


Abstract
Single-stranded (ss) gapped regions in bacterial genomes (gDNA) are formed on W- and C-strands during replication, repair, and recombination. Using non-denaturing bisulfite treatment to convert C to U on ssDNA, combined with deep sequencing, we have mapped gDNA gap locations, sizes, and distributions in Escherichia coli for cells grown in mid-log phase in the presence and absence of UV irradiation, and in stationary phase cells. The fraction of ssDNA on gDNA is similar for W- and C-strands, ∼1.3% for log phase cells, ∼4.8% for irradiated log phase cells, and ∼8.5% for stationary phase cells. After UV irradiation, gaps increased in numbers and average lengths. A monotonic reduction in ssDNA occurred symmetrically between the DNA replication origin of (OriC) and terminus (Ter) for log phase cells with and without UV, a hallmark feature of DNA replication. Stationary phase cells showed no OriC → Ter ssDNA gradient. We have identified a spatially diverse gapped DNA landscape containing thousands of highly enriched ‘hot’ ssDNA regions along with smaller numbers of ‘cold’ regions. This analysis can be used for a wide variety of conditions to map ssDNA gaps generated when DNA metabolic pathways have been altered, and to identify proteins bound in the gaps.
INTRODUCTION
Single stranded (ss)DNA is generated as intermediate structures in genomic DNA (gDNA) in a variety of biochemical pathways including DNA replication, repair, recombination, and RNA transcription. In eukaryotes, ssDNA regions in gDNA have been measured by labeling the nascent DNA strand with bromodeoxyuridine (BrdU) followed by ChIP-sequencing (1–3). BrdU incorporation in wild-type Escherichia coli is inefficient (4–6), thus limiting its utility in measuring ssDNA in the E. coli genome. Electron microscopic imaging has proved valuable in visualizing ssDNA gapped regions (7), but it has not been used for a whole genome analysis.
In this paper, we have investigated asynchronous populations of cells growing exponentially in the presence and absence of UV irradiation, and cells growing in stationary phase. Escherichia coli gDNA has been treated with sodium bisulfite under non-denaturing conditions to detect ssDNA gaps throughout the genome. We show that this powerful technique, when used in conjunction with next-generation DNA sequencing allows an extensive analysis of ssDNA gaps in gDNA, including a determination of the amount of ssDNA present, its location along the entire genome, its distribution on W- and C- strands and on leading- and lagging-DNA replication strands. ssDNA gaps have been mapped with close to single nt precision within 10 nt windows, for gapped regions from 50 nt up to ∼500 nt long.
The data display a spatially diverse landscape of ssDNA distributed throughout gDNA. Notably, the amount of ssDNA is similar on leading- and lagging-replication strands for the three growth conditions; however, the number and distribution of gaps exhibit distinct differences for each growth condition. We have emphasized the mapping and characterization of chromosomal regions containing high ssDNA concentrations, termed ‘hot’ regions, and regions with a paucity of ssDNA, ‘cold’ regions. A future high-resolution application for the method described here could use a standard ChIP-Seq approach to identify individual proteins located within ssDNA gaps. Whereas ssDNA gaps are routinely associated with DNA replication and DNA repair processes such as nucleotide excision repair, others can be traced to replisome collisions with DNA lesions or other barriers. Such collisions can result in fork collapse, fork regression, and other repair outcomes (8–15). In a process first documented by Howard-Flanders, Rupp, Clark and others (16–19), replisomes sometimes do not halt DNA synthesis at a template lesion. Instead, they skip over the lesion, leaving it behind in a single strand gap, often called a post-replication gap (20). The past five decades have witnessed only limited progress in understanding the mechanism and molecular outcomes of lesion skipping. We still do not know how often post-replication gaps are formed, how they are formed, how large they are, or what types of lesions or DNA structures are most proficient in triggering their formation. Our current study begins to address this gap in knowledge.
MATERIALS AND METHODS
Materials
MG1655 E. coli strain is from our lab collection. Control 203 nt ssDNA fragment was prepared as described previously (21). Mung Bean Nuclease (10 000 units/ml), Monarch PCR & DNA clean-up kit were purchased from New England Biolabs. DNase-free RNAse A (100 mg/ml) was from Qiagen, Proteinase K, sodium metabisulfite and hydroquinone were from Sigma-Aldrich. Accel-NGS Methyl-Seq DNA Library kit was purchased from Swift Biosciences, MI.
E. coli cells
MG1655 cells were grown in LB broth in the presence of 0.2% glucose at 37 °C. Mid-log cells were harvested when OD600 reached 0.5. Cells from an overnight culture (OD600 ∼ 3.0) were collected as stationary cells. Cells were washed twice with TBS buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl) and cell pellets were stored at −80°C. To prepare UV-irradiated cells, 20 ml of the mid-log cells were washed and re-suspended in 20 ml ice-cold 100 mM MgSO4. 10 ml of cells in a sterile plastic Petri dish were irradiated with 254 nm UV light at 100 J/m2 using a CL-1000 UV crosslinker (UVP, Inc. Upland, California). Immediately after UV-treatment, cells were centrifuged at 4000 × g for 7 min at 4°C and resuspended in 20 ml LB. After incubation at 37 °C for 10 min, UV-treated cells were washed twice with TBS buffer by centrifugation at 4000 × g for 7 min at 4°C. The cell pellets were stored at −80°C and used for genomic DNA purification. Titration of viable cells before and after UV-treatment showed that only 0.2% of cells survived after an exposure to a high dose of UV-irradiation (100 J/m2), and 1.8% of irradiated cells survived after 10 min incubation at 37°C. Cell survival was measured by plating aliquots of UV-irradiated cells (1:100 dilution in LB) immediately after UV-exposure. Plating aliquots of the same cells after 10 min incubation in LB at 37°C was used to determine cell survival 10 min post UV-irradiation.
Genomic DNA purification
Escherichia coli cells (equivalent of 5 ml culture at OD600 = 0.5) were lysed with lysozyme (2 mg/ml) in 200 μl of lysis buffer (20 mM Tris, pH 8.0, 5 mM EDTA, 100 mM NaCl) for 10 min at 37°C. Cellular RNA was removed by treatment with RNAse A (100 μg/ml) for 20 min at 37°C. Cellular proteins were then digested by addition of Proteinase K (final concentration 0.5 mg/ml) and SDS (final concentration 1%) and incubated for 2 h at 37°C. Genomic DNA (gDNA) was purified by twice extracting with phenol:chloroform:isoamyl alcohol (25:24:1) and precipitated with ethanol. Purified gDNA were resuspended in TE buffer (10 mM Tris–HCl, pH 8.0; 0.1 mM EDTA) and stored at −20°C.
Bisulfite treatment of control ssDNA and gDNA
Non-denaturing bisulfite treatment was carried out by incubation of ssDNA (1 μg) or purified gDNA (5 μg) with a freshly prepared solution of 5 M sodium bisulfite and 20 mM hydroquinone at 37 °C. The presence of hydroquinone helps to protect DNA from degradation during bisulfite treatment (22). Following incubation for 18 h, bisulfite-treated DNAs were washed three times with water using Amicon Ultra 10 K Centrifugal Filter Device (Millipore) and desulphonated in 0.3 M NaOH solution for 20 min at room temperature. After removal of NaOH by the Amicon Centrifugal Filter Device, the DNA solution was buffer-exchanged to 10 mM Tris pH 8.0 using Bio-spin P6 column (Bio-Rad, CA, USA).
Illumina library preparation and genome sequencing
Bisulfite-treated gDNA (1 μg) was sheared to an average size of ∼200–250 bp by sonication using a Covaris S2 instrument. 10 ng of sheared DNA was used directly in Illumina's sequencing library preparation. For a subset of experiments, sheared gDNA (200 ng) was incubated with Mung Bean (MB) nuclease (0.5 units) for 30 min at 30°C and purified using a Monarch PCR & DNA ‘clean-up’ kit. For Illumina's sequencing library preparation, Accel-NGS Methyl-Seq DNA Library kit (Swift Biosciences, MI) and 10 ng of sheared gDNA (or control DNA) were used to prepare Illumina's sequencing library according to the company's protocol with 9 PCR amplification cycles. This library preparation kit relies on enzymatic attachment of adaptors directly on bisulfite-treated ssDNA by the company's Adaptase technology. The libraries were purified by two rounds of SPRI bead clean up, quantified by Qubit and qPCR, and subjected to Illumina's sequencing (2 × 150 bp paired-end) on a Mini-Seq or a Next-Seq 550 instrument using High Output Reagent Kits (300-cycles).
Sequencing data analysis
Illumina sequencing data were analyzed by CLC Genomic Workbench (v. 21.0) (Qiagen). High quality sequencing reads were imported and trimmed 25 bases from the 5′-end and 20 nt from the 3′- end. Sequencing reads having a length of 101–105 nt long were used to analyze ssDNA gaps. To quantify genomic ssDNA content, bisulfite-treated reads were aligned and mapped to the MG1655 reference genome (Accession number NC_000913) using a Bisulfite Sequencing (BS-seq) directional protocol with a length fraction setting of 0.8 and similarity fraction setting of 0.9. This BS-seq mapping involved in silico conversion of all reads. All cytosine residues within the first reads of a pair were converted to thymine (CT conversions) and all guanine residues in the second reads of a pair were converted to adenine (complement of CT conversions). The MG1655 reference genome was also converted into two different in silico versions. For the CT-converted genome all C residues were replaced by T, and for the GA-converted genome, all G residues were converted to A. The in silico converted reads were then independently mapped to the CT- and GA-converted reference genomes and the better score of the two mappings was reported as final mapping result. Reads that mapped non-specifically (i.e. reads aligned at more than one genome position with equally good scores) were ignored and not used for analysis. Non-specific match reads often represent genes with multiple copies in E. coli chromosome such as seven copies of ribosomal 23S, 16S and 5S, numerous insertion sequences, Rhs elements and tufA and tufB genes. Counts and percentages of mismatches between the mapped reads and the reference for each base were calculated using a ‘QC for read mapping’ tool in the CLC genomic workbench.
To identify sequencing reads that represent ssDNA gaps on the W-strand (or C-strand), the reads were serially aligned and mapped to the CT-converted (or GA-converted) reference genomes using a standard ‘Map reads to reference’ tool in the CLC program, with a similar fraction setting of 0.95 and match length fraction settings varying from 1.0 to 0.1. In the first mapping round, all reads were first aligned and mapped to the CT-converted (or GA-converted) reference genomes with the match length fraction setting as 1.0. Reads that mapped to the reference genome were extracted and used to identify ssDNA gaps of >100 nt on the W-strand. Unmapped reads were subjected to a second mapping to the CT-converted (or GA-converted) reference genomes using the match length setting at 0.9. Reads that mapped to the reference genome in the second round were extracted and used to identify ssDNA gaps of 90–100 nt on the W-strand. Subsequent read mappings were carried out in similar manner with a stepwise length fraction setting at 0.8, …, 0.5 to extract reads that represent ssDNA gap sizes from 80–90 nt, 70–80 nt, 60–70 nt, 50–60 nt, respectively. Although gaps from 10 to 50 nt were identified, the presence of small numbers of C residues did not allow a clear distinction to be made between C to U conversions emanating from bona fide genomic ssDNA gaps and sporadic C to U conversions that may have occurred during bisulfite treatment of genomic dsDNA. Some reads in genomic regions with low GC content mapped to the CT-converted (or GA-converted) reference genomes, but do not represent ssDNA gaps. These reads were removed by their alignment to the unconverted MG1655 reference genome.
Sequencing reads having their entire length aligned and mapped to the CT-converted or GA-converted reference genomes were used to analyze the spatial distribution of ssDNA gaps on W- or C-chromosomal strands, respectively. Hot and cold genomic regions on W- and C-strands were identified by the ‘Whole Genome Coverage Analysis’ tool in the CLC Genomic Workbench, using 50 bp as the minimum length and 0.01 as the ‘P-value threshold’ value. This tool identifies regions in ssDNA read mapping (CT-converted reference genome for the W-strand, and GA-converted reference genome for the C-strand) with ‘unexpectedly’ high or low ssDNA read coverage. The coverage analysis involves the examination of the coverage in each of the positions in the ssDNA read mapping and marks the ones with coverage in the lower or upper tails of the estimated Poisson distribution. Regions with consecutive positions marked as having high or low ssDNA coverage are designated as ‘hot’ or ‘cold’ regions.
RESULTS
We have identified the genome-wide locations of ssDNA gapped intermediates in the E. coli chromosome and have analyzed their properties and possible functions. Three cell populations have been analyzed: cells grown exponentially in the presence and absence of UV radiation, and cells grown to stationary phase. We have measured the locations, sizes, and distributions of ssDNA gaps on the W and C chromosomal strands, and on leading- and lagging- replication strands.
The technical process entails the following steps (Figure 1A). First, genomic (g)DNA is isolated and then incubated with sodium bisulfite under non-denaturing conditions. This initial step results in efficient conversion of C → U in ssDNA (∼95%), with little C deamination in genomic dsDNA (∼5%). The gDNA is then mechanically sheared to yield 200–250 bp fragments, suitable for in Illumina's next-gen sequencing. Genomic ssDNA gaps on the W-strand are identified as sequencing reads containing tracks with all C residues converted to T; ssDNA gaps on the C-strand are identified as sequencing reads containing tracks with all G residues converted to A. In an experiment performed in parallel, sheared gDNA was treated with Mung Bean (MB) Nuclease. MB Nuclease specifically digests ssDNA (23) released from genomic ssDNA gaps, but does not act on sheared dsDNA fragments. This experiment allows for correction of background bisulfite-induced C → U deamination taking place in genomic dsDNA under non-denaturing conditions. The analytical mapping of ssDNA gaps entails the alignment of sequencing reads (101–105 nt) to E. coli reference genomes with all C residues converted to T, which identifies the W-strand, and with all G residues converted to A, which identifies the C-strand. The locations, sizes and biological features of the gapped ssDNA regions are described and evaluated below.
Figure 1.

Detection of genomic ssDNA gaps by non-denaturing bisulfite treatment and DNA sequencing. (A) Schematic representation of ssDNA gap detection. Purified genomic DNA was incubated with sodium bisulfite at 37°C for 18 h, followed by mechanical shearing to ∼200–250 bp fragments. After Illumina whole genome sequencing, ssDNA gaps on the W- or C-strands were identified as reads containing sequence tracks with all C → T or G → A conversions. (B–D) Representative sequencing reads from a bisulfite-treated ssDNA control (B), fragmented genomic dsDNA (C) and genomic ssDNA gaps (D). Reads derived from ssDNA gaps have either all C converted to T (green highlight) or G converted to A (red highlight) when aligned to a MG1655 reference sequence (bottom line without highlight). Data aggregations for reads are shown at the top of the sequence reads.
Measuring the ssDNA gap content in the E. coli chromosome in exponentially dividing cells ± UV and during stationary phase
We measured the efficiency of C to U conversion by non-denaturing bisulfite treatment using a 203 nt long control ssDNA substrate, which corresponds approximately to the average size of sheared gDNA. Following incubation with sodium bisulfite, the ssDNA was purified and sequenced (see Materials and Methods). All sequencing reads contained C → T conversions essentially at all C template positions (Figure 1B). Bisulfite-generated C deamination occurs at ∼95% efficiency under DNA non-denaturing conditions (Table 1, ‘Bisulfite-treated ssDNA control’). The observed low background of other than C → T mutations (less than 0.2% for C → A/G, G → C/T, A → N or T → N) possibly result from a combination of PCR-generated mutations, spontaneous hydrolytic deamination occurring during DNA processing, and Illumina sequencing errors.
Table 1.
Mismatches detected in bisulfite-treated genomic DNA
| Detected mismatches (%) | |||||||
|---|---|---|---|---|---|---|---|
| Sample | C → T | C → A/G | G → A | G → C/T | A → N* | T → N | Number of sequenced bases |
| Bisulfite-treated ssDNA control | |||||||
| Expt 1 | 94.80 | 0.06 | 0.13 | 0.06 | 0.17 | 0.07 | 174 549 412 |
| Expt 2 | 94.80 | 0.06 | 0.13 | 0.07 | 0.17 | 0.06 | 167 138 560 |
| gDNA without bisulfite treatment | |||||||
| Mid-log | 0.04 | 0.06 | 0.04 | 0.06 | 0.11 | 0.11 | 3 415 108 394 |
| Bisulfite-treated gDNA | |||||||
| Mid-log | |||||||
| Expt 1 | 6.29 | 0.07 | 6.28 | 0.07 | 0.11 | 0.11 | 9 028 568 963 |
| Expt 2 | 6.37 | 0.07 | 6.37 | 0.07 | 0.12 | 0.12 | 563 946 106 |
| MB Nuclease | 4.98** | 0.15 | 5.06** | 0.15 | 0.26 | 0.26 | 334 177 870 |
| UV-irradiated | |||||||
| Expt 1 | 8.78 | 0.07 | 8.77 | 0.07 | 0.11 | 0.11 | 2 486 784 904 |
| Expt 2 | 10.86 | 0.07 | 10.82 | 0.07 | 0.12 | 0.12 | 536 444 294 |
| MB Nuclease | 5.96** | 0.12 | 6.04** | 0.12 | 0.21 | 0.21 | 296 677 061 |
| Stationary | |||||||
| Expt 1 | 11.26 | 0.06 | 11.23 | 0.06 | 0.11 | 0.11 | 1 866 168 393 |
| Expt 2 | 15.65 | 0.07 | 15.67 | 0.07 | 0.13 | 0.13 | 575 894 587 |
| MB Nuclease | 5.90** | 0.14 | 6.02** | 0.14 | 0.24 | 0.24 | 359 640 871 |
* A → N and T → N denote all three mismatches at A and T bases.
** Significant decrease (P < 0.0001, Chi-square test) in the levels of C → T and G → A mutations, compared to samples not treated with Mung Bean Nuclease (MB Nuclease).
We have mapped ssDNA gaps in gDNA and have analyzed their content in asynchronous populations of E. coli during exponential growth (mid-log phase cells), mid-log phase cells irradiated with a high UV dose (100 J/m2), and cells in stationary phase. Optimization of sequencing reads to obtain high quality scores were achieved by trimming 25 nt from the 5′-end and 20 nt from the 3′-end, and then filtered to select for uniform lengths of 101–105 nt. The reads were mapped to an MG1655 reference genome using a bisulfite sequencing alignment algorithm, which maps all reads regardless of whether C → T conversion occurred in individual reads (see Materials and Methods). The maps contained sequencing reads derived from genomic dsDNA (e.g. Figure 1C), as well as reads derived from genomic ssDNA gaps (e.g. Figure 1D). Typical individual sequencing reads derived from genomic dsDNA contained one or a few sporadic C → T or G → A mutations (Figure 1C). In contrast, sequencing reads from the ssDNA control substrate, and from genomic ssDNA gaps contained either C → T conversion or G → A conversion at nearly all C or G sites, respectively (Figure 1B, D). Bisulfite-induced C → U deamination was highly efficient on ssDNA (∼95%), but it also occurred on genomic dsDNA using the same non-denaturing conditions, albeit at a low frequency (5%) (see MB Nuclease-treated gDNA, Table 1). Non-specific C → U deaminations on gDNA were likely caused by bisulfite action on transient ssDNA generated by local conformational fluctuations within dsDNA (24,25). Transient disruption of base pairing in dsDNA occurs at temperatures well below the DNA melting temperature resulting in the formation of short ssDNA bubbles containing one or several bases (24,25) that possibly can be deaminated at C sites by sodium bisulfite.
The whole genome distribution of mapped reads showed a distinct pattern for mid-log and UV-irradiated cells with highest levels of reads located at the replication origin (OriC). The levels of mapped reads gradually declined moving bidirectionally away from OriC, with the fewest reads located at the replication termination site (Ter) (Figure 2A, top two graphs). A symmetric and monotonic reduction in mapped reads at increasing distances from OriC is a hallmark of chromosomal replication in cells growing asynchronously in exponential phase. In contrast to mid-log and UV-irradiated cells, mapped reads for stationary cells appeared to be uniformly distributed along the entire length of E. coli chromosome (Figure 2A, bottom graph).
Figure 2.

Whole genome distribution of aligned sequencing reads. (A) Aggregated coverage graphs of all mapped reads for mid-log, UV-irradiated mid-log, and stationary cells. Three blue shades indicate the average read coverage values at the genome positions (blue color), the maximum coverage values (dark blue color), and the minimum coverage values (light blue color). The origin of replication (OriC), Dif and replication termination sites TerA, TerB, TerC, TerD are indicated by dashed vertical lines. Approximate chromosomal boundaries of four macrodomains (Ori MD – green, Ter MD – cyan, Left MD – blue, Right MD – red) are indicated at the bottom. (B) Estimated ssDNA gap content on the top and bottom strands. The values represent the averages and standard deviations from two independent experiments.
A genome-wide analysis of base substitution mutations at C, G, A and T in aligned reads revealed that only C → T and G → A mutations were specifically increased in bisulfite-treated gDNA, ranging from 6.3% for mid-log cells to 15.7% for stationary cells (Table 1). For each experiment, C → T and G → A mutations were detected at similar levels. The other mutations (C → A/G, G → C/T, A → N or T → N) were detected at levels comparable with the corresponding mutations in non-bisulfite treated gDNA (Table 1), suggesting that these mutations arose as a combination of PCR and sequencing errors. The removal of ssDNA genomic fragments by MB Nuclease prior to sequencing led to a significant reduction (P < 0.0001) in both C → T and G → A mutations compared to non-MB Nuclease treated samples (Table 1). Since bisulfite treatment of ssDNA gaps present on the top W strand resulted in sequencing reads with C → T conversions and ssDNA gaps on the C strand – reads with G → A conversions, the excess of C → T and G → A mutations above background mutation levels can be used to estimate the genomic content of ssDNA that mapped to W- and C-strands, respectively. Using C → T and G → A levels in MB Nuclease-treated gDNA as the background values for spontaneous bisulfite-induced C deamination in genomic dsDNA, the level of genomic ssDNA gaps is estimated to be ∼1.4% for mid-log cells, ∼4.8% for UV-irradiated cells and ∼8.5% for stationary cells (Figure 2B). ssDNA gaps appeared to be present at about equal levels on W and C strands within each of the three cell populations (Figure 2B).
The E. coli genome contains two oppositely replicating halves designated as replichore 1, replicating clockwise on the right side of OriC, and replichore 2, replicating counterclockwise on the left side of OriC (26). An analysis of ssDNA gap contents showed that the leading- and lagging-strands in each replichore contained similar levels of ssDNA distributed throughout the gDNA (Table 2). Thus, the percentage of ssDNA on the E. coli genome is essentially the same on the W and C strands (Figure 2B), and it remains the same when considering leading- and lagging-strands separately (Table 2).
Table 2.
Comparison of ssDNA gap contents on the leading and lagging strands
| ssDNA gap content (%)a | ||
|---|---|---|
| Leading strand | Lagging strand | |
| Mid-log cells | ||
| Replichore 1 (OriC right) | 1.4 ± 0.1 | 1.2 ± 0.1 |
| Replichore 2 (OriC left) | 1.5 ± 0.1 | 1.2 ± 0.1 |
| UV-irradiated cells | ||
| Replichore 1 (OriC right) | 4.7 ± 1.1 | 4.8 ± 1.7 |
| Replichore 2 (OriC left) | 5.0 ± 1.5 | 4.5 ± 1.4 |
| Stationary cells | ||
| Replichore 1 (OriC right) | 8.3 ± 2.8 | 8.4 ± 3.5 |
| Replichore 2 (OriC left) | 8.7 ± 3.1 | 8.2 ± 3.1 |
aValues represent the averages and standard deviations from two independent experiments.
ssDNA gap distribution and lengths throughout the E. coli chromosome
Read mappings were carried out to analyze ssDNA gap sizes on W and C strands. To obtain reads that derived from ssDNA gaps on the W strand, reads were directly mapped to an in-silico CT-converted MG1655 genome (i.e. a genome containing all C residues changed to T). Reads that fully aligned to the CT-converted genome were identified as ssDNA reads since the entire sequences were derived from ssDNA gaps on the W strand. For reads that partially aligned to the CT-converted genome, only the aligned regions of the reads represent ssDNA gaps. Considering the average bisulfite-induced C → T conversion rate is 95% (Table 1), the alignment parameter ‘match fraction’ (the minimum percentage identity between the aligned region and the reference sequence) for all mappings was set at 0.95.
In the first round of mapping, the ‘length fraction’ (the minimum percentage of the total alignment length that exactly matches the reference sequence) was set at 1.0, i.e., the mapped reads are identical in length to the reference. Reads that did not conform to the 100% length match criteria in the first round were used in subsequent mapping rounds with length fractions reduced in steps going from 0.9 to 0.5. Since all reads were processed to have similar lengths between 101–105 nt, these mappings correspond to ssDNA gap sizes in windows of 10 nt, ranging from 50 nt to >100 nt (Figure 3). The same ssDNA gap analysis was carried out for the C strand, where an in-silico GA-converted MG1655 genome with all G bases changed to A was used as the reference for read mappings. In this paper, the ssDNA gap resolution is in a range of 50–100 nt, which is set by the length fraction that align precisely to the CT-converted or GA-converted reference genome within each read (101 to 105 nt). For gaps <50 nt, the presence of small numbers of C residues did not allow a distinction to be made between C to U conversions occurring in gDNA gaps and sporadic C to U conversions by bisulfite-treatment of genomic dsDNA.
Figure 3.
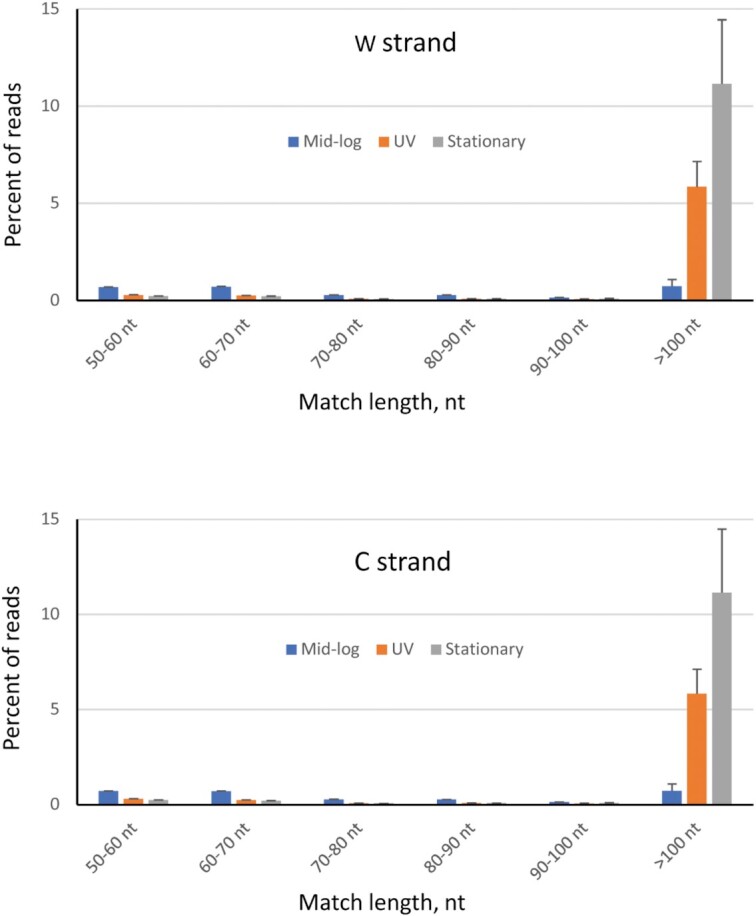
Distribution of sequencing reads containing an aligned region corresponding to ssDNA on W- and C-strands. CT- and GA-converted MG1655 genomes were used as reference genomes to identify ssDNA gap on W- and C-strands, respectively. Bar colors indicate mid-log cells (blue color), UV-irradiated mid-log cells (orange color), and stationary cells (gray color). Each bar represents the average percentage of reads containing an aligned region corresponding to ssDNA gaps with the indicated gap size (± standard deviation), from 2 independent experiments.
We have measured the distribution of ssDNA gap sizes on W- and C-strands for each cell population (Figure 3). For mid-log phase cells, relatively low frequencies of reads containing a region corresponding to genomic ssDNA gaps were observed, mainly in size ranges of 50–60 nt, 60–70 nt and larger than 100 nt (∼0.7% each). In contrast, gaps larger than 100 nt on W- and C-strands occurred with a much higher frequency in UV-irradiated cells (∼5.8%) and stationary cells (∼11%). Reads that aligned along their entire length to the CT- or GA-converted reference genome represent ssDNA gaps >100 nt on W- and C-strands, respectively. Although individual reads were within a 101–105 nt range, ssDNA gaps >105 nt can nevertheless be detected if both the forward reads (5′-end of a sheared fragment) and reverse reads (3′-end of a sheared fragment) map to the same CT- or GA-converted reference genome. The gDNA fragment is partially single stranded when one read in a pair mapped to one of the two reference genomes. The entire gDNA fragment is fully single stranded when both reads in a pair mapped to the same reference genome. For mid-log phase cells, 60% of the read pairs show both reads mapped to CT- or GA-converted genomes (Supplementary Figure S1A). However, for UV-irradiated and stationary phase cells, essentially all read pairs (98%) have both forward and reverse reads mapped to one of the genomes. Although the sheared gDNA fragments have an average length of 200–250 bp, the distribution of lengths for the mapped pairs reveals the presence of ssDNA for up to 400 nt for mid-log phase cells and up to 500 nt for UV irradiated mid-log and stationary phase cells (Supplementary Figure S1B). In summary, longer (>100 nt) ssDNA gaps were predominantly found to occur in UV irradiated mid-log and stationary phase cells, whereas numbers of ssDNA gaps >100 nt were far smaller in mid-log phase cells.
An analysis of the spatial distribution of ssDNA gaps on W and C strands on the E. coli chromosome
Sequencing reads that have their entire length aligned to the CT-converted or GA-converted reference genomes correspond to ssDNA gaps on W- or C-chromosomal strands, respectively. The whole genome coverage maps built from these reads were used to analyze the spatial distribution of ssDNA gaps. The distribution of ssDNA chromosomal gaps during mid-log phase growth has been determined with W- and C-strands combined (Figure 4A, top panel) and for each strand separately (Figure 4A, middle and bottom panels). The same gap analysis depicting UV irradiated mid-log and stationary phase cells is shown in Supplementary Figure S2. In mid-log phase cells ± UV, the average ssDNA content declined moving from OriC to Ter (Figure 4A and Supplementary Figure S2, ‘light blue shading’). Superimposed on the average ssDNA profiles were multiple peaks and troughs containing gDNA regions with increased and decreased ssDNA content, respectively (Figure 4A and Supplementary Figure S2, ‘dark blue shading’). A ‘zoomed-in’ profile illustrates the presence of a genomic DNA region containing greater and lower than average ssDNA levels on W- and C-strands (Figure 4B).
Figure 4.

Whole genome distribution of ssDNA gaps in E. coli. (A) Aggregated coverage graphs of mapped ssDNA reads for mid-log cells for both W- and C-strands (top panel), W-strand (middle panel) and C-strand (bottom panel). Blue color displays the average coverage values at the genome positions, dark blue – the maximum coverage values and light blue – the minimum coverage values. The origin of replication (OriC), Dif and replication termination sites TerA, TerB, TerC, TerD are indicated by dashed vertical lines. Approximate chromosomal boundaries of four macrodomains (Ori MD – green, Ter MD – cyan, Left MD – blue, Right MD – red) are indicated at the bottom. (B) A zoom-in genomic segment containing a hot ssDNA region and a cold ssDNA region. Each line below the aggregated graph represents an aligned ssDNA read. Green indicates the C to T conversion and red indicates G to A conversions in the aggregated data and in individual ssDNA reads.
The observation of approximately equal numbers of ssDNA reads that mapped to CT- and GA-converted genomes (Figure 4A, Supplementary Figures S1 and S2), implies the presence of similar amounts of ssDNA present on W- and C-strands. A finer-grained analysis identified chromosomal regions in which ssDNA is strongly enriched (‘hot’ regions) and regions containing a paucity of ssDNA (‘cold’ regions). The hot and cold regions are those for which the read-map coverage lies in the upper and lower tails of a Poisson distribution (Materials and Methods). Using 50 bp as a minimum length and P-value cut-off = 0.01, we identified 1301 hot ssDNA regions and 333 cold ssDNA regions on the W-strand, and 1335 hot ssDNA regions and 339 cold ssDNA regions on C-strand for mid-log cells (Table 3). The hot and cold regions have average lengths of 108 and 81 bp, respectively. For UV-irradiated cells, there were 2200 hot ssDNA regions and 1300 cold regions identified on W- and C-strands, and for stationary cells, there were 935 to 984 hot and cold regions contained on W- and C-strands. A detailed analysis for the numbers of hot and cold regions, their average and maximum sizes, and distances separating them is shown in Tables 3, 4 and in Supplementary Table S1.
Table 3.
Hot and cold genomic regions for ssDNA gaps on the W- and C-strand
| Hot ssDNA regions | Cold ssDNA regions | |||
|---|---|---|---|---|
| W-strand | C-strand | W-strand | C-strand | |
| Mid-log cells | ||||
| Number of regions | 1301 | 1335 | 333 | 339 |
| Average size (± SD), bp* | 108 ± 69 | 107 ± 56 | 81 ± 30 | 85 ± 34 |
| Maximum size, bp | 1165 | 677 | 205 | 264 |
| Median distance between regions, bp | 1059 | 1066 | 5590 | 5795 |
| UV-irradiated cells | ||||
| Number of regions | 2188 | 2198 | 1266 | 1297 |
| Average size (± SD), bp | 135 ± 100 | 132 ± 101 | 115 ± 66 | 115 ± 72 |
| Maximum size, bp | 1198 | 1767 | 700 | 781 |
| Median distance between regions, bp | 376 | 393 | 1309 | 1221 |
| Stationary cells | ||||
| Number of regions | 984 | 962 | 953 | 935 |
| Average size (± SD), bp | 105 ± 111 | 103 ± 118 | 109 ± 63 | 110 ± 65 |
| Maximum size, bp | 1966 | 2505 | 693 | 742 |
| Median distance between regions, bp | 2470 | 2930 | 2740 | 1916 |
*Values represent the averages and standard deviations (SD) from two independent experiments.
Table 4.
Hot and cold genomic regions for ssDNA gaps on the leading and lagging strands
| Hot ssDNA regions | Cold ssDNA regions | |||
|---|---|---|---|---|
| Leading strand | Lagging strand | Leading strand | Lagging strand | |
| Mid-log cells | ||||
| Number of regions | 1081 | 1555 | 393 | 279 |
| Average size (± SD), bpa | 103 ± 56 | 111 ± 67 | 83 ± 33 | 84 ± 29 |
| Maximum size, bp | 597 | 1165 | 264 | 220 |
| Median distance between regions, bp | 1219 | 970 | 4988 | 6295 |
| UV-irradiated cells | ||||
| Number of regions | 2286 | 2100 | 1255 | 1308 |
| Average size (± SD), bp | 136 ± 104 | 131 ± 95 | 117 ± 74 | 113 ± 64 |
| Maximum size, bp | 1767 | 1198 | 781 | 578 |
| Median distance between regions, bp | 372 | 394 | 1224 | 1282 |
| Stationary cells | ||||
| Number of regions | 1111 | 835 | 888 | 1000 |
| Average size (± SD), bp | 103 ± 110 | 105 ± 121 | 111 ± 67 | 109 ± 61 |
| Maximum size, bp | 2505 | 1966 | 699 | 742 |
| Median distance between regions, bp | 2354 | 3171 | 3123 | 2617 |
aValues represent the averages and standard deviations (SD) from two independent experiments.
The distribution of hot and cold ssDNA regions throughout the E. coli genome
We determined the distribution of hot and cold regions that lie within 10 kb segments along the E. coli chromosome (Figure 5). For mid-log cells, hot regions are highly concentrated in the vicinity of OriC for W- and C-strands, averaging 8–10 regions per 10 kb segment (Figure 5A). A gradual decrease in the density of hot regions occurs moving bidirectionally away from OriC toward Ter, which contains 0–2 regions per 10 kb (Figure 5A, top two panels). This gradient in ssDNA hot regions correlates with the numbers of replication forks, being greatest at replication origins in exponentially growing E. coli and diminishing roughly monotonically toward Ter. A tabulation of the data from Figure 5A shows that there are about 1.4-fold more hot regions identified on the lagging-strand (1555) compared to the leading-strand (1081) (Table 4). The average lengths of hot and cold regions are roughly similar on both strands (103–111 bp). The cold ssDNA distribution exhibited a reverse pattern with the highest density in the vicinity of Ter and lowest density near OriC (Figure 5A, bottom two panels). Cold ssDNA regions are favored by about 1.4-on the leading-strand (393) compared to the lagging-strand (279) (Table 4). For cells growing in mid-log phase, it seems possible, maybe even likely, that the reciprocal distribution of hot and cold regions could arise from the presence of lagging-strand Okazaki fragments. If these patterns are associated with normal DNA replication, the result suggests that the start and stop points for the generation of Okazaki fragments and ssDNA regions on the leading strand template are not entirely random.
Figure 5.

Spatial distribution of hot and cold ssDNA genomic regions. Distribution of hot and cold genomic regions on W- and C-strands for mid-log (A), UV-irradiated mid-log (B), and stationary cells (C). Each bar in the graphs represents the number of hot or cold ssDNA regions in a 10 kb genomic segment. The origin of replication (OriC), Dif and replication termination sites TerA, TerB, TerC, TerD are indicated by dashed vertical lines. Approximate chromosomal boundaries of four macrodomains (Ori MD – green, Ter MD – cyan, Left MD – blue, Right MD – red) are indicated at the bottom. A sketch at the top indicates the leading and lagging strands for each of the replichores on the left and the right of OriC.
It is perhaps surprising that UV-irradiated cells exhibit even more accentuated distributions of hot and cold ssDNA regions (Figure 5B). Hot regions are highly concentrated at OriC and sharply declined bidirectionally within ∼1000 kb distance. Hot regions are virtually absent in the second half of each replichore, i.e. ∼1000 kb on both sides of Ter. Conversely, cold region densities were peaked at Ter and sharply decreased over a distance of ∼1000 kb on both sides (Figure 5B). A distinctive feature of irradiated vs. non-irradiated rapidly dividing cells is that in contrast to mid-log phase cells, which had a ∼1.4-fold excess of ssDNA present on the lagging-strand in the absence of UV (Table 4), hot regions with ssDNA were observed at similar levels on leading- and lagging strands in UV irradiated cells (Table 4). In contrast, the non-dividing stationary phase cells show no indication of an ssDNA gradient between OriC and Ter, and indeed an approximately uniform distribution of hot ssDNA regions was observed for leading- and lagging strands throughout the entire E. coli chromosome (Figure 5C, Table 4). We note however the presence of numerous, ‘spiked’ deviations from the average for hot and cold ssDNA regions that were observed throughout the genome for each class of cells (Figure 5).
The E. coli chromosome has been shown to be organized as a ring composed of four macrodomains (MD): Ori MD, Ter MD, left MD and right MD, and two-less structured regions (27,28). DNA recombination occurs facilely within macrodomains, whereas recombination between different macrodomains is far more restricted (28,29). We find that hot ssDNA regions are concentrated within the Ori MD in mid-log phase cells (Figure 5A, B). Conversely, cold ssDNA regions tend to concentrate within the Ter MD (Figure 5A, B).
An examination of two nicking motifs contained in hot ssDNA gapped regions
We have probed hot ssDNA gapped regions to identify RecBCD-mediated Chi recombination hot spot motifs (5′-GCTGGTGG) (30) and 5′-GATC postreplication mismatch repair MutH nicking motifs (31). There are 1–3% hot ssDNA regions that were found to contain Chi sites (Table 5). Therefore, RecBCD-mediated recombination events are unlikely to be responsible for generating ssDNA in hot genomic regions. GATC sites, which are required for MutH-MutL mediated nicking on the unmethylated strand of hemi-methylated DNA during mismatch repair, occur at a frequency of 1/243 bp in the E. coli genome (31). Since the average lengths of hot genomic regions lies in a range of 80–130 bp (Table 3), then ∼33–53% of the hot gaps could, by chance, contain a GATC site. These sites were present in 28% of mid-log phase cells, 42% of UV irradiated mid-log phase cells, and 32% of stationary phase cells (Table 5). All are present at close to the predicted average frequencies suggesting that perhaps MMR might be implicated in the generation of hot regions of ssDNA throughout the gDNA.
Table 5.
Characteristics of Hot and Cold ssDNA regions
| Percent of regions (%) | ||
|---|---|---|
| Hot ssDNA regions | Cold ssDNA regions | |
| Mid-log cells | ||
| With a chi site | 1.6 | 2.0 |
| With a GATC site | 28.0 | 28.7 |
| In ORFs | 83.1 | 84.2 |
| In intergenic regions | 16.9 | 15.8 |
| UV-irradiated cells | ||
| With a Chi site | 3.2 | 2.1 |
| With a GATC site | 42.2 | 29.2 |
| In OFRs | 94.6 | 85.2 |
| In intergenic regions | 5.4 | 14.8 |
| Stationary cells | ||
| With a Chi site | 1.6 | 1.8 |
| With a GATC site | 32.3 | 24.7 |
| In OFRs | 94.3 | 79.4 |
| In intergenic regions | 5.7 | 20.6 |
Protein coding genes occupy a sizable part of the genome (87.8%); stable RNA accounts for ∼0.8%, with 11.4% occurring as intergenic DNA, including regulatory regions and a variety of additional functions (26). We determined the locations of hot and cold regions in open reading frames (ORFs) and in intergenic DNA (Table 5). For mid-log cells, hot and cold regions are distributed essentially randomly between ORFs and intergenic regions. Small departures from randomness in hot and cold ssDNA regions were observed for UV-irradiated and stationary phase cells.
DISCUSSION
This study provides a comprehensive first look at the ssDNA landscape in the E. coli genome. A diverse pattern of ssDNA regions is distributed throughout the E. coli genome, present to a similar extent on W- and C-strands, and on leading- and lagging- strands. The gDNA gap content depends on cell growth conditions. The ss/ds DNA ratio is approximately 1.3% for log phase cells, which increased to 4.8% in the presence of high UV radiation and reached 8.5% for unirradiated stationary phase cells (Table 2). Rapidly dividing log phase cells ± UV contained a marked ssDNA gradient, high proximal to OriC and decreasing bidirectionally reaching a minimum at Ter (Figures 4A, 5A and B, Supplementary Figure S2). In contrast, the distribution of ssDNA gaps is essentially flat in non-dividing stationary phase cells (Figure 5C, Supplementary Figure S2). There were large numbers of short gaps in unirradiated log phase cells. Gap sizes, along with numbers, increased substantially in UV irradiated cells (Figure 3). As DNA synthesis largely halts for a period after UV irradiation (32–34), the large increase is not readily explained by increased replisome encounters with UV lesions, as discussed below. The patterns we establish in the current work provide a launching point for a wide variety of studies to explore gap generation as a part of DNA metabolism.
Two classic papers published in the mid and late ‘60s described the formation of ssDNA gaps in the E. coli chromosome (35,36). In the first, Rupp and Howard-Flanders used sucrose gradient ultracentrifugation to measure the size distributions of chromosomal ssDNA gaps in UV-irradiated E. coli (35). In the second, Okazaki et al. used radioactive pulse-chase experiments in combination with sucrose gradient ultracentrifugation, to show that replication occurred in short fragments (subsequently named Okazaki fragments) on the lagging replication strand, which were then joined into full-length chromosomal DNA (36). Notably, Okazaki's model for discontinuous lagging-strand synthesis and continuous leading-strand synthesis (i.e. semi-discontinuous DNA replication) did not rule out that interrupted, i.e. discontinuous DNA synthesis could also occur on the leading strand.
During the ensuing 50 years, genetic and biochemical studies have identified numerous proteins used during DNA replication, repair and recombination that are involved in gap formation. Gaps have been shown to be formed in chromosomal DNA in response to exogenous DNA damage, and during normal DNA processes in unstressed cells. However, a way to measure the distribution of ssDNA gaps in the E. coli chromosome has not been previously reported. Here, we present a method of detection of genomic ssDNA gaps by whole genome sequencing of bisulfite-treated gDNA (Figure 1A).
For the three cell populations, ssDNA gap levels appeared to be similar on the W- and C-strands (Figure 2B). Despite different modes of DNA synthesis (continuous for the leading-strand and discontinuous for the lagging-strand), ssDNA gap content was found to be similar on the leading- and lagging-strands, specific for each growth condition (Table 2). The roughly equal numbers of full-length ssDNA sequencing reads that aligned and mapped to CT-converted and GA-converted genomes (Figures 3 and 4A, Supplementary Figure S1A) provide additional support that ssDNA gaps are present at similar levels on the leading and lagging strands.
The genome-wide distribution of ssDNA gap intermediates was analyzed by mapping full-length ssDNA reads to W- and C-strands at each location along the E. coli chromosome (Figure 4A, Supplementary Figure S2). Average ssDNA levels in mid-log cells ± UV show a gradual decrease going from OriC to Ter (Figure 4A, Supplementary Figure S2, blue color shades) with essentially the same amount of ssDNA on the W-strand and on the C-strand (Figure 4A, middle and bottom read-map graphs). However, the ssDNA is distributed non-uniformly along the E. coli chromosome with the presence of localized hot regions enriched for ssDNA, and cold regions that contain much lower levels of ssDNA relative to the genome average (Figure 4B). A compilation of hot ssDNA regions shows strong similarities for W- and C-strands (Table 3). For mid-log cells, there are ∼1300 hot regions distributed throughout the E. coli genome having an average length ∼ 108 bp with a median separation between the regions of ∼ 1060 bp. Compared to hot regions, there are much fewer cold regions, 333–339 regions per strand with an average size of 81–85 bp. Hot regions are concentrated around OriC with very few found in the vicinity of the Ter site. In contrast, many more cold regions are located proximal to the Ter site (Figure 5A). When leading- and lagging-strands were examined individually, hot regions appeared to be 1.4-fold higher on the lagging- strand, compared to the leading-strand. Conversely, there are ∼1.4-fold more cold regions on the leading-strand compared to the lagging-strand (Table 4). The differences in distribution of hot and cold regions on the leading- and lagging-strands might be attributable to discontinuous lagging-strand synthesis and near-continuous leading-strand synthesis. Alternatively, the observed strand bias might be due to a difference in orientation of gene transcription relative to the direction chromosome replication. Both co-directional and head-on collisions between replications forks and RNA polymerases can cause stalling or collapse of the replication forks leading to ssDNA formation with more detrimental effects implicated for head-on collision events (37–41). We examined whether hot genomic ssDNA regions occurred more frequently in genes transcribed in an opposite direction of replication forks (head-on), than in genes transcribed in the same direction of replication forks (co-directional) (Supplementary Table S2). The analysis showed that hot ssDNA regions are present at similar frequencies in ‘head-on’ genes and in ‘co-directional’ genes for all three cell populations (Supplementary Table S2).
Post UV-irradiated cells were harvested 10 min after exposure to UV light (100 J/m2). Based on previous UV exposure experiments (32,34), we estimate that this UV dose generated approximately 4000 cyclobutane–pyrimidine dimers per E. coli chromosome, or an average of one cyclobutane–pyrimidine dimer per 2.2 kb on either the W- or C-strand. The presence of a high number of replication-blocking lesions is likely to be responsible for killing 99.8% of the cells immediately after irradiation, while only ∼1.8% of the cells appeared to survive at 10 min post UV irradiation (see Materials and Methods). Compared to non-irradiated cells, UV-treated cells showed a ∼8-fold increase in long ssDNA gaps (>100 nt) on W- and C-strands (5.8% versus 0.7%) (Figure 3). In contrast to non-UV-irradiated cells, leading- and lagging-strands in UV-irradiated cells appeared to contain roughly similar numbers of hot (2286 versus 2100) and cold (1255 versus 1308) regions (Table 4).
Where are the long ssDNA gaps coming from? The long ssDNA gaps seen during normal exponential growth are likely to reflect two processes, lesion-skipping by the DNA polymerase to generate post-replication gaps and mismatch repair. Recent work (42) has reinforced the idea that post-replication gaps are generated in virtually every replication cycle during growth in rich media. Mismatch repair should also occur multiple times in each replication cycle (43). The two processes would both generate longer stretches of ssDNA and together they likely account for the long gaps seen in the log phase cells. Based on the appearance of many pyrimidine dimers after UV irradiation, one might expect additional lesion-skipping and many new post-replication gaps as suggested 50 years ago by the work of Rupp and Howard-Flanders (18). However, replication largely halts for about 20 min after UV irradiation (32–34) with the possible exception of some extension of the leading strand (44). The replisome is not progressing and thus not encountering large numbers of pyrimidine dimers. Most of the genomic breaks uncovered by Rupp and Howard-Flanders may reflect transient strand breaks created by nucleotide excision repair. Something is nevertheless occurring that is generating larger amounts of ssDNA, with lengths that cannot be explained by excision repair. We note that the lengths of ssDNA we can document are limited, by the paired reads method, to about 500 nucleotides. The lengths of ssDNA being generated may be much longer. Somewhat extensive DNA unwinding and/or nucleolytic degradation may be occurring, at the replisome or elsewhere, a phenomenon not detected by other methods and that requires further research to explain. In stationary phase cells, the numbers of long ssDNA regions increase still further. We speculate that random genomic nicks and strand breaks may get enlarged by nucleolytic digestion in an environment where ATP-dependent DNA repair processes are constrained by metabolic limitation but nuclease action is not.
Hot ssDNA gaps might reflect sites of frequent replication fork stalling or collapse, high levels of recombination, and R-loop formation during transcription. The range of lengths for hot regions is sufficient to accommodate the many cellular processes involved in ssDNA generation, going from a low of ∼1–12 kb ssDNA gaps for base and nucleotide excision repair (45,46), to a high of ∼50–2 kb ssDNA gaps associated with homologous recombination, post-replication mismatch repair, replication fork – transcription bubble collisions, and the generation of lagging-strand Okazaki fragments during DNA replication (9,38,41,47). The low percentage of hot regions containing Chi recombination hotspots in the three cell populations (Table 5) suggests that RecBCD-mediated recombination is unlikely to be responsible for generating ssDNA in these regions. The distribution of hot ssDNA regions in mid-log cells ±UV is also consistent with the involvement of chromosome replication in generating ssDNA gaps. Notably, the distribution of hot ssDNA regions exhibits strong spatial polarity, highest in the vicinity of OriC, decreasing sharply for a distance of ∼1000 kb on both leading- and lagging-strand DNA, and barely detectable in the vicinity of Ter (Figure 5A, B). The approximate symmetry centered around OriC on leading- and lagging-strands, for W- and C-strands, is consistent with the possibility that ssDNA in the hot regions occur principally as DNA replication intermediates generated in cells growing asynchronously in log phase. Conversely, the distribution of cold ssDNA regions has the opposite spatial polarity pattern on W- and C-leading and lagging strands, with the number of cold ssDNA regions peaking at Ter site and gradually decreasing toward OriC (Figure 5A, B). We have found that hot regions in UV-irradiated cells are strongly prevalent in regions surrounding OriC. Previous studies have shown that E. coli essentially halts ongoing replication for at least 15–20 min following UV-irradiation (32–34). Nevertheless, OriC continues to fire at its normal rate in a DnaA-dependent manner (34,48). Active origins of replication in UV-irradiated cells would lead to enrichment of replication forks around OriC regions, which could also contribute to the strong localization of hot ssDNA regions in the vicinity of OriC (Figure 5B).
An earlier publication used a Na bisulfite-deep sequencing-informatics approach to characterize R-loops in E. coli gDNA (49). Unlike the earlier study, our DNA was treated with RNase to remove most of the RNA and R-loops. Based on our analysis of hot- and cold-gapped regions (Figures 4 and 5, Table 4), we have identified potential R-loop structures for the purpose of estimating their contribution to gapped ssDNA regions (Supplementary Table S3). For mid-log phase cells, there were 1.7-fold more hot ssDNA regions on the transcribed non-template strands (∼700) compared to template strands (∼400), suggesting that R-loops make a significant contribution to ssDNA gaps. Since RNA:DNA hybrids are insensitive to exposure to bisulfite, an almost exact reciprocal relationship was observed for cold ssDNA regions, which showed 1.7-fold more ssDNA regions on the template strands (∼170) compared to non-template strands (∼100). Based on the data in Supplementary Table S3, we estimate that R-loops contribute about 12 to 15% of the ssDNA observed in both hot- and cold regions in mid-log phase cells. In contrast, there appeared to be no significant transcription strand bias for UV irradiated and stationary phase cells (Supplementary Table S3).
Okazaki's model for discontinuous lagging-strand DNA replication (36) was compatible with the possibility that discrete DNA fragments might also be present on the leading-strand, arising perhaps as replication intermediates, or possibly not. Between 1968 and 2019, many leading-strand discontinuities were traced, not to replication intermediates, but instead to intermediates arising during DNA damage repair, e.g. BER, NER, MMR (50,51), and to pathways used to rescue stalled replication forks (20). In 2019, Kuzminov and colleagues showed that by eliminating BER, NER, MMR, and especially RER (removal of misincorporated ribonucleotides) the leading strand is then synthesized in a long continuous stretch (∼80 kb) (44), which corresponds to ∼3.5% of the distance between OriC and Ter. However, when repair pathways are present, there are large numbers of leading strand discontinuities observed for cells grown in the absence of exogenous DNA damage (44). Interruptions in synthesis that result in RNA-initiated repriming can lead to the generation of ssDNA gaps on the leading strand (18,20,52). The origin of these leading strand gaps is not known, and, apart from the generation of Okazaki fragments, the same holds true for regions containing an excess of lagging strand gaps.
We’ve described a straightforward method to map ssDNA gapped regions on the E. coli chromosome with near-single nt precision. A new insight obtained from the current data is the detection of high concentrations of ssDNA gapped regions (i.e. hot ssDNA regions) on the leading strand that lie close to the replication origin OriC in actively dividing cells (Figure 5A, B). The presence of ssDNA on the leading strand at the replication fork is consistent with a biochemical characterization of model replication forks reported by Manhart and McHenry (53), which used DNA–protein crosslinking to identify the presence of SSB bound simultaneously on lagging- and leading-strands during primosome assembly in proximity with an E. coli replication fork. The ssDNA gap distributions observed for mid-log cells ± UV (Figure 5A, B) are compatible with replication-restart models that involve replisome reassembly. For example, the median distance between hot ssDNA regions in heavily UV-irradiated cells (∼372 bp, Table 4) could reflect the need for frequent replication fork reassembly in response to the presence of high numbers of inadvertent blocks to DNA synthesis. Furthermore, the average and maximum lengths of the leading strand hot ssDNA regions in mid-log cells ±UV (Table 4) are sufficient to accommodate SSB assembly.
In summary, the bisulfite sequencing method presented here can be used to map ssDNA gaps generated during different stages of cell growth. It can be used to map ssDNA gaps in situations where replication, recombination, repair pathways have been modified, and in the presence of exogenous agents that damage DNA. Several general patterns of ssDNA generation have been revealed in the current study, setting a critical baseline for future work. The method is amenable to identifying proteins bound in the ssDNA gaps using ChIP-Seq analysis. Although at present the origins of chromosomal ssDNA gaps remain elusive, we hope to build on the current results and utilize this method as one important tool to explore genomic gap creation in many different DNA metabolism contexts.
DATA AVAILABILITY
Raw next-generation sequencing data have been deposited to SRA database (Submission ID: SUB10466715; BioProject ID: PRJNA769733).
Supplementary Material
Contributor Information
Phuong Pham, Departments of Biological Sciences and Chemistry, University of Southern California, Los Angeles, CA 90089-2910, USA.
Yijun Shao, Departments of Biological Sciences and Chemistry, University of Southern California, Los Angeles, CA 90089-2910, USA.
Michael M Cox, Department of Biochemistry, University of Wisconsin-Madison, Madison, WI 53706-1544, USA.
Myron F Goodman, Departments of Biological Sciences and Chemistry, University of Southern California, Los Angeles, CA 90089-2910, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of General Medical Sciences [1RM1GM130450 to M.M.C., M.F.G.]; National Institute of Environmental Health Sciences [R35ES028343 to M.F.G.]; Y.S. was supported by a USC Provost's Undergraduate Research Fellowship. Funding for open access charge: National Institutes of General Medical Sciences [1RM1GM130450].
Conflict of interest statement. None declared.
REFERENCES
- 1. Karnani N., Taylor C.M., Malhotra A., Dutta A.. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol. Biol. Cell. 2010; 21:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hyrien O. Peaks cloaked in the mist: the landscape of mammalian replication origins. J. Cell Biol. 2015; 208:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu C., Gan H., Han J., Zhou Z.X., Jia S., Chabes A., Farrugia G., Ordog T., Zhang Z.. Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol. Cell. 2014; 56:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hanawalt P.C. Grossman L., Moldave K.. Methods in Enzymology. 1967; 12:Academic Press; 702–708. [Google Scholar]
- 5. Coote J.G., Binnie C.. Tolerance to bromodeoxyuridine in a thymidine-requiring strain of bacillus subtilis. J. Gen. Microbiol. 1986; 132:481–492. [DOI] [PubMed] [Google Scholar]
- 6. Titz B., Hauser R., Engelbrecher A., Uetz P.. The escherichia coli protein YjjG is a house-cleaning nucleotidase in vivo. FEMS Microbiol. Lett. 2007; 270:49–57. [DOI] [PubMed] [Google Scholar]
- 7. Lopes M., Foiani M., Sogo J.M.. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006; 21:15–27. [DOI] [PubMed] [Google Scholar]
- 8. Cox M.M. Historical overview: searching for replication help in all of the rec places. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8173–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cox M.M. The nonmutagenic repair of broken replication forks via recombination. Mutat. Res. 2002; 510:107–120. [DOI] [PubMed] [Google Scholar]
- 10. Branzei D., Szakal B.. Building up and breaking down: mechanisms controlling recombination during replication. Crit. Rev. Biochem. Mol. Biol. 2017; 52:381–394. [DOI] [PubMed] [Google Scholar]
- 11. Heller R.C., Marians K.J.. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 2006; 7:932–943. [DOI] [PubMed] [Google Scholar]
- 12. Yeeles J.T., Poli J., Marians K.J., Pasero P.. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013; 5:a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kowalczykowski S.C. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 2000; 25:156–165. [DOI] [PubMed] [Google Scholar]
- 14. Cox M.M., Goodman M.F., Kreuzer K.N., Sherratt D.J., Sandler S.J., Marians K.J.. The importance of repairing stalled replication forks. Nature. 2000; 404:37–41. [DOI] [PubMed] [Google Scholar]
- 15. Kuzminov A. Recombinational repair of DNA damage in escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 1999; 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Howard-Flanders P. Repair by genetic recombination in bacteria: overview. Basic Life. Sci. 1975; 5A:265–274. [DOI] [PubMed] [Google Scholar]
- 17. Rothman R.H., Clark A.J.. The dependence of postreplication repair on uvrB in a recF mutant of Escherichia coli K-12. Mol. Gen. Genet. 1977; 155:279–286. [DOI] [PubMed] [Google Scholar]
- 18. Rupp W.D., Howard-Flanders P.. Discontinuities in the DNA synthesized in an excision-defective strain of escherichia coli following ultraviolet irradiation. J. Mol. Biol. 1968; 31:291–304. [DOI] [PubMed] [Google Scholar]
- 19. Rupp W.D., Wilde C.E. 3rd, Reno D.L., Howard-Flanders P.. Exchanges between DNA strands in ultraviolet-irradiated escherichia coli. J. Mol. Biol. 1971; 61:25–44. [DOI] [PubMed] [Google Scholar]
- 20. Marians K.J. Lesion bypass and the reactivation of stalled replication forks. Annu. Rev. Biochem. 2018; 87:217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pham P., Malik S., Mak C., Calabrese P.C., Roeder R.G., Goodman M.F.. AID-RNA polymerase II transcription-dependent deamination of IgV DNA. Nucleic. Acids. Res. 2019; 47:10815–10829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raizis A.M., Schmitt F., Jost J.P.. A bisulfite method of 5-methylcytosine mapping that minimizes template degradation. Anal. Biochem. 1995; 226:161–166. [DOI] [PubMed] [Google Scholar]
- 23. Laskowski M. Sr Purification and properties of the mung bean nuclease. Methods Enzymol. 1980; 65:263–276. [DOI] [PubMed] [Google Scholar]
- 24. Peyrard M., Cuesta-Lopez S., James G.. Nonlinear analysis of the dynamics of DNA breathing. J. Biol. Phys. 2009; 35:73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phelps C., Lee W., Jose D., von Hippel P.H., Marcus A.H.. Single-molecule FRET and linear dichroism studies of DNA breathing and helicase binding at replication fork junctions. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:17320–17325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blattner F.R., Plunkett G. 3rd, Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F.et al.. The complete genome sequence of escherichia coli K-12. Science. 1997; 277:1453–1462. [DOI] [PubMed] [Google Scholar]
- 27. Duigou S., Boccard F.. Long range chromosome organization in escherichia coli: the position of the replication origin defines the non-structured regions and the right and left macrodomains. PLoS Genet. 2017; 13:e1006758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valens M., Penaud S., Rossignol M., Cornet F., Boccard F.. Macrodomain organization of the escherichia coli chromosome. EMBO J. 2004; 23:4330–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Niki H., Yamaichi Y., Hiraga S.. Dynamic organization of chromosomal DNA in escherichia coli. Genes Dev. 2000; 14:212–223. [PMC free article] [PubMed] [Google Scholar]
- 30. Stahl F., Myers R.. Old and new concepts for the role of chi in bacterial recombination. J. Hered. 1995; 86:327–329. [DOI] [PubMed] [Google Scholar]
- 31. Marinus M.G., Lobner-Olesen A.. DNA methylation. EcoSal Plus. 2014; 6: 10.1128/ecosalplus.ESP-0003-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Courcelle C.T., Chow K.H., Casey A., Courcelle J.. Nascent DNA processing by RecJ favors lesion repair over translesion synthesis at arrested replication forks in escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:9154–9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Courcelle J., Hanawalt P.C.. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated escherichia coli. Mol. Gen. Genet. 1999; 262:543–551. [DOI] [PubMed] [Google Scholar]
- 34. Rudolph C.J., Upton A.L., Lloyd R.G.. Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev. 2007; 21:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Howard-Flanders P., Rupp W.D., Wilkins B.M., Cole R.S.. DNA replication and recombination after UV irradiation. Cold Spring Harb. Symp. Quant. Biol. 1968; 33:195–207. [DOI] [PubMed] [Google Scholar]
- 36. Okazaki R., Okazaki T., Sakabe K., Sugimoto K., Sugino A.. Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains. Proc. Natl. Acad. Sci. U.S.A. 1968; 59:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lang K.S., Hall A.N., Merrikh C.N., Ragheb M., Tabakh H., Pollock A.J., Woodward J.J., Dreifus J.E., Merrikh H.. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell. 2017; 170:787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu B., Alberts B.M.. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science. 1995; 267:1131–1137. [DOI] [PubMed] [Google Scholar]
- 39. Liu B., Wong M.L., Tinker R.L., Geiduschek E.P., Alberts B.M.. The DNA replication fork can pass RNA polymerase without displacing the nascent transcript. Nature. 1993; 366:33–39. [DOI] [PubMed] [Google Scholar]
- 40. Mirkin E.V., Mirkin S.M.. Mechanisms of transcription-replication collisions in bacteria. Mol. Cell. Biol. 2005; 25:888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pomerantz R.T., O’Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010; 327:590–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Romero Z.J., Chen S.H., Armstrong T., Wood E.A., van Oijen A., Robinson A., Cox M.M.. Resolving toxic DNA repair intermediates in every e. coli replication cycle: critical roles for RecG, uup and radD. Nucleic Acids Res. 2020; 48:8445–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schaaper R.M. Base selection, proofreading, and mismatch repair during DNA replication in escherichia coli. J. Biol. Chem. 1993; 268:23762–23765. [PubMed] [Google Scholar]
- 44. Cronan G.E., Kouzminova E.A., Kuzminov A.. Near-continuously synthesized leading strands in escherichia coli are broken by ribonucleotide excision. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krokan H.E., Bjoras M.. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013; 5:a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sancar A., Tang M.S.. Nucleotide excision repair. Photochem. Photobiol. 1993; 57:905–921. [DOI] [PubMed] [Google Scholar]
- 47. Modrich P., Lahue R.. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996; 65:101–133. [DOI] [PubMed] [Google Scholar]
- 48. Wendel B.M., Hollingsworth S., Courcelle C.T., Courcelle J.. UV-induced DNA damage disrupts the coordination between replication initiation, elongation and completion. Genes Cells. 2021; 26:94–108. [DOI] [PubMed] [Google Scholar]
- 49. Leela J.K., Syeda A.H., Anupama K., Gowrishankar J.. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tye B.K., Chien J., Lehman I.R., Duncan B.K., Warner H.R.. Uracil incorporation: a source of pulse-labeled DNA fragments in the replication of the escherichia coli chromosome. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Amado L., Kuzminov A.. Low-molecular-weight DNA replication intermediates in escherichia coli: mechanism of formation and strand specificity. J. Mol. Biol. 2013; 425:4177–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yeeles J.T., Marians K.J.. Dynamics of leading-strand lesion skipping by the replisome. Mol. Cell. 2013; 52:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Manhart C.M., McHenry C.S.. Identification of subunit binding positions on a model fork and displacements that occur during sequential assembly of the escherichia coli primosome. J. Biol. Chem. 2015; 290:10828–10839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw next-generation sequencing data have been deposited to SRA database (Submission ID: SUB10466715; BioProject ID: PRJNA769733).