at concentrations selective for their primary targets for four days and cell viability was measured with a CellTiter-Glo assay. D, NCI-H1693 cells were treated with combinations of MLN8237 and AZD1152 at their selective for two days and cell death was measured with a CaspaseGlo 3/7 assay. E, F, Identical assays were repeated with GSK1070916 treatments. G, NCI-H1693 cells were treated with combinations of Aurora A and B/C selective inhibitors for seven days and cell growth was measured with a crystal violet staining assay. H, NCI-H1693 cells were treated with serial concentrations of MLN8237 in combination with serial dilutions of AZD1152 for four days and cell viability was measured with a CellTiter-Glo assay. I, J, Identical assays were repeated with GSK1070916 treatments. Data from CellTiter-Glo and CaspaseGlo 3/7 assays are presented as means of triplicate biological replicates with s.d. Some error bars are smaller than the data symbols. Statistical significance in the figures was assessed by one-way ANOVA and post hoc Dunnett’s multiple comparison tests. ** indicates statistical significance (P<0.01).
Based on these observations, we hypothesized that pan-Aurora kinase inhibitors would be non- or minimally effective on NCI-H1693 cells when compared with the first response phases of MLN8237, VX-689, AZD1152 and GSK1070916. We performed biochemical assays identical to the ones performed with the four Aurora A or B and C selective inhibitors. We observed that pan-Aurora inhibitors, VX-680/Tozasertib (VX-680 henceforth) and PHA-739358/Danusertib (PHA-739358 henceforth) have a resistance response pattern resembling the second phase of multiphasic response of the first four selective inhibitors (Supplementary Fig. S1A–H).
To determine how frequently NSCLC cell lines survived simultaneous inhibition of all Aurora kinases, we measured the dose-responses to MLN8237 in a panel of 77 NSCLC and 4 immortalized human bronchial epithelial cell (HBEC) lines. More than fifty percent of these cell lines had a multiphasic camel-back drug response pattern with more cell survival and growth at the pan-Aurora kinase inhibitory phase in comparison to the responses at lower Aurora A-selective doses (Fig. 5A). In addition, the rest of tested NSCLC lines were either uniformly sensitive or uniformly resistant to the effective doses of MLN8237 with a sigmoidal or monophasic response, respectively (Fig. 5A). An identical screen with VX-689 produced results similar to those obtained with the screen of MLN8237 (Fig. 5B). In these studies, Erlotinib, an EGFR-targeted therapy agent and FDA-approved drug for NSCLCs, was used as control in parallel to MLN8237 and VX-689, on the same day at identical conditions and no multiphasic “camelback” response pattern was observed (Fig. 5C). Overall, our data indicated that the multiphasic response pattern against Aurora A inhibitors is common for NSCLC cell lines, and more cells survived and continued to grow when all Aurora kinases were inhibited in comparison to selective Aurora A inhibition.
Figure 5. Survey of a NSCLC line panel reveals that more than 50% of cell lines have more cancer cell survival and growth at higher concentrations of Aurora kinase A inhibitors.
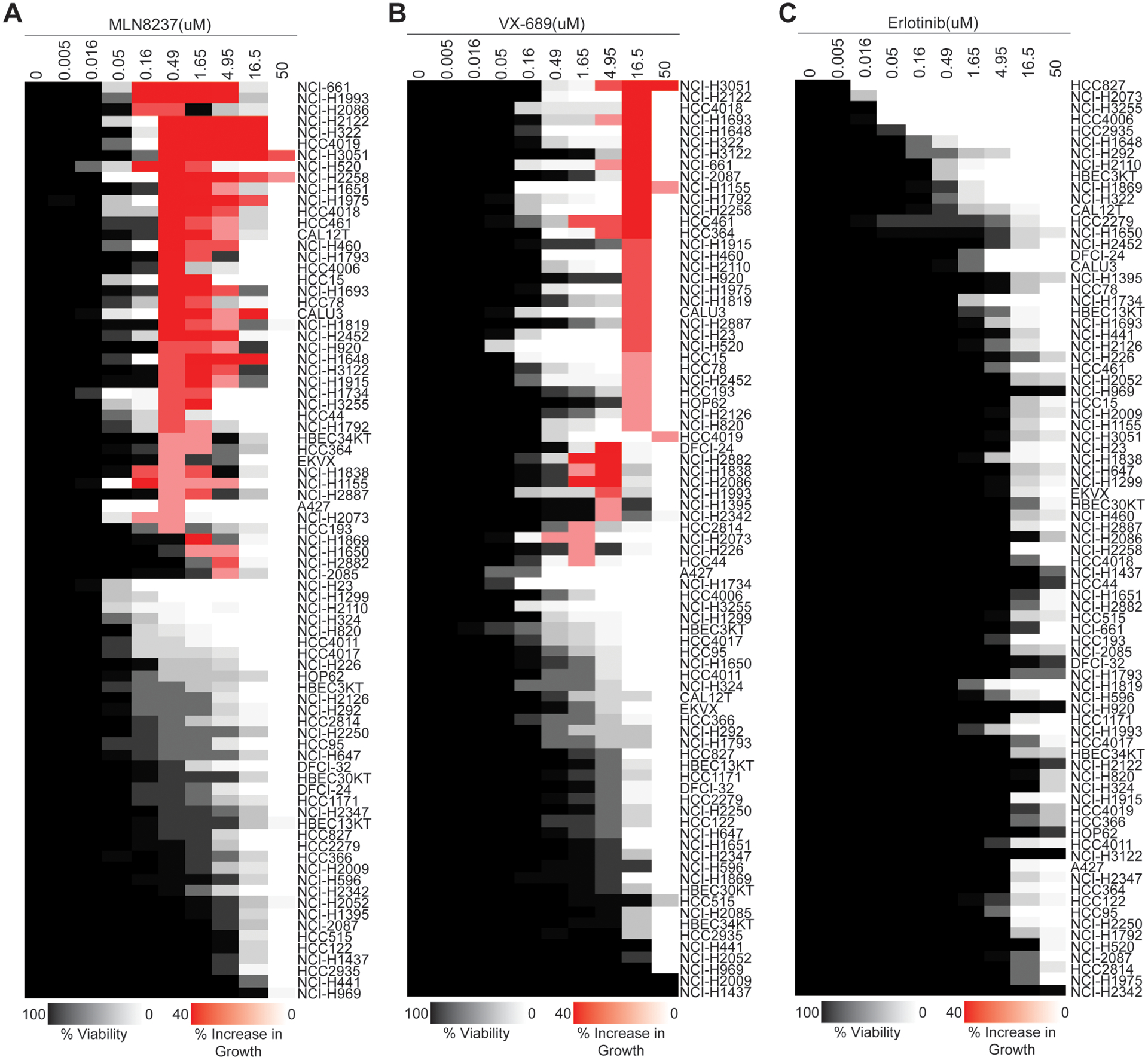
A, A panel of NSCLC and HBEC lines was treated with MLN8237, B, VX-689 or C, Erlotinib. The results of dose–response experiments are shown in heatmap format. Decrease in viability in comparison to the untreated control is shown in shades of gray and white. Increase in viability in comparison to the cytotoxicity induced at lower concentrations is colored in shades of red.
Inactivation of Aurora kinase signaling allows cells to develop into polyploid giant cancer cells (PGCCs)
To determine what type of growth cells adopt after the loss of Aurora kinase signaling, we examined cellular morphology and DNA content after inhibiting Aurora kinases individually or in combination. After the inhibition of Aurora kinases with the non-selective high concentration of MLN8237, we observed that NCI-H1693 cells grow substantially in size instead of numbers and therefore, cellular ATP levels increase similar to untreated proliferating cells (Fig. 6A–B, Supplementary Fig. S2A–C, Supplementary Fig. S3A). Lamin proteins A and C are structural components of the nuclear membrane and therefore, they are direct reporters of the nuclear envelope boundaries. For this reason, NCI-H1693 cells were stained with specific antibodies against Lamin A/C four days after the inhibition of Aurora kinases and visualized with immunofluorescence. The nuclear morphology of these cells together with enlarged size and elevated DNA content, demonstrated that these cells develop into multinucleated polyploid giant cancer cells (Fig. 6B, Supplementary Fig. S2A–C, Supplementary Fig. S3), a cancer cell type frequently found in drug-resistant tumor populations and high-grade tumors. The average area of cells treated with pan-Aurora kinase inhibitor for four days increased and cells had much more nuclear DNA in comparison to the untreated ones (Fig. 6C), which has reached to 4N or more genomic content in ~98% of total population of treated cells (Supplementary Fig. S3B–C). Polyploid genome was also compartmentalized in multiple nuclei in these cells (Fig. 6B–C, Supplementary Fig. S2A–C). Cell numbers were not increasing (Fig. 6D) in the treated samples, but were balanced by increasing the cellular size. Therefore, total ATP levels present in the treated wells remained similar to the wells with untreated cells. DNA replication also continued (Fig. 6D). Increasing DNA content and cell size are characteristics of an endoreplication (47). Overall, our findings suggested that after inhibiting Aurora kinase signaling, cells might omit all or most of mitosis, continue with intact Gap (G) and Synthesis (S) phases of cell cycle, and develop into polyploid giant cancer cells (Fig. 6E).
Figure 6. Loss of Aurora kinase signaling allows cells to grow into polyploid giant cancer cells.
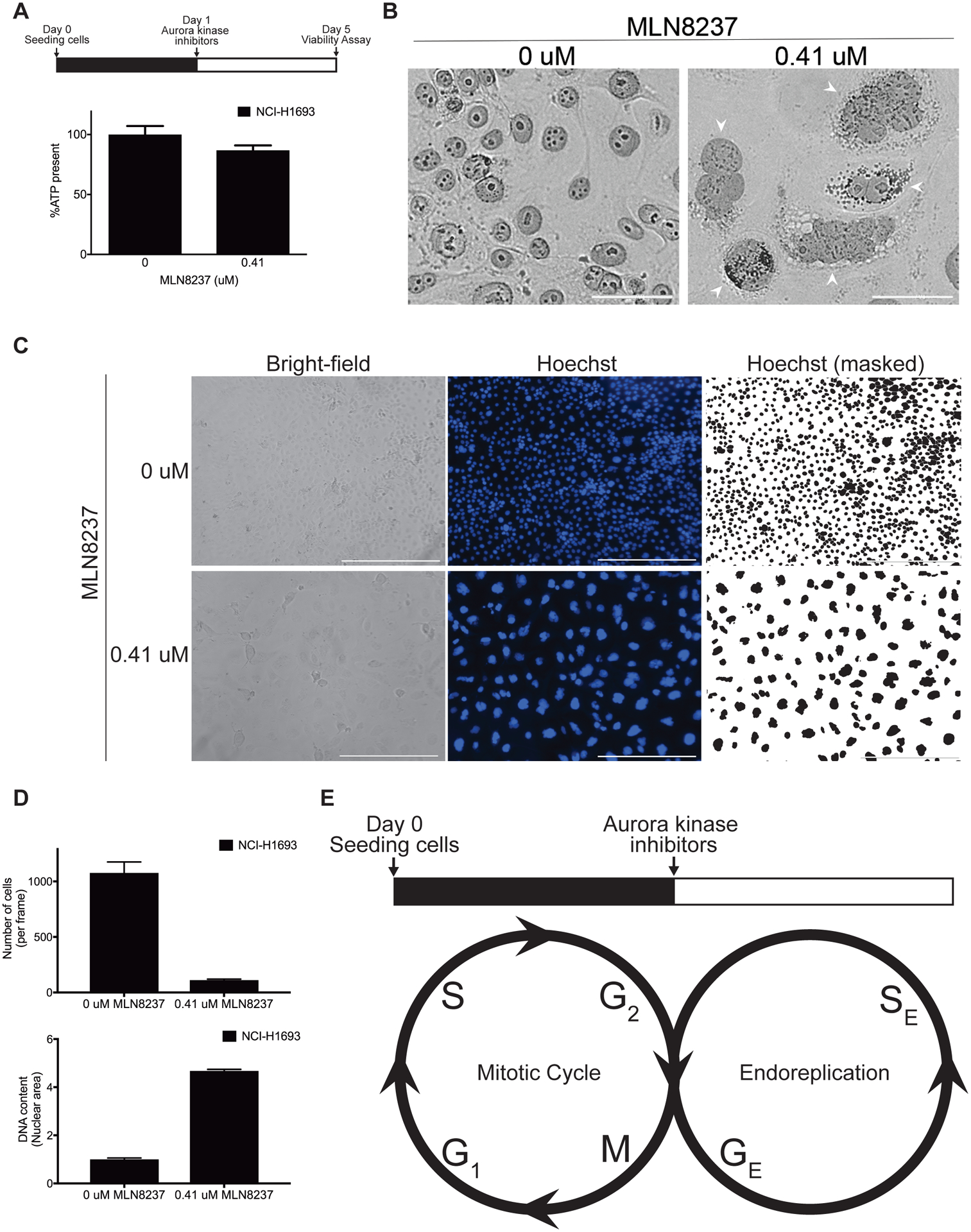
A, NCI-H1693 cells were treated with 0.41 μM of MLN8237 for four days and the ATP present was measured with a CellTiter-Glo assay. B, NCI-H1693 cells were treated with DMSO or 0.41 μM of MLN8237 for four days, stained with monoclonal antibodies against Lamin A/C and visualized with bright field or fluorescent microscopy. To provide detailed views of nuclear morphology (Arrowheads), merged images filtered for magenta light wavelengths were presented (Scale bar, 50 μM). C, NCI-H1693 cells were treated with 0.41 μM of MLN8237 for four days and visualized with bright-field or fluorescent microscopy at 10X magnification. Hoechst reagent was used to visualize the nuclei (Scale bar, 400 μM). D, The numbers of cells and their mean nuclear area from treated and untreated control samples are graphed. E, A proposed model of an endoreplication induced with the loss of Aurora kinase signaling. GE: Growth (G) phase in endoreplication, SE: Synthesis (S) phase in endoreplication.
Induced polyploid giant cancer cells (PGCCs) resist antimitotic agents
If an endoreplication variant avoids mitosis when Aurora kinase signaling is turned off, we hypothesized that mitotic machinery including spindle-related components and regulators should partially or completely be dispensable in the induced PGCCs. Thus, cells would be non-responsive to antimitotic agents under these conditions instead of causing additive or synergistic effects whereas perturbing interphase cellular machineries should remain cytotoxic (Fig. 7A). We tested NCI-H1693 cells with 17 anti-cancer or cytotoxic agents including 9 acting during interphase (Gemcitabine, Carboplatin, Cisplatin, Etoposide, Topotecan, Doxorubucin, Pemetrexed, Methotrexate, 5-Fluorouracil, Aphidicollin and Bafilomycin A) and 8 inhibiting mitosis (Paclitaxel, Docetaxel, Vinorelbine, Vinblastine, PLK1 inhibitor BI-6727 (Volasertib), CENPE inhibitor GSK923295, EG5 inhibitors Ispinesib and VS-83). For the cell viability assessments, the EC100 of each agent was used (Supplementary Table S1). Aurora kinase inhibition prevented toxicity from all antimitotic drugs whereas cells were sensitive to interphase perturbagens regardless of the presence of Aurora inhibitors (Fig. 7B). In addition to individual treatments with these anti-cancer agents, we also added a cocktail of the eight antimitotic agents to our survey and observed that PGCCs generated by pan-Aurora kinase inhibition were able to avoid death even in that extreme condition (Fig. 7B). In parallel to the cell viability assays, we stained NCI-H1693 cells with crystal violet to check the status of cell growth on day 7 in comparison to the cell density on Day 0. Our results confirmed that NCI-H1693 cells survived and continued to grow in the presence of all antimitotics and their cocktail, but not the anti-interphase perturbagens (Fig. 7C). Lastly, we quantified the extent of tolerance of the polyploid giant cancer cells to individual antimitotic agents by performing cell viability assays with serial concentrations of eight antimitotic drugs in the presence and absence of pan-Aurora kinase inhibitors. We observed at least 4 logarithmic (log) magnitude of drug resistance to Paclitaxel, 5 logs to Docetaxel (Fig. 7D), and at least 2 logs of significant resistance to others, which include the Polo-like kinase (PLK) and kinesin spindle protein (KSP) inhibitors (Supplementary Fig. S4A–F). In parallel to cell viability assays, we verified that the cells surviving the co-treatment with antimitotic agents and Aurora kinase inhibitors are uniformly composed of PGCCs with immunofluorescence (Supplementary Fig. S4G).
Figure 7. Inhibition of Aurora kinase signaling provides resistance to antimitotic toxins, but not interphase toxins.
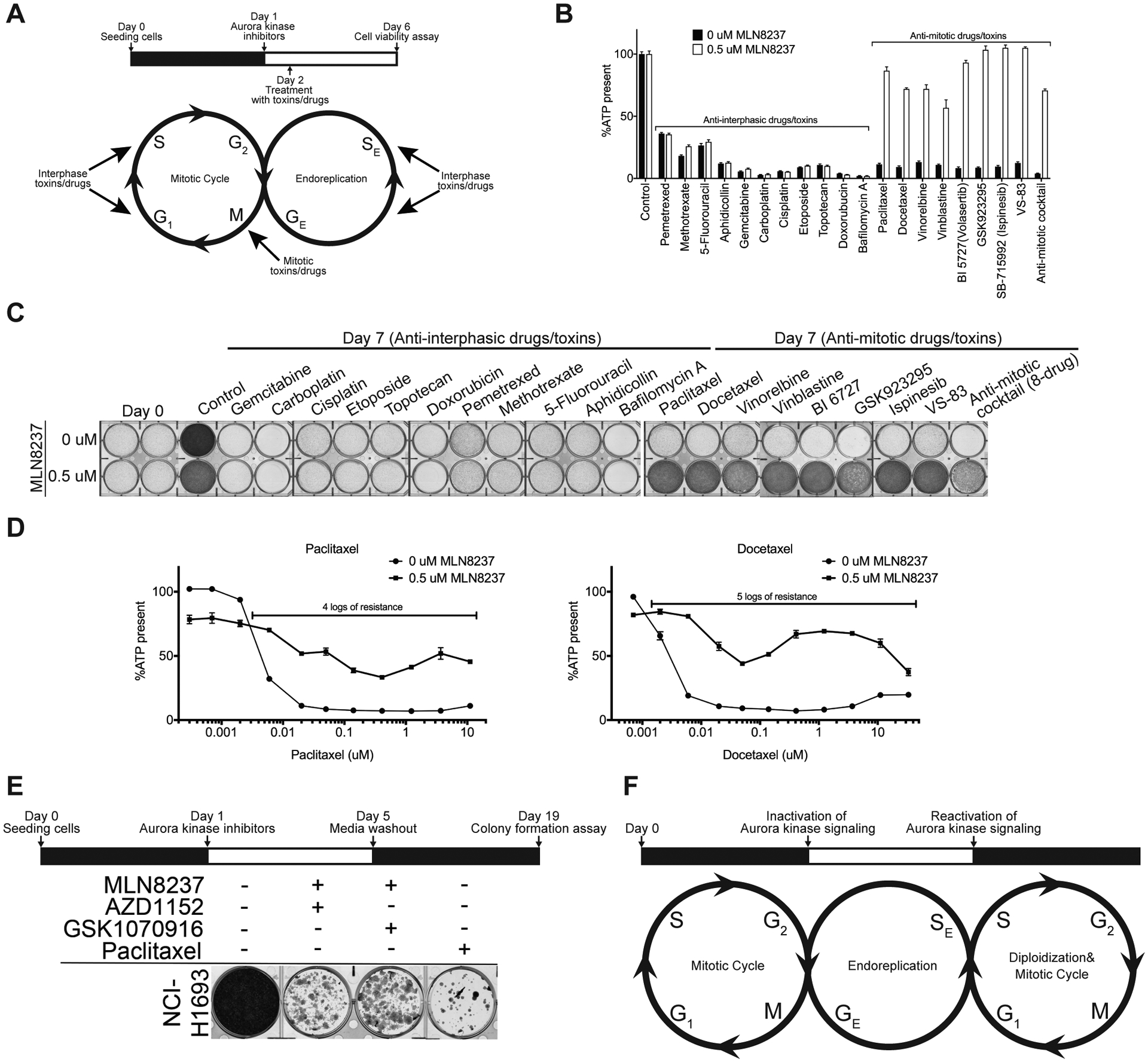
A, NCI-H1693 cells were co-treated with DMSO or 0.5 μM of MLN8237 and IC100 concentrations of interphase or mitotic perturbagens. B, Cell viability and growth were assessed with a CellTiter-Glo assay (after four days of treatment) or C, crystal violet staining assay (after seven days of treatment), respectively. D, NCI-H1693 cells were co-treated with DMSO or 0.5 μM of MLN8237 and serial concentrations of Paclitaxel or Docetaxel. Cell viability was measured with a CellTiter-Glo assay. E, NCI-H1693 cell line was treated with 0.1 μM of MLN8237 and 0.2 μM AZD1152 or 20 nM GSK1070916 for four days. Treatment media were replaced with drug-free media for 14 days. Colony formation was measured with a crystal violet staining assay. F, Proposed model of the cellular life cycle in the presence and absence of Aurora kinase signaling. GE: Growth (G) phase in endoreplication, SE: Synthesis (S) phase in endoreplication. Data for CellTiter-Glo assays are means of triplicate biological replicates with s.d. Some error bars are smaller than the data symbols.
Finally, we investigated whether the NCI-H1693 PGCCs that had switched to endoreplication could reenter the proliferative cell cycle and regenerate daughter cells. Four days after inhibiting Aurora kinase signaling with combinations of Aurora A inhibitor, MLN8237 and Aurora B, C inhibitors AZD1152 or GSK1070916 at concentrations selective for their primary targets, the media with the combination treatments was washed out and NCI-H1693 cells were allowed to grow in the absence of any drug treatments for 14 days. NCI-H1693 cells recovered from the combination treatment and were able to form colonies, indicating the ability of cells to reenter mitotic cell cycle (Fig. 7E, Supplementary Fig. S3E). In comparison, NCI-H1693 cells treated with Paclitaxel had significantly fewer colonies. These observations indicate that loss of Aurora kinase signaling initiates an endoreplication, which maintains growth in cell size instead of increases in cell numbers. These cells grow into polyploid giant cancer cells and can re-enter a proliferative cell cycle when Aurora kinase signaling is restored (Fig. 7F).
Identification of the target and mechanism of action for biologically active molecules remains a major task for drug discovery. To explore the possible use of a chemically-induced endoreplication as a high-throughput assay to identify antimitotic chemicals, NCI-H2122 cells, one of the NSCLC lines with the most dramatic multiphasic camelback response to MLN8237 (Fig. 1E), was used to profile 12 cytotoxic chemicals in the presence or absence of 0.5 uM of MLN8237, a concentration that induces endoreplication (Supplementary Fig. S5A, B). The chemicals used in this test had been identified as toxic for subsets of our NSCLC cell line panel in a previous high-throughput screen, but their mode of action was unknown (48). Of the 12 chemicals, MLN8237-induced NCI-H2122 PGCCs were protected from one, SW172170 (Supplementary Fig. S5C). To determine if SW172170, but not the other chemical probes, had an antimitotic mechanism of action as predicted by our assay, 48 hours after treating with the compounds, we checked for increased phosphorylation of Histone H3 as a sign of mitotic arrest. SW172170 was the only compound showed increase in Histone H3 phosphorylation levels (Supplementary Fig. S5D). As an orthogonal assay, NCI-H2122 cells treated with SW172170 as a single agent were stained for DNA and tubulin. These cells had condensed DNA and multipolar spindles, which are the typical characteristics of mitotic arrest and catastrophe (Supplementary Fig. S5E). This demonstrates that chemical inhibition of Aurora kinases in a cell line that responds by endoreduplicating can be used as a screen for antimitotic mode of action and therefore, can be employed as a tool to assess the cell cycle phase (interphase vs mitosis), in which a toxic chemical acts.
Discussion
Tumors and tumor-derived cell lines contain polyploid giant cancer cells with markedly increased genomic content, often enclosed within multiple nuclei. Their frequency increases after exposure to therapeutic agents. These PGCCs are not dormant, and form macroscopic colonies and tumors in immunodeficient mice (8–10). Their daughter cells harbor stemness, self-renewal and mitotic capacities (11–13). Coherent with these attributes, a significant number of recent studies with different biological systems (tumor-derived cell lines, animal models and specimens from cancer patients) have underlined the crucial roles played by giant cancer cells in tumorigenesis, metastasis, immunosuppression and disease recurrence after cancer therapy (3, 4, 49). PGCCs contribute to solid tumor heterogeneity and are important histological properties of malignant tumors used in pathological diagnosis. Our findings demonstrate that inactivation of Aurora kinases A, B and C combinatorially is a signal to switch to an endoreplication that forms multinucleated polyploid giant cancer cells.
Our results indicate that in NSCLC cells Aurora kinases function together to keep the cancer cell in a proliferative cell cycle. In many cell types, only Aurora A and B kinases are expressed and the extent to which Aurora C plays a role in this activity is not determined by our work. In a majority of NSCLC cell lines we tested, chemical inhibition of all family members of Aurora kinases allowed the cells to adopt an alternative cell cycle where cells continue to replicate DNA, become multinucleate and grow in size, but do not divide. In this state, cells are resistant to all antimitotic drugs tested because the mitotic machinery is no longer engaged. This phenotype is observed with non-small cell lung cancer cells and confirmed by the genetic depletion of Aurora kinases (Supplementary Fig. S3D). These PGCCs do not overexpress senescence-associated stress proteins (SASPs) p16, p21 and p27 (Supplementary Fig. S3F), and are able to reenter proliferative cell cycle although the magnitude of polyploidy affects the colony formation efficacy (Supplementary Fig. S3G–H). Our findings together suggest that Aurora kinases may operate as a cell cycle switch and their off status generates PGCCs tolerant to antimitotic therapy in those cell lines where the alternative cell cycle is available. This phenomenon in other cancer lineages and normal cells is also of interest and remains to be investigated.
In nature there are numerous examples of alternative cell cycles, called endoreplications (also known as endoreduplication, endocycle or endomitosis), that generate healthy polyploid cells for specialized functions. Endoreplication is essential for the proper development of specialized cell types in mammalian systems like megakaryocytes, placental trophoblast cells, hepatocytes and cardiomyocytes (50). The level of ploidy varies dramatically among tissues, organs and species. Mammalian placental trophoblast cells can exhibit ploidy levels of 4096 times the haploid number of chromosomes (4096C). Megakaryocytes generate nuclei with DNA content up to 128C. The highest recorded level for hepatocytes and cardiomyocytes is 32C. Polyploidy can be also stress- and injury-induced to allow improved cell longevity in the incidences such as tissue injury, chemotoxic exposure or mitotic catastrophe (51).
Cancers can take advantage of endoreplications by creating heterogeneity of cell types, some of which are more drug resistant than the parental cells (52). It is possible that some cancer cells do this by adopting an alternate cell cycle program similar to those available to specialized cell types. In the brain, a blood-brain barrier is formed by endothelial cells and these specialized cell types are known to be decorated with efflux pumps and upregulate drug metabolizing enzymes (53, 54). The glial cells forming the barrier in fruit fly Drosophila melanogaster was shown to require polyploidization for proper differentiation (55, 56). In cancers, drug resistance mechanisms to lower the cytosolic drug concentrations with these two systems are well-documented and could originate from cancer cells that were capable of growing in endothelial cell-like endocycles.
How mitotic cycles are molecularly routed towards endoreplications is largely unknown. However, a recent study on cells from Drosophila melanogaster uncovered a role for Aurora kinase B in endoreplication and its morphogenesis (57). Reduced expression of Aurora kinase orthologues in plants was also shown to control the switch from meristematic cell proliferation to endoreduplication and differentiation (58). Furthermore, Aurora kinase A was found downregulated during the megakaryocyte endomitosis and differentiation in mice (59). In our experiments, chemically inhibiting Aurora kinases A or B alone did not robustly induce endoreplication in human lung cancer cells, but led to growth arrest and reduced cell numbers. Whether this is a difference between animal, plant and insect cells, or cancer and normal cells remains to be determined.
Centrosome amplification and multipolarization were previously shown to be responsible of forming multinucleate cells. However, PGCCs formed after the inhibition of Aurora kinases exhibit defects in centrosomal amplification and structure (Supplementary Fig. S6A–B). In these cells, a single centrosome was detected and therefore, our data suggest that multinucleation in these cells is independent of centrosome amplification. However, whether centrosomes play a non-canonical role in multinucleation still remains to be explored. Secondly, we observe that after the substantial cytotoxicity (Fig. 4C–F, Supplementary Fig. S7A–B) NCI-H1693 cells surviving the treatments with Aurora A or B, C inhibitors at concentrations selective to their primary targets also contained polyploid giant cells, but predominantly in mononucleated state (Supplementary Fig. S7A, C).
Unexpected adverse interactions in clinical trials with combination therapies occur and mostly the reasons remain unknown. Currently, 60 clinical trials with MLN8237, 10 with VX-689, 12 with AZD1152, 14 with GSK1070916, 11 with VX-680 and 8 with PHA-739358 have been reported in various cancer types and among those many are performed in combination with other antimitotic drugs such as taxanes and vinca alkaloids. Among the concluded trials, some revealed that targeted mitotic inhibitors, including Aurora kinase inhibitors, were less effective than expected (60, 61). Secondly, enlargement of cells in non-responsive cancers or tumor models after treatments with various chemo- or -targeted therapies has been reported (52). Increased cell size was also observed in studies with Aurora kinase-targeted therapies including MLN8237 (62–64). However, the cause for this unexpected cell enlargement remained unknown. Our study presents a potential mechanistic explanation for these clinical results.
Although paclitaxel has been suggested to cause cytotoxicity in tumors through effects on cells in interphase, our findings indicate that a major drug resistance mechanism for this class of anticancer agents involves avoidance of its antimitotic effects.
Lastly, the multiphasic dose-response pattern that we observed with inhibitors of Aurora kinase signaling is not appropriately recognized by the standard approaches for drug dose-response curve fitting. It was recently reported that the failure rate of the classical Hill equation is more than 25% in large scale screens (65). In experiments investigating the response of NSCLC cell panels to synthetic chemicals (48), we have observed that dose-responses poorly represented by sigmoidal curves are quite common. Some of these are undoubtedly due to chemical solubility or other experimental artifacts, but our current report indicates that some indicate a biological response that is not considered by the assumptions underlying most commonly used curve fitting programs.
In summary, our data suggest that Aurora kinase signaling functions at the intersection between proliferative cell cycle and endoreplication. Switching to endoreplication variants creates growing polyploid giant cancer cells tolerant to antimitotic therapy. These PGCCs also retain the capability to reenter the proliferative cell cycle after the treatments are terminated.
Supplementary Material
Significance:
These findings provide a novel insight about how cancer cells respond to Aurora kinase inhibitors and identify a new mechanism responsible for resistance to these agents and other antimitotic drugs.
Acknowledgements
We thank Bruce Posner and the personnel of the Simmons Comprehensive Cancer Center High-throughput Screening Core Facility for aid in dose-response experiments on the NSCLC cell line panel. The human tumor-derived cell panel was created by Drs. John D. Minna and Adi F. Gazdar. We thank our colleague Iryna Zubovych for critical comments. This work was supported by grants CA176284 and CA221978 to MGR.
Footnotes
Conflict of Interest Disclosure: The authors declare no potential competing interests.
References
- 1.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94. [DOI] [PubMed] [Google Scholar]
- 2.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–34. [DOI] [PubMed] [Google Scholar]
- 3.Mirzayans R, Murray D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int J Mol Sci. 2020;21(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Niu N, Zhang J, Qi L, Shen W, Donkena KV, et al. Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer. Curr Cancer Drug Targets. 2019;19(5):360–7. [DOI] [PubMed] [Google Scholar]
- 5.The Liu J. “life code”: A theory that unifies the human life cycle and the origin of human tumors. Semin Cancer Biol. 2020;60:380–97. [DOI] [PubMed] [Google Scholar]
- 6.Bharadwaj D, Mandal M. Senescence in polyploid giant cancer cells: A road that leads to chemoresistance. Cytokine Growth Factor Rev. 2020;52:68–75. [DOI] [PubMed] [Google Scholar]
- 7.Diaz-Carballo D, Saka S, Klein J, Rennkamp T, Acikelli AH, Malak S, et al. A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity. Cancer Res. 2018;78(9):2318–31. [DOI] [PubMed] [Google Scholar]
- 8.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437(7061):1043–7. [DOI] [PubMed] [Google Scholar]
- 9.Weihua Z, Lin Q, Ramoth AJ, Fan D, Fidler IJ. Formation of solid tumors by a single multinucleated cancer cell. Cancer. 2011;117(17):4092–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene. 2014;33(1):116–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez-Sanchez LM, Jimenez C, Valverde A, Hernandez V, Penarando J, Martinez A, et al. CoCl2, a mimic of hypoxia, induces formation of polyploid giant cells with stem characteristics in colon cancer. PLoS One. 2014;9(6):e99143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solari F, Domenget C, Gire V, Woods C, Lazarides E, Rousset B, et al. Multinucleated cells can continuously generate mononucleated cells in the absence of mitosis: a study of cells of the avian osteoclast lineage. J Cell Sci. 1995;108 ( Pt 10):3233–41. [DOI] [PubMed] [Google Scholar]
- 13.Sundaram M, Guernsey DL, Rajaraman MM, Rajaraman R. Neosis: a novel type of cell division in cancer. Cancer Biol Ther. 2004;3(2):207–18. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S, Mercado-Uribe I, Hanash S, Liu J. iTRAQ-based proteomic analysis of polyploid giant cancer cells and budding progeny cells reveals several distinct pathways for ovarian cancer development. PLoS One. 2013;8(11):e80120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fei F, Zhang D, Yang Z, Wang S, Wang X, Wu Z, et al. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J Exp Clin Cancer Res. 2015;34:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S, Zhang D, Yang Z, Zhang X. Tumor Budding, Micropapillary Pattern, and Polyploidy Giant Cancer Cells in Colorectal Cancer: Current Status and Future Prospects. Stem Cells Int. 2016;2016:4810734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niu N, Zhang J, Zhang N, Mercado-Uribe I, Tao F, Han Z, et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis. 2016;5(12):e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu N, Mercado-Uribe I, Liu J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene. 2017;36(34):4887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogden A, Rida PC, Knudsen BS, Kucuk O, Aneja R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett. 2015;367(2):89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–4. [DOI] [PubMed] [Google Scholar]
- 21.Finkin S, Aylon Y, Anzi S, Oren M, Shaulian E. Fbw7 regulates the activity of endoreduplication mediators and the p53 pathway to prevent drug-induced polyploidy. Oncogene. 2008;27(32):4411–21. [DOI] [PubMed] [Google Scholar]
- 22.Puck TT, Marcus PI. Action of x-rays on mammalian cells. J Exp Med. 1956;103(5):653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lv H, Shi Y, Zhang L, Zhang D, Liu G, Yang Z, et al. Polyploid giant cancer cells with budding and the expression of cyclin E, S-phase kinase-associated protein 2, stathmin associated with the grading and metastasis in serous ovarian tumor. BMC Cancer. 2014;14:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mittal K, Donthamsetty S, Kaur R, Yang C, Gupta MV, Reid MD, et al. Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer. Br J Cancer. 2017;116(9):1186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amend SR, Torga G, Lin KC, Kostecka LG, de Marzo A, Austin RH, et al. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate. 2019;79(13):1489–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bigner SH, Mark J, Burger PC, Mahaley MS Jr., Bullard DE, Muhlbaier LH, et al. Specific chromosomal abnormalities in malignant human gliomas. Cancer Res. 1988;48(2):405–11. [PubMed] [Google Scholar]
- 27.Fei F, Zhang M, Li B, Zhao L, Wang H, Liu L, et al. Formation of Polyploid Giant Cancer Cells Involves in the Prognostic Value of Neoadjuvant Chemoradiation in Locally Advanced Rectal Cancer. J Oncol. 2019;2019:2316436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sirois I, Aguilar-Mahecha A, Lafleur J, Fowler E, Vu V, Scriver M, et al. A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability. Mol Cancer Res. 2019;17(12):2492–507. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Zheng M, Fei F, Li C, Du J, Liu K, et al. EMT-related protein expression in polyploid giant cancer cells and their daughter cells with different passages after triptolide treatment. Med Oncol. 2019;36(9):82. [DOI] [PubMed] [Google Scholar]
- 30.Tagal V, Wei S, Zhang W, Brekken RA, Posner BA, Peyton M, et al. SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat Commun. 2017;8:14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64(24):9027–34. [DOI] [PubMed] [Google Scholar]
- 32.Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. 2014;9(3):e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bury L, Coelho PA, Simeone A, Ferries S, Eyers CE, Eyers PA, et al. Plk4 and Aurora A cooperate in the initiation of acentriolar spindle assembly in mammalian oocytes. The Journal of cell biology. 2017;216(11):3571–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. The Journal of cell biology. 2001;155(7):1109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gopalan G, Chan CS, Donovan PJ. A novel mammalian, mitotic spindle-associated kinase is related to yeast and fly chromosome segregation regulators. The Journal of cell biology. 1997;138(3):643–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17(2):93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(17):4764–74. [DOI] [PubMed] [Google Scholar]
- 38.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371(Pt 1):199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408(3):297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walter AO, Seghezzi W, Korver W, Sheung J, Lees E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene. 2000;19(42):4906–16. [DOI] [PubMed] [Google Scholar]
- 42.Bolton MA, Lan W, Powers SE, McCleland ML, Kuang J, Stukenberg PT. Aurora B kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Molecular biology of the cell. 2002;13(9):3064–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. The Journal of cell biology. 2001;152(4):669–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, et al. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Molecular and cellular biology. 2002;22(3):874–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson K, Lai Z, McDonald OB, Stuart JD, Nartey EN, Hardwicke MA, et al. Biochemical characterization of GSK1070916, a potent and selective inhibitor of Aurora B and Aurora C kinases with an extremely long residence time1. Biochem J. 2009;420(2):259–65. [DOI] [PubMed] [Google Scholar]
- 46.Mortlock AA, Foote KM, Heron NM, Jung FH, Pasquet G, Lohmann JJ, et al. Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. Journal of medicinal chemistry. 2007;50(9):2213–24. [DOI] [PubMed] [Google Scholar]
- 47.Lee HO, Davidson JM, Duronio RJ. Endoreplication: polyploidy with purpose. Genes Dev. 2009;23(21):2461–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMillan EA, Ryu MJ, Diep CH, Mendiratta S, Clemenceau JR, Vaden RM, et al. Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell. 2018;173(4):864–78 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J The dualistic origin of human tumors. Semin Cancer Biol. 2018;53:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edgar BA, Zielke N, Gutierrez C. Endocycles: a recurrent evolutionary innovation for post-mitotic cell growth. Nat Rev Mol Cell Biol. 2014;15(3):197–210. [DOI] [PubMed] [Google Scholar]
- 51.Ovrebo JI, Edgar BA. Polyploidy in tissue homeostasis and regeneration. Development. 2018;145(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puig PE, Guilly MN, Bouchot A, Droin N, Cathelin D, Bouyer F, et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol Int. 2008;32(9):1031–43. [DOI] [PubMed] [Google Scholar]
- 53.Arvanitis CD, Ferraro GB, Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77(4):491–502. [DOI] [PubMed] [Google Scholar]
- 55.Unhavaithaya Y, Orr-Weaver TL. Polyploidization of glia in neural development links tissue growth to blood-brain barrier integrity. Genes Dev. 2012;26(1):31–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Von Stetina JR, Frawley LE, Unhavaithaya Y, Orr-Weaver TL. Variant cell cycles regulated by Notch signaling control cell size and ensure a functional blood-brain barrier. Development. 2018;145(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rotelli MD, Policastro RA, Bolling AM, Killion AW, Weinberg AJ, Dixon MJ, et al. A Cyclin A-Myb-MuvB-Aurora B network regulates the choice between mitotic cycles and polyploid endoreplication cycles. PLoS Genet. 2019;15(7):e1008253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petrovska B, Cenklova V, Pochylova Z, Kourova H, Doskocilova A, Plihal O, et al. Plant Aurora kinases play a role in maintenance of primary meristems and control of endoreduplication. New Phytol. 2012;193(3):590–604. [DOI] [PubMed] [Google Scholar]
- 59.Goldenson B, Kirsammer G, Stankiewicz MJ, Wen QJ, Crispino JD. Aurora kinase A is required for hematopoiesis but is dispensable for murine megakaryocyte endomitosis and differentiation. Blood. 2015;125(13):2141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benada J, Macurek L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules. 2015;5(3):1912–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manchado E, Guillamot M, Malumbres M. Killing cells by targeting mitosis. Cell Death Differ. 2012;19(3):369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kogiso M, Qi L, Braun FK, Injac SG, Zhang L, Du Y, et al. Concurrent Inhibition of Neurosphere and Monolayer Cells of Pediatric Glioblastoma by Aurora A Inhibitor MLN8237 Predicted Survival Extension in PDOX Models. Clin Cancer Res. 2018;24(9):2159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haruki T, Papari-Zareei M, Zhang W, Zhang Y, Stastny V, Minna J, et al. Relationship Between MYC Family Status and Sensitivity to Aurora Kinase Inhibitors in Neuroendocrine and Other Lung Cancer Cell Lines. J Thorac Oncol. 2017;12(11):S2044–S. [Google Scholar]
- 64.Lee JW, Parameswaran J, Sandoval-Schaefer T, Eoh KJ, Yang DH, Zhu F, et al. Combined Aurora Kinase A (AURKA) and WEE1 Inhibition Demonstrates Synergistic Antitumor Effect in Squamous Cell Carcinoma of the Head and Neck. Clin Cancer Res. 2019;25(11):3430–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Di Veroli GY, Fornari C, Goldlust I, Mills G, Koh SB, Bramhall JL, et al. An automated fitting procedure and software for dose-response curves with multiphasic features. Sci Rep. 2015;5:14701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.