


Abstract
We report the properties of two mutations in the exonuclease domain of the Saccharomyces cerevisiae DNA polymerase ϵ. One, pol2-Y473F, increases the mutation rate by about 20-fold, similar to the catalytically dead pol2-D290A/E290A mutant. The other, pol2-N378K, is a stronger mutator. Both retain the ability to excise a nucleotide from double-stranded DNA, but with impaired activity. pol2-Y473F degrades DNA poorly, while pol2-N378K degrades single-stranded DNA at an elevated rate relative to double-stranded DNA. These data suggest that pol2-Y473F reduces the capacity of the enzyme to perform catalysis in the exonuclease active site, while pol2-N378K impairs partitioning to the exonuclease active site. Relative to wild-type Pol ϵ, both variants decrease the dNTP concentration required to elicit a switch between proofreading and polymerization by more than an order of magnitude. While neither mutation appears to alter the sequence specificity of polymerization, the N378K mutation stimulates polymerase activity, increasing the probability of incorporation and extension of a mismatch. Considered together, these data indicate that impairing the primer strand transfer pathway required for proofreading increases the probability of common mutations by Pol ϵ, elucidating the association of homologous mutations in human DNA polymerase ϵ with cancer.
INTRODUCTION
The normal division of labor at the nuclear DNA replication fork in Saccharomyces cerevisiae is orchestrated by three DNA polymerases (Pols) (1,2). DNA polymerase α primes replication of Okazaki fragments, after which DNA polymerases δ and ϵ primarily replicate the lagging strand and the leading DNA strand, respectively (3–6). Replication is template-directed, ensuring that both DNA strands are accurately replicated prior to cell division. The high fidelity of nuclear DNA replication is achieved by three highly coordinated processes. The first is selection of the incoming nucleotide (7–9). Pols δ and ϵ, but not Pol α, have exonuclease catalytic activity that can excise mismatches during ongoing replication. This proofreading can occur intrinsically, when a mismatch made at either polymerase active site transitions as frayed single-stranded DNA to the exonuclease active site for removal without intervening dissociation of the enzyme. The exonuclease active site of Pol δ can also bind directly to single-strand DNA, which potentially allows extrinsic proofreading of a mismatch made by any of the three major replicases (10–12) The third process is DNA mismatch repair, which occurs after replication, and defects in which are strongly associated with disease, including cancer (13,14) and references therein).
Replicative DNA polymerases share a common architecture likened to that of a right hand, with fingers, thumb and palm subdomains composing the polymerase catalytic core (15) (Figure 1). Pol ϵ also has a unique P-domain that enhances processivity, i.e. the number of nucleotidyl transfer reactions occurring during a single pol-DNA binding event (16–18). Common to both Pols δ and ϵ are domains that contain the 3′-exonuclease that can proofread replication errors. The exonuclease active site is separated from the polymerase active site by ∼30–40Å (15–18), a distance that compels 3–4 nucleotides of the nascent DNA strand to be unpaired in order to permit excision of a 3′-terminal nucleotide. Structural comparison indicates that like the polymerase catalytic core, the exonuclease domains of Pols δ and ϵ share a common architecture (15,16) containing exonuclease (Exo) I-III motifs composed of residues found in the N-terminal region of the primary structure (19) (Figure 1).
Figure 1.

Pol2 Catalytic Core and Exo I, II, and III Motifs Illustrating the Exonuclease Active Site. (A) Cartoon structural representation of Pol2 (6FWK), bound to duplex DNA (dark purple); N-terminal domain (light yellow), P-domain (bright purple) and exonuclease domain (green) with polymerase domain in shades of blue - thumb (slate blue), fingers (deep teal) and palm (sky blue) sub-domains. (B) Exonuclease domain of Pol2 with DNA (bright purple template & magenta nascent strand) from a structure of RB69 DNA polymerase with primer bound in the exonuclease domain (1CLQ). Modeled by alignment of the exonuclease domains. Conserved Exo I-III motif residues represented as sticks. (C) As in B, showing only Exo I, Exo II and Exo III motifs from Pol2. Terminal and penultimate nucleotides of the nascent strand shown as sticks. Metal ions depicted as dark grey spheres. (A–C) The orientation of the exonuclease active site is aligned to the proposed transition state from Pol-I (28, 26). (D) Sequence alignment of Exo I–III motifs. Highlighted in grey are the conserved carboxylates, in black are the residues which are the focus of this study, labeled in (C).
The Exo I, II and III motifs are highly conserved in proofreading polymerases in both A and B families from viral, prokaryotic and eukaryotic sources (19–23). These motifs combine to form the DEDD active site belonging to a super family of nucleases that includes RNases and other DNases (24,25). Figure 1D describes these motifs, and highlights the conservation of the four carboxylates for a subset of proofreading DNA polymerases. The transition state architecture has been proposed from structures of A-family Escherichia coli DNA Pol I bound with DNA in the exonuclease active site, and describes a common two-metal ion mechanism for nucleotidyl transfer reactions (26–28). Using structural data for Pol I (26–28) and the B family polymerases from the bacteriophages Phi29 (29) and RB69 (30), we have modeled DNA in the exonuclease active site of a recently published structure of S. cerevisiae Pol2 (31), the catalytic subunit of Pol ϵ (Figure 1). For ease of interpretation, we present this model of the Exo I–III motifs in an orientation which mimics that of the proposed transition state for Pol I (26,28) (Figure 1C).
Dozens of mutations in the exonuclease domains of Pols δ and ϵ have been identified by DNA sequence analysis of tumors in cancer patient genomes (32–36) and references therein). The consequences of these mutations range in prognosis from drivers of hypermutation to passenger mutations in microsatellite-unstable tumors of individuals carrying mismatch repair deficiencies characterized by poor survival (13,14) and references therein). For mutations located in the exonuclease domain, the mutator effects have generally been attributed to defective exonucleolytic proofreading resulting in increased mutation rates. This is most apparent in the tumor-associated Pol δ D316G/H/N and Pol ϵ D275A/G/V mutations that alter essential exonuclease active site catalytic residues (13,14,34,36,37). Certain other mutations in the exonuclease domain are hypermutators that increase the mutation frequency beyond that of a defect in exonuclease activity alone (13,38). For example, among the most common exonuclease domain mutations identified in human tumors are changes of a highly conserved proline to arginine or histidine in Pol ϵ, and to leucine in Pol δ (13,32,33,35,36). In a Canr forward mutation assay in S. cerevisiae, the pol2-P301R mutation generates the highest mutation rate reported for a single pol2 point mutation in the exonuclease domain (38). The pol2-P301R mutation increases the mutation rate by 150-fold over wild type Pol ϵ, exceeding that observed for the exonuclease deficient variant pol2-04 (pol2-D290A/E292A) (39,40) and see further discussion below). This is but one example of numerous such mutations (38) and references therein), illustrating that much work still needs to be done to understand possible mechanisms to account for elevated mutation rates in diseased individuals resulting from reduced replication fidelity.
Here, we describe mutational studies and complementary biochemical experiments for S. cerevisiae homologs of two additional exonuclease domain mutations identified in Pol ϵ in human tumors, POLE-N363K and POLE-Y458F. The POLE-Y458F mutation was identified as an A:T transversion in a patient with pancreatic cancer (41). The POLE-N363K mutation was identified as an A:C transversion in pancreatic, colorectal and endometrial cancers, as well as in large cell glioblastoma (14,42–44). Pedigree analysis of families carrying these autosomal dominant mutations demonstrate high penetrance for cancer (14,42–44). In contrast to the pol2-P301R mutation mentioned above, these peptides are distal to the polymerase-exonuclease domain interface, residing in the putative DNA binding cleft proximal to the exonuclease active site. The Asn and Tyr residues reside in the Exo II and Exo III motifs, respectively, and are highly conserved in proofreading DNA pols (Figure 1D).
Structural and biochemical studies of the viral homologues for Pol2-N378 have described this residue as critical to the process of primer strand transfer from the polymerase active site into the exonuclease active site (21,45,46). It has been proposed that this residue induces a torsion on the backbone of the DNA substrate, enhancing the chemical transformation and removal of the terminal nucleotide (46,47). Mutations that change this residue change both the rate and sequence specificity for exonucleolytic degradation of single and double-stranded DNA substrates (21). Similar studies examining the role of the viral and bacterial homologues for pol2-Y473 have identified this residue as an organizational component of the exonuclease active site (28,29,48,49), interacting with the terminal phosphate upon which chemistry is performed and the activated hydroxide ion that is poised for attack of the phosphodiester linkage. Additionally, a recent Pol ϵ structure reveals that the phenolic oxygen interacts with the A-site catalytic metal ion (31). Mutational studies suggest this tyrosine is not only responsible for organization of the active site for chemical transformation, but it is additionally involved in the stability of the primer terminus in the exonuclease active site (48), with a Y:A mutation stabilizing the dwell time of the primer terminus in the exonuclease active site under non-catalytic conditions.
The purpose of the present study is to examine the S. cerevisiae homologs for two variants identified in human cancer, pol2-N378K and pol2-Y473F. We demonstrate that these variants are indeed strong mutators, with similar but slightly different mutational specificity. We then present biochemical evidence which demonstrate, using yeast homologues of cancer-associated mutations in the exonuclease domain of human Pol ϵ, loss of DNA replication fidelity can occur through multiple mechanisms.
MATERIALS AND METHODS
Generation of mutant strains
All bacterial cloning experiments utilized the Escherichia coli strain DH10B. Bacteria were grown with NZYM (MP Biomedicals) media supplemented with chloramphenicol (25 μg/ml). For standard growth, yeast were propagated on synthetic complete (SC) solid media (6.7 g/L Difco yeast nitrogen base without amino acids, 2% wt/vol dextrose, 2 g/L SC amino acid mix (Bufferad), and 2% agar) or liquid YPD media (1% wt/vol Bacto yeast extract, 2% wt/vol Bacto peptone, and 2% wt/vol dextrose). To select for transformation with the hygromycin B phosphotransferase gene (HPH) expression cassette, we used solid YPD media supplemented with hygromycin (200 μg/ml).
We engineered diploid strains using CRISPR/CAS9 using the Modular Cloning (MoClo) Yeast Toolkit (YTK), which utilizes Golden Gate Cloning technology (50). All oligonucleotides used in these experiments can be found in Table S1. Goldengate cloning allows multiple DNA fragments with different mutations to be combined into a single vector in one assembly reaction. For each mutation to be introduced, primers were designed to produce two adjacent PCR products with 5′ overhangs with BsmBI Type II restriction endonuclease sites. BsmBI digestion of the two PCR products creates unique complementary four nucleotide overhangs from within the POL2 coding sequence, which form ‘scarless’ junctions following ligation during the assembly reaction. We used the ‘Assembly Wizard’ tool in the cloud-based online informatics program ‘Benchling’ to design a coherent set of unique junctions for each assembly reaction so that only a single circular plasmid would emerge. For a PCR template we used a cloned 5′ fragment of the POL2 gene that had previously been engineered to remove pre-existing BsmBI and BsaI sites (pAJH250). The template had also been modified to include two different sgRNA cleavage-resistant sites. The first of these is found at the 5′ end of the gene (Table S1, see lower case nts in Pol2prt1f). The second is targeted by SgRNA_pol2a, which anneals to nucleotides 148527–148546 of chromosome 14. To make this site cleavage-resistant, we introduced silent mutations (lower case) between nts 148430–148544 (5′-aGaGTcACcACgAAt-3′). Given the locations of the desired mutations for this study (N378K and Y473F) we wanted the ability to utilize a third sgRNA recognition site targeted by SgRNA_POL2_149726, which anneals to nts149723-149742 of chromosome 14. To make this site cleavage-resistant, we designed two oligonucleotides (POL2SgRNA_149726_RTf and POL2SgRNA_149726_RTr) that introduced silent mutations that inactivated the PAM site and created a convenient DraI site for restriction fragment length polymorphism-PCR (RFLP-PCR). For each of the candidate pol2 mutator mutations, we designed primer pairs (pol2N378Kf, pol2N378Kr; and pol2Y473Ff, pol2Y473Fr) that introduced the mutations as well as additional silent mutations to facilitate RFLP-PCR detection (Table S1). One additional design feature of the repair templates should be noted. In keeping with the yeast MoClo convention, the finalized pol2 repair template can be linearized by BsaI digestion, due to nested restriction sites engineered into the pol2prt1f and pol2prt2rb primers.
To create the three mutator repair templates, we combined three PCR fragments (pcrA, pcrB, and pcrC) with pYTK001. To create the pcrA fragments for the assemblies, we amplified the DNA with POL2prt1f and the reverse primer for each candidate mutator mutation (e.g. pol2N378Kr). To create the pcrB fragments, the forward primer for each allele (e.g. pol2N378Kf) was used with POL2SgRNA_149726_RTr. Finally, to amplify the pcrC fragment (used for each of the three assemblies) we used POL2SgRNA_149726_RTf and POL2prt2rb. All Golden Gate assembly reactions utilized 20 fm of pYTK001 and 40 fm of each pcrA, pcrB, and pcrC fragment in a 20 ul reaction in 1x T4 DNA ligase buffer using 1 μl of BsmBI (10 units) and 1 μl of T4 DNA ligase (400 units) from New England Biolabs. Fully assembled plasmids were recovered by transformation of E. coli and sequenced confirmed.
To introduce the mutations into yeast we used a dual CAS9/sgRNA expression vector, built using the same basic strategy as Shaw et al. (51). Strains were derived from Δ|(−2)|-7B-YUNI300 (MATa CAN1 his7-2 leu2Δ::kanMX ura3Δ trp1-289 ade2-1 lys2ΔGG2899-2900) (52), commonly termed Δ7. The approach assembles the final dual expression vector in yeast using homologous recombination-directed ‘gap repair’ using linearized DNA fragments from two plasmids: a CAS9 ‘dummy’ dual expression vector (pAJH103, Herr et al., in preparation) and an sgRNA expression cassette vector (derived from pAJH001, Herr et al., in preparation). The CAS9 dummy dual expression vector carries the CAS9 coding sequence flanked by a strong promoter and termination sequence, a 2-micron origin of replication, a selectable marker (HPH), and two 500 bp homology arms flanking a removable dummy sequence (GFP), bounded by BsmBI sites that facilitate gap creation. The sgRNA cassette is flanked by the same homology arms but bounded by BsaI sites. pAJH001 carries internal BsmBI sites that permit cloning of sgRNA sequences (see Table S1) as described for pYTK050 (50). Following digestion with the appropriate enzymes the two digests are co-transformed into yeast, allowing gap repair to occur, creating a stably replicated 2-micron plasmid. The key difference in our approach from that of Shaw et al. was the use of homology arms based on the mouse Adh1 gene to direct the gap repair (Herr et al. in preparation).
All yeast transformations were carried out using a high efficiency transformation procedure (53). For most transformations we diluted an overnight culture of yeast 1:50 in 50mls YPD and cultured the cells ∼ 4–5 hours at 30°C to obtain a concentration of 1–2 × 107 cells/ml. We harvested the cells by centrifugation and suspended the cells in 25 mls 0.1 M Lithium Acetate. We then spun the cells down and suspended them 1 ml 0.1 M Lithium Acetate. We counted the cells and adjusted the concentration to 109 cells/ml. We set up transformations in PCR tubes by first preincubating 107 cells (10 μls) for 30 minutes at room temperature with 1 ul freshly denatured Salmon Sperm DNA (10mg/ml) and 10 μls of the DNA to be transformed (10 ng of the linearized CAS9/sgRNA dummy vector digest; 20 ng of the sgRNA_POL2_149726 expression cassette digest; and 200 ng of the linearized repair template). After the preincubation step, we mixed the cells again and then added 79 uls of a mixture containing 60μl 50% PEG 3350, 9μl 1 M Lithium Acetate, and 10μl DMSO, gently mixed the cells by pipetting, and incubated them for 30 minutes at 30°C. We then shifted the cell suspensions to 42°C in a thermocycler for 14 minutes, mixing the cells gently halfway through. We centrifuged the cells, removed the supernatant, suspended them in 5mM CaCl, and incubated them for an additional 10 minutes at room temperature, before plating serial dilutions of the transformation mixes onto selection plates to ensure that single colonies were obtained.
Protein Mutagenesis, Expression and Purification
Expression plasmid PJL1 (POL2 and POL2-D290A/E292A) (54) was mutagenized using Agilent SDM XL kit with the following primer pairs,
N378K-F_5′-gccaatcgaaaaagtcacccttgaaggtggatataacagtg-3′
N378K-R_5′-cactgttatatccaccttcaagggtgactttttcgattggc-3′
Y473F-F 5′-ctgcatcggaaacagaaaattcggaaaggtgctgtg-3′
Y473F-R 5′-cacagcacctttccgaattttctgtttccgatgcag-3′.
These plasmids were combined with the Pol ϵ accessory subunits (DPB2, DPB3, DPB4) plasmid, pJL6, in a two-step yeast transformation. Protein expression and purification generally follow established methods (18,54,55). Briefly, holoenzyme expression induced by addition of 4% galactose and grown for 6 hours. Pelleted cells resuspended in 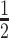 equivalent volume of sterile water and frozen by slow addition into liquid nitrogen. Frozen cells lysed by freezer mill and thawed in equal volume of buffer (150mM tris-acetate pH7.8, 50 mM NaOAc, 3 mM EDTA, 1 mM DTT, 10 mM NaHSO3 1 μM pepstatin A, 1 μM leupeptin, 5 mM benzamide, 300 μM PMSF. 175mM ammonium sulfate) was added to solubilized lysate and allowed to mix at 4°C for 5 minutes, after which 40 μL/mL of 10% polymin-p was added and allowed to stir for 15 minutes. Lysate was cleared by centrifugation and supernatant was precipitated with 0.3 g/mL ammonium sulfate powder and ultra-centrifugation. Supernatant was discarded and pellet resuspended in buffer containing 25 mM hepes pH 7.6, 10% glycerol, 1.5 mM EDTA, 0.005% NP40, 1 mM DTT, 5 μM pepstatin A, 5 μM leupeptin. Sample was batch bound to 1 mL glutathione sepharose beads (GE) equilibrated in the same buffer with the addition of 300mM NaOAc. Slurry was batch washed by gravity column with 15 mL fresh equilibration buffer, followed by 20 mL wash buffer containing 750 mM NaOAc. Sample was eluted with wash buffer containing 20 mM glutathione at 4°C overnight into a fresh tube containing 40U PreScission protease. 4 subunit Pol ϵ was then purified by FPLC in two steps. First using a 1 mL MonoQ column with 10 mL linear gradient of elution buffers containing 800 mM NaOAc and 1200 mM NaOAc. Then using a 1mL MonoS column by 15 mL linear gradient of elution buffers containing 200 and 800 mM NaOAc. Samples were concentrated by Amicon Ultra4 10K centrifugal concentrators.
equivalent volume of sterile water and frozen by slow addition into liquid nitrogen. Frozen cells lysed by freezer mill and thawed in equal volume of buffer (150mM tris-acetate pH7.8, 50 mM NaOAc, 3 mM EDTA, 1 mM DTT, 10 mM NaHSO3 1 μM pepstatin A, 1 μM leupeptin, 5 mM benzamide, 300 μM PMSF. 175mM ammonium sulfate) was added to solubilized lysate and allowed to mix at 4°C for 5 minutes, after which 40 μL/mL of 10% polymin-p was added and allowed to stir for 15 minutes. Lysate was cleared by centrifugation and supernatant was precipitated with 0.3 g/mL ammonium sulfate powder and ultra-centrifugation. Supernatant was discarded and pellet resuspended in buffer containing 25 mM hepes pH 7.6, 10% glycerol, 1.5 mM EDTA, 0.005% NP40, 1 mM DTT, 5 μM pepstatin A, 5 μM leupeptin. Sample was batch bound to 1 mL glutathione sepharose beads (GE) equilibrated in the same buffer with the addition of 300mM NaOAc. Slurry was batch washed by gravity column with 15 mL fresh equilibration buffer, followed by 20 mL wash buffer containing 750 mM NaOAc. Sample was eluted with wash buffer containing 20 mM glutathione at 4°C overnight into a fresh tube containing 40U PreScission protease. 4 subunit Pol ϵ was then purified by FPLC in two steps. First using a 1 mL MonoQ column with 10 mL linear gradient of elution buffers containing 800 mM NaOAc and 1200 mM NaOAc. Then using a 1mL MonoS column by 15 mL linear gradient of elution buffers containing 200 and 800 mM NaOAc. Samples were concentrated by Amicon Ultra4 10K centrifugal concentrators.
Mutation Rates (Forward Mutation Assay and Fluctuation Analysis)
Freshly dissected tetrads of yeast Δ7 strains homozygous for their POL2 variation and URA3 in orientation 1 were used for each fluctuation analysis and forward mutation study, performed as recently described (56). Briefly, at least 20 independent cultures from single colonies were inoculated in 5 ml YPDA rich liquid media with 100 μg/ml supplemental adenine. The cultures were incubated at 30 °C until saturation (usually ∼3 days for cells with no growth defect). The cultures then were diluted and plated on to non-selective synthetic media and selective media (containing 5-fluoroorotic acid (for selection of ura3 mutants). Mutations were calculated using Drake's formula (57). New data collected on pol2-D290A/E292A and WT-pol2 was used as an internal control for experiments and combined with previously published rates data (9,58).
Exonuclease Activity
Triplicate reactions were in normal buffer conditions; 5% glycerol, 1 mM DTT, 1 x TE (pH8), in nuclease free water. 10 nM Enzyme and 10 nM DNA1ii were preincubated in buffer for 10 minutes at 30°C then initiated with 11 mM MgCl2. After 5 seconds, reactions were quenched with equal volume stop solution (20 mM EDTA, 95% formamide, 0.1% bromophenol blue). Individual replicates were analyzed by 12% denaturing PAGE, scanned for Cy-3 fluorescence by Typhoon, and quantified by ImageQuant. Replicates were averaged and standard deviations are reported.
Exonuclease Processivity
Duplicate reactions were in normal buffer conditions; 5% glycerol, 1 mM DTT, 1 x TE (pH8), in nuclease free water. 50 nM Enzyme and 50 nM DNA1ii were preincubated in buffer for 10 minutes at 30°C then initiated with 11 mM MgCl2 in the presence of 2 μg/mL heparin. Heparin is a long poly anion that acts as a molecular trap when the enzyme dissociates. Thus, these plots describe the termination pattern of the bands in the gel. After 2 minutes, reactions were quenched with equal volume stop solution (20 mM EDTA, 95% formamide, 0.1% bromophenol blue). Individual replicates were analyzed by 12% denaturing PAGE, scanned for Cy-3 fluorescence by Typhoon, and quantified using ImageQuant. Quantification of bands considers the band volume at each nucleotide of the labeled oligo. Termination probability at each base is calculated as (band intensity)/(sum of that band + all subsequent bands). Duplicate reactions we averaged and plotted as a function of sequence. The substrate and terminal product for these reactions were omitted in the plots for clarity.
Polymerase-Exonuclease Kinetic Balance Assay
Using normal buffer conditions, 5% glycerol, 1 mM DTT, 1 x TE (pH8), in nuclease free water, triplicate reactions were preincubated for 10-minutes at 30°C in the presence of fold-molar ratios of cellular dNTP concentrations (dA 16 μM, dC 14 μM, dG 12 μM and dT 30 μM (59)) with 5 nM Enzyme and 5 nM DNA1i. DNA contained primer:template ratio of 1:1.25 to ensure all labeled DNA was duplex. 1-minute reactions were initiated with 11 mM MgCl2 in the presence of 2 μg/mL heparin to prevent multiple turn over reactions that deplete dNTP pools. For each enzyme the concentrations tested for triplicate repeat reactions were as follows: WT – 0.01, 0.05, 0.1, 0.5 -fold; N378K – 0.1 (x10−3), 0.5 (x10−3), 0.1 (x10−3), 0.5 (x10−3) -fold; Y473F – 0.05 (x10−3), 0.01 (x10−3), 0.5 (x10−3), 0.1 (x10−3) -fold. Reactions were quenched with equal volume stop solution (20mM EDTA, 95% formamide, 0.1% bromophenol blue). Individual replicates were analyzed by 12% denaturing PAGE, scanned for Cy-3 fluorescence by Typhoon, and quantified using ImageQuant. Based on an averaged plot of the two activities, exonuclease and polymerase, the intersection point was estimated and is described as a fold-cellular dNTP concentration dependent switch point.
Polymerase Activity
Using normal buffer conditions (5% glycerol, 1 mM DTT, 1 x TE (pH8), in nuclease free water), triplicate reactions were preincubated for 10-minutes at 30°C in the presence of cellular dNTP concentrations (dA 16 μM, dC 14 μM, dG 12 μM and dT 30 μM (59)) with 10nM Enzyme and 10 nM DNA1i. 1-minute reactions were initiated with 11 mM MgCl2 in the presence of 2 μg/mL heparin to prevent multiple turn over reactions that deplete dNTP pools. Reactions were quenched with equal volume stop solution (20 mM EDTA, 95% formamide, 0.1% bromophenol blue). Individual replicates were analyzed by 12% denaturing PAGE, scanned for Cy-3 fluorescence by Typhoon, and quantified using ImageQuant. Product accumulation was calculated as the sum of every band larger than the substrate, while full length product accumulation was calculated as the percentage of product at the full length of 35 added nucleotides. Polymerase termination probability was determined as described for the exonuclease reactions. All plots describe averages from triplicates and error shown in standard deviation.
T•T Mismatch Incorporation and Extension Assay
Using normal buffer conditions, 5% glycerol, 1 mM DTT, 1 x TE (pH8), in nuclease free water, triplicate reactions were preincubated for 10-minutes at 30°C in the presence of 50 μM dTTP with 10 nM Enzyme and 10 nM DNA2. DNA contained primer:template ratio of 1:1.25 to ensure all labeled DNA was duplex. 1-minute reactions were initiated with 11 mM MgCl2. Reactions were quenched with equal volume stop solution (20 mM EDTA, 95% formamide, 0.1% bromophenol blue). Replicates were analyzed by 12% denaturing PAGE, scanned for Cy-3 fluorescence by Typhoon, and quantified using ImageQuant. Quantification was done as described in figure legend.
We have evaluated solution structures of the WT pol epsilon and its N378K and Y473F variants using molecular dynamics simulations. The initial structure of the wild type was obtained from the X-ray crystal coordinated of the pdb ID: 4M8O. The missing residues (91–107, 226–231, 669–675) in the crystal coordinates were introduced using Modeller-9.2 (60). Variant systems were created by mutating N378 and Y473 residues to Lys and Phe, respectively using Coot-0.8 (61). Crystallographic water molecules, metal ions (Mg2+ and Zn2+), the fragment of DNA bound to pol epsilon, and the incoming nucleoside triphosphate (dATP) were kept in the simulations. After introducing protons using Molprobity (62) counter ions were added, each system was solvated in a box of water, and each solvent box was selected so that box boundaries were at least 12 Å from the closest protein atom (total atoms in each system ranging from 184 781 to 184 791). Randomly selected water molecules were converted to Na+ and Cl– ions so that the final medium was a 100 mM NaCl solution. Prior to equilibration, the following molecular dynamics simulation protocol was executed; all systems were subjected to (i) a quick 10 000 step minimization, (ii) 2-ns belly dynamics runs with fixed peptide at 200 K, (iii) minimization, (iv) low temperature constant pressure dynamics with fixed protein to assure a reasonable starting density (about 4ns), (v) minimization, (vi) step-wise slow heating molecular dynamics at constant volume (so far, all simulations were performed under position constraints for heavy atoms in biomolecules) and (vii) constant volume molecular dynamics for 50 ns with the slow release of position constraints on heavy atoms. All final unconstrained trajectories were calculated at 300 K under the constant volume (for 500 ns with time step 1 fs) using the PMEMD module of Amber18 (63) to accommodate long range interactions. All protein parameters were taken from the FF14SB force field. All calculations were triplicated with the second and third simulations were started with the conformations selected at 20 and 30 ns time intervals of the primary simulation (these structures were then subjected to re-equilibration before their production runs). Root mean square deviations and positional fluctuations represented by B-factors were calculated using the CPPTRAJ utility program provided by Amber18. The B-factors were calculated from the last 300 ns segment of each trajectory.
RESULTS
Mutator effects of pol2-N378K and pol2-Y473F variants
We began this study by testing if the pol2-N378K and pol2-Y473F variants were indeed mutators. To achieve this, we determined the rate and specificity of mutations generated by the pol2-N378K and pol2-Y473F variants relative to wild-type (WT) Pol2 and its exonuclease deficient derivative, pol2-04 (pol2-D290A/E292A). Consistent with previous observations (9,58), pol2-D290A/E292A increases the rate of mutations in the URA3 reporter gene by about 20-fold relative to WT Pol2 (Table 1 and Figure 2). The overall mutation rate in the pol2-Y473F variant resembles that of pol2-D290A/E292A (Figure 2). The overall mutation rate of the pol2-N378K is somewhat higher, being a statistically significant 43-fold higher than for WT Pol2 and 2-fold higher than the rates for the pol2-Y473K and pol2-D290A/E292A mutants. Sequence analysis of URA3 mutants indicates that single base mismatches predominate in the variant polymerase strains (Table 1). Particularly notable are ‘hot spot’ locations where multiple mutations were observed, most notably A•T to T•A transversions at base pairs 279 and 686 in the ura3 gene (Table 2 and Supplementary Figure S1). These two hotspots were also observed in previous studies of the pol2D290A/E292A mutant (Table 1, (9,58). Because Pol ϵ primarily replicates the leading DNA strand, and here URA3 is in orientation 1 relative to the nearest replication origin, ARS306, we infer that these substitutions are generated by insertion of dTTP opposite a template thymine by Pol ϵ. Other types of substitutions are also generated by both variants, notably G•C to G•A and G•T substitutions. Among the two variants, the mutation rates for some of these substitutions are similar, but they differ in other instances, with rates for pol2-N378K being higher (Table 2). Notably, the G•T substitution at position 345 stands out as distinct to the Pol ϵ variants, but is not observed in the WT or pol2-D290A/E292A strains. The rate of mutation at this locus is remarkably high for the pol2-N378K mutant. Further studies will address the complexities of why this spot becomes a mutational hotspot in these strains.
Table 1.
Mutation Rates & Classes of Mutations as determine by fluctuation analysis (Figure 2)
| Pol2 | URA3R (10−7) | 95% CI | No. mutants | Transitions | Transversions | Insertions | Deletions |
|---|---|---|---|---|---|---|---|
| WT | 0.18 | 0.14–0.23 | 211 | 0.04 | 0.06 | 0 | 0.01 |
| D290A/E292A | 3.7 | 3–5.6 | 206 | 0.56 | 2.5 | 0.18 | 0.27 |
| N378K | 7.8 | 4.9–12 | 181 | 1.1 | 6.6 | 0.26 | 0.43 |
| Y473F | 3.3 | 2.6–4.2 | 189 | 0.37 | 2.1 | 0.28 | 0.6 |
Figure 2.

Mutation rates (forward mutation assay and fluctuation analysis). Absolute (×10–7) mutation rates determined by yeast fluctuation analysis and forward mutation assays. Error bars show 95% confidence interval. Values reported above the error bars are the fold increase in mutation rate relative to WT.
Table 2.
Rates of mutations (×10–7) for specific hotspots in the URA3 gene, calculated as the reporter gene mutation rate multiplied by the fraction of the hotspot occurrence in all colonies sequenced
| Pol2 | T97C | ΔA160 | A279T | G345T | C461A | G679T | A686T |
|---|---|---|---|---|---|---|---|
| WT | ≤0.001 | ≤0.001 | ≤0.001 | ≤0.001 | ≤0.001 | ≤0.001 | ≤0.001 |
| D290A/E292A | ≤0.001 | 0.2 | 0.6 | 0.1 | ≤0.001 | ≤0.02 | 1.2 |
| N378K | 0.2 | 0.1 | 1 | 1.5 | 0.1 | 0.2 | 1.8 |
| Y473F | 0.1 | 0.4 | 0.6 | 0.5 | 0.1 | 0.1 | 0.3 |
Exonuclease activity of the N378K and Y473F variants on primer-template DNA
To begin investigating the mechanisms that underlie the observed mutation rates, we examined digestion of a primer strand when bound to its cognate template (DNA1ii Supplementary Figure S2), in the absence of the dNTPs required for DNA synthesis. This experiment is used to determine if the ability to perform exonucleolysis on a fully pair DNA duplex is altered by the mutations. As anticipated, WT Pol ϵ efficiently performs exonucleolysis (Figure 3) and does so in a processive fashion (Figure 3C and Supplementary Figure S3). The two variant enzymes did perform digestion, but their macroscopic rates are reduced by ∼40-fold (Figure 3B). Both variants also reduced the processivity of double-stranded DNA digestion, with N378K Pol ϵ completely terminating digestion at only half the chain length of WT Pol ϵ, and Y473F Pol ϵ terminating digestion after excising only one, two or three nucleotides (Figure 3C). In electrophoretic mobility shift assays, the variant enzymes exhibit enhanced binding relative to the wild type enzyme (Supplementary Figure S4), suggesting that the differences are not due to a reduced capacity of the enzyme to bind duplex DNA.
Figure 3.

Exonuclease activity assays. (A) Representative PAGE image for reactions described in b, with reactions in the same order as bar plot. (B) Log bar-plots describing the amount of exonuclease product formed during 5 s reactions in the presence of either double-stranded (DS) or single-stranded (SS) DNA. (C) Average termination probability during 1-min reactions for each enzyme in the presence of heparin trap. Sequence describes the nucleotide encountered by the enzyme starting with the 3′ end on left. Termination probability is determined by calculating (band intensity)/(sum of that band + all subsequent bands in the reaction). Data shown for DNA1ii (Supplementary Figure S2).
Exonuclease activity of the N378K and Y473F variants on single-stranded DNA
Next, we compared the exonuclease activities of the two variant polymerases in-vitro using oligonucleotide substrates mimicking a region of the URA3 gene (Supplementary Figure S2, DNA1ii). Using purified four-subunit holoenzymes (54), we examined the ability of each holoenzyme to digest single-stranded DNA. While this may not be a meaningful substrate biologically, single-stranded DNA provides a handle to test the capacity of the exonuclease active site to perform catalysis without the necessity of DNA first being bound to the polymerase active site. Wild-type Pol ϵ exhibited highly active exonuclease activity on single-stranded DNA (Figure 3). In contrast, while both variant enzymes do digest ssDNA (Figure 3A, B), their digestion rates differ significantly, with the rate for the N378K variant exhibiting 40% of wild-type Pol ϵ activity, while the Y473F variant reduced to only 4% of WT Pol ϵ (Figure 3A, B). Moreover, the processivity of single-stranded DNA digestion also differed. Under single hit conditions, in the presence of heparin trap, Pol2-N378K is capable of being as processive on single-stranded DNA as WT Pol ϵ, but Y473F is much less processive, excising only one or two nucleotides before dissociating (Figure 3C and Supplementary Figure S3). These data indicate that the extents to which each mutation alters exonuclease activity and processivity on single-stranded DNA are unique.
The kinetic balance between proofreading and DNA polymerization
During DNA replication a natural balance between DNA synthesis and proofreading occurs. We set out to examine the competition between DNA synthesis and proofreading for these Pol ϵ variants. These reactions contained increasing concentrations of dNTPs present in the relative ratios reported in wild-type yeast cells (59). At low dNTP concentrations, exonuclease activity dominates (Figure 4), and as dNTP concentrations increase, the balance shifts from exonucleolysis to polymerization. The two exonuclease domain mutations perturb this kinetic balance, decreasing the concentration of dNTPs required to overcome exonucleolytic activity relative to WT Pol ϵ by 27-fold and 200-fold, for N378K and Y473F Pol ϵ, respectively (Figure 4). This observation indicates that both variants are significantly less likely to perform exonucleolytic cleavage in the presence of normal cellular dNTP concentrations.
Figure 4.

Kinetic Balance Assay. Representative gel for the reaction products from the polymerase exonuclease balance assay. Above, the Pol ϵ variant and fold dNTP concentrations for each lane are described. The estimated concentration of dNTP required for 50% product from each catalytic activity (substrate annotated with*, polymerase product above and exonuclease product below) is described as the Kinetic Switch. Inset plots the dNTP concentration dependent kinetic switch for each Pol ϵ enzyme; wild type (blue), N378K (green), Y473F (red). The y-axis is fold cellular dNTP where the concentrations for 1-fold are – dA 16 μM, dC 14 μM, dG 12 μM and dT 30 μM (59).
Polymerase activity in the absence of exonuclease activity
To further dissect the mechanism behind the mutagenicity observed for pol2-N378K and Y473F variants, we directly tested their ability to synthesize DNA by combining these mutations with the exonuclease deficient version of the enzyme, pol2-D290A/E292A. In the presence of a heparin trap and established normal-cellular dNTP concentrations, we observed 2.4-fold more polymerase product for pol2-N378K/D290A/E292A and 1.5-fold more polymerase product for pol2-Y473F/D290A/E292A relative to the exonuclease-inactive pol2-D290A/E292A version of the enzyme (Figure 5), with no significant change in the sequence dependent termination probability as a function of the mutations (Figure 5A, D). However, there is a marked increase in the amount of full-length product formed by pol2-N378K (Figure 5A, C). These data indicate that the polymerase function of pol2-N378K is enhanced relative to both pol2-D290A/E292A and pol2-Y473F.
Figure 5.

Polymerase Activity. Average reaction products during 1-min reactions by each enzyme on duplex DNA1i in the presence of heparin trap and cellular concentrations of dNTPs (59). (A) Representative PAGE image for reaction. (B) Bar plot of average total reaction product. (C) Bar plots describing the average full-length product. (D) Termination probability at each nucleotide in the template listed left to right, 3′ to 5′ respectively, determined by calculating (band intensity)/(sum of that band + all subsequent bands). Error bars describe standard deviation (B–D).
Mismatch extension activity of N373K and Y473F
To explore the possibility that the pol2-N378K variant enhances the rate of errors at sites commonly mutated by pol2-D290A/E292A, we tested the ability of the variants to make and extend a mismatch on a paired primer-template duplex. We utilized a substrate to test the incorporation of a T•T mismatch (DNA2 in Supplementary Figure S2) because this is the most common mutation observed in the forward mutation assays with these enzymes (Table 2 and Supplementary Figure S1). The probabilities of T•T mismatch incorporation for pol2D290A/E292A and pol2-Y473F/290A/E292A are nearly identical in this context (Figure 6A, B). In contrast, pol2-N378K/D290A/E292A produced twice as much mismatched product under these conditions (Figure 6A, B). The design of the substrate is such that after misincorporation of T across T, the mismatched end can be further extended by incorporation of a properly paired T•A (Figure 6A and Supplementary Figure S2). The probability of mismatch extension by pol2-N378K/D290A/E292A relative to pol2D290A/E292A is increased nearly 1.5-fold (Figure 6A, C), and described by the schematic in Figure 6D. Thus, the pol2-N378K/D290A/E292A variant increases the probability of both incorporating and extending a mismatch.
Figure 6.

T•T Mismatch Incorporation and Extension Assay. (A) Denaturing PAGE image demonstrating the product accumulation of dTMP incorporation for DNA2 by variants of Pol ϵ. Prime (P) and Template (T) terminal base pair of substrate labeled with resulting incorporated dTMP T:X base pairs described. (B) Graphical representation for the probability of incorporating a T calculated by dividing total product generated by each reaction (T:T + T:A + T:G). (C) Graphical representation for the probability of mismatch extension product generated by each reaction (T:A + T:G)/(T:T + T:A + T:G). (D) Schematic representation of reactions.
DISCUSSION
The results presented here provide insight into the different mechanisms by which mutations in the exonuclease domain of yeast Pol ϵ reduce the fidelity of nuclear DNA replication. We discuss the results for each variant and then briefly consider their implications.
The role of the phenolic oxygen of residue 473
The mutation rate observed for the pol2−Y473F variant demonstrates this mutation reduces the ability of the enzyme to edit mistakes that evade the initial process of nucleotide selectivity by Pol ϵ (Figure 2). The spectrum of mutations identified in the URA3 reporter gene, and their respective rates, generally mimic those observed for pol2- D290A/E292A. Combined with the reduced rates of exonucleolysis observed with both single-stranded and double-stranded DNA, the data suggest that this amino acid substitution impairs the ability to perform exonucleolytic cleavage. Prior studies with other related polymerases (see Introduction) show that this residue is located near the exonuclease active site and that it interacts with the terminal phosphate (27,28) and the catalytic metal ion (26,27,31), that it coordinates the hydroxide ion for attack of the phosphodiester linkage (49), and that it governs the stability of the primer terminus in the exonuclease active site (48). Also noteworthy, in structures of Pol-I, RB69 and Phi29, this tyrosine is rotated away from the active site and the phenolic oxygen is H-bonded to the backbone or R-groups of an unstructured loop (26,29,30,49). These facts suggest additional levels of regulation and promiscuity in the interacting partners of the phenolic oxygen.
In a recent structure of Pol ϵ liganded to both the A and B-site metal ions (31), the phenolic oxygen coordinates the A-site metal ion required to lower the energy barrier for the exonucleolytic cleavage reaction. Our exonuclease experiments demonstrate that without this oxygen, coordination of the substrate for catalysis is significantly impaired but is not totally eliminated (Figure 3A). In additional structures of both Pol ϵ (16) and Pol δ (64), this conserved residue coordinates a structurally conserved water molecule whose exchange might act to guide the substrate and or metal ion(s) into the exonuclease active site. Additionally, rather than interacting with structurally proximal partners that are distal in sequence, in the structures of Pol ϵ (16) and Pol δ (64), this tyrosine is within hydrogen bond distance with the backbone of the conserved Exo III carboxylate, a helical neighbor residing four amino acids away and whose activity is critical to catalysis. Considering our data and the results described for related DNA polymerases, it appears then that this tyrosine is involved in multiple steps of exonucleolysis, including substrate binding, active site assembly across the transition state and ejection of the substrate and or product from the exonuclease active site.
The role of the conserved asparagine at position 378
The mutation rate and biochemical properties conferred by the N378K mutation imply that changing this single base pair in the Pol2 gene disrupts the exonucleolytic capacity of pol ϵ by changing the balance between exonucleolysis and polymerization, without eliminating the intrinsic ability of the exonuclease to excise a nucleotide. Compared to the catalytically dead pol2-D290A/E292A mutant, the average URA3 mutation rate of pol2-N378K is two-fold higher, with specific mismatches generated at even higher rates, and the polymerization and exonucleolytic properties of N3787K Pol ϵ are different from D290A/E292A Pol ϵ. Unlike the exonuclease deficient pol2-D290A/E292A enzyme that fails to digest DNA, the N378K variant retains the ability to digest both single-stranded and double-stranded DNA, but does so with reduced total activity. Moreover, the N378K variant binds to duplex DNA more avidly, its polymerase activity is more active, and it switches from exonucleolysis to polymerization at 27-fold lower dNTP concentration.
These results are generally consistent with data described for Phi29 DNA polymerase and E. coli Pol I. A change in the homologous asparagine in Phi29 DNA polymerase was found to perturb the pathway of primer strand transfer between the polymerase and exonuclease active sites, and it also affects the rate of processive exonucleolysis (21,45). Those observations led the authors to suggest that the charge on the asparagine is a determinant of single-stranded DNA binding and exonucleolytic activity. Observations with the equivalent asparagine to alanine change in E. coli Pol I also describe a general decrease in exonuclease activity on both single and double-stranded DNA (48). To better understand the consequence of the mutations, we employed molecular dynamics to estimate the contributions of these mutations to the catalytic core dynamics and describe the structural consequence of each mutant. The results (Supplementary Figure S5) indicate no major structural changes in the mobility and relative Cα proximity of the Pol2 catalytic core as a function of the mutations. The structure suggests that K378 does not directly interact with the exonuclease active site. Rather, in the time scale of our estimations, the lysine resides in the putative path taken by the primer strand as it transfers from the polymerase active site to the exonuclease active site, and therefore may act as a physical barrier to the primer strand entering the exonuclease active site.
Collectively then, the data suggest that the N378K variant does not simply inactivate the exonuclease catalytic activity of pol ϵ per se, as does the change in pol2D290A/E292A. Rather, the N378K change interferes with the ability of Pol ϵ to proofread replication errors by altering two other properties. One is to prevent the mismatch made at the polymerase active site from fraying sufficiently to enter the exonuclease active site. The other, and perhaps related change is to increase the ability of the polymerase active site to more efficiently create (Figure 5B) and then extend (Figure 5C) a mismatch. These properties are consistent with the possibility that the N378K change alters the exonuclease domain such that it interferes with the primer strand transfer pathway, stimulating the activity of the polymerase catalytic core, including the ability to create and extend mismatches. In doing so, a mismatch will be more readily fixed into duplex DNA rather than being proofread, thereby accounting for the strong mutator phenotype.
Extrinsic proofreading
The human equivalent of yeast N378K Pol ϵ is one of several hypermutators associated with cancer that has been identified in the putative DNA-binding cleft of the exonuclease domain of Pol ϵ. Impairing the pathway of primer strand transfer could stimulate polymerase activity by creating a barrier to the exonuclease active site, therefore stabilizing the nascent strand in the polymerase active site. This mechanism is of particular interest in the context of extrinsic proofreading, whereby the dissociation of Pol ϵ (12) (or Pol α, see (10,11) after making an error allows the exonuclease active site of Pol δ to bind directly to and then excise a mismatch (Figure 7). We suggest that the N378K mutation suppresses the extrinsic proofreading activity of Pol δ by promoting the ability of N378K Pol ϵ to create and then extend mismatches without dissociation. This same mechanism may contribute to some of the mutability of the Y473F variant, which shares with N378K a greater than an order of magnitude reduction in the dNTP concentration required to switch from predominantly exonucleolysis to polymerization. Our data demonstrate that the hypermutator phenotype associated with mutations in the DNA-binding cleft of the exonuclease domain in DNA polymerases may be driven not only by changes in the balance between the DNA synthesis and proofreading capacities of the enzyme but also by the processivity of the enzyme, reducing the probability for extrinsic proofreading. The competition between inter and intramolecular proofreading being regulated by the intrinsic processivity of the enzyme was first suggested in studies of T5 DNA polymerase and DNA Pol I in the 1980s (65–67). Our data and proposed model for Pol ϵ (Figure 7) are in strong agreement with data describing how DNA travels between the two active sites for DNA Pol I (67).
Figure 7.

Mismatch Editing and Extension. Model describing the process of mismatch incorporation, editing and extension by Pol ϵ. This model includes the possibility of extrinsic proofreading by Pol δ. Proposed and measured mechanisms from these experiment for attenuating various branchpoints in the DNA synthesis and editing pathways are described in the accompanying table.
Concluding remarks
In principle, there are numerous mechanisms by which DNA replication fidelity can be reduced (see insert to Figure 7 for the subset tested here). It is now clear that concomitantly perturbing two or more of these mechanisms can result in the hypermutator phenotypes now associated with cancer. The present study highlights the likelihood that replication fidelity can be diminished by reducing proofreading through inactivation of exonuclease activity directly, as in pol2-D290A/E292, or indirectly by reducing partitioning of a mismatch to the exonuclease active site, thereby promoting mismatch extension and/or suppressing extrinsic proofreading. We also demonstrate that mutations in the exonuclease domain can have pleiotropic effects on DNA binding, polymerase activity and processivity by both active sites. The distinct mutation rates and biochemical properties for the two Pol2 mutations described herein provide evidence for a potential mechanism for the hypermutator phenotype, namely, increasing the intrinsic processivity of the DNA polymerase. Knowledge of the mechanisms that are responsible for mutations, exactly how they operate, and their consequences for developing chemotherapies, are all questions for future studies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Matt Longley, Scott Lujan and Zhixiong Zhou for their thoughtful edits. We also want to acknowledge the generosity of Erik Johansson for supplying the Pol ϵ expression plasmids, pJL1 and pJL6.
Contributor Information
Joseph M Dahl, Genome Integrity Structural Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, DHHS, Research Triangle Park, NC 27709, USA.
Natalie Thomas, Genome Integrity Structural Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, DHHS, Research Triangle Park, NC 27709, USA.
Maxwell A Tracy, Department of Laboratory Medicine and Pathology, UW Medicine, Seattle, WA 98195, USA.
Brady L Hearn, Department of Laboratory Medicine and Pathology, UW Medicine, Seattle, WA 98195, USA.
Lalith Perera, Genome Integrity Structural Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, DHHS, Research Triangle Park, NC 27709, USA.
Scott R Kennedy, Department of Laboratory Medicine and Pathology, UW Medicine, Seattle, WA 98195, USA.
Alan J Herr, Department of Laboratory Medicine and Pathology, UW Medicine, Seattle, WA 98195, USA.
Thomas A Kunkel, Genome Integrity Structural Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, DHHS, Research Triangle Park, NC 27709, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Division of Intramural Research of the National Institutes of Health; National Institute of Environmental Health Sciences [Z01 ES065070 to T.A.K., Z01 ES043010 to L.P.]; National Institute of General Medical Sciences [R01GM118854-01 to A.J.H.]. Funding for open access charge: National Institute of Environmental Health Sciences [Z01 ES065070].
Conflict of interest statement. None declared.
REFERENCES
- 1. Garg P., Burgers P.M.. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit. Rev. Biochem. Mol. Biol. 2005; 40:115–128. [DOI] [PubMed] [Google Scholar]
- 2. Johnson A., O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 2005; 74:283–315. [DOI] [PubMed] [Google Scholar]
- 3. Nick McElhinny S.A., Gordenin D.A., Stith C.M., Burgers P.M., Kunkel T.A. Division of labor at the eukaryotic replication fork. Mol. Cell. 2008; 30:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burgers P.M.J., Kunkel T.A.. Eukaryotic DNA replication fork. Annu. Rev. Biochem. 2017; 86:417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lujan S.A., Williams J.S., Kunkel T.A.. DNA polymerases divide the labor of genome replication. Trends Cell Biol. 2016; 26:640–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kunkel T.A., Burgers P.M.J.. Arranging eukaryotic nuclear DNA polymerases for replication: specific interactions with accessory proteins arrange Pols alpha, delta, and in the replisome for leading-strand and lagging-strand DNA replication. Bioessays. 2017; 39: 10.1002/bies.201700070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson K.A. The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta. 2010; 1804:1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kunkel T.A. DNA replication fidelity. J. Biol. Chem. 2004; 279:16895–16898. [DOI] [PubMed] [Google Scholar]
- 9. St Charles J.A., Liberti S.E., Williams J.S., Lujan S.A., Kunkel T.A.. Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA Repair (Amst.). 2015; 31:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pavlov Y.I., Frahm C., McElhinny S.A.N., Niimi A., Suzuki M., Kunkel T.A.. Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr. Biol. 2006; 16:202–207. [DOI] [PubMed] [Google Scholar]
- 11. Nick McElhinny S.A., Pavlov Y.I., Kunkel T.A. Evidence for extrinsic exonucleolytic proofreading. Cell Cycle. 2006; 5:958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou Z.-X., Lujan S.A., Burkholder A.B., Charles J.S., Dahl J., Farrell C.E., Williams J.S., Kunkel T.A.. How asymmetric DNA replication achieves symmetrical fidelity. Nat. Struct. Mol. Biol. 2021; 28:1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Campbell B.B., Light N., Fabrizio D., Zatzman M., Fuligni F., de Borja R., Davidson S., Edwards M., Elvin J.A., Hodel K.P.et al.. Comprehensive analysis of hypermutation in human cancer. Cell. 2017; 171:1042–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rayner E., van Gool I.C., Palles C., Kearsey S.E., Bosse T., Tomlinson I., Church D.N.. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer. 2016; 16:71–81. [DOI] [PubMed] [Google Scholar]
- 15. Johansson E., Dixon N.. Replicative DNA polymerases. Cold Spring Harb. Perspect. Biol. 2013; 5:a012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hogg M., Osterman P., Bylund G.O., Ganai R.A., Lundstrom E.B., Sauer-Eriksson A.E., Johansson E.. Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon. Nat. Struct. Mol. Biol. 2014; 21:49–55. [DOI] [PubMed] [Google Scholar]
- 17. Asturias F.J., Cheung I.K., Sabouri N., Chilkova O., Wepplo D., Johansson E.. Structure of Saccharomyces cerevisiae DNA polymerase epsilon by cryo-electron microscopy. Nat. Struct. Mol. Biol. 2006; 13:35–43. [DOI] [PubMed] [Google Scholar]
- 18. Chilkova O., Jonsson B.H., Johansson E.. The quaternary structure of DNA polymerase epsilon from Saccharomyces cerevisiae. J. Biol. Chem. 2003; 278:14082–14086. [DOI] [PubMed] [Google Scholar]
- 19. Bernad A., Blanco L., Lazaro J.M., Martin G., Salas M.. A conserved 3′—-5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell. 1989; 59:219–228. [DOI] [PubMed] [Google Scholar]
- 20. Soengas M.S., Esteban J.A., Lazaro J.M., Bernad A., Blasco M.A., Salas M., Blanco L.. Site-directed mutagenesis at the Exo III motif of phi 29 DNA polymerase; overlapping structural domains for the 3′-5′ exonuclease and strand-displacement activities. EMBO J. 1992; 11:4227–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Vega M., Lazaro J.M., Salas M., Blanco L.. Primer-terminus stabilization at the 3′-5′ exonuclease active site of phi29 DNA polymerase. Involvement of two amino acid residues highly conserved in proofreading DNA polymerases. EMBO J. 1996; 15:1182–1192. [PMC free article] [PubMed] [Google Scholar]
- 22. Blanco L., Bernad A., Salas M.. Evidence favouring the hypothesis of a conserved 3′-5′ exonuclease active site in DNA-dependent DNA polymerases. Gene. 1992; 112:139–144. [DOI] [PubMed] [Google Scholar]
- 23. Braithwaite D.K., Ito J.. Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 1993; 21:787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zuo Y., Deutscher M.P.. Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res. 2001; 29:1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Horio T., Murai M., Inoue T., Hamasaki T., Tanaka T., Ohgi T.. Crystal structure of human ISG20, an interferon-induced antiviral ribonuclease. FEBS Lett. 2004; 577:111–116. [DOI] [PubMed] [Google Scholar]
- 26. Steitz T.A., Steitz J.A.. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:6498–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Freemont P.S., Friedman J.M., Beese L.S., Sanderson M.R., Steitz T.A.. Cocrystal structure of an editing complex of Klenow fragment with DNA. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:8924–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beese L.S., Steitz T.A.. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991; 10:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kamtekar S., Berman A.J., Wang J., Lazaro J.M., de Vega M., Blanco L., Salas M., Steitz T.A.. Insights into strand displacement and processivity from the crystal structure of the protein-primed DNA polymerase of bacteriophage phi29. Mol. Cell. 2004; 16:609–618. [DOI] [PubMed] [Google Scholar]
- 30. Shamoo Y., Steitz T.A.. Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell. 1999; 99:155–166. [DOI] [PubMed] [Google Scholar]
- 31. Parkash V., Kulkarni Y., Ter Beek J., Shcherbakova P.V., Kamerlin S.C.L., Johansson E.. Structural consequence of the most frequently recurring cancer-associated substitution in DNA polymerase epsilon. Nat. Commun. 2019; 10:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cancer Genome Atlas Research, N., Kandoth C., Schultz N., Cherniack A.D., Akbani R., Liu Y., Shen H., Robertson A.G., Pashtan I., Shen R.et al.. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cancer Genome Atlas Research, N Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Palles C., Cazier J.B., Howarth K.M., Domingo E., Jones A.M., Broderick P., Kemp Z., Spain S.L., Guarino E., Salguero I.et al.. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013; 45:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Church D.N., Briggs S.E., Palles C., Domingo E., Kearsey S.J., Grimes J.M., Gorman M., Martin L., Howarth K.M., Hodgson S.V.et al.. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013; 22:2820–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bellido F., Pineda M., Aiza G., Valdes-Mas R., Navarro M., Puente D.A., Pons T., Gonzalez S., Iglesias S., Darder E.et al.. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016; 18:325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barbari S.R., Kane D.P., Moore E.A., Shcherbakova P.V.. Functional analysis of cancer-associated DNA polymerase epsilon variants in Saccharomyces cerevisiae. G3 (Bethesda). 2018; 8:1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kane D.P., Shcherbakova P.V.. A common cancer-associated DNA polymerase epsilon mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 2014; 74:1895–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xing X., Kane D.P., Bulock C.R., Moore E.A., Sharma S., Chabes A., Shcherbakova P.V.. A recurrent cancer-associated substitution in DNA polymerase epsilon produces a hyperactive enzyme. Nat. Commun. 2019; 10:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hansen M.F., Johansen J., Bjornevoll I., Sylvander A.E., Steinsbekk K.S., Saetrom P., Sandvik A.K., Drablos F., Sjursen W.. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam. Cancer. 2015; 14:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rohlin A., Zagoras T., Nilsson S., Lundstam U., Wahlstrom J., Hulten L., Martinsson T., Karlsson G.B., Nordling M.. A mutation in POLE predisposing to a multi-tumour phenotype. Int. J. Oncol. 2014; 45:77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vande Perre P., Bonnet D., Toulas C., Hamzaoui N., Selves J., Corsini C., Chipoulet E., Guimbaud R.. Germline mutation c.1089C >A of POLE could be associated with an increased risk of colorectal cancer and glioblastoma. Eur. J. Hum. Genet. 2018; 26:595–596. [DOI] [PubMed] [Google Scholar]
- 44. Vande Perre P., Siegfried A., Corsini C., Bonnet D., Toulas C., Hamzaoui N., Selves J., Chipoulet E., Hoffmann J.S., Uro-Coste E.et al.. Germline mutation p.N363K in POLE is associated with an increased risk of colorectal cancer and giant cell glioblastoma. Fam. Cancer. 2019; 18:173–178. [DOI] [PubMed] [Google Scholar]
- 45. Lieberman K.R., Dahl J.M., Wang H.. Kinetic mechanism at the branchpoint between the DNA synthesis and editing pathways in individual DNA polymerase complexes. J. Am. Chem. Soc. 2014; 136:7117–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Del Prado A., Rodriguez I., Lazaro J.M., Moreno-Morcillo M., de Vega M., Salas M.. New insights into the coordination between the polymerization and 3′-5′ exonuclease activities in varphi29 DNA polymerase. Sci. Rep. 2019; 9:923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lam W.C., Thompson E.H., Potapova O., Sun X.C., Joyce C.M., Millar D.P.. 3′-5′ exonuclease of Klenow fragment: role of amino acid residues within the single-stranded DNA binding region in exonucleolysis and duplex DNA melting. Biochemistry. 2002; 41:3943–3951. [DOI] [PubMed] [Google Scholar]
- 48. Lam W.C., Van der Schans E.J., Joyce C.M., Millar D.P.. Effects of mutations on the partitioning of DNA substrates between the polymerase and 3′-5′ exonuclease sites of DNA polymerase I (Klenow fragment). Biochemistry. 1998; 37:1513–1522. [DOI] [PubMed] [Google Scholar]
- 49. Wang C.X., Zakharova E., Li J., Joyce C.M., Wang J., Konigsberg W.. Pre-steady-state kinetics of RB69 DNA polymerase and its exo domain mutants: effect of pH and thiophosphoryl linkages on 3′-5′ exonuclease activity. Biochemistry. 2004; 43:3853–3861. [DOI] [PubMed] [Google Scholar]
- 50. Lee M.E., DeLoache W.C., Cervantes B., Dueber J.E.. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth Biol. 2015; 4:975–986. [DOI] [PubMed] [Google Scholar]
- 51. Shaw W.M., Yamauchi H., Mead J., Gowers G.F., Bell D.J., Oling D., Larsson N., Wigglesworth M., Ladds G., Ellis T.. Engineering a model cell for rational tuning of GPCR signaling. Cell. 2019; 177:782–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pavlov Y.I., Shcherbakova P.V., Kunkel T.A.. In vivo consequences of putative active site mutations in yeast DNA polymerases alpha, epsilon, delta, and zeta. Genetics. 2001; 159:47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gietz R.D., Schiestl R.H.. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007; 2:31–34. [DOI] [PubMed] [Google Scholar]
- 54. Sabouri N., Johansson E.. Translesion synthesis of abasic sites by yeast DNA polymerase epsilon. J. Biol. Chem. 2009; 284:31555–31563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abdulovic A.L., Hile S.E., Kunkel T.A., Eckert K.A.. The in vitro fidelity of yeast DNA polymerase delta and polymerase epsilon holoenzymes during dinucleotide microsatellite DNA synthesis. DNA Repair (Amst.). 2011; 10:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou Z.X., Williams J.S., Kunkel T.A.. Studying ribonucleotide incorporation: strand-specific detection of ribonucleotides in the yeast genome and measuring ribonucleotide-induced mutagenesis. J. Vis. Exp. 2018; 10.3791/58020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Drake J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Williams J.S., Clausen A.R., Nick McElhinny S.A., Watts B.E., Johansson E., Kunkel T.A.. Proofreading of ribonucleotides inserted into DNA by yeast DNA polymerase ϵ. DNA Repair (Amst.). 2012; 11:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McElhinny Nick, Watts S.A., Kumar B.E., Watt D., Lundstrom D.L., Burgers E.B., Johansson P.M., Chabes E., Kunkel T.A. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:4949–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sali A., Blundell T.L.. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993; 234:779–815. [DOI] [PubMed] [Google Scholar]
- 61. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 62. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Case D.A., Ben-Shalom I.Y., Brozell S.R., Cerutti D.S., Cheatham T.E.I., Cruzeiro V.W.D., Darden T.A., Duke R.E., Ghoreishi D., Gilson M.K.et al.. 2018; San Francisco: University of California; Amber 2018. [Google Scholar]
- 64. Johansson E., Majka J., Burgers P.M.. Structure of DNA polymerase delta from Saccharomyces cerevisiae. J. Biol. Chem. 2001; 276:43824–43828. [DOI] [PubMed] [Google Scholar]
- 65. Das S.K., Fujimura R.K.. Mechanism of 3′ to 5′ exonuclease associated with phage T5-induced DNA polymerase: processiveness and template specificity. Nucleic. Acids. Res. 1980; 8:657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Das S.K., Fujimura R.K.. Mechanism of primer-template-dependent conversion of dNTP leads to dNMP by T5 DNA polymerase. J. Biol. Chem. 1980; 255:7149–7154. [PubMed] [Google Scholar]
- 67. Joyce C.M. How DNA travels between the separate polymerase and 3′-5′-exonuclease sites of DNA polymerase I (Klenow fragment). J. Biol. Chem. 1989; 264:10858–10866. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.