


Abstract
Tyrosine site-specific recombinases (SSRs) represent a versatile genome editing tool with considerable therapeutic potential. Recent developments to engineer and evolve SSRs into heterotetramers to improve target site flexibility signified a critical step towards their broad utility in genome editing. However, SSR monomers can form combinations of different homo- and heterotetramers in cells, increasing their off-target potential. Here, we discover that two paired mutations targeting residues implicated in catalysis lead to simple obligate tyrosine SSR systems, where the presence of all distinct subunits to bind as a heterotetramer is obligatory for catalysis. Therefore, only when the paired mutations are applied as single mutations on each recombinase subunit, the engineered SSRs can efficiently recombine the intended target sequence, while the subunits carrying the point mutations expressed in isolation are inactive. We demonstrate the utility of the obligate SSR system to improve recombination specificity of a designer-recombinase for a therapeutic target in human cells. Furthermore, we show that the mutations render the naturally occurring SSRs, Cre and Vika, obligately heteromeric for catalytic proficiency, providing a straight-forward approach to improve their applied properties. These results facilitate the development of safe and effective therapeutic designer-recombinases and advance our mechanistic understanding of SSR catalysis.
INTRODUCTION
Cre is a tyrosine site-specific recombinase (SSR), which forms a homotetramer that stringently catalyzes recombination of DNA between loxP target sites (1). The loxP sequence is composed of two 13 bp palindromic half-sites flanking an 8 bp spacer region where recombination occurs (Figure 1A). The Cre/loxP system is commonly used for genomic modifications because of its precision and robustness within a variety of organisms (2). However, application of Cre/loxP across organisms requires pre-introduction of loxP target sequences into the host genome, which can be time-consuming and cumbersome (3,4). To overcome this limitation and to broaden the use of SSRs, directed molecular evolution has been utilized to produce Cre variants with altered DNA specificities for predefined (pseudo)-symmetric DNA target sites naturally occurring within host genomes (5–10). The practicality of this approach was first demonstrated where Tre, an evolved Cre-type enzyme, was generated to specifically recognize and excise HIV-1 proviral DNA from the human genome (6). Tre and other evolved SSRs represent the potential for site-specific recombinases to be adapted for therapeutic applications (7,11,12).
Figure 1.
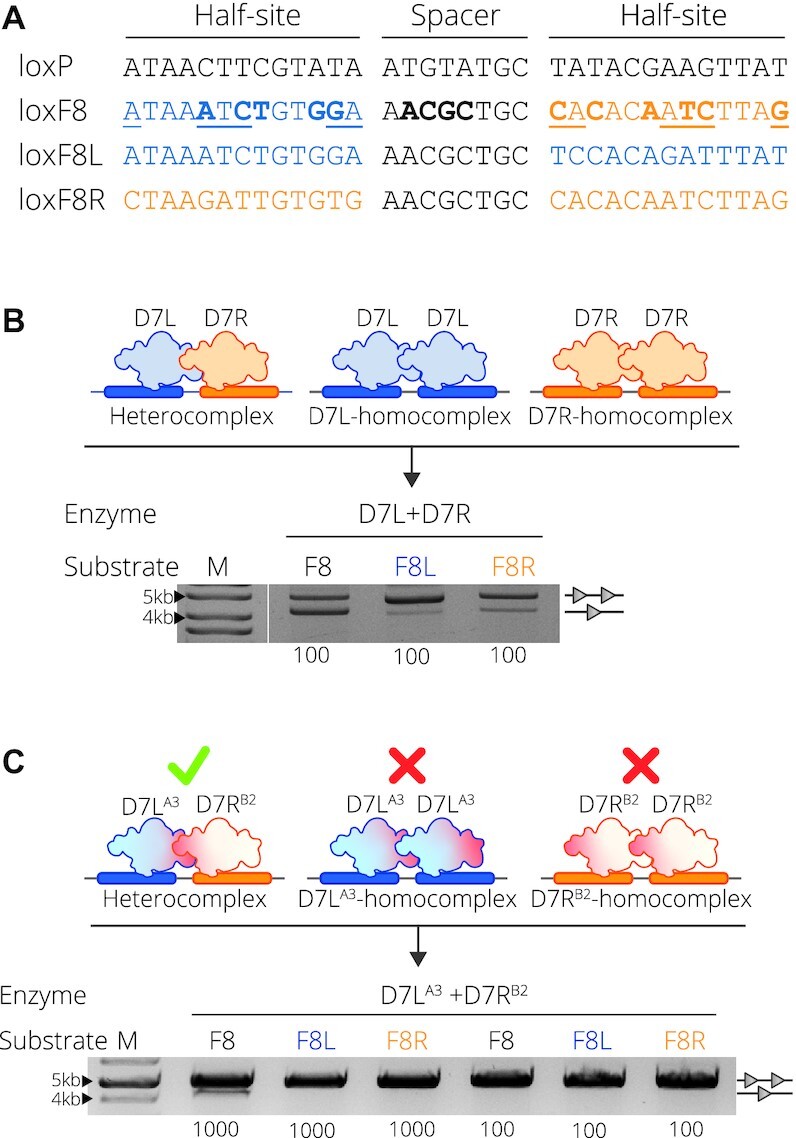
D7 target site substrates and recombination activity. (A) Full target sequences of loxP, loxF8, loxF8L (blue) and loxF8R (orange) separated into the two half-sites flanking the spacer sequence. The asymmetric loxF8 site is displayed with the left half-site as blue and the right half site as orange correlating to the full symmetric sites loxF8L and loxF8R, respectively. Underlined nucleotides in the sequence represent asymmetric positions and the bold nucleotides represent positions differing from the loxP sequence. (B) Complex formation and resulting activity of the D7L and D7R monomers. Upper panel: D7L (blue recombinase) and D7R (orange recombinase) heterocomplex formation with the loxF8 asymmetric site (blue and orange rectangles) and the homocomplex of D7L and D7R with the symmetric sites of loxF8L (blue rectangles) and loxF8R (orange rectangles), respectively. Lower panel: Bacterial assay of recombination activity of co-expressed D7L and D7R (Enzyme) on target sites loxF8, loxF8L and loxF8R (indicated with Substrate F8, F8L and F8R, respectively) all induced with a concentration of 100 μg/ml l-Arabinose (listed along the bottom of the gel image). Recombination is signified by the band aligned with the single triangle and non-recombined events are indicated by two triangles. M = 10 kb ladder (GeneRuler DNA Ladder Mix 10 kb). (C) Upper panel: Schematic of the desired recombination events when mutations (indicated in red) are applied to the molecules. Mutations from the group A3 are applied to D7L and B2 is applied to D7R indicated by the superscript (20). Desired recombination is depicted as such: when both mutated monomers are present, they are able to form an active heterocomplex (green check) with the asymmetric loxF8 site (blue and orange rectangles); in isolation they cannot form functional homotetrameric complexes (red X) with symmetric sites. Lower panel: recombination activity of D7LA3 and D7RB2 on the three substrates. Concentration (μg/ml) of L-Arabinose used for induction is indicated along the bottom. M = 10 kb ladder (GeneRuler DNA Ladder Mix 10kb).
Significant progress has been made to engineer novel tyrosine SSRs capable of recombination on a range of DNA substrates (13–15), including non-symmetric sites (8,9,16). Approaches focusing on the modular nature of the recombinases have been effectively employed to alter specificity in both serine and tyrosine recombinases. With the use of protein fusions to zinc-finger or TAL-effector domains, fused serine recombinases demonstrated an altered specificity to target novel sequences (17–19). For tyrosine SSRs, the recombination of non-symmetric target sites was first demonstrated by co-expression of wild-type Cre and a Cre variant that together catalyzed recombination between artificial asymmetric loxP-loxM7 sites, demonstrating proof of concept that engineered heterospecific SSRs can be generated (16). This general principle has recently been extended to achieve recombination between asymmetric target sequences naturally occurring in the human genome by combining two evolved Cre variants (8). The Cre-type molecules, each with unique half-site specificities, were first generated through directed evolution on their respective symmetric sites. The distinct variants were then expressed together forming a functional heterotetramer capable of specifically excising a DNA fragment flanked by the desired asymmetric human target sites (8). More recently, this approach was used to correct a chromosomal inversion causing a genetic human disorder (Preprint) (9). By combining two designer-recombinases targeting the asymmetric loxF8 sequence located on the human X-chromosome, Lansing et al. showed that the heterotetramer (D7) could efficiently correct the genomic int1h inversion to reestablish factor VIII expression in patient-derived cells (Preprint) (9). The D7 SSR is composed of two unique Cre-type subunits, D7L and D7R, each evolved to bind to their corresponding half site, loxF8L and loxF8R, respectively (Figure 1A).
Using more than one recombinase with different target specificities inherently carries risks. By using several Cre-derived recombinases with different specificities, there is an increased possibility of subunit assembly into undesired functional complexes, including homotetramers, and heterotetramers of unequal monomer combinations, that could cause recombination events at non-target sites. To mitigate these potential off-target effects, approaches to assure that only SSR heterotetramers with the desired monomer combination have catalytic activity are critical for increasing their safety in therapeutic applications (Preprint) (9). To improve the applied properties of the heterospecific D7/loxF8 recombinase system, we set out to identify mutations that render the monomers functionally inactive in isolation, while they can form active heterotetramers when co-expressed in cells.
MATERIALS AND METHODS
Plasmid construction
Previously described plasmids containing the target sites of loxF8, loxF8L and loxF8R were used for evolution (pEVO-loxF8, pEVO-loxF8L and pEVO-loxF8R, respectively) (Supplementary Figure S1A) (Preprint) (9). To verify the presence of the vector within the bacteria, the vector contains a chloramphenicol resistance gene driven by the CAT promoter. The mutations published by Zhang et al. (20) were introduced into the sequence of both D7 subunits through DNA fragments synthesis (Twist Bioscience) with mutations A3 (K25R, D29R, R32E, D33L, Q35R, R337E, E123L) applied to D7L and mutations B2 (E69D, R72E, L76E, E308R) applied to D7R (Preprint) (9). The synthesized fragments were inserted into the pEVO vectors in two cloning steps using standard restriction enzyme digest and ligation (enzymes purchased from NEB). D7LA3 was cloned with the restriction sites SacI and XhoI. D7RB2 was subsequently cloned to pEVO-D7LA3 with restrictions sites BsrGI and XbaI (SacI-HF, XhoI, BsrGI-HF and XbaI restriction enzymes purchased from NEB). Once the correct sequences were confirmed by Sanger sequencing with primers p1 and p2 (Supplementary Table S1), both molecules were subcloned into the three different pEVO vectors containing the target sites of loxF8, loxF8L and loxF8R with the restriction enzymes SacI-HF and SbfI-HF (NEB) (Supplementary Figure S1A).
The vector used for library analysis, pEVO-lacZ, was adapted from a previously described plasmid (7) (Supplementary Figure S2A). Symmetric target sites loxF8L (top strand 5′-3′ ATAAATCTGTGGAGCATACATTCCACAGATTTAT) and loxF8R (top strand 5′-3′ CTAAGATTGTGTGGCATACATCACACAATCTTAG) were added with the spacer sequence of loxP to prevent the recombination of the symmetric sites with the loxF8 site. The symmetric target sites flank two strong transcriptional terminators (21). Upon recombination, the removal of the terminator sequences allows for the transcription of the lacZα fragment, which is driven by the constitutive- polycistronic CAT promoter and translation is initiated by an additional ribosomal binding site (RBS) (Supplementary Figure S2). Cells expressing the lacZα fragment were identified on plates containing 20 mg/ml X-Gal (Sigma-Aldrich).
Library design
The starting library was constructed by targeting the A3 and B2 positions with an adapted method of incorporating synthetic oligonucleotides via gene reassembly (ISOR) (22). To reduce library complexity, we limited diversity to a subset of positions located along the largest monomer-monomer interface: from A3, positions 25, 29, 32 and 33; from B2, positions 69, 72 and 76 (Supplementary Figure S3A), while the remaining positions were unchanged to encode for the original D7 amino acid sequence. At each of the diversified positions, mutations were limited to a subset of amino acids previously predicted for the interface redesign (D, E, H, I, K, M, N, Q, S, A, G, L, P, T, V, R) (20). The mutations were incorporated via oligonucleotides designed with a combination of degenerate codons (VNS, GHW and MDG) to encode for a more even distribution of amino acids in the library compared to the standard NNN or NNS degenerate codons (primers 5–20 Supplementary Table S1). VNS encodes 16 possible amino acids (D, E, H, I, K, M, N, Q, S, A, G, L, P, T, V, R), GHW encodes four possible amino acid variants (D, E, A, V) and MDG encodes five amino acid variants (K, L, M, Q, R). This results in an expression library of 60 000 possible residue combinations for D7L and 4000 residue combinations for D7R.
Substrate-linked directed evolution
Positive selection pressure for variants active on the asymmetric site (loxF8) and negative selection pressure for the variants inactive on the symmetric sites (loxF8L and loxF8R) were achieved through a modified method of substrate-linked directed evolution (5) (Supplementary Figure S3A–D). Each cycle of evolution involves the diversification of the two libraries through error prone PCR (MyTaq DNA Polymerase, Bioline) and selection of the mutant D7L and D7R pairs with the desired activity on the presented target site.
To begin the evolution cycles, the diversified libraries are first cloned together into the pEVO containing the target site with the restriction sites of SacI (located directly upstream of the D7L recombinase sequence) and SbfI (located downstream of the D7R recombinase sequence) (SacI-HF and SbfI-HF were purchased from NEB) (Supplementary Figure S3A, B). The vector is then transformed into electrocompetent XL1-Blue Escherichia coli and grown overnight in LB/arabinose to induce recombinase expression. The vectors are subsequently purified from the over-night culture (GeneJET Plasmid-Miniprep-Kit, Thermo Scientific) where the sample now contains a mixture of recombined and non-recombined vectors.
Selection iterated between positive and negative selection. Positive selection selects for variants active on loxF8 (Supplementary Figure S3 B (5a. Selection for active variants)) and negative selection selects variants displaying no activity on loxF8L or loxF8R (Supplementary Figure S3 B (5b. Selection for inactive variants)). To perform positive selection for loxF8 recombination, the purified plasmid was digested with enzymes NdeI and AvrII (NEB) to linearize all non-recombined variants (Supplementary Figure S3C). A PCR was then performed with primers p1 and p3 to amplify only the clones that performed recombination (Supplementary Figure S3C). Negative selection was achieved by having a primer that could bind between the symmetric target sites (primer p4) amplifying only those recombinases that have not carried out recombination (Supplementary Figure S3D). The selected variants (either active variants resulting from the positive selection, or inactive variants resulting from the negative selection) were then carried on to the next cycle by cloning into a fresh pEVO vector with the desired target sequences.
To evolve variants active on the asymmetric loxF8 site and inactive on the symmetric loxF8L and loxF8R sites, selection was cycled between the three target sequences after each evolution iteration. i.e. round one of evolution would select for the variants active on loxF8. Round two of evolution would select for variants inactive on the loxF8L site and round three of evolution would select for variants inactive on the loxF8R site (Figure 2A and Supplementary Figure S3B). Recombination efficiency was monitored through the plasmid-based activity assay (Supplementary Figure S1B).
Figure 2.

Directed evolution and analyses of obligate D7 SSRs. (A) Selection scheme to evolve D7L (blue) and D7R (orange) paired variants that are active on the asymmetric loxF8 target site (blue and orange rectangles), but inactive on the symmetric loxF8L (blue rectangles) or loxF8R (orange rectangles) target sites. The circular arrow indicates the 26 iterative selection cycles that were employed. (B) Comparison of recombination activity of the start library and the end library after 26 cycles of directed evolution and selection. Employed vectors with the target sites loxF8 (F8), loxF8L (F8L, blue) and loxF8R (F8R, orange) are shown. Bands of non-recombined plasmids are indicated by a line with two triangles, while recombined bands are marked by a line with one triangle. Concentration (μg/ml) of l-Arabinose used for recombinase induction is indicated along the bottom. M = 10 kb ladder (GeneRuler DNA Ladder Mix 10 kb). (C) Mutation analysis of candidate obligate D7 SSRs. The frequency of altered residues at a given position is shown in percent for the D7L (blue) and D7R (orange) clones. Positions with a mutation frequency over 20% are highlighted and labelled (blue for residue positions in D7L and orange for residue positions in D7R). The amino acid most commonly occurring in the highlighted positions are listed to the side of the bar. The height represents the occurrence of the amino acid compared to other variants in that position. (D) Activity of D7L and D7R recombinases expressed in isolation compared to activity of D7LK201R and D7RQ311R, respectively, on target sites loxF8, loxF8L and loxF8R. Bands of non-recombined plasmids are indicated by a line with two triangles, while recombined bands are marked by a line with one triangle. Concentration (μg/ml) of l-Arabinose used for recombinase induction is indicated along the bottom. M = 10 kb ladder. (E) Activity of D7L + D7R compared to D7LK201R+ D7RQ311R on the asymmetric site loxF8 and symmetric sites loxF8L and loxF8R. Bands of non-recombined plasmids are indicated by a line with two triangles, while recombined bands are marked by a line with one triangle. Concentration (μg/ml) of L-Arabinose used for recombinase induction is indicated along the bottom. M = 10 kb ladder. (F) Quantification and reproducibility of recombination in E. coli. Recombination (in %) calculated from measuring band intensities as shown in D/E on indicated target sites are shown for D7L + D7R (light gray) and D7LK201R + D7RQ311R (dark gray). Recombination as percentage along the y-axis. Bacterial assays were done in triplicates (n = 3) plotted as points on the bar graphs, the error bars indicate the standard deviation from the mean and statistical relevance of the triplicates was assessed using an unpaired t-test, non-corrected for multiple comparisons. (ns): P > 0.5, (*): P ≤ 0.05, (**): P ≤ 0.01, (***): P ≤ 0.001, (****): P ≤ 0.0001.
Recombinase activity assay: plasmid-based and blue-white screen
To visualize the recombination activity of recombinase clones or recombinase libraries on the target site of interest, a plasmid-based assay was used as previously described (5,8,9) (Supplementary Figure S1B). To quantify the recombinase activity, the band intensities were determined with GelAnalyzer 19.1 (GelAnalyzer 19.1 (www.gelanalyzer.com)) using the ladder bands as a point of reference. Experimental variation between lanes was normalized by measuring band percentage where the value of each band is expressed as a percentage of the sum of all the bands in that lane. The quantified recombination was plotted in R 4.0.3 (R Core Team, 2018) with dplyr v1.0.7 (https://dplyr.tidyverse.org/) and visualized with ggplot2 v3.3.5 (https://ggplot2.tidyverse.org/). Bacterial test digests were done in triplicates (n = 3) and statistical relevance of the triplicates was assessed using an unpaired t-test, non-corrected for multiple comparisons.
For activity analysis and selection of active variants of the final library, a blue-white activity screen was used (Supplementary Figure S2A–C). The library was ligated to the pEVO-LacZ plasmid with SacI and SbfI restriction sites (SacI-HF and SbfI-HF restriction enzymes purchased from NEB). The plasmid pEVO-LacZ contains restriction enzyme sites NdeI and AvrII flanked by the on-target asymmetric loxF8 site (NdeI and AvrII restriction enzymes purchased from NEB). Upon on-target recombination, the restriction sites are excised. To select for recombinase variants that are active on the loxF8 site, the purified plasmids are digested with NdeI and AvrII (NEB), linearizing the plasmids containing inactive recombinase pairs. The resulting digest is either transformed or amplified with primers outside of the restriction sites (primers p1 and p3), removing the variants inactive on loxF8. Additionally, located upstream of the LacZα fragment, the pEVO-LacZ plasmid has two transcriptional terminators that are flanked by the symmetric off-target loxSYM sites (either loxF8L or loxF8R). Upon off-target recombination the transcriptional terminators are excised, permitting transcription of the LacZα fragment, turning the colony blue indicated on X-Gal plates. Therefore, the remaining white colonies either represent an inactive recombinase pair or a pair that is highly specific for loxF8.
To eliminate the inactive recombinases, the library was induced overnight with low levels of arabinose (10 ug/ml l-Arabinose, Sigma-Aldrich) prior to the blue-white screening. The purified plasmids were digested with NdeI and AvrII (NEB), both unique restriction sites flanked by the loxF8 target sites (Supplementary Figure S2 A), to linearize plasmids with recombinase pairs that are inactive on loxF8. Remaining undigested plasmids were then retransformed and induced with a higher (100 ug/ml l-Arabinose, Sigma-Aldrich) level of arabinose to allow for sensitive detection of low-level symmetric site activity. Once plated on X-Gal and IPTG (Sigma-Aldrich) indicator plates containing chloramphenicol and arabinose (Sigma-Aldrich), the activity of the recombinase variant was read as blue or white. White colonies were then selected, where white indicates that the colony carries a mutant displaying activity on the loxF8 site and no activity on the symmetric site (Supplementary Figure S2B). Subsequently a colony PCR was performed with primers binding upstream of the symmetric site (p23) and downstream of the asymmetric site (p24) to amplify over the target sites to confirm the proper activity on both sites. A band of size 1.7 kb indicated the desired activity profile (Supplementary Figure S2C).
Sequencing analysis
Clones selected from the blue/ white screen were further analyzed to determine the mutational changes in the monomer subunits that had evolved in selected recombinase pairs of D7L and D7R. To determine which mutations were occurring at the highest frequency for each mutated recombinase, the sequences of the D7L mutants and the D7R mutants were compared to the respective D7L and D7R sequences. The sequencing data was handled in R 4.0.3 (R Core Team, 2018) with dplyr v1.0.7 (https://dplyr.tidyverse.org/) and visualized with ggplot2 v3.3.5 (https://ggplot2.tidyverse.org/).
Expression in mammalian cells and recombination efficiency in mammalian cells
A fluorescence-based reporter assay was used to determine the recombination properties of the obligate monomers as described elsewhere (Preprint) (9). 350.000 HEK293T cells were seeded per well on a 12-well plate the day before transfection. Combinations of mRNA, each encoding for one recombinase variant (i.e. D7L, D7R, D7LK201R or D7RQ311R) along with mRNA encoding a blue fluorescent protein (BFP) were co-transfected into a HEK293T reporter cell line containing integrated lox sites flanking a puromycin resistance cassette (Supplementary Figure S4A). Upon recombination of these sites a downstream monomeric red fluorescent protein (mCherry) is expressed. 48 h after transfection the recombinase activity was quantified via FACS using the MACSQuant VYB flow cytometer (Miltenyi Biotec). Recombination was quantified by determining the percentage of cells displaying red fluorescence within the blue fluorescence population. Cell populations were grouped with forward scatter (FSC) and side scatter (SSC) to group the total cell population and then the single cells. Cells expressing BFP were grouped with forward scatter and BFP (Supplementary Figure S4B). From the BFP population, cells expressing mCherry were grouped with forward scatter and mCherry expression (Supplementary Figure S4C). The recombination efficiency was plotted with GraphPad Prism version 8 (GraphPad Software, La Jolla California USA, www.graphpad.com) and statistical relevance of the biological triplicates was assessed using an unpaired t-test, non-corrected for multiple comparisons.
HEK293T cell culture
HEK293T cells were cultured using DMEM (Gibco) supplemented with 10% FBS (Capricorn Scientific) and 1% Penicillin–Streptomycin (ThermoFisher) in a 12-well format. When reaching a confluency of 90% the cells were split. Each well was washed once with PBS and 100 μl of Trypsin (Gibco) was added. After incubation for 3 min at 37°C the detached cells were collected in a 15 ml tube. Cells were counted with the Countess 3 FL Automated Cell Counter (ThermoFisher) and seeded at a density of 75.000 cells/well in 1 ml medium for maintenance. For transfection the cells were seeded at a density of 350.000 cells/well in 1 ml medium.
mRNA transfection
HEK293T cells were transfected 24 h after seeding. For each transfection reaction a 1.5 ml tube was prepared with a total of 300 ng of mRNA (100 ng tagBFP mRNA and 200 ng recombinase mRNA) (Supplemental Figure S4 A). 100 μl Opti-MEM I Reduced Serum Media was mixed with 1.5 μl Lipofectamine MessengerMax (ThermoFisher) and added to the mRNA sample. The mixture was briefly vortexed at maximum speed and incubated for 15 min at RT. In the meantime, the medium of the cells was replaced with fresh medium. The transfection mixture was then added to the cells. The next day the medium was changed and the cells were analyzed 2 days post transfection.
PCR-based genomic inversion detection
The inversion of the loxF8 locus after treating HEK293T cells with the D7 recombinase dimer was detected as described previously (Preprint) (9).
In vitro transcription (IVT)
mRNA was produced using the HiScribe™ T7 ARCA mRNA Kit (NEB) and purified with the Monarch RNA Cleanup Kit (NEB) following the manufacture's manual. The D7 recombinase dimer and tagBFP templates for the IVT were generated as previously described (Preprint) (9). The templates for the different Cre variants were generated using primer p21 and primer p22 (Supplementary Table S1). mRNA aliquots of 4 μg were stored at -80°C for up to 6 months.
Molecular modeling and dynamics simulation
The synaptic three-dimensional (3D) structure of the Cre/loxPA5′(TS)A1P complex used for the modelling was taken from the Brookhaven protein data bank (PDB ID 3C29, 2.2 Å, DOI: 10.2210/pdb3C29/pdb). Given the symmetry of this homotetrameric complex, and for simplification of the in-silico studies, only two Cre monomers (chains A and B) and one loxP molecule (chains C and D) were considered. Missing side chain atoms of residue K201 not resolved in the crystal structure were modeled as follows: first, a lysine side chain was modeled in PyMOL v2.2.0 (Schrödinger, LLC), and a rotamer search was applied in order to find an energetically favorable conformer (later on further refined by molecular dynamics (MD); see below). Rotamer selection was guided by the 3D disposition of the K201 residue in another crystal structure available in the PDB that includes density for this side chain (i.e. the pre-cleavage Cre/loxP complex structure PDB ID 1Q3U (2.9 Å)). PyMOL was used to model the corresponding mutations on Cre/loxPA′(TS)A1P to obtain the structure of the wild-type (wt) system (Figure 5A, B) and the following Cre mutants: CreK201R(A)-K201R(I), CreQ311R(A)-Q311R(I), CreK201R(A)-Q311R(I), CreQ311R(A)-K201R(I), denoting by (A) and (I) the active and inactive monomer, respectively (Figure 5C, Supplementary Figure S5C–F). The structures of Crewt and mutants in complex with loxP were energy refined by MD simulations in AMBER20 (23) (https://ambermd.org/) using ff14SB and bsc1 force fields for protein and DNA, respectively. Details of the MD protocol applied are given in Supplementary Materials (MD Section). MD trajectories were recorded every 10 ps, visualized with VMD (24) and evaluated in terms of intermolecular H-bonds by using the CPPTRAJ module implemented in AMBER. A hydrogen bond occupancy >10% with a distance acceptor-donor cutoff of 3.5 Å and a 120° angle were taken as criteria for dynamic hydrogen bond formation in the last 100 ns of each MD simulation. Data analysis was performed with Origin2019b (Origin 2019b (2019) OriginLab, Northampton, MA, available: http://www.originlab.com). Figures were created in PyMOL v2.2.0 (Schrödinger, LLC).
Figure 5.

Structural details of the studied recombinase/DNA system. (A) MD-refined structure of the Cre/loxP X-ray complex (PDB ID 3C29). The loxP DNA (dark gray cartoon in ladder representation) is shown in complex with two Cre monomers (active (A) and inactive (I) in orange and blue cartoons, respectively). Residues K201 and Q311 are depicted in CPK. The dashed box marks the region zoomed in and shown in detail in panel C. (B) Sequence of the loxP site with top (TS) and bottom strand (BS) labelled. Arrows indicate cleavage points by the catalytic tyrosine (Y324) in each DNA strand for the selected PDB structure, with the first cut indicated by the red arrow. Bases in the spacer region are numbered at the bottom. (C) Detail of the superimposition of the MD-refined structures (taken from last 50 ns of simulation) of wild-type Cre/loxP and mutant CreQ311R(A)-K201R(I)/loxP. In the wild-type complex, Cre active (A) monomer is shown in orange, inactive (I) in blue and the DNA in dark gray, whereas for the mutant, the active monomer is shown in ochre, the inactive in gray-blue and the DNA in light gray. For clarity, cartoon representations are shown with transparency, and side chains of relevant residues are in balls and sticks and labelled. Intermolecular H-bonds are depicted with dashed lines (ochre and light gray for the active and inactive monomers, respectively). The green check mark at the bottom of the panel indicates that the CreQ311R(A)-K201R(I) mutant is active. (D) Reversing the mutations yields an inactive D7 recombinase. Recombination activity of D7L + D7R compared to D7LK201R+ D7RQ311R and D7LQ311R+ D7RK201R on target the site loxF8 is shown. Bands of non-recombined plasmids are indicated by a line with two triangles, while recombined bands are marked by a line with one triangle. Concentrations of l-Arabinose (μg/ml) are listed along the bottom. M = GeneRuler DNA Ladder Mix 10kb.
RESULTS
Monomer–monomer interface mutations reduce recombination activity
To form obligate Cre-type SSR heterotetrameric complexes for asymmetric substrates, previous work has focused on redesigning the protein-protein interface of the interacting monomers (16,25). In particular, Zhang et al. have engineered an obligate Cre heterotetramer to recombine the artificial asymmetric loxM7/loxP target sequence (20). Key positions for the interface redesign were selected from predicted mutations that would form an alternative interaction surface between the mutated Cre molecules. To investigate whether the same interface mutations can be adopted for other heterospecific Cre-type SSRs, we tested the effect of the mutations on the D7 designer-recombinase that is composed of two unique Cre-type subunits, D7L and D7R, capable of targeting the asymmetric sequence of loxF8 (Preprint) (9). We generated D7-variants by mutating the two subunits to potentially form an obligate heterotetrameric D7 recombinase. Therefore, the D7L variant (D7LA3) was generated by introducing the published obligate interface mutations K25R, D29R, R32E, D33L, Q35R, E123L and R337E, whereas the D7R variant (D7RB2) harbored the mutations E69D, R72K, L76E and E308R (20).
To assess the effects of the obligate mutations, it is necessary for the mutants to form catalytically incompatible homotetramers. This can be observed by the loss of recombination on the symmetric sites loxF8L and loxF8R that require the formation of D7L and D7R homotetramers, respectively. Additionally, the desired obligate activity would result in recombination of the asymmetric loxF8 site comparable to that of the original D7 recombinase. In order to compare the recombination efficiency before and after the applied mutations, first, non-mutated D7L and D7R were inserted together to a DNA plasmid harboring target substrates (either the loxF8, loxF8L or loxF8R target site) and expressed in bacteria (Figure 1B and Supplementary Figure S1B). As expected, co-expression of both monomers, D7L and D7R, resulted in efficient recombination on the asymmetric loxF8 target site (Figure 1B). Recombination was also observed for both the symmetric loxF8L and loxF8R target sites (Figure 1B), presumably due to functional homotetramer formation of their corresponding evolved recombinase (Figure 1B). Introduction of the interface mutations (D7LA3 and D7RB2) should prevent the homotetramer assembly and, thereby, block recombination on the symmetric loxF8L and loxF8R sites, while the different subunits should still be able to form active heterotetramers on the asymmetric loxF8 sequence (Figure 1C). Indeed, co-expression of D7LA3 and D7RB2 did not lead to detectable recombination from vectors carrying the symmetric loxF8L or loxF8R sequences (Figure 1C), indicating that these mutations prevented the formation of active homotetramers. Unfortunately, recombination on the asymmetric loxF8 site also had no detectable recombination events at a recombinase induction level of 100 μg/ml l-Arabinose, with low (∼9%) recombination activity detectable at substantially increased SSR expression level (1000 μg/ml L-Arabinose, Figure 1C). Hence, the applied mutations worked in principle, but also led to reduction in recombination activity of the D7 heterotetramer. Therefore, we sought to recover the activity by searching for combinations of amino acid changes more suitable for an obligate D7 system.
Substrate-linked directed evolution to evolve obligate D7 recombinases with high activity
To search for beneficial residue changes we began by building two targeted libraries for the D7L and D7R recombinase variants around the previously described (20) mutated interface positions (A3 – K25R, D29R, R32E, D33L, Q35R, E123L and R337E, and B2 – E69D, R72K, L76E and E308R) involved in the protein-protein interface where the remaining non targeted amino acid positions were kept as the original D7 sequence. The libraries where then applied to the well-established substrate-linked directed evolution (SLiDE) procedure (5,22). The two D7L and D7R starting libraries were cloned together into the evolution vector (pEVO) to begin iterative positive selection for activity on the asymmetric site (loxF8) and negative selection on the symmetric sites (loxF8L and loxF8R) through a modified version of SLiDE (Figure 2A and Supplementary Figure S3A–D). By evolving the two libraries together instead of in parallel, we were able to select for functional pairs with the desired activity profile. After 26 cycles of SLiDE, we detected a marked increase in recombination activity for the asymmetric site and negligible recombination on the symmetric sites (Figure 2B), indicating that heterodimeric pairs of recombinases with desirable features had evolved.
To eliminate any carry over of inactive recombinase variants that had leaked through selection, single variant pairs were assessed by using a blue-white colony screen. The selection plasmid (pEVO-LacZ) allowed for simultaneous identification of variants that did not recombine the symmetric sites while showing activity on the asymmetric loxF8 site (Supplementary Figure S2). 75 white colonies were selected, and the encoded recombinase pairs were sequenced. Surprisingly, none of the hypermutated amino acid positions (69, 72 and 76) were found to be enriched in the evolved D7R recombinases. In contrast, the glutamine at position 311 was changed in 41% (31 of 75 variants) of the sequenced D7R recombinases, where 87% (27 of 31) of the mutants displayed an arginine at this position (Figure 2C, Supplementary Tables S2 and S3). These results indicate that the initial targeted residues did not lead to the desired outcome and, instead, a single substitution that had occurred randomly during evolution was preferred (Q311R), presumably preventing recombination on the symmetric loxF8R site while maintaining activity when co-expressed with D7L variants on the asymmetric loxF8 sequence.
Sequencing the D7L-derived clones uncovered five positions that were mutated in more than 20% of the sequenced clones (positions 25, 29, 201, 282 and 305, Figure 2C, Supplementary Tables S2 and S3). Positions 25 and 29 most likely derived from the initial targeted residues involved in protein-protein interactions, while positions 201, 282 and 305 were not targeted in the start library, implying that they arose through random mutations and selection during directed evolution.
The most surprising result was the frequently mutated position 201, which was found to be changed in 48% (36 out of 75) of the clones, with all harboring an arginine at this position rather than a lysine (Figure 2C, Supplementary Tables S2 and S3). What makes this alteration so interesting is that lysine 201 is highly conserved throughout the tyrosine SSR family (26) and it has been described as essential for the catalytic activity of Cre (27–29) by facilitating DNA cleavage during recombination, coordination of activity of the four monomers in the tetramer, and Holliday junction formation (27,30). Hence, recombinases with alterations at position 201 would not be expected to function. On the basis of these observations, we decided to further explore if mutating the catalytic K201 residue in D7L would inactivate the SSR when expressed as a monomer and, if that was the case, recombinase activity could be rescued by the presence of the paired Q311R mutation on the D7R monomer.
To determine the effects on recombination of the mutations applied to each monomer, we first evaluated their ability to recombine their original targets when expressed in isolation as monomers. Sole expression of D7LK201R or D7RQ311R on the symmetric loxF8L or loxF8R sites, respectively, did not lead to detectable recombination, demonstrating that these mutations inactivate the enzymes when expressed in isolation (Figure 2D). Furthermore, D7LK201R or D7RQ311R were inactive on the asymmetric loxF8 site when expressed in isolation (Figure 2D). In sharp contrast, when D7LK201R and D7RQ311R were co-expressed, efficient recombination on the asymmetric loxF8 was observed (Figure 2E, F), whereas no recombination was detectable on the symmetric loxF8L and loxF8R sites (Figure 2E-F). Importantly, loxF8 sites were recombined at a similar rate compared to the original D7 clone (Figure 2F), indicating that the two mutations did not considerably compromise activity of the overall recombinase complex. To test if D7LK201R together with D7RQ311R could form other active heterotetramers, we co-expressed the mutants across a panel of substrates with different half-site arrangements of loxF8 (Supplementary Figure S6A). None of these combinations showed appreciable recombination in comparison to the original loxF8 sequence (Supplementary Figure S6B). Therefore, by pairing one mutation applied to each recombinase monomer we obtained an obligate D7 SSR complex with comparable activity to the original D7 SSR.
Combined D7LK201R and D7RQ311R support obligate recombination in mammalian cells
Because the D7 recombinase is targeted for applications within the human genome, our next step was to examine the activity of the obligate D7LK201R + D7RQ311R complex in human cells. To allow straight-forward quantification, we measured recombination efficiency in a HEK293T reporter cell line (Preprint) (9). The reporter cell line was co-transfected with mRNA carrying the recombinases along with an mRNA coding for tagBFP to monitor transfection efficiencies (Figure 3A). Transfection of the obligate D7LK201R + D7RQ311R molecules revealed a recombination efficiency of 83% compared to 80% of wild-type D7 (Figure 3B, C), implying that the introduction of the obligate mutations did not compromise recombinase activity in human cells. Importantly, transfecting each mutant monomer subunit in isolation did not yield in loxF8 target site recombination (Figure 3B, C).
Figure 3.

Obligate D7LK201R+ D7RQ311R activity in mammalian cells. (A) Schematic presentation of mRNA expression constructs and the reporter HEK293T cell line. Employed mRNAs with indicated features (5′cap and polyA tail) expressing a nuclear localization signal (NLS) fused to the recombinase and the tagBFP mRNA are shown. The stable reporter cell line harbors two loxF8 sites (blue/orange triangles) that flank a puromycin selection gene (puro). Once successfully excised by recombination, mCherry is expressed from the SFFV promoter (arrow). (B) Recombination assays in mammalian cells. Representative brightfield and mCherry fluorescent images of reporter HEK293T cells transfected with indicated recombinases. The 100 μm scale bar is indicated. (C) Quantification of the recombination efficiencies 48 h after transfection of HEK293T reporter cells with indicated recombinases, analyzed by FACS (Error bar represents the SD of experiments performed in biological triplicates, n = 3). Recombination efficiency determined by percent of cells expressing mCherry of the total of BFP positive cells. (†) indicates normalization to BFP signal. (*) and (****) indicate P < 0.05 and P < 0.0001, respectively. (D) Scheme of the genomic inversion detection PCR. The WT orientation is indicated by the amplicon produced by primers colored in blue and gray. The inverted orientation is indicated by primers in orange and gray. Exon 1 of the factor VIII gene and the genomic loxF8 target sites are indicated. (E) Gel showing PCR results with primers detecting the WT orientation (top panel) and primers detecting the inverted orientation (bottom panel). Results for transfections with indicated mRNAs are shown. Controls included HEK293T cells that were transfected with BFP mRNA only (non-treated), int1h patient DNA carrying the inversion of exon1 (Inverted-Ctrl), non-treated cells (WT-Ctrl) and water only (H2O). M = GeneRuler DNA Ladder Mix 10 kb (F) Sequences of predicted off-target sites in the human genome with high sequence similarity to loxF8L (HG1L and HG2L) and loxF8R (HG1R and HG2R). Bold nucleotides indicate nucleotides differing from the loxF8 target sequence and underlined nucleotides indicate asymmetry. (G) Bacterial assay for recombination activity of D7L + D7R compared to the activity of D7LK201R+ D7RQ311R on the indicated human off-target sites. Recombination activity on loxF8 is shown as control. Bands of non-recombined plasmids are indicated by a line with two triangles, while recombined bands are marked by a line with one triangle. 100 (μg/ml) of l-Arabinose used for recombinase induction is indicated along the bottom. M = GeneRuler DNA Ladder Mix 10 kb.
The D7 recombinase was generated to correct the genomic int1h inversion frequently found in hemophilia A patients (31). The enzyme recognizes two loxF8 sequences that are found on the human X-chromosome at a distance of 140 kb from one another. The first site is present in intron 1 of the factor VIII gene, and the second site is located 130 kb upstream of the factor VIII transcription start site (9,32,33). The D7 SSR has been shown to efficiently invert the displaced exon 1 sequence flanked by the loxF8 target sites upon expression in human cells (Preprint) (9). To confirm the ability of the D7LK201R + D7RQ311R variants to act on these sites at the endogenous locus, we extracted genomic DNA from HEK293T cells transfected with D7LK201R and D7RQ311R mRNAs and ran a PCR based assay designed to detect the inversion of exon 1 (Figure 3D). Indeed, expression of D7LK201R + D7RQ311R led to inversion of the genomic fragment (Figure 3E), demonstrating that the obligate mutations did not interfere with the enzyme's activity to recombine this disease-causing inversion.
To evaluate if the D7LK201R + D7RQ311R heterodimer improved target site specificity, we analyzed its activity on four predicted human off-target sites (Figure 3F) by employing a plasmid-based activity assay (Supplementary Figure S1B). Consistent with previous data (10), the wild-type D7 recombinase displayed no detectible activity on 3 of the 4 sequences, but it showed activity on the HG2L off-target site (Figure 3G). In comparison, the obligate D7LK201R + D7RQ311R complex showed no detectable activity on all four of the predicted off-target sites (Figure 3G), demonstrating its improved applied properties. Together, these results show that the D7LK201R + D7RQ311R heterodimer promotes target site specificity while maintaining comparable recombination efficiency for the loxF8 target site in mammalian cells.
K201R and Q311R mutations render Cre and Vika recombinases obligate
To explore a more general applicability and obtain insights into the molecular mechanism of this obligate SSR system, we investigated the phenotype of the corresponding mutations in two naturally occurring homotetrameric SSR complexes, namely Cre/loxP and Vika/vox (34). To test the system, CreK201R and CreQ311R were generated. The obligate mutations were also incorporated into Vika at positions 219 and 330 according to the conserved sequences seen in its sequence alignment with Cre (34), giving rise to two mutant monomers, namely VikaK219R and VikaQ330R. Their activity was analyzed on an excision substrate in E. coli. When CreK201R or CreQ311R were expressed in isolation, no recombination was observed on the loxP targets (Figure 4A). In sharp contrast, co-expression of the two variants rendered the system recombination competent, suggesting that this combination of mutations can convert recombinases to catalytically obligate systems. To test whether both mutations introduced into the same monomer result in recombination, we generated the double mutant CreK201R-Q311R. Expression of CreK201R-Q311R did not lead to detectable recombination (Figure 4A), indicating that the mutations have to be present on different monomers to allow for the formation of active SSR complexes.
Figure 4.

Application of obligate mutations to natural recombinase systems. (A) Introduction of point mutations renders Cre recombinase obligate. Activity on loxP of both mutations applied to a single subunit, CreK201R-Q311R; co-expression of CreK201R and CreQ311R; or expression as a single mutated subunit of CreK201R or CreQ311R in isolation are shown. The concentration of L-Arabinose (μg/ml) is listed along the bottom. Bands of non-recombined plasmids are indicated by a line with two triangles, while recombined bands are marked by a line with one triangle. M = GeneRuler DNA Ladder Mix 10kb. Quantified recombination of bacterial assay. Recombinases Cre, CreK201R+ CreQ311R, CreK201R and CreQ311R recombination (%) activity on loxP target site. Recombination as percentage along the y-axis. Bacterial assays are done in triplicates (n = 3) plotted as points on the bar graphs. (B) Recombination activity on the vox target site of Vika compared to Vika with the obligate correlating mutations of K291R (VikaK219R) in one subunit and Q330R (VikaQ330R) in the second mutated subunit. Bands of non-recombined plasmids are indicated by a line with two triangles, while recombined bands are marked by a line with one triangle. Concentration (μg/ml) of L-Arabinose used for recombinase induction is indicated along the bottom. M = 10 kb ladder. Quantified recombination of bacterial assay. Recombinases Vika, VikaK219R + VikaQ330R, VikaK219R and VikaQ330R recombination (%) activity on vox target site. Recombination as percentage along the y-axis. Bacterial assays are done in triplicates (n = 3) plotted as points on the bar graphs. (C) Scheme of mRNAs and the employed HEK293T loxP reporter cell line. mRNAs with indicated features (5′cap and polyA tail) expressing a nuclear localization signal (NLS) fused to the recombinase and the tagBFP mRNA are shown. The stable reporter cell line harbors two loxP sites (gray triangles) that flank a puromycin selection gene (puro). Once successfully excised by recombination, mCherry is expressed from the SFFV promoter (arrow). (D) Evaluation of recombination efficiency by microscopy. Bright field and fluorescent mCherry cell images for cells transfected with indicated mRNAs are shown. Scale bar = 100 μm. (E) | FACS-based quantification of recombination efficiency. The percentage of mCherry-positive cells is shown for indicated mRNA transfections. (†) indicates normalization to BFP signal. Error bars represent the standard deviation from the mean of experiments performed in biological triplicates (n = 3) and statistical relevance of the triplicates was assessed using an unpaired t-test, non-corrected for multiple comparisons. (ns): P > 0.5, (*): P ≤ 0.05, (**): P ≤ 0.01, (***): P ≤ 0.001, (****): P ≤ 0.0001.
Co-expression of VikaK219R and VikaQ330R on the vox target sequence displayed a similar activity profile as seen in Cre (although with some loss of activity), whereas no recombination was observed when the mutated monomers were expressed in isolation (Figure 4B).
We next evaluated if the obligate Cre system could function efficiently in mammalian cells and maintain the recombination profile seen in bacteria. A HEK293T red fluorescent reporter cell line was transfected with SSR mRNAs to evaluate recombination activity (Figure 4C). When the Cre mutants were expressed alone, negligible recombination activity was detected (Figure 4D, E), signifying that CreK201R and CreQ311R expressed in isolation are inactive. In sharp contrast, co-expression of CreK201R and CreQ311R yielded a recombination efficiency of 80%, only marginally lower than the 90% recombination efficiency observed with wild-type Cre on loxP (Figure 4D, E). Together, these results demonstrate that the targeted mutations are not only applicable to the heterotetrametric D7/loxF8 complex, but can also be applied to obtain obligate systems of naturally occurring SSRs.
Molecular modeling and dynamics simulation reveal underlying mechanism driving obligate heterotetramer activity
In order to obtain insights into the molecular mechanism rendering the Cre recombinase system obligate, we generated 3D molecular models of CreK201R and CreQ311R bound to loxP based on the synaptic Cre/loxP co-crystal structure (PDB ID 3C29, Figure 5A). In this particular crystal structure, the active Cre monomer is poised to cleave the top strand (TS) of the loxP target site (Figure 5B) as observed in other crystallographic structures (i.e. PDB ID 1Q3U and 1NZB) (35). MD simulations of our models and comparative H-bond analysis at the catalytic site of the wild-type system and the investigated mutants revealed the molecular bases for catalysis in the obligate heterodimer and shed light on why the single mutants, CreK201R and CreQ311R are inactive (Supplementary Figure S5). For CreK201R, the catalytic R201 was dramatically shifted in the inactive monomer, which facilitated its interaction with the DNA backbone at base A2 of the bottom strand (BS) instead of base T5 on the top strand (TS) as it is observed in Crewt, which likely compromises recombination proficiency. Furthermore, the catalytic Y324 of the active monomer was found partially displaced from phosphate base T3′ TS and established van der Waals contacts with the phosphate base A4′ (Supplementary Figure S5 C). For the CreQ311R mutant, in the inactive monomer we observed that the catalytic Y324 was displaced (Supplementary Figure S5A), while K201 lost important interactions with the DNA backbone at the TS. Furthermore, in the active monomer, the catalytic residue H289, known to play an important role in recombination catalysis (36), lost the H-bond interaction with the phosphate at base A4′ TS in the catalytic site, compromising the catalytic activity of this mutant (Supplementary Figure S5D). For the Cre variant containing the K201R and Q311R mutations in the active and inactive monomer, respectively (CreK201R(A)-Q311R(I)), Y324 in the inactive monomer was found displaced and interacting with the phosphate at base C3 on the BS, while K201 in the same inactive monomer had moved dramatically to also interact with the DNA backbone at base C3 on the BS, and no recognition of T5 at the TS was observed (Supplementary Figure S5E), indicating that this mutational configuration does not constitute an active recombinase.
Lastly, the analysis of the CreQ3111R(A)-K201R(I) model (i.e. Q311R and K201R mutations introduced in the active and inactive monomer, respectively) showed that this configuration would be in agreement with an active enzyme, as all catalytic residues were properly positioned to allow recombination (Figure 5C, Supplementary Figure S5F). Hence, our theoretical models explained the obligate nature of these two mutations and their catalytic influence. Furthermore, they predicted the importance of their respective combination order (i.e. active versus inactive monomer) for constituting an active enzyme.
This structure-based prediction prompted us to reevaluate the D7 heterodimer and investigate whether the mutations could be swapped between D7L and D7R. According to the results obtained from the modelling and MD simulation, reversing the order of the mutations at residues 201 and 311 might not be compatible with recombination activity. Indeed, testing the mutant D7LQ311R + D7RK201R revealed that this swapped combination is inactive (Figure 5D). Together, these results provide a molecular explanation for the obligate nature and suggest that engineering of residues implicated in recombination catalysis is a powerful approach to improve the applied properties of SSRs.
DISCUSSION
By altering the DNA-specificity of Cre through engineering and directed evolution, distinct SSR variants can be generated that together recombine asymmetric target sequences as heterotetramers (5,7–9). The generation of such heterotetrameric SSR systems substantially broadens the potential sequences that can be targeted within genomes. However, possible combinations of different subunits could lead to active SSR byproducts capable of catalyzing off-target recombination. Previously, prevention of homotetramer formation was achieved through structure-guided redesign of several residues implicated in the protein-protein interface between the different recombinase monomers (20). Hence, this approach to generate obligate SSR systems may be difficult to implement if structural data of the enzyme is not available, and thus may not be easily adaptable to distantly related recombinases. Employing directed molecular evolution, we discovered that obligate SSR systems can be generated by mutating residues implicated in recombination catalysis. Importantly, this novel way of generating obligate SSRs only required the alteration of one conserved residue within each distinct SSR monomer. This simplified approach could potentially be applied to different engineered or natural SSRs even without prior knowledge of their 3D structures. The performed MD-based studies helped us to establish a working hypothesis and gave insights on plausible molecular aspects rendering the recombinase system functionally active. Nevertheless, additional aspects on the molecular mechanism triggering the catalytic reaction remain open and could be the focus of future investigations.
The selection and identification of mutations that can transform naturally occurring SSRs into a functionally obligate system, as demonstrated for the wild-type Cre and Vika recombinases, could be applied in more sophisticated genetic and synthetic biology studies. Numerous conditional knockout mouse models that have been generated are based on the Cre/loxP system (37,38). Typically, animals carrying the floxed allele are crossed with mice expressing Cre from a tissue specific promoter to achieve inactivation of the gene in a particular organ or cell-type. This approach could be further refined by expressing CreK201R and CreQ311R from two different promoters. Here, deletion of the gene would only happen in cells where both promoters are active and therefore improve cell-type resolution. In a similar fashion, further enhancement of precision for genetic lineage tracing studies (39) could be achieved by employing the obligate CreK201R and CreQ311R system. Likewise, obligate SSRs might allow for the generation of more refined circuits for synthetic biology, where SSRs are frequently used to build biosensors and biological machines (40,41).
In our study, we focused on the characterization of one mutation pair (an SSRK201R monomer in combination with an SSRQ311R monomer). Yet, our SLiDE screen identified other combinations that might also constitute simple obligate SSRs. Future studies will be required to investigate whether these mutational pairings are equally suitable to represent efficient and specific obligate SSR systems and if they also function by influencing recombination catalysis. In particular, the observed changes at residue 282 of D7L represent an interesting candidate in this regard, because this residue is located within the J-K loop, which is known to make several contacts with the DNA (42) and might therefore impact recombination catalysis.
In summary, this work provides a straightforward approach to reduce off-target recombination and improved specificity of engineered and wild-type SSRs. However, further studies are required to evaluate the applied advantages of the obligate SSRK201R plus SSRQ311R monomer combinations. In particular, we demonstrate the enhanced specificity of the D7LK201R and D7RQ311R system at one off-target site. Additional efforts utilizing more unbiased assays (43) would be beneficial to further investigate the improved properties of these obligate mutations. Furthermore, the obligate mutations should be tested in additional natural and engineered SSRs to probe its robustness and general applicability. Finally, the obligate system will have to show its improvements in animal models before therapeutic implementation could be considered.
DATA AVAILABILITY
Quality assessment of the molecular models was done with PROCHECK ((44). Coordinates are available upon request. Flow cytometry data were deposited in FlowRepository (ID FR-FCM-Z3PT).
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Buchholz laboratory and Carla Guillén-Pingarrón for fruitful discussions. The High-Performance Computing Center of the Technische Universität Dresden (ZIH TUD) provided computational infrastructure.
Contributor Information
Jenna Hoersten, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
Gloria Ruiz-Gómez, Structural Bioinformatics, BIOTEC TU Dresden, Tatzberg 47-51, 01307 Dresden, Germany.
Felix Lansing, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
Teresa Rojo-Romanos, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
Lukas Theo Schmitt, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
Jan Sonntag, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
M Teresa Pisabarro, Structural Bioinformatics, BIOTEC TU Dresden, Tatzberg 47-51, 01307 Dresden, Germany.
Frank Buchholz, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Union [ERC 742133, H2020 UPGRADE 825825]; German Research Council [DFG BU 1400/7-1 and PI 600/4-1]; Bundesministerium für Bildung und Forschung GO-Bio (BMBFGO-Bio) [031B0633]. Funding for open access charge: TU Dresden.
Conflict of interest statement. Technical University (Technische Univeristät) Dresden has filed a patent application based on this work, in which J.H., F.L. and F.B. are listed as inventors.
REFERENCES
- 1. Duyne G.D.V. Cre recombinase. Microbiol. Spectr. 2015; 3:119–138. [DOI] [PubMed] [Google Scholar]
- 2. Meinke G., Bohm A., Hauber J., Pisabarro M.T., Buchholz F.. Cre recombinase and other tyrosine recombinases. Chem. Rev. 2016; 116:12785–12820. [DOI] [PubMed] [Google Scholar]
- 3. Anastassiadis K., Schnütgen F., Melchner H., Stewart A.F.. Chapter nine gene targeting and site-specific recombination in mouse ES cells. Methods Enzymol. 2013; 533:133–155. [DOI] [PubMed] [Google Scholar]
- 4. Monetti C., Nishino K., Biechele S., Zhang P., Baba T., Woltjen K., Nagy A.. PhiC31 integrase facilitates genetic approaches combining multiple recombinases. Methods. 2011; 53:380–385. [DOI] [PubMed] [Google Scholar]
- 5. Buchholz F., Stewart A.F.. Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat. Biotechnol. 2001; 19:1047–1052. [DOI] [PubMed] [Google Scholar]
- 6. Sarkar I., Hauber I., Hauber J., Buchholz F.. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007; 316:1912–1915. [DOI] [PubMed] [Google Scholar]
- 7. Karpinski J., Hauber I., Chemnitz J., Schäfer C., Paszkowski-Rogacz M., Chakraborty D., Beschorner N., Hofmann-Sieber H., Lange U.C., Grundhoff A.et al.. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 2016; 34:401–409. [DOI] [PubMed] [Google Scholar]
- 8. Lansing F., Paszkowski-Rogacz M., Schmitt L.T., Schneider P.M., Romanos T.R., Sonntag J., Buchholz F.. A heterodimer of evolved designer-recombinases precisely excises a human genomic DNA locus. Nucleic Acids Res. 2020; 48:472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lansing F., Mukhametzyanova L., Rojo-Romanos T., Iwasawa K., Kimura M., Paszkowski-Rogacz M., Karpinski J., Grass T., Sonntag J., Schneider P.M.et al.. Correction of a factor VIII genomic inversion with designer-recombinases. 2020; bioRxiv doi:02 November 2020, preprint: not peer reviewed 10.1101/2020.11.02.328013. [DOI] [PMC free article] [PubMed]
- 10. Santoro S.W., Schultz P.G.. Directed evolution of the site specificity of Cre recombinase. Proc National Acad Sci. 2002; 99:4185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hauber I., Hofmann-Sieber H., Chemnitz J., Dubrau D., Chusainow J., Stucka R., Hartjen P., Schambach A., Ziegler P., Hackmann K.et al.. Highly significant antiviral activity of HIV-1 LTR-specific Tre-recombinase in humanized mice. PLoS Pathog. 2013; 9:e1003587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bolusani S., Ma C.-H., Paek A., Konieczka J.H., Jayaram M., Voziyanov Y.. Evolution of variants of yeast site-specific recombinase Flp that utilize native genomic sequences as recombination target sites. Nucleic Acids Res. 2006; 34:5259–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Soni A., Augsburg M., Buchholz F., Pisabarro M.T.. Nearest-neighbor amino acids of specificity-determining residues influence the activity of engineered Cre-type recombinases. Sci. Rep.-UK. 2020; 10:13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abi-Ghanem J., Chusainow J., Karimova M., Spiegel C., Hofmann-Sieber H., Hauber J., Buchholz F., Pisabarro M.T.. Engineering of a target site-specific recombinase by a combined evolution- and structure-guided approach. Nucleic Acids Res. 2013; 41:2394–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shah R., Li F., Voziyanova E., Voziyanov Y.. Target-specific variants of Flp recombinase mediate genome engineering reactions in mammalian cells. FEBS J. 2015; 282:3323–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saraf-Levy T., Santoro S.W., Volpin H., Kushnirsky T., Eyal Y., Schultz P.G., Gidoni D., Carmi N.. Site-specific recombination of asymmetric lox sites mediated by a heterotetrameric Cre recombinase complex. Bioorgan. Med. Chem. 2006; 14:3081–3089. [DOI] [PubMed] [Google Scholar]
- 17. Gaj T., Mercer A.C., Sirk S.J., Smith H.L., Barbas C.F.. A comprehensive approach to zinc-finger recombinase customization enables genomic targeting in human cells. Nucleic Acids Res. 2013; 41:3937–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olorunniji F.J., Rosser S.J., Stark W.M.. Purification and in vitro characterization of zinc finger recombinases. Methods Mol. Biol. 2017; 1642:229–245. [DOI] [PubMed] [Google Scholar]
- 19. Voziyanova E., Li F., Shah R., Voziyanov Y.. Genome targeting by hybrid Flp-TAL recombinases. Sci. Rep.-UK. 2020; 10:17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang C., Myers C.A., Qi Z., Mitra R.D., Corbo J.C., Havranek J.J.. Redesign of the monomer–monomer interface of Cre recombinase yields an obligate heterotetrameric complex. Nucleic Acids Res. 2015; 43:9076–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Y.-J., Liu P., Nielsen A.A.K., Brophy J.A.N., Clancy K., Peterson T., Voigt C.A.. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods. 2013; 10:659–664. [DOI] [PubMed] [Google Scholar]
- 22. Herman A., Tawfik D.S.. Incorporating synthetic oligonucleotides via gene reassembly (ISOR): a versatile tool for generating targeted libraries. Protein Eng. Des. Sel. 2007; 20:219–226. [DOI] [PubMed] [Google Scholar]
- 23. Case D.A., Belfon K., Ben-Shalom I.Y., Brozell S.R., Cerutti D.S., Cheatham T.E. III, Cruzeiro V.W.D., Darden T.A., Duke R.E., Giambasu G.et al.. AMBER 2020. J. Chem. Inf. Model. 2020; 53:1689–1699. [Google Scholar]
- 24. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graphics. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 25. Gelato K.A., Martin S.S., Liu P.H., Saunders A.A., Baldwin E.P.. Spatially directed assembly of a heterotetrameric Cre-Lox synapse restricts recombination specificity. J. Mol. Biol. 2008; 378:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esposito D., Scocca J.J.. The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res. 1997; 25:3605–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gibb B., Gupta K., Ghosh K., Sharp R., Chen J., Duyne G.D.V.. Requirements for catalysis in the Cre recombinase active site. Nucleic Acids Res. 2010; 38:5817–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin S.S., Chu V.C., Baldwin E.. Modulation of the active complex assembly and turnover rate by protein-DNA interactions in Cre-LoxP recombination. Biochemistry-us. 2003; 42:6814–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo J., Liu Q., Morihiro K., Deiters A.. Small-molecule control of protein function through Staudinger reduction. Nat. Chem. 2016; 8:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abi-Ghanem J., Samsonov S.A., Pisabarro M.T.. Insights into the preferential order of strand exchange in the Cre/loxP recombinase system: impact of the DNA spacer flanking sequence and flexibility. J. Comput. Aid Mol. Des. 2015; 29:271–282. [DOI] [PubMed] [Google Scholar]
- 31. Park C.-Y., Kim D.H., Son J.S., Sung J.J., Lee J., Bae S., Kim J.-H., Kim D.-W., Kim J.-S.. Functional correction of large factor VIII gene chromosomal inversions in hemophilia a patient-derived iPSCs using CRISPR-Cas9. Cell Stem. Cell. 2015; 17:213–220. [DOI] [PubMed] [Google Scholar]
- 32. Oldenburg J., Pezeshkpoor B., Pavlova A.. Historical review on genetic analysis in hemophilia A. Semin. Thromb. Hemost. 2014; 40:895–902. [DOI] [PubMed] [Google Scholar]
- 33. Lannoy N., Hermans C.. Principles of genetic variations and molecular diseases: applications in hemophilia A. Crit. Rev. Oncol. Hemat. 2016; 104:1–8. [DOI] [PubMed] [Google Scholar]
- 34. Karimova M., Abi-Ghanem J., Berger N., Surendranath V., Pisabarro M.T., Buchholz F.. Vika/vox, a novel efficient and specific Cre/loxP-like site-specific recombination system. Nucleic Acids Res. 2013; 41:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ennifar E., Meyer J.E.W., Buchholz F., Stewart A.F., Suck D. Crystal structure of a wild-type Cre recombinase– lox P synapse reveals a novel spacer conformation suggesting an alternative mechanism for DNA cleavage activation. Nucleic Acids Res. 2003; 31:5449–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kachroo A.H., Ma C.-H., Rowley P.A., Maciaszek A.D., Guga P., Jayaram M.. Restoration of catalytic functions in Cre recombinase mutants by electrostatic compensation between active site and DNA substrate. Nucleic Acids Res. 2010; 38:6589–6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chandras C., Zouberakis M., Salimova E., Smedley D., Rosenthal N., Aidinis V.. CreZOO—the European virtual repository of Cre and other targeted conditional driver strains. Database. 2012; 2012:bas029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murray S.A., Eppig J.T., Smedley D., Simpson E.M., Rosenthal N.. Beyond knockouts: cre resources for conditional mutagenesis. Mamm. Genome. 2012; 23:587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He L., Li Y., Li Y., Pu W., Huang X., Tian X., Wang Y., Zhang H., Liu Q., Zhang L.et al.. Enhancing the precision of genetic lineage tracing using dual recombinases. Nat. Med. 2017; 23:1488–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lapique N., Benenson Y.. Digital switching in a biosensor circuit via programmable timing of gene availability. Nat. Chem. Biol. 2014; 10:1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamanishi M., Matsuyama T.. A modified Cre- lox genetic switch to dynamically control metabolic flow in Saccharomyces cerevisiae. ACS Synth. Biol. 2012; 1:172–180. [DOI] [PubMed] [Google Scholar]
- 42. Petyuk V., McDermott J., Cook M., Sauer B.. Functional mapping of Cre recombinase by pentapeptide insertional mutagenesis. J. Biol. Chem. 2004; 279:37040–37048. [DOI] [PubMed] [Google Scholar]
- 43. Bessen J.L., Afeyan L.K., Dančík V., Koblan L.W., Thompson D.B., Leichner C., Clemons P.A., Liu D.R.. High-resolution specificity profiling and off-target prediction for site-specific DNA recombinases. Nat. Commun. 2019; 10:1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M.. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993; 26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Quality assessment of the molecular models was done with PROCHECK ((44). Coordinates are available upon request. Flow cytometry data were deposited in FlowRepository (ID FR-FCM-Z3PT).