

Abstract
PURPOSE
Apolipoprotein B mRNA-Editing Catalytic Polypeptide-like (APOBEC) enzymes are mutagenic factors contributing to tumor progression and therapy resistance. However, the effects of APOBEC-induced protein changes have not been systematically assessed. Here, we describe the effects of APOBEC on the coding sequence in primary and metastatic estrogen receptor–positive (ER+)/human epidermal growth factor receptor 2–negative (HER2–) breast cancer (BC).
METHODS
We determined the enrichment of amino acid (AA) changes resulting from APOBEC mutagenesis in 323 primary BC tumors and 424 metastatic breast cancer (mBC) lesions via comparison with a simulated mutational genomic landscape not under selection pressure. We subsequently explored genes with recurrent APOBEC-associated AA changes and investigated the clonality of individual APOBEC-associated mutations. Using public sequencing data from an independent primary BC and mBC cohort, we further confirm our findings by reporting genes having these enriched AA changes in an APOBEC context.
RESULTS
Our analysis demonstrated that several APOBEC-derived AA changes are significantly enriched compared with a simulated AA change distribution drawn at random. Among the enriched AA changes, Glutamate(E)>Lysine(K) and Glutamate(E)>Glutamine(Q) were mostly found at hotspots in oncogenes, whereas termination codons (Glutamine[Q]>STOP[X] and Serine[S]>STOP[X]) occurred in tumor suppressor genes and, mostly, not at hotspot locations. These mutations are found in genes contributing to BC initiation, eg, introduction of termination codons in TP53, MAP3K1, and CDH1 (in lobular BC) and two oncogenic hotspots in PIK3CA (p.E542K and p.E545K). In endocrine-resistant BC, we observed APOBEC-induced termination codons at ER-modulating genes, KMT2C, ARID1A, NF1, and ZFHX3. Finally, in mBC, compared with single PIK3CA mutations, dual PIK3CA mutations occurred more frequently in an APOBEC context (Fisher's exact P < .001).
CONCLUSION
Our results show that APOBEC mutagenesis recurrently targets various known drivers of BC initiation, progression, and endocrine resistance.
BACKGROUND
Apolipoprotein B mRNA-Editing Catalytic Polypeptide-like (APOBEC) enzymes are important mutagenic factors in multiple human cancers. APOBEC enzymes cause two single base-pair substitution (SBS) signatures that are characterized by C-to-T transitions (SBS2) and C-to-G transversions (SBS13) in predominantly, but not restricted to, TCW trinucleotide contexts (W = A or T).1 APOBEC mutagenesis affects the evolutionary history of tumors by initiating the expansion of subclones. Consequently, APOBEC mutagenesis can contribute to tumor progression and/or therapy resistance.2 Indeed, in ER+/human epidermal growth factor receptor 2–negative (HER2–) tumors, the number of APOBEC mutations is increased in metastatic breast cancer (mBC) when compared with primary breast cancer (pBC).3,4 Although this increase might in part be explained by either functional advantages or selection under treatment, recent work has shown that a subset of APOBEC mutations may be the result of improved spatial availability of the target base enabled by the loop structure within a DNA hairpin.5
CONTEXT
Key Objective
Apolipoprotein B mRNA-Editing Catalytic Polypeptide-like (APOBEC) mutagenesis and subsequent differential selection of mutated genes may play an important role in the evolutionary history of different cancer types, including breast cancer (BC). Here, we systematically assessed the effects of APOBEC-induced protein changes in estrogen receptor–positive (ER+)/human epidermal growth factor receptor 2–negative (HER2–) BC in primary and metastatic diseases.
Knowledge Generated
We demonstrate that APOBEC is a driving force of genomic evolution in ER+/HER2– BC by clonal enrichment of focal oncogenic mutations in PIK3CA and tumor-suppressive alterations in various genes that are involved in BC tumor formation and resistance to endocrine therapy.
Relevance
This study provides better knowledge on the effects of APOBEC on the etiology of ER+/HER2– breast tumors. The insight that APOBEC induces mutations that hyperactivate PI3K signaling and mutations in genes that are associated with resistance to endocrine treatment could affect clinical management of tumors with high APOBEC contribution.
The most likely process whereby favorable traits are selected will involve amino acid (AA) changes in the derived protein, altering its functionality. Since APOBEC is thought to be enzymatically capable of mutating any C>T or C>G in a TW context, all AA changes that can occur via this process are a priori equally likely to occur if they are not selected by environmental pressure, such as endocrine treatment. However, specific AA changes have been reported; for instance, mutations in PIK3CA (eg, p.E545K and p.E542K) are present in an APOBEC context and are found frequently in multiple cancer types with high APOBEC activity.6,7 Recently, additional clonal drivers and subclonal passengers within APOBEC-derived hotspots were identified in patients with bladder cancer.8 The effects of APOBEC mutagenesis on the full coding sequence and its significance in patients with ER+/HER2– breast cancer (BC), however, have thus far not been systematically assessed.
Here, we determine which of the APOBEC-derived codon changes are significantly enriched in ER+/HER2– pBC and mBC using whole-genome sequencing (WGS) data from previously described cohorts.3,9 This enabled us to identify genomic alterations that are likely subjected to differential selection in ER+/HER2– pBC and mBC. Subsequently, we describe in which genes or at which hotspots these AA changes occur most frequently in pBC, in mBC, and in an mBC cohort showing intrinsic or acquired resistance to hormonal therapy, using sequencing data from independent previously described cohorts.3,9,10 Using this approach, we provide an overview of the effects of APOBEC mutagenesis on the coding sequence in BC and subsequently describe how these alterations evolve during tumor progression and/or under endocrine treatment.
METHODS
The full methods are described in the Data Supplement. Briefly, we determined which APOBEC-associated AA changes are more frequently observed in pBC and mBC when compared with a distribution of random draws of a mutational repertoire of a genome not under selection pressure. This implies that these enriched AA changes in pBC or mBC are under differential selection (Fig 1). For defining all possible AA changes that can result from APOBEC-related single-base substitutions (SBS2/SBS13) on the coding strand of the genome (GRCh37), we created a catalog of all possible APOBEC-induced codon changes (Fig 1, top right) that reflect all the possible AA changes if the complete transcriptome would be mutated by APOBEC, here defined as C>T and C>G in a TCW context. By bootstrapping (1,000 random draws with replacement) from this catalog, we can estimate how often AA changes can occur by chance (eg, approximately 17% of the random draws are E>Q). We subsequently used WGS data from 323 ER+/HER2– pBC samples9 and from 424 ER+/HER2– mBC samples3 to get the actual observed AA changes. As we assumed that APOBEC mutagenesis occurs at random, we performed the bootstrap with 100,000 iterations (ie, we repeated the bootstrap 100,000 times), resulting in a null distribution for each AA change (Fig 1, bottom middle). The fraction of times that the randomly drawn relative frequency of an AA change was at least as high as the actual observed relative frequency was used to determine the P value. We performed three comparisons: pBC versus the catalog of all possible APOBEC-induced codon changes, mBC versus the catalog, and pBC versus mBC. Specifically, for the first two comparisons, the observed frequency of AA changes resulting from APOBEC activity in the pBC and mBC cohort was compared with the randomly drawn sets from the catalog of expected mutations. The third comparison aimed to identify which AA changes occurred more frequently in mBC than in pBC. For this, we used the actually observed AA changes caused by APOBEC in pBC as the catalog of all possible AA changes from which we again used the bootstrap method with 100,000 iterations to identify enriched AA changes in mBC versus pBC. In addition, we used targeted sequencing data from an endocrine-resistant cohort previously described by Razavi et al10 to compare the presence of APOBEC mutations in an ER+/HER2– mBC population irrespective of the treatment line to patients with mBC who are resistant to endocrine therapy.
FIG 1.
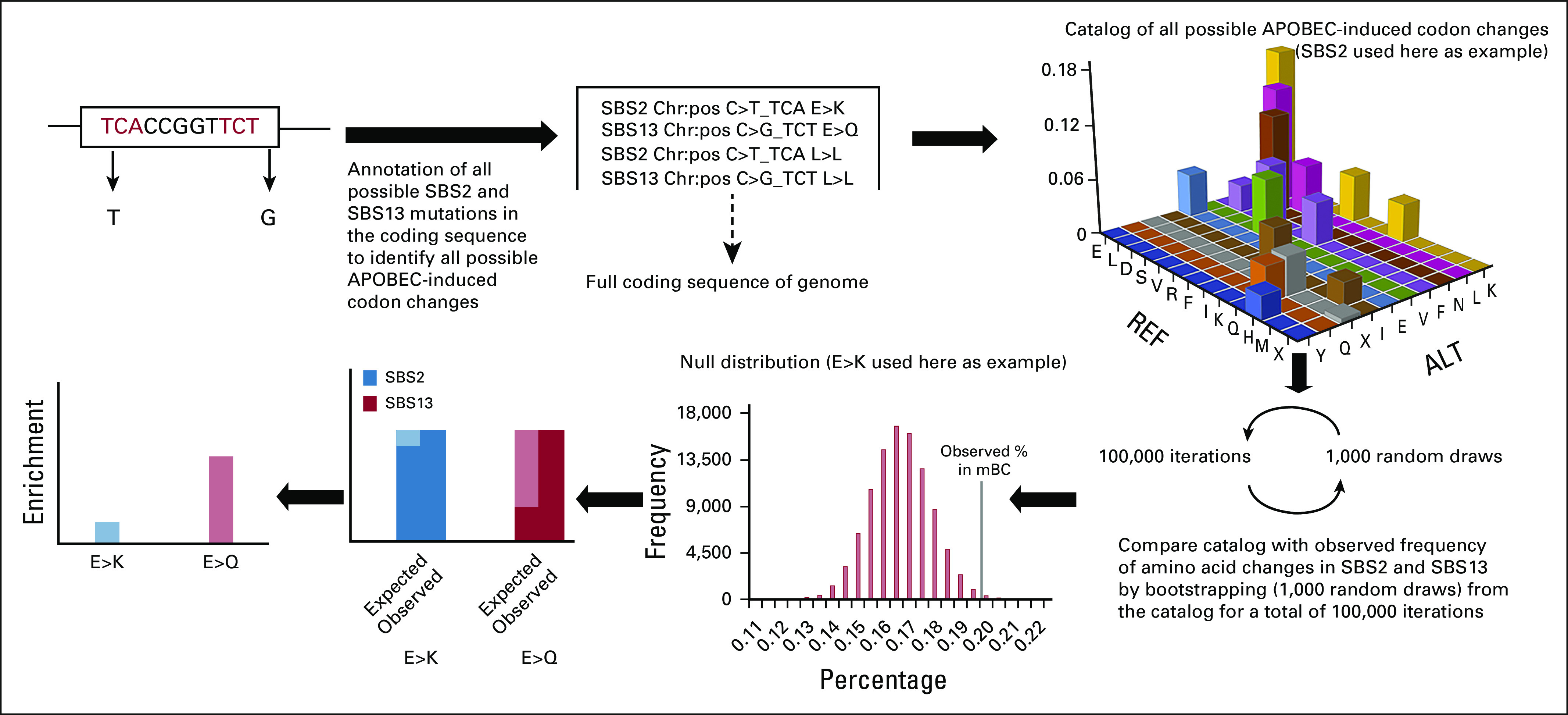
Overview of the methodology used to determine enrichment of APOBEC-derived AA changes. To directly compare which AA changes occurred more frequently in mBC than in pBC, the catalog of all possible APOBEC-induced codon changes was replaced by observed APOBEC-induced codon changes in pBC. AA, amino acid; ALT, alternative base; APOBEC, Apolipoprotein B mRNA-Editing Catalytic Polypeptide-like; C, cysteine; E, glutamate; F, phenylalanine; G, glycine; K, lysine; L, leucine; mBC, metastatic breast cancer; N, asparagine; pBC, primary breast cancer; Q, glutamine; REF, reference base; S, serine; SBS, single base-pair substitution; T, threonine; V, valine; X, STOP (stop codon).
Single-stranded DNA loops that are part of hairpins are known to be highly targeted by APOBEC enzymes.5 To investigate whether hairpins affect our statistical approach, we matched genomic locations of hairpins using annotation of optimal APOBEC3A sites.5
We determined whether identified APOBEC-derived mutations in mBC3 were either clonal or subclonal by using WGS data as described before.11
Statistical Evaluation
Statistical tests other than the bootstrap/iterate analysis described above are specified throughout the results and were two-sided and considered statistically significant when P < .05. IBM SPSS Statistics 25 (ICM Corp, Armonk, NY) was used for the statistical analyses.
Ethics Approval and Consent to Participate
pBC cohort: Data were publicly available.9
mBC cohort: For our analyses, we selected patients with mBC who were included under the protocol of the Center for Personalized Cancer Treatment (CPCT) consortium (CPCT-02 Biopsy Protocol, ClinicalTrial.gov identifier: NCT01855477), which was approved by the medical ethics committee of the University Medical Center Utrecht, the Netherlands. A detailed description of the consortium and whole patient cohort has been described recently.4
Razavi cohort: Data were publicly available.10
RESULTS
Identification of APOBEC-Derived Enriched Codon Changes
Our approach to determine enrichment of APOBEC-derived AA changes is presented in Figure 1. In short, we established a catalog of all possible changes that APOBEC is capable of producing in the complete coding sequence of the human genome (Fig 1, top right). By randomly drawing from this catalog, we simulate the distribution of codon changes in a genome when no selection pressure is present. By comparing the actual observed codon changes in pBC and mBC with the randomly drawn sets, specific enriched AA changes are identified that are likely selected for (synonymous events are included here in the definition of AA change, and all single-letter AAs are listed in the abbreviations list). We analyzed WGS data from 323 pBC tumors9 and 424 metastatic biopsies from patients with mBC; all patients included were ER+/HER2–.3 In pBC, AA changes E>K, Q>X, S>L, and F>F (SBS2) and L>L, Q>E, L>F, L>V, S>C, and K>N (SBS13) were significantly enriched compared with randomly drawn sets, whereas E>K, L>L, Q>X, and S>L (SBS2) and E>Q, L>L, Q>E, and L>F (SBS13) were enriched in mBC (P < .05). When using the data from pBC as the catalog of possible AA changes to draw random sets from, E>K, E>Q, and S>X were enriched in mBC relative to those already observed in pBC (Fig 2).
FIG 2.

Enrichment of AA changes (difference between observed frequency and a random sample) in pBC and mBC and in mBC with pBC as a random sample for SBS2 and SBS13 (*P ≤ .05, **P ≤ .01, and ***P ≤ .001). Only AA changes with a frequency ≥ 1% in a random sample are shown. AA, amino acid; C, cysteine; E, glutamate; F, phenylalanine; H, histidine; I, isoleucine; K, lysine; L, leucine; mBC, metastatic breast cancer; mBC, metastatic breast cancer; N, asparagine; pBC, primary breast cancer; Q, glutamine; R, arginine; S, serine; SBS, single base-pair substitution; T, threonine; V, valine; X, STOP (stop codon); Y, tyrosine. APOBEC drives early, metastatic, and endocrine-resistant disease in ER+/HER2– breast cancer
As previously described,5 loop structures in hairpins are prone to APOBEC mutagenesis. In total, 1.53% of all APOBEC-derived mutations that were identified in mBC were located within a hairpin, an enrichment when compared with the frequency that is expected on the basis of the catalog of all possible APOBEC-induced AA changes (0.18%, chi-square P < .001). To elucidate whether the presence of codons in hairpins biased the observed enrichment of the above, we repeated the bootstrap procedure excluding hairpin locations from the catalog of all possible APOBEC-induced changes that were randomly sampled from. This analysis revealed that all the abovementioned enriched AA changes remained significant. In addition, the relative frequency of all possible APOBEC AA changes in the catalog was not different with and without hairpin locations (Data Supplement).
Presence and Clonality of APOBEC-Derived Codon Changes in Genes or Hotspots
We investigated at which hotspots or in which genes the enriched APOBEC-derived AA changes occurred recurrently in pBC, mBC, and endocrine-resistant mBC, implying that they are subjected to differential selection, Table 1. We also investigated whether these mutations were clonal. We performed the latter analysis only in the 424 mBC cases for which WGS data were available since the lower sequence coverage in the pBC cohort (median coverage pBC cohort: 40×; median coverage mBC cohort: 110×) likely yields inferior results.
TABLE 1.
Results of Enriched Amino Acid Changes in pBC and mBC

From all APOBEC-derived mutations in mBC, the majority (90%) was clonal. However, in general, the fraction of APOBEC mutations was not different among all exonic clonal and subclonal mutations that were identified within genes (Fisher's exact P = .895, Fig 3).
FIG 3.

Fraction of APOBEC mutations among clonal and subclonal mutations. Only exonic mutations within a gene were included in this analysis. APOBEC, Apolipoprotein B mRNA-Editing Catalytic Polypeptide-like.
Helical PIK3CA mutations (eg, p.E535K and p.E542K) were the most frequently identified E>K mutations in patients with primary, metastatic, and endocrine-resistant BC (Data Supplement). A subset of patients had dual nonsynonymous mutations in PIK3CA, of which the majority (93%) was clonal. Although the prevalence of those dual PIK3CA mutations was lower among patients with pBC (n = 11 of 323, 3%) than among patients with mBC (n = 31 of 424, 7%) in the WGS cohorts (Fisher's exact P = .024), this difference was not confirmed in the data set from Razavi. Strikingly, dual PIK3CA mutations occurred more frequently in an APOBEC context when compared with single PIK3CA mutations in the metastatic setting (Fisher's exact P < .001), whereas this difference was not observed in pBC (Fisher's exact P = 1, Fig 4) and also in the data set from Razavi.
FIG 4.
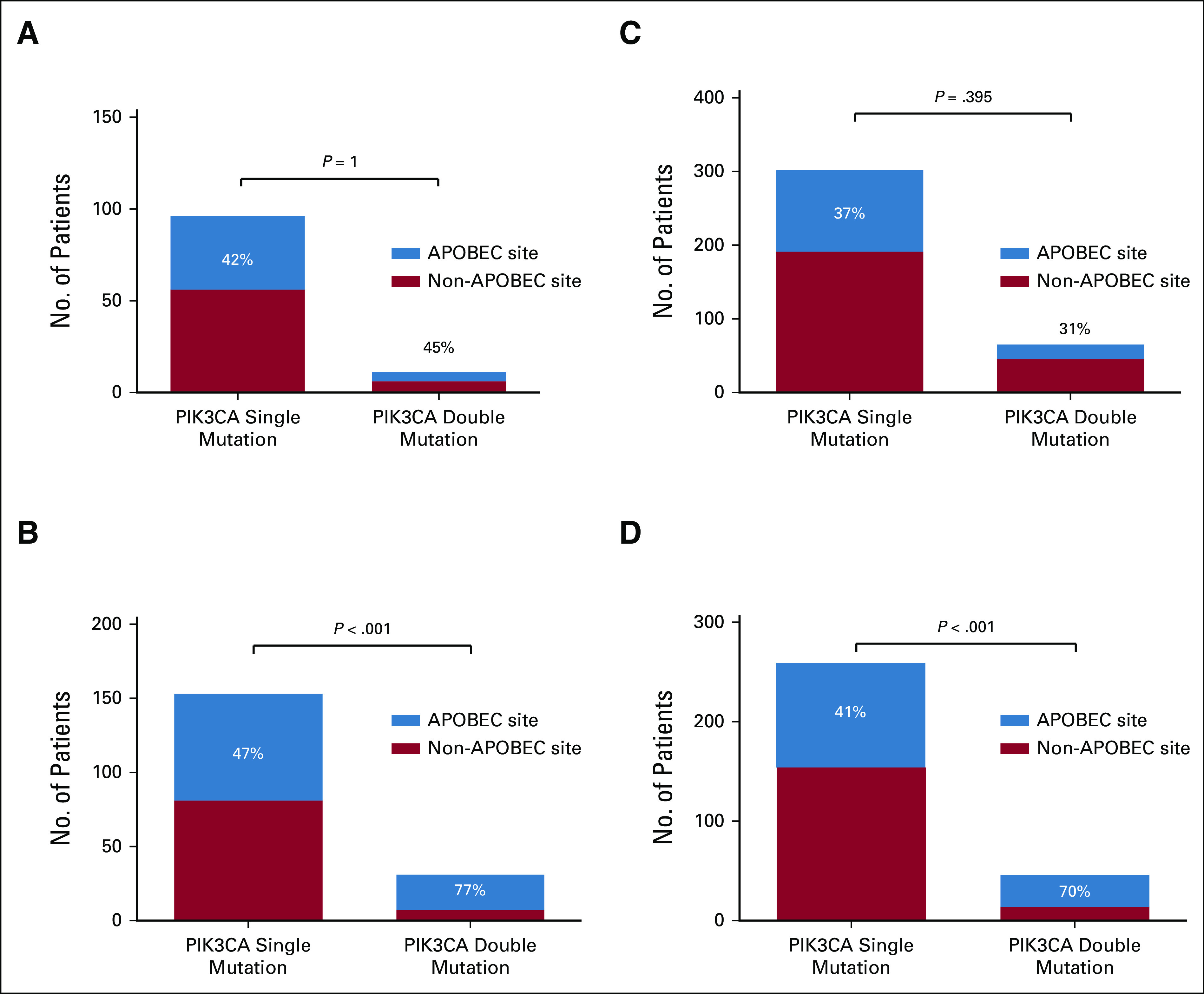
Prevalence of PIK3CA mutations in pBC and mBC: (A) pBC, BASIS cohort; (B) mBC, CPCT-02 cohort; (C) pBC, Razavi cohort; and (D) mBC, Razavi cohort. The Fisher's exact test was used to determine differences between the fraction of APOBC mutations between single and dual PIK3CA mutations. For patients with dual PIK3CA mutations, at least one mutation occurring at an APOBEC site was required to be classified as ‘APOBEC site’. APOBEC, Apolipoprotein B mRNA-Editing Catalytic Polypeptide-like; mBC, metastatic breast cancer; pBC, primary breast cancer.
In pBC, APOBEC-induced inactivating Q>X mutations occurred most frequently in CDH1 (16%) of all Q>X mutations, NCOR1 (10%), TP53 (10%), and MAP3K1 (11%), all well-known drivers of pBC.9 In fact, APOBEC-derived Q>X mutations in CDH1 were the sole inactivating mutation in 6% of all lobular pBC tissues. In mBC, those inactivating mutations in CDH1 (0.6%), TP53 (0.5%), and MAP3K1 (0.5%) remained, although their relative contribution to the total number of APOBEC-induced mutations decreased. Additional widespread inactivating mutations were identified in KMT2C, ZHX3, NF1, PTEN, and RB1 (Fig 5 and Data Supplement). All these mutations were clonal, apart from a single mutation in PTEN and ZFHX3. S>X mutations did not have a high prevalence in pBC and were only recurrently detected in ARID1A (11% of all S>X mutations), BRCA2 (11%), and KMT2C (11%) in mBC, Figure 5 and Data Supplement.
FIG 5.

Genes with mutations that induce stop codons (Q>X or S>X) in an APOBEC context (SBS2 or SBS13). Only genes with a mutation frequency of ≥ 0.5% in the metastatic setting are shown. APOBEC, Apolipoprotein B mRNA-Editing Catalytic Polypeptide-like; mBC, metastatic breast cancer; pBC, primary breast cancer; Q, glutamine; S, serine; SBS, single base-pair substitution; X, STOP (stop codon).
Besides some recurrent clonal mutations in PIK3CA and ESR1 p.E380Q, we did not identify additional recurrent E>Q mutations (Data Supplement). Moreover, we did not find any genes having recurrent L>L, L>F, L>V, S>C, or K>N mutations. To understand why synonymous L>L mutations were unexpectedly found enriched in mBC, we studied whether L>L mutations were coenriched with other enriched codon changes. Therefore, we defined the number of driver mutations resulting from APOBEC mutagenesis for each sample: Q>X in CDH1, NF1, TP53, KMT2C, ARID1A, MAP3K1, and RB1 and E>K in PIK3CA. We associated the fraction of L>L mutations per total number of SBS2 mutations with the number of SBS2-induced driver mutations and observed a significant correlation, suggesting evidence of coenrichment between L>L mutations and other SBS2-induced drivers (Jonckheere-Terpstra test for ordered alternatives: P = .001, Data Supplement). Since no recurrent driver genes in SBS13 were identified, apart from few E>Q mutations in ESR1 and PIK3CA, we were unable to perform a similar analysis for E>Q or L>L in a SBS13 context.
DISCUSSION
APOBEC enzymes are important mutagenic factors driving a subgroup of ER+/HER2-BCs. We used an alternative approach that has been used thus far, in which we draw random sets from all possible codon changes induced by APOBEC, enabling the identification of those codon changes that are enriched in pBC and mBC. Besides confirmation of previously described APOBEC effects, our analysis described the evolution of APOBEC effects over time and pinpointed specific biologic effects of APOBEC mutagenesis in BC such as the presence of dual activating mutations in PIK3CA and the widespread accumulation of inactivating stop codons in tumor suppressor genes, including genes modulating the estrogen signaling pathway. The latter genes were previously reported to be involved in endocrine resistance, mutually exclusive to ESR1 mutations.
Although several studies have identified APOBEC driver mutations occurring in TCN motifs rather than only in TCW motifs,8 we did not include TCN motifs. First, TCW is the vastly dominant contribution12 in the APOBEC signatures. Second, since TCN motifs also comprise the TCG motif, which is mainly age-related,12 we expected to falsely attribute certain mutations to APOBEC activity by including TCN motifs. Thus, the definition that we used to characterize a variant as APOBEC results in enriched AA changes that are very likely attributable to APOBEC activity (ie, highly specific). Also, we only analyzed data from ER+/HER2–tumors. Although APOBEC mutations have been identified in ER-negative BC, which were mainly caused by APOBEC3B,13 a significant enrichment of APOBEC mutations in mBC when compared with pBC was only observed in the ER+/HER2 subtype. In addition, a validation cohort like the cohort from the study by Razavi et al10 was only available for ER+ tumors.
Recent findings have suggested that hairpin structures are susceptible to APOBEC mutagenesis and favor enrichment of subclonal passenger mutations.5 In our analysis, we show that APOBEC-driven AA changes were rarely located within hairpins in mBC (1.53% of all APOBEC mutations), but this percentage was significantly higher than the frequency that is expected on the basis of the catalog of all possible APOBEC-induced AA changes (0.18%). However, further analysis showed no specifically enriched AA changes in relation to the hairpins (Data Supplement). We conclude that hairpin structures make it somewhat easier for APOBEC to mutate DNA, without showing a selection preference for a specific AA change. In addition, the majority of APOBEC mutations were clonal, as was also seen in bladder cancer.8 Taken together, both the clonal nature and the finding that most APOBEC mutations were not located in putative hairpin regions suggest that the enriched APOBEC-induced codon changes are likely driving events. The sequencing depth of our mBC cohort (100×) might have hampered the detection of subclonal events; thus, the significance of subclonal enriched APOBEC AA changes remains unclear in our analysis.
We summarize the AA changes (including synonymous events) that were enriched in pBC and mBC in Table 1 and categorized the implications that the AA changes might have during evolution of BC. First, we identified codon changes that were significantly enriched in pBC but not in mBC (F>F, L>V, S>C, and K>N), suggesting that these do not provide additional selective advantage for the tumor cell during disease progression. We also did not find genes in which these codon changes were recurrently present, so their significance remains unclear.
The next category includes codon changes that were enriched in mBC only, suggesting that these are late selection events: S>X and L>L. S>X mutations were found recurrently within several genes such as ARID1A, BRCA2, and KMT2C, in which a third of all S>X mutations were found. L>L mutations were the only synonymous mutations that were found enriched in mBC. We did not find evidence supporting a biologic effect of the L>L mutations as they were not recurrently observed in specific genes, nor did we find evidence for enrichment of L>L mutations within hairpins as L>L remained significantly enriched when we bootstrapped from a catalog without hairpin locations. However, although synonymous mutations are considered silent, recent studies have suggested that they might affect mRNA splicing, stability, and translation.14 In addition, we demonstrated that L>L mutations in SBS2 were in part coenriched with other selected APOBEC drivers in a SBS2 context identified in our analysis. Thus, L>L mutations in a SBS2 context may be passengers that are coselected.
Two AA changes were further enriched in mBC when compared with pBC: E>K and E>Q. As described by us and others,6,15 APOBEC-derived E>K changes recurrently occur in the helical domain (p.E542K and p.E545K) of PIK3CA in both pBC and mBC. Like kinase domain mutations, which are not APOBEC-related, PIK3CA helical domain mutations are known drivers in BC. Differences in proliferative characteristics between helical and kinase domain mutations affecting clinical outcomes have been suggested16,17 and also disputed.18,19 A subset of patients had dual nonsynonymous PIK3CA mutations. In mBC, dual nonsynonymous PIK3CA mutations occurred more frequently at APOBEC sites than single PIK3CA mutations, a difference that was not observed in pBC. Independent validation in a second cohort10 suggests that APOBEC-derived PIK3CA mutations accumulate over time, a phenomenon that is likely explained by their added selective advantage for tumor cells. Indeed, dual PIK3CA mutations are known to hyperactivate PI3K signaling, which is further supported by their enhanced sensitivity to the recently approved PI3Kα selective inhibitors.20
An E>Q change was the most strongly enriched SBS13-derived change. However, a direct explanation for their enrichment could not be clearly identified nor were they enriched through hairpins. Except for ESR1 p.E380Q and PIK3CA p.E453Q, E>Q changes were not recurrent. ESR1 p.E380Q activates ER to a lesser extent than other activating ESR1 mutations unrelated to APOBEC, which may explain the absence of strong selection in endocrine-resistant mBC of this particular mutation compared with other activating ESR1 mutations.21 Apart from rare mutations in ESR1 and PIK3CA, we did observe compound heterozygous clonal mutations in MAP3K13, FANCA, TP53, and SPEN supportive of a tumor-suppressive function.
Finally, the last category consists of four AA changes, which showed a continuous selection: these were enriched in both pBC and mBC when compared with the random sample, but their enrichment was not augmented in mBC when compared with pBC. Of those AA changes, S>L, Q>E, and L>F were not recurrent at genes nor at hotspots, and we were unable to elucidate the precise reason for their enrichment in BC. By contrast, Q>X was recurrently present in several genes, albeit at different positions. Only in PTEN and TP53, some positions were frequently affected by APOBEC mutagenesis. The facts that Q>X is enriched in both early and late BC and that S>X appears as a late selection event indicate that another major effect of APOBEC mutagenesis on the coding sequence is the introduction of stop codons, likely resulting in subsequent gene inactivation. Genes with Q>X mutations in pBC and mBC were observed in known tumor suppressor genes CDH1, TP53, NCOR1, and MAP3K1, likely persistently driving BC. Importantly, our finding that in 6% of all lobular BCs, APOBEC-derived inactivation of CDH1 is the sole event suggests that APOBEC mutagenesis is directly responsible for a subset of lobular BCs.22 In mBC, Q>X and S>X mutations emerged in genes that have been associated with endocrine resistance like KMT2C, ARID1A, ZFHX3, and NF1. Thus, complete loss of these loci is frequently a result of APOBEC activity and under differential selection during endocrine treatment. This finding is in line with their established biologic role, ie, loss of ER activity modulators KMT2C, ARID1A, NF1, and ZFHX3 worsens outcomes on antiestrogen therapy.23-27 Of note, our analyses did show that S>X and Q>X were often found inside hairpin structures (Fig 3), possibly rendering these results as less likely driving events. However, of the 65 S>X and Q>X changes located inside hairpins, only a single mutation was present in the genes discussed above (p.Q2054X in KMT2C and eight other mutations in this gene outside hairpins).
There are some limitations to our approach. Although mitigated by the sample size, we did not use paired pBC and mBC samples. Instead, for our primary analysis, we used WGS data originating from two cohorts (BASIS cohort and CPCT cohort), in which the tumors were sequenced at a different depth. This might have introduced detection bias of low frequent, subclonal events, mainly in the pBC cohort. However, most findings were confirmed in different data sets.8,10 In addition, our cohorts were heterogeneous with regard to the treatment that patients had received. This has potentially complicated the attribution of certain APOBEC effects to the environmental pressure of given treatments.
In conclusion, using an approach on the basis of enrichment of AAs, which has not been used thus far, we demonstrated that the coding sequence of ER+/HER2– BC evolves as a result of APOBEC mutagenesis by introducing accumulating focal oncogenic mutations in PIK3CA and tumor-suppressive alterations at nonhotspot locations. These alterations drive early, metastatic, and endocrine-resistant BC. Given the APOBEC-related hyperactivation of the PI3K pathway, BC patients with high APOBEC contribution might substantially benefit from the recently introduced PI3K inhibitors.28 This would be a first step toward selecting optimal treatment for patients with BC tumors with profound APOBEC mutagenesis who are also prone to develop resistance to endocrine treatment by inactivation of negative modulators of ER activity.
Stefan Sleijfer
Honoraria: Skyline Diagnostics
Consulting or Advisory Role: Philips Research (Inst)
Research Funding: AB Science (Inst), Sanofi (Inst), Merck KGaA (Inst), Lilly (Inst)
Patents, Royalties, Other Intellectual Property: Test on circulating tumor cells in prostate cancer
Travel, Accommodations, Expenses: AstraZeneca
John W. M. Martens
Consulting or Advisory Role: Novartis/Pfizer
Research Funding: Merck Serono (Inst), CytoTrack, GlaxoSmithKline
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented at the virtual European Breast Cancer Conference (EBCC-12), October 2-3, 2020.
SUPPORT
M.K.B. Research funding: Dutch Cancer Society (no. NKB-EMCR-2016-108154). S.S. Research funding: Cancer Genomics Netherlands (CGC.nl), funded by the Netherlands Organization for Scientific Research (NWO). J.W.M.M. Consulting honorarium: Novartis, Research funding: Cancer Genomics Netherlands (CGC.nl), funded by the Netherlands Organization for Scientific Research (NWO), Research funding: Dutch Cancer Society (No. NKB-EMCR-2016-10270).
M.K.B. and M.S. contributed equally to this work.
DATA SHARING STATEMENT
pBC cohort: The somatic mutations for the BASIS cohort were extracted from the European Genome-phenome Archive (accession code EGAS00001001178).
mBC cohort: The WGS and corresponding clinical data were requested from the Hartwig Medical Foundation and provided under data request No. DR-068. Both WGS and clinical data are freely available for academic use from the Hartwig Medical Foundation through standardized procedures; request forms can be found at https://www.hartwigmedicalfoundation.nl.
Razavi cohort: Breast cancer data (primary, n = 869 and metastatic, n = 965) can be downloaded from the Data Supplement: https://www.sciencedirect.com/science/article/pii/S1535610818303684?via%3Dihub.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: Manouk K. Bos, Marcel Smid
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I =Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Stefan Sleijfer
Honoraria: Skyline Diagnostics
Consulting or Advisory Role: Philips Research (Inst)
Research Funding: AB Science (Inst), Sanofi (Inst), Merck KGaA (Inst), Lilly (Inst)
Patents, Royalties, Other Intellectual Property: Test on circulating tumor cells in prostate cancer
Travel, Accommodations, Expenses: AstraZeneca
John W. M. Martens
Consulting or Advisory Role: Novartis/Pfizer
Research Funding: Merck Serono (Inst), CytoTrack, GlaxoSmithKline
No other potential conflicts of interest were reported.
REFERENCES
- 1.Swanton C, McGranahan N, Starrett GJ, et al. APOBEC enzymes: Mutagenic fuel for cancer evolution and heterogeneity Cancer Discov 5704–7122015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non–small-cell lung cancer N Engl J Med 3762109–21212017 [DOI] [PubMed] [Google Scholar]
- 3.Angus L, Smid M, Wilting SM, et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies Nat Genet 511450–14582019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertucci F, Ng CKY, Patsouris A, et al. Genomic characterization of metastatic breast cancers Nature 569560–5642019 [DOI] [PubMed] [Google Scholar]
- 5. Buisson R, Langenbucher A, Bowen D, et al. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019;364:eaaw2872. doi: 10.1126/science.aaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson S, Chakravarthy A, Su X, et al. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development Cell Rep 71833–18412014 [DOI] [PubMed] [Google Scholar]
- 7.Glaser AP, Fantini D, Wang Y, et al. APOBEC-mediated mutagenesis in urothelial carcinoma is associated with improved survival, mutations in DNA damage response genes, and immune response Oncotarget 94537–45482017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi M-J, Meng X-Y, Fontugne J, et al. Identification of new driver and passenger mutations within APOBEC-induced hotspot mutations in bladder cancer. Genome Med. 2020;12:85. doi: 10.1186/s13073-020-00781-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences Nature 53447–542016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razavi P, Chang MT, Xu G, et al. The genomic landscape of endocrine-resistant advanced breast cancers Cancer Cell 34427–4382018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Priestley P, Baber J, Lolkema MP, et al. Pan-cancer whole-genome analyses of metastatic solid tumours Nature 575210–2162019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer Nature 500415–4212013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jalili P, Bowen D, Langenbucher A, et al. Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat Commun. 2020;11:2971. doi: 10.1038/s41467-020-16802-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma Y, Miladi M, Dukare S, et al. A pan-cancer analysis of synonymous mutations. Nat Commun. 2019;10:2569. doi: 10.1038/s41467-019-10489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannataro VL, Gaffney SG, Sasaki T, et al. APOBEC-induced mutations and their cancer effect size in head and neck squamous cell carcinoma Oncogene 383475–34872019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pang H, Flinn R, Patsialou A, et al. Differential enhancement of breast cancer cell motility and metastasis by helical and kinase domain mutations of class IA phosphoinositide 3-kinase Cancer Res 698868–88762009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbareschi M, Buttitta F, Felicioni L, et al. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas Clin Cancer Res 136064–60692007 [DOI] [PubMed] [Google Scholar]
- 18. Pereira B, Chin SF, Rueda OM, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dimitrios Z, Luc te M, Roger LM, et al. Tumor PIK3CA genotype and prognosis in early-stage breast cancer: A pooled analysis of individual patient data J Clin Oncol 36981–9902018 [DOI] [PubMed] [Google Scholar]
- 20.Vasan N, Razavi P, Johnson JL, et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors Science 366714–7232019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spoerke JM, Gendreau S, Walter K, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. 2016;7:11579. doi: 10.1038/ncomms11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bukholm IK, Nesland JM, Kåresen R, et al. E-cadherin and alpha-, beta-, and gamma-catenin protein expression in relation to metastasis in human breast carcinoma J Pathol 185262–2661998 [DOI] [PubMed] [Google Scholar]
- 23.Gala K, Li Q, Sinha A, et al. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERalpha enhancer function Oncogene 374692–47102018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu G, Chhangawala S, Cocco E, et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer Nat Genet 52198–2072020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng ZY, Anurag M, Lei JT, et al. Neurofibromin is an estrogen receptor-alpha transcriptional Co-repressor in breast cancer Cancer Cell 37387–402 e72020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong X-Y, Sun X, Guo P, et al. ATBF1 inhibits estrogen receptor (ER) function by selectively competing with AIB1 for binding to the ER in ER-positive breast cancer cells J Biol Chem 28532801–328092010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z, Yamashita H, Toyama T, et al. ATBF1-A messenger RNA expression is correlated with better prognosis in breast cancer Clin Cancer Res 11193–1982005 [PubMed] [Google Scholar]
- 28.André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer N Engl J Med 3801929–19402019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
pBC cohort: The somatic mutations for the BASIS cohort were extracted from the European Genome-phenome Archive (accession code EGAS00001001178).
mBC cohort: The WGS and corresponding clinical data were requested from the Hartwig Medical Foundation and provided under data request No. DR-068. Both WGS and clinical data are freely available for academic use from the Hartwig Medical Foundation through standardized procedures; request forms can be found at https://www.hartwigmedicalfoundation.nl.
Razavi cohort: Breast cancer data (primary, n = 869 and metastatic, n = 965) can be downloaded from the Data Supplement: https://www.sciencedirect.com/science/article/pii/S1535610818303684?via%3Dihub.