

 This work is licensed under a
This work is licensed under a Abstract
Summary
Adrenocortical carcinoma is a rare disease with poor prognosis whose clinical heterogeneity can at times present a challenge to accurate and timely diagnosis. We present the case of a patient who presented with extensive pulmonary lesions, mediastinal and hilar lymphadenopathy and an adrenal mass in whom the oncological diagnosis was initially uncertain. Through the use of immunohistochemistry, biochemistry and genomic testing, an accurate diagnosis of adrenocortical carcinoma was ultimately made which resulted in more directed treatment being administered. The use of multidisciplinary input and genomics to aid in diagnosis and prognosis of adrenocortical carcinoma is discussed.
Learning points
Adrenocortical carcinomas can present a diagnostic challenge to clinicians given it is a rare malignancy with significant clinical heterogeneity.
Specialist multidisciplinary team input is vital in the diagnosis and management of adrenocortical carcinomas.
Hormonal testing is recommended in the diagnostic workup of adrenal masses, even in the absence of overt clinical signs/symptoms of hormone excess.
Immunostaining for the highly sensitive and specific steroidogenic factor-1 is vital for accurate diagnosis.
Genomics can provide prognostic utility in management of adrenocortical carcinoma.
Patient Demographics: Adult, Male, Asian - Indian, Australia
Clinical Overview: Adrenal, Tumours and neoplasia
Related Disciplines: Oncology
Publication Details: Error in diagnosis/pitfalls and caveats, January, 2022
Background
Adrenocortical carcinoma (ACC) is a rare malignancy, with a reported annual incidence of 0.5–2.0 cases per million (1). Most cases are sporadic, though some are associated with an underlying hereditary syndrome, such as Li-Fraumeni syndrome (with TP53 gene mutation), MEN1, Lynch Syndrome (with mismatch repair gene mutations), familial adenomatous polyposis and Beckwith–Wiedeman syndrome (2). Outcomes for patients diagnosed with ACC are generally poor with 5-year survival rates of <30% (1).
Patients with ACC often present either with mass effect symptoms (30–40% of patients) or with clinical features of hormone excess (50–60% of patients) (2). However, ACC demonstrates significant clinical, morphological, and molecular heterogeneity, which can make accurate diagnosis difficult in some cases. Histologically, ACC typically immunostains for steroidogenic factor 1 (SF1), MelanA, alpha-inhibin, calretinin and synaptophysin, without staining for cytokeratins, S100 and chromogranin A (3, 4). However, other than SF1 (sensitivity 98.6%, specificity 100%) (3) most markers have poorer sensitivity and specificity for ACC.
The era of molecular genomic testing has yielded significant developments in our understanding of ACC pathophysiology and prognostication, with certain molecular patterns improving our ability to differentiate ACC from adenomas and other molecular patterns providing prognostic utility in ACC (2). It is also feasible that molecular testing may prove diagnostically useful in malignancies of unknown primary or in differentiating ACC from other malignancies metastatic to the adrenal gland (hepatocellular carcinoma, renal carcinoma) or other primary tumours of the adrenal glands such as phaeochromocytoma.
In this case report, we describe the utility of immunohistochemistry and multidisciplinary care as an aid to accurate diagnosis in a case of ACC with initial diagnostic uncertainty due to concomitant pulmonary and mediastinal involvement. Additionally, the prognostic utility of molecular testing is described.
Case presentation
A 34-year-old male presented to an external hospital with severe pleuritic right chest pain and right upper quadrant abdominal pain, and a preceding 2-month history of night sweats and dyspnoea without weight loss. Additionally, there were no symptoms or signs of clinical hormone excess, such as a Cushingoid appearance, hypertension, hyperglycaemia or weight gain. He was an ex-smoker with a past medical history of glucose-6-phosphate dehydrogenase deficiency, asthma and gastro-oesophageal reflux disease, managed with salbutamol and pantoprazole.
Investigation
His initial investigations included a CT scan demonstrating bilateral lung nodules, pathologically enlarged mediastinal and hilar lymph nodes up to 30 mm in size and a heterogeneous large right adrenal lesion (55 × 80 × 50 mm) with internal areas of necrosis (Fig. 1). He underwent two endobronchial biopsies of a right upper lung lobe lesion. These were both reported to show limited quantities of a partly necrotic and nested proliferation of epithelioid cells with moderate amounts of eosinophilic cytoplasm, round hyperchromatic nuclei and small nucleoli. No glandular or squamous differentiation was seen. There were occasional mitotic figures. Tumour cells expressed synaptophysin, with some very weak pancytokeratin (AE1/AE3) and inhibin staining. They were negative for TTF1, p40, MelanA and S100. Ki67 proliferative index was estimated at 20–25%. A provisional diagnosis of non-small cell carcinoma with neuroendocrine differentiation was made, although it was noted that confident histopathological assessment was difficult on the limited quantities of partly necrotic tumour. Following case review at an oncology multidisciplinary meeting, a lung primary with adrenal metastasis was favoured over metastatic adrenocortical carcinoma. Carboplatin and etoposide (CE) regimen was recommended as first-line therapy and commenced in January 2020.
Figure 1.

Initial CT chest and adrenal imaging. Panel A: CT chest demonstrating bilateral lung nodules and hilar lymphadenopathy. Panel B: Non-contrast CT adrenal demonstrating heterogeneous right adrenal mass with an area of necrosis and high density. Panels C and D: Portal venous phase CT adrenal demonstrating right adrenal mass.
The patient was subsequently referred to our centre (a quaternary cancer centre) following disease progression after two cycles of CE for consideration of additional therapies. Review of the original pathology confirmed the previously reported findings with the exception of Ki67 index, which was thought to be under-estimated due to technical artefact. A repeat Ki67 proliferative index was scored as 60–70%. Further immunostaining showed that the tumour cells did not express CK7, CK19, INSM1, chromogranin or SSTR2. Serum chromogranin A and neuron-specific enolase (NSE) were elevated (Table 1). A gallium-68 DOTATATE PET/CT scan investigating for possible neuroendocrine tumour did not demonstrate significant DOTATATE uptake in the thoracic or adrenal lesions whilst FDG PET/CT in March 2020 (Fig. 2, Panel A) showed intense uptake in the pulmonary nodules, mediastinal nodes and adrenal mass. A confident diagnosis could not be made, but a neuroendocrine neoplasm was thought unlikely. The possibility of a non-small cell carcinoma, potentially of lung origin, was raised. A limited targeted next-generation sequencing panel showed wild-type EGFR, KRAS, BRAF and MET. ALK immunohistochemistry was negative, and ROS1 and RET were not rearranged by fluorescence in situ hybridisation. Atezolizumab/bevacizumab was added to CE after two cycles given the presumed lung carcinoma. However, restaging FDG PET/CT in April 2020 after four cycles of CE demonstrated disease progression in lungs and adrenal gland (Fig. 2, Panel B), though clinically his night sweats had resolved and general improvement was noted in his clinical state.
Table 1.
Patient’s baseline serum and urine biochemistry results.
| Hormone | Result | Reference range |
|---|---|---|
| Testosterone | 9.9 | 8.3–30.2 nmol/L |
| DHEA-S | 58.91 | 1.30–16.10 umol/L |
| Oestradiol | 126 | 40–160 pmol/L |
| Progesterone | 3.7 | <0.6 nmol/L |
| Cortisol | 630 | 100–535 nmol/L |
| 11-deoxycortisol | 13.6 | 0.2–4.6 nmol/L |
| ACTH | 2.3 | 7.2–63.3 ng/L |
| Aldosterone | 89 | <654 pmol/L |
| Renin | 136.8 | 2.8–39.9 mIU/L |
| Aldosterone:renin | <1 | |
| Chromogranin A | 3490 | 27–94 µg/L |
| Neuron-specific enolase | 26 | <12 µg/L |
| 24-h urine catecholamines | ||
| Adrenaline | 118 | <111 nmol/day |
| Noradrenaline | 225 | <476 nmol/day |
| Dopamine | 2483 | <2601 nmol/day |
DHEA-S: dehydroepiandrosterone sulfate, ACTH: adrenocorticotropic hormone
Figure 2.

Serial FDG PET/CT scan images. FDG PET/CT scan demonstrating right adrenal mass, bilateral lung nodules and hilar nodal metastases (panel A), with significant interval progression despite treatment with carboplatin and etoposide (panels B and C), and finally significant metabolic regression of all lesions on mitotane and streptozocin (panel D).
A repeat CT-guided lung biopsy was performed. Immunostaining on this showed the tumour cells strongly expressed vimentin, CD56 and SF-1 (NR5A1) (Fig. 3). Negative stains included calretinin, arginase-1, PAX8, PSA, desmin, NUTM1, ERG, CD30 and OCT3/4. Tumour cells were mismatch repair-proficient. TP53 was strongly expressed, and RB1 expression was absent in tumour cells. The expression of synaptophysin, CD56, inhibin, vimentin and SF-1 favoured metastatic ACC; the absence of TTF-1, ALK and NUTM1 expression or EGFR, KRAS, BRAF, MET, ROS1 and RET mutation suggested lung adenocarcinoma was unlikely. Further investigations at our hospital included a 24-h urine collection which did not demonstrate marked catecholamine excess and serum biochemistry which revealed normal range testosterone and oestrogen, with elevated DHEA-S, progesterone, cortisol, 11-deoxycortisol and suppressed ACTH (Table 1) consistent with excess adrenal hormone production. Renin was elevated; however, this measurement was taken whilst the patient was on an angiotensin-converting enzyme (ACE) inhibitor.
Figure 3.
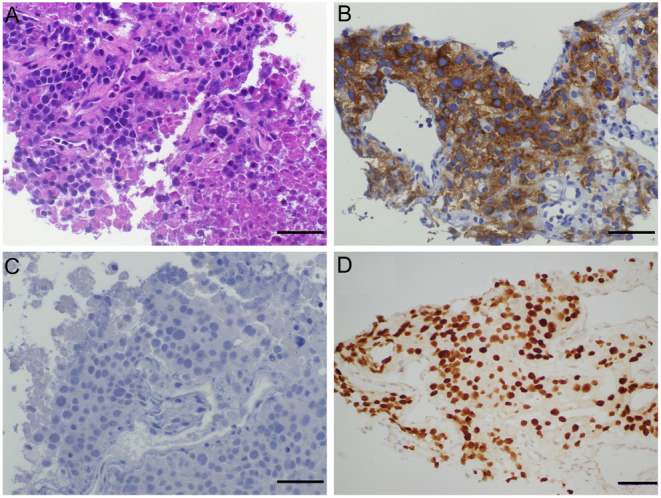
Immunohistochemistry of lung biopsy. Tumour tissue immunohistochemistry demonstrating nested proliferation of tumour cells with round hyperchromatic nuclei on haematoxylin and eosin stain (panel A), strong diffuse positive staining for synaptophysin (panel B), negative stain for INSM1 (panel C) and diffuse strong nuclear expression of SF-1 (panel D). Scale bar = 50 µm.
Whole-genome sequencing (WGS) was performed on the lung biopsy specimen, which also favoured ACC over lung large cell neuroendocrine carcinoma (LCNEC). WGS demonstrated low tumour mutational burden (<4 mutations/megabase) and somatic mutations in TP53, RB1, CTNNB1, ATRX (Table 2), all with additional loss of heterozygosity as demonstrated by high allele frequencies. No pathogenic germline mutation was detected. Features supporting the diagnosis of ACC included: CTNNB1 mutations are rare in LCNEC (2%) and common in ACC (21%) (cBioPortal GENIE cohort); ATRX mutations are more common in ACC (12%) than LCNEC (4%) (5, 6). In addition, a high number of chromosomal breaks, high tumour purity (99%) and whole-genome doubling were detected, similar to other reported ACC samples (6). Notably, this tumour harboured co-existent TP53 and CTNNB1 mutations, which has previously been reported to occur mutually exclusively (7).
Table 2.
Genomic testing results.
| Gene | Mutation | Type | Allele frequency (%) |
|---|---|---|---|
| TP53 | ENST00000269305.8:c.490A>G:exon5:p.K164E | Somatic | 98 |
| RB1 | ENST00000267163.5:c.1442_1470del:exon16:p.I481fs | Somatic | 98 |
| CTNNB1 | ENST00000349496.11:c.133_135del:exon3:p.S45del | Somatic | 97 |
| ATRX | ENST00000373344.10:c.1834A>T:exon9:p.K612* | Somatic | 97 |
| Other key genomic features | |||
| Low mutation burden (2.38 mutations per megabase) | |||
| High number of chromosomal breaks | |||
| Whole-genome doubling | |||
* indicates the amino acid change leads to a stop codon.
Treatment
Further progression was evident on FDG-PET/CT in June 2020 (Fig. 2, Panel C). Based on this, treatment was altered to mitotane and streptozocin (as patient had already received CE as part of his initial chemotherapy it was felt that mitotane-etoposide, doxorubicin, cisplatin (EDP) was unlikely to add clear benefit). Mitotane was up-titrated to 4.5 g/day over the first month targeting mitotane blood levels of 14–20 mg/L (8), and concomitant replacement dose hydrocortisone was commenced.
Outcome and follow-up
Unfortunately, after an initial partial metabolic response with mitotane and streptozocin on FDG PET/CT performed in August 2020 (Fig. 2, Panel D), 2 months after change in therapy, he subsequently developed disease progression and subsequently progressed through several lines of therapy including docetaxel, gemcitabine/mitotane, doxorubicin/mitotane, and mitotane/gemcitabine/docetaxel and is now having supportive care only.
Discussion
ACC is a rare malignancy with poor prognosis and high recurrence rates even after complete surgical resection. To date, systemic therapies for advanced ACC have demonstrated low response rates. Even more recent advances with the First International Randomized Trial in Locally Advanced and Metastatic Adrenocortical Carcinoma Treatment (FIRM-ACT) study (8) establishing mitotane with etoposide, doxorubicin and cisplatin as first-line therapy, median progression-free survival is still only 5 months in those with advanced ACC.
This case highlights the difficulties and importance of accurate diagnosis of malignancies affecting the adrenal gland, particularly when there are no overt clinical features of hormone excess and the initial imaging reveals extra-adrenal disease. It also demonstrates the need for a multidisciplinary team consisting of oncologists, pathologists, radiologists, nuclear physicians, endocrinologists and endocrine surgeons experienced in evaluating adrenal lesions as the differential diagnosis is broad and includes primary malignancies originating in the adrenal gland as well as extra-adrenal primary tumours with metastatic spread to the adrenal glands. It is prudent to exclude hormonal excess early in the diagnostic process, and involvement of an experienced endocrinologist is essential for biochemical evaluation when investigating and managing patients with large adrenal masses.
Our case highlights the diagnostic utility of the immune marker SF-1 in obtaining the correct diagnosis. In our case, limited amounts of the initial biopsy specimen as well as elevated biochemical markers such as chromogranin A and NSE (likely a result from double-dose proton-pump inhibitor use) potentially led to an initial misclassification. Furthermore, oncocytic ACC is characterised by nests of large pleomorphic cells with large nuclei and variable architectural features that may overlap with other malignancies (4), including large cell neuroendocrine carcinoma. Further review of the pathology specimen as well as a repeat biopsy for additional immunohistochemical evaluation at a quaternary cancer centre and an external pathologist specialising in ACC led to the revised diagnosis in our case. Immunostaining for SF-1 should be performed in all cases with a dominant adrenal mass. If SF-1 staining was performed earlier in the diagnostic workup of our case, it may have led to an earlier diagnosis and therefore earlier ACC-specific therapy. Additionally, early biochemical assessment of patients presenting with adrenal lesions is important and may guide diagnosis as well as direct management if biochemical adrenal hormone excess is identified. Furthermore, both urine and serum steroid profile assessments may be utilised to aid in the diagnosis of ACC, with urinary steroid profiles achieving greater than 90% sensitivity and specificity for identification of ACC (9, 10).
The rapidly evolving contribution of genomics to the ACC landscape has provided a wealth of new information to clinicians managing patients with ACC. ACC ranks amongst the top five cancers with the highest levels of chromosomal aneuploidy, the top ten cancers with the highest proportion of whole-genome doubling and amongst the bottom ten cancers in terms of single-nucleotide mutation frequency (2). Despite tumour molecular heterogeneity, there have been certain recurrent molecular alterations that have been identified. These include genes affecting activation of the Wnt/β-catenin pathway – ZNRF3 (frequency of mutation: 16–21%), CTNNB1 (16–33%), APC (8%), MEN1 (7%); genes affecting p53 apoptosis/Rb1 cell cycle pathway – TP53 (16–21%), CDKN2A (11–15%), RB1 (7%), CDK4 (2%); genes affecting chromatin remodelling suggesting a role for epigenetic deregulation in tumorigenesis – ATRX (7%), DAXX (6%) and genes involved in chromosome maintenance – TERT (14%), TERF2 (7%) (6, 7, 11, 12). In addition to the genes commonly involved in ACC (including TP53, RB1, CTNNB1, ATRX which were affected in our case), many other characteristic molecular features of ACC were also seen in our case. ACC has higher tumour purity (82%) relative to other tumours (6), and in our case, tumour purity was 99%. Furthermore, ACC typically demonstrates low tumour mutational burden, with large pan-genomic studies finding an average of 0.6–0.9 mutations per megabase (6, 11), with our case demonstrating <4 mutations/megabase.
A few of the genomic characteristics seen in our case have been associated with poor prognosis in large pan-genomic studies. Zheng et al. (6) categorised ACC into three groups based on patterns of chromosomal alterations, with a ‘chromosomal’ subgroup being defined by whole chromosome or chromosomal arm gains/deletions, a ‘noisy’ subgroup with high number of chromosomal breaks and finally a ‘quiet’ subgroup with very few copy number alterations. The high number of chromosomal breaks seen in our case were reminiscent of the ‘noisy’ subgroup, which is also associated with the worst survival. Additionally, whole-genome doubling which was seen in our case, and which occurs in 68% of ‘noisy’ subtype, 51% of ‘chromosomal’ subtype, and 0% of the ‘quiet’ subtype, has been associated with tumour aggressiveness (6).
Our case highlights the complexities of ACC diagnosis and management, necessitating a multidisciplinary team approach. The SF-1 immunomarker aids diagnostic certainty in cases of ACC, and the evolving field of genomics can provide useful prognostic information in such patients.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
The whole-genome sequencing was available through enrolment in the PRECISION program funded by the Victorian Comprehensive Cancer Centre (VCCC).
Patient consent
Written informed consent was obtained from the patient for publication of manuscript and images.
Author contribution statement
R W, J L and N S were involved in the conception, drafting and revision of the manuscript. B S contributed to revisions of the manuscript. S L and C K contributed to revisions of the manuscript and interpretation of genomic data. O P and A G provided immunohistochemistry images for publication and aided with interpretation of immunohistochemistry results within the manuscript.
References
- 1.Abiven G, Coste J, Groussin L, Anract P, Tissier F, Legmann P, Dousset B, Bertagna X, Bertherat J. Clinical and biological features in the prognosis of adrenocortical cancer: poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. Journal of Clinical Endocrinology and Metabolism 2006912650–2655. ( 10.1210/jc.2005-2730) [DOI] [PubMed] [Google Scholar]
- 2.Crona J, Beuschlein F. Adrenocortical carcinoma – towards genomics guided clinical care. Nature Reviews: Endocrinology 201915548–560. ( 10.1038/s41574-019-0221-7) [DOI] [PubMed] [Google Scholar]
- 3.Sbiera S, Schmull S, Assie G, Voelker HU, Kraus L, Beyer M, Ragazzon B, Beuschlein F, Willenberg HS, Hahner S.et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. Journal of Clinical Endocrinology and Metabolism 201095E161–E171. ( 10.1210/jc.2010-0653) [DOI] [PubMed] [Google Scholar]
- 4.Erickson LA, Rivera M, Zhang J. Adrenocortical carcinoma: review and update. Advances in Anatomic Pathology 201421151–159. ( 10.1097/PAP.0000000000000019) [DOI] [PubMed] [Google Scholar]
- 5.Derks JL, Leblay N, Lantuejoul S, Dingemans AC, Speel EM, Fernandez-Cuesta L. New insights into the molecular characteristics of pulmonary carcinoids and large cell neuroendocrine carcinomas, and the impact on their clinical management. [published correction appears in J Thorac Oncol 2018 13(8) 1229]. Journal of Thoracic Oncology 201813752–766. ( 10.1016/j.jtho.2018.02.002) [DOI] [PubMed] [Google Scholar]
- 6.Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G.et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. [published correction appears in Cancer Cell 2016 30(2) 363]. Cancer Cell 201629723–736. ( 10.1016/j.ccell.2016.04.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Juhlin CC, Goh G, Healy JM, Fonseca AL, Scholl UI, Stenman A, Kunstman JW, Brown TC, Overton JD, Mane SM.et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. Journal of Clinical Endocrinology and Metabolism 2015100E493–E502. ( 10.1210/jc.2014-3282) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, Schade-Brittinger C, Lacroix A, Jarzab Bet al. Combination chemotherapy in advanced adrenocortical carcinoma. New England Journal of Medicine 20123662189–2197. ( 10.1056/NEJMoa1200966) [DOI] [PubMed] [Google Scholar]
- 9.Taylor DR, Ghataore L, Couchman L, Vincent RP, Whitelaw B, Lewis D, Diaz-Cano S, Galata G, Schulte KM, Aylwin Set al. A 13-steroid serum panel based on LC-MS/MS: use in detection of adrenocortical carcinoma. Clinical Chemistry 2017631836–1846. ( 10.1373/clinchem.2017.277624) [DOI] [PubMed] [Google Scholar]
- 10.Arlt W, Biehl M, Taylor AE, Hahner S, Libé R, Hughes BA, Schneider P, Smith DJ, Stiekema H, Krone N.et al. Urine steroid metabolomics as a biomarker tool for detecting malignancy in adrenal tumors. Journal of Clinical Endocrinology and Metabolism 2011963775–3784. ( 10.1210/jc.2011-1565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, René-Corail F.et al. Integrated genomic characterization of adrenocortical carcinoma. Nature Genetics 201446607–612. ( 10.1038/ng.2953) [DOI] [PubMed] [Google Scholar]
- 12.Lippert J, Appenzeller S, Liang R, Sbiera S, Kircher S, Altieri B, Nanda I, Weigand I, Gehrig A, Steinhauer S.et al. Targeted molecular analysis in adrenocortical carcinomas: a strategy toward improved personalized prognostication. Journal of Clinical Endocrinology and Metabolism 20181034511–4523. ( 10.1210/jc.2018-01348) [DOI] [PubMed] [Google Scholar]