

Abstract
The incidence of syphilis has risen worldwide in the last decade in spite of being an easily treated infection. The causative agent of this sexually transmitted disease is the bacterium Treponema pallidum subspecies pallidum (TPA), very closely related to subsp. pertenue (TPE) and endemicum (TEN), responsible for the human treponematoses yaws and bejel, respectively. Although much focus has been placed on the question of the spatial and temporary origins of TPA, the processes driving the evolution and epidemiological spread of TPA since its divergence from TPE and TEN are not well understood. Here, we investigate the effects of recombination and selection as forces of genetic diversity and differentiation acting during the evolution of T. pallidum subspecies. Using a custom-tailored procedure, named phylogenetic incongruence method, with 75 complete genome sequences, we found strong evidence for recombination among the T. pallidum subspecies, involving 12 genes and 21 events. In most cases, only one recombination event per gene was detected and all but one event corresponded to intersubspecies transfers, from TPE/TEN to TPA. We found a clear signal of natural selection acting on the recombinant genes, which is more intense in their recombinant regions. The phylogenetic location of the recombination events detected and the functional role of the genes with signals of positive selection suggest that these evolutionary processes had a key role in the evolution and recent expansion of the syphilis bacteria and significant implications for the selection of vaccine candidates and the design of a broadly protective syphilis vaccine.
Keywords: recombination, selection, phylogenetic congruence, treponematoses, genome analysis
Introduction
Although genetic variation plays a central role in microbial evolution, some microorganisms have notably low levels of genetic variability, including some of the most virulent human pathogens, such as Mycobacterium tuberculosis, Bacillus anthracis, or Yersinia pestis (Didelot and Maiden 2010; Achtman 2012; Gagneux 2018). Treponema pallidum subsp. pallidum (TPA), the bacterium responsible for syphilis (Singh and Romanowski 1999), was also a deadly pathogen with devastating health consequences until the advent of penicillin in the mid-20th century, which enabled treatment and led to a significant reduction in incidence despite recent increases. Interestingly, TPA displays strikingly low levels of sequence diversity, lower than that of other genetically monomorphic pathogens such as those mentioned above (Didelot and Maiden 2010; Achtman 2012; Gagneux 2018).
The evolutionary forces of mutation, recombination, and natural selection that generate patterns of genetic diversity in such monomorphic pathogens are of great interest. In-depth analyses of these patterns provide insights into evolutionary histories and enable the prediction of future evolutionary trajectories. Understanding how these forces have shaped current population genetic structure in particular species is also useful for epidemiological purposes, as information on the dominant strains in outbreaks and drug resistance, among others, can be used to guide infection management (Achtman 2008).
The increasing application of high-throughput sequencing (HTS) technologies has generated a wealth of complete microbial genomes that, in turn, has allowed the comparisons required to characterize the extent of genetic variation in microbial pathogens, including monomorphic ones. HTS has also enabled the tracking of the rise and spread of antibiotic resistance (Davies and Davies 2010), the detection of changes in pathogenicity and virulence (Liu et al. 2006; Sánchez-Busó et al. 2014), and outbreak investigations (Sánchez-Busó et al. 2014; Francés-Cuesta et al. 2021). However, genomic information derived from mapping to a reference genome in HTS studies is highly dependent on the reference selected (Valiente-Mullor et al. 2021).
Together with other processes responsible for generating patterns of genetic diversity, it is imperative to examine recombination, which generates diversity (Awadalla 2003; Lefébure and Stanhope 2007), and natural selection, which shapes it by either maintaining or driving some alleles to fixation or extinction. Particularly the role of recombination is critical, because inferences on the evolutionary history of a species must take into account both vertical and nonvertical inheritance, assessing the contribution and consequences of each type of processes (Anisimova et al. 2003; Didelot and Maiden 2010). Since reconstructing the evolutionary history of an organism is useful for detecting loci under selection, these analyses would also be affected (Joseph et al. 2011). However, the detection of recombining loci and those evolving under natural selection is a challenging task because natural selection may quickly remove allelic variants arising from recombination events, or drive them to fixation, which poses additional difficulties for accurately identifying recombinant loci (Didelot and Maiden 2010).
The study of recombination and natural selection in T. pallidum has been hindered by its low levels of genetic variability and lack of identification of molecular mechanisms for horizontal gene transfer (Šmajs et al. 2012), which has led researchers to refer to it as a clonal species (Achtman 2008; Šmajs et al. 2012). Nonetheless, several studies have pointed at the occurrence of recombination or exchange of genetic material in TPA (Gray et al. 2006; Harper et al. 2008; Pětrošová et al. 2012; Čejková et al. 2013; Staudová et al. 2014; Arora et al. 2016; Tong et al. 2017; Marks et al. 2018; Pospíšilová et al. 2018; Schuenemann et al. 2018; Strouhal et al. 2018; Majander et al. 2020; Mikalová et al. 2020) and the presence of natural selection acting in these bacteria (Čejková et al. 2012; Giacani, Brandt, et al. 2012; Giacani, Chattopadhyay, et al. 2012; Kumar, Caimano, et al. 2018). For example, genes coding for TPA integral membrane proteins appear to be involved in genetic changes (Staudová et al. 2014; Arora et al. 2016; Mikalová et al. 2017), while also being subjected to human host selective pressures (Kumar, Caimano, et al. 2018). Overall, the growing body of evidence suggests that the role of recombination may be underestimated (Staudová et al. 2014; Arora et al. 2016; Mikalová et al. 2017).
Here, we have used a large data set of 75 genomes—all those available at the start of the analysis—across the three subspecies of T.pallidum, namely TPA, T. pallidum subsp. pertenue (TPE), and T. pallidum subsp. endemicum (TEN), to explore how intersubspecies recombination and natural selection have shaped the current diversity patterns observed in TPA. To prevent the problems arising from using one single reference for mapping, we have used reference genomes from the major lineages of T. pallidum. Our findings provide insights into how lateral gene transfer is a source for change and adaptation in this pathogen, which is responsible for the re-emerging syphilis infections.
Results
Reference-Based Alignments
The SNP calling results for each sample and reference are listed in supplementary table 1, Supplementary Material online. The resulting multiple sequence alignments spanned a total of 1,139,633 bp (NIC-mapped data set, supplementary file 1, Supplementary Material online, in 10.5281/zenodo.5160123), 1,139,569 bp (SS14-mapped data set, supplementary file 2, Supplementary Material online, in 10.5281/zenodo.5160123), and 1,139,744 bp (CDC2-mapped data set, supplementary file 3, Supplementary Material online, in 10.5281/zenodo.5160123), respectively.
Recombination Events in T. pallidum
To determine the effects of recombination as a force of genetic diversity and differentiation, we applied the phylogenetic incongruence method (PIM) procedure to the three multiple alignments obtained from different genomes as reference for mapping. Firstly, we performed a likelihood mapping test to ascertain which genes had some phylogenetic signal. For the NIC-mapped data set, 380 out of the 978 genes showed a phylogenetic signal and were retained for the ensuing analyses (supplementary table 2, Supplementary Material online). The remaining genes were discarded because for all the quartets considered the distribution of the corresponding likelihoods fell in the central zone of the triangle. Using the SS14-mapped and CDC2-mapped data sets, this step yielded 498 and 535 genes with some phylogenetic signal, respectively (supplementary tables 3 and 4, Supplementary Material online).
Next, for each gene retained in the likelihood mapping analyses, we tested the phylogenetic congruence between trees, comparing the tree obtained from the gene alignment and the tree obtained from the complete genome alignment using the SH and ELW topology tests. For the NIC-mapped data set, only 44 genes showed reciprocal incongruence. In contrast, 62 and 75 genes displayed reciprocal incongruence in the SS14-mapped and CDC2-mapped data sets, respectively. Overall, 90 genes displayed reciprocal incongruence in at least one of the three data sets, whereas only 29 genes did so for all three data sets (supplementary table 5, Supplementary Material online). Moreover, all the reciprocal tests, that is, the complete genome alignment tested with the tree derived from it and with the trees derived from each gene alignment, yielded the same result for the three data sets: acceptance of the complete genome tree with probability or weight equal to 1 and rejection of the alternative tree with null probability or weight.
We checked for the presence of a minimum of three consecutive homoplasic SNPs contributing to the reciprocal incongruence results in these 90 genes. Only 12 genes had three or more consecutive homoplasic SNPs and the rest were not considered further. It is worth noting that the recombination event observed in tp0164 displayed reciprocal incongruence in the topology analyses of the NIC-mapped data set only. However, the recombination event was detected in the SNP alignment for all three data sets.
Our analyses identified only one recombination event per gene (table 1 and supplementary figs. 1–12, Supplementary Material online) except for genes tp0136 and tp0865, for which seven and four events were detected, respectively (supplementary fig. 13, Supplementary Material online). The average length of the recombinant region was around 470 bp (471.85, median = 391), with a minimum of 12 bp and a maximum of 1,900 bp. The average number of SNPs encompassed in these events was 15.38, with a minimum of 3 and a maximum of 45 nucleotides. In total, the identified recombination events account for 294 out of the 927 SNPs (31.72%) found among the TPA strains analyzed here (table 1).
Table 1.
Recombination Events in Treponema pallidum Detected Using PIM with the NIC-Mapped Data Set.
| Gene_Event | Start | End | Size (bp) | SNPs | Origin | Receptor | Function (UniProt) |
|---|---|---|---|---|---|---|---|
| TP0136_1 | 158,092 | 158,104 | 13 | 4 | TPE | SW6 | Adhesin allowing binding to fibronectin |
| TP0136_2 | 158,138 | 158,149 | 12 | 4 | Seattle 81-4 | SW6 | |
| TP0136_3 | 158,149 | 158,167 | 19 | 3 | TPE | Seattle 81-4 | |
| TP0136_4 | 158,271 | 158,336 | 66 | 6 | TPE | Nichols clade | |
| TP0136_5 | 158,346 | 158,364 | 18 | 7 | TEN | Nichols clade | |
| TP0136_6 | 158,915 | 158,976 | 62 | 3 | TPE-TEN | Nichols clade | |
| TP0136_7 | 159,312 | 159,323 | 12 | 5 | TPE-TEN | Nichols clade | |
| TP0164 | 187,064 | 187,177 | 113 | 4 | TPE-TEN | NE20, SEA86 cluster | troB, iron/zinc/manganeseABC superfamily ATP-binding cassette transporter, ABC protein |
| TP0179 | 198,040 | 198,428 | 391 | 9 | TPE-TEN | Nichols clade | Hypothetical protein |
| TP0326 | 347,027 | 347,956 | 929 | 32 | TEN | SS14 clade excluding Mexico A | β-barrel assembly machinery A (BamA) ortholog and rare outer membrane protein |
| TP0462 | 492,772 | 493,605 | 834 | 43 | TPE-TEN | NE20, SEA86 cluster | Hypothetical protein |
| TP0488 | 522,981 | 523,620 | 640 | 41 | TEN | Mexico A | Mcp2, methyl-accepting chemotaxis protein |
| TP0515 | 555,872 | 557,771 | 1,900 | 17 | TPE-TEN | Nichols clade | LptD homolog |
| TP0548 | 593,563 | 594,215 | 653 | 45 | TPE-TEN | Nichols clade | OMP—FadL family |
| TP0558 | 606,171 | 606,591 | 421 | 4 | TPE-TEN | SS14 clade | NiCoT family nickel–cobalt transporter, high affinity |
| TP0865_1 | 945,224 | 945,542 | 319 | 13 | TPE-TEN | NE20, SEA86 cluster | OMP—FadL family |
| TP0865_2 | 945,224 | 945,542 | 319 | 13 | TPE-TEN | Seattle 81-4 | As above |
| TP0865_3 | 945,830 | 946,298 | 469 | 18 | TEN | NE20, SEA86 cluster | As above |
| TP0865_4 | 945,830 | 946,298 | 469 | 18 | TEN | Seattle 81-4 | As above |
| TP0967 | 1,051,257 | 1,052,302 | 1,046 | 15 | TPE-TEN | Seattle 81-4 | TolC-homolog |
| TP0968 | 1,052,414 | 1,053,617 | 1,204 | 22 | TEN | Seattle 81-4 | TolC-homolog |
Note.—For each recombination event (denoted as gene_event), we report the start and end positions of the event (coordinates according to the Nichols strain), its size in base pairs, the number of SNPs detected in the event, the donor (origin) cluster/strain, the recipient (receptor) cluster/strain (supplementary figs. 1–12, Supplementary Material online, for additional details), and the functional significance of the gene according to UniProt.
We found an additional recombination event in the tp1031 (tprL) gene which was not among the 12 recombination events detected by PIM despite showing reciprocal incongruence in the topology tests in the SS14-mapped and CDC2-mapped data sets. Nevertheless, tp1031 was classified as a gene with a high number of SNPs and, also, without signs of positive selection acting on it (see subsection below). A detailed analysis of this gene revealed 18 SNPs present only in SS14 clade strains (positions 745–872) (supplementary fig. 14, Supplementary Material online). This high variation found in the SS14 clade seems to be the result of a transfer from another Treponema subspecies or species not identified in the public databases.
In addition, we checked for intragenomic recombination in tp0897 (also known as tprK) (table 2). The most similar genome fragments to those included in the variable regions of tprK correspond to coding and intergenic regions between tp0126a and tp0138. However, none of these putative donors for the variable regions in tprK was detected by PIM (table 1).
Table 2.
Summary of BlastN Searches of Variable Sequences (Defined in Centurion-Lara et al. [2004]) in the tp0897 (tprK) Gene (Deposited in GenBank and Reported in supplementary table 4 of Pinto et al. [2016]) Using as Query the 75 Complete Treponema pallidum Genome Sequences Analyzed in This Study.
| Variable Region | Unique Sequences | Matches |
|---|---|---|
| V1 | 31 | tp0126d, tp0126c, tp0129, tp0130 |
| V2 | 54 | tp0126c, tp0128, tp0129, tp0130 |
| V3 | 63 | tp0126c, IR-tp0127-tp0128, tp0128, tp0129, IR-tp0129-tp0130, IR-tp0130-tp0131 |
| V4 | 29 | tp0126a, IR-tp0126d-tp0126c, IR-tp0129-tp0130, |
| V5 | 80 | tp0126a, IR-tp0126d-tp0126c, tp0126c, IR-tp0128d-tp0129, tp0129, IR-tp0129-tp0130, tp0130 |
| V6 | 155 | tp0126a, IR-tp0126d-tp0126c, IR-tp0127-tp0128, tp0128, tp0129, IR-tp0129-tp0130 |
| V7 | 102 | tp0126d, tp0126c, tp0127, tp0129, tp0130, IR-tp0136-tp0138 |
Most Recombination Events Have Occurred between Subspecies
Next, for each of the three data sets, we removed the 12 recombinant genes and tp0897 from the multiple alignment, in order to build a nonrecombinant genome phylogeny. As the three tree topologies were almost identical (supplementary figs. 15 and 16, Supplementary Material online), we selected the phylogenetic tree for the NIC-mapped data set (fig. 1) to represent the 21 recombination events detailed in supplementary figures 1–12, Supplementary Material online. The SS14-mapped and CDC2-mapped data sets were not considered for the remaining analyses. All but one recombination event corresponded to intersubspecies transfers, from TPE/TEN to TPA. The only exception was the event tp0136_2 (table 1), which corresponded to an intrasubspecies transfer (within TPA), specifically, to a transfer from the Nichols to the SS14 clade (table 1 and supplementary figs. 1–12, Supplementary Material online). These recombination events between the clades further support a geographically close common history of the TPA and TPE lineages, which cannot be concluded from the geographical distribution of extant lineages (Majander et al. 2020).
Fig. 1.

Maximum likelihood tree obtained from the NIC-based data set without the 12 recombinant genes detected using PIM and the tp0897 gene (resulting alignment length 1,117,857 bp). The different subspecies corresponding to yaws (TPE), bejel (TEN), and the Nichols and SS14 clades of syphilis (TPA) are indicated in the figure. Nodes with bootstrap support values larger than 70% are indicated by red circles. Representative sequences used in the analysis of recombination by Gubbins and ClonalFrameML are labeled in red color.
Among the intersubspecies transfers, 11 originated from the TPE/TEN clade: these events included the most frequent donor-recipient combination, which involved the entire Nichols clade as recipient (five events). The assignment of an event to the TPE/TEN clade does not necessarily mean that the actual source of the event was an ancestor of TPE and TEN strains; it could have also been a more recent descendant of any of these two subspecies that shared the same variants. Consequently, we cannot ascertain the exact phylogenetic location of the donor genome. Additionally, we detected six events originating from the TEN clade and three events originating from the TPE. Interestingly, strains in the Nichols clade were the most frequent recipient of external DNA, involving particular strains or clusters within (nine events) or the ancestor of the entire clade (in seven events). Only one event involving tp0558 had the whole SS14 clade as recipient, whereas there were two events in tp0136 (one event originated in TPA), one in tp0488 and another in tp0326, that had SW6, Mexico A and all SS14 clade but one (Mexico A) strain in this clade as recipients, respectively.
The effect of recombination on the phylogenetic reconstruction of T. pallidum can be seen by comparing the topologies of two maximum likelihood (ML) trees: that obtained with all genes included in the alignment of the NIC-mapped data set (1,139,633 bp), and that obtained after excluding the recombinant genes plus tp0897 from the alignment (1,117,857 bp) (supplementary fig. 17, Supplementary Material online). As expected, the most remarkable differences between the two topologies involved those strains and clusters frequently implicated in recombination events. In the Nichols clade, Seattle 81-4 occupies a basal position in the nonrecombinant tree, whereas this position corresponds to NE20 and SEA86 in the complete genome-based tree. In fact, this difference in the topology is responsible for the doubling of recombination events inferred in locus tp0865, in which identical events had to be postulated for Seattle 81-4 and for the NE20-SEA86 cluster. In tp0865, two different recombinant regions were detected which, based on the phylogeny derived from the nonrecombinant core genome of T. pallidum, corresponded to four different transfers (supplementary fig. 10, Supplementary Material online). An alternative explanation, requiring only two recombination events, would involve separate transfers for each region from the source clade (TPE/TEN and TPE, respectively) to the Nichols clade, with a subsequent loss of the transferred fragments in the sister clade to NE20-SEA86 in the ML phylogenetic tree (fig. 1). These explanations rely on the assumption that the phylogenetic reconstruction after removal of recombinant regions is accurate. But there are other processes that can obscure phylogenetic reconstruction (Brocchieri 2001; Maddison and Knowles 2006; Degnan et al. 2009). Thus, it is important to take into account that there might be alternative topologies providing a simpler explanation for the observation of four recombination events in tp0865.
The BAL3 strain also displays a dramatic topological change from a sister branch to the SS14 clade in the complete genome tree to a well-supported position within the Nichols clade in the nonrecombinant tree. For the SS14 clade, there are numerous minor differences between the two trees. Nonetheless, the basal position of Mexico A is retained in both. Interestingly, the relationships of all strains from the yaws (TPE) and bejel (TEN) clades are consistent in both trees.
Recombination Detection with Alternative Tools
The two alternative tools detected more recombination events than PIM (supplementary tables 6 and 7 and figs. 18 and 19, Supplementary Material online). Gubbins detected 58 potential recombination events spanning 57 genes, whereas ClonalFrameML identified 92 potential events spanning 44 genes. Ten of the 12 recombinant genes detected by PIM (tp0136, tp0179, tp0326, tp0462, tp0488, tp0515, tp0548, tp0865, tp0967, tp0968) were also detected by ClonalFrameML and Gubbins. Additionally, Gubbins detected tp0558 but did not detect tp0164, whereas for ClonalFrameML, the pattern was reversed (fig. 2). The average length of the recombinant regions detected by Gubbins and ClonalFrameML was 2,199 bp (range 33–12,566 bp) and 470 bp (range 4–3,508), respectively.
Fig. 2.
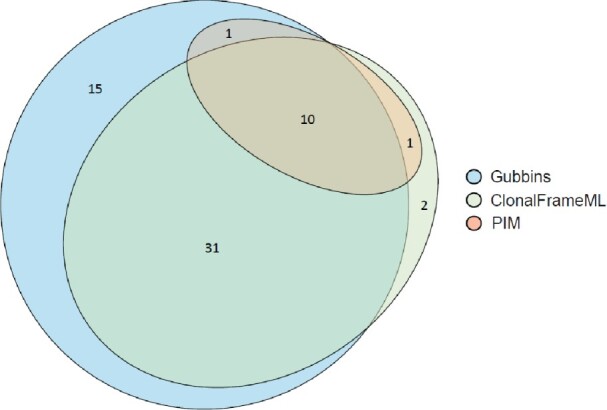
Euler diagram showing a summary of the number of recombinant genes detected by PIM, Gubbins, and ClonalFrameML. About ten of the 12 genes detected using PIM were also detected by ClonalFrameML and Gubbins, and one additional gene was detected simultaneously by each of these methods and PIM. Gubbins and ClonalFrameML detected 46 and 33 additional recombinant genes, respectively.
Of the 57 recombinant genes detected by Gubbins, 46 were not detected by PIM. Of these, ten did not pass the phylogenetic signal test, 23 did not pass the phylogenetic congruence tests, eight did not pass the polyphyletic SNP distribution, tp0897 or tprK is a hypervariable gene, tp1031 (tprL) gene is discussed as an additional recombinant gene with a likely donor from an unidentified Treponema species, and three genes (tp0857, tp0863, and tp1030) were not tested because several of its sequences contain a high number of Ns (supplementary table 6, Supplementary Material online).
Of the 44 total recombinant genes detected by ClonalFrameML, 33 were not detected by PIM. Of these, two did not pass the phylogenetic signal test, 21 did not pass the phylogenetic congruence tests, and seven did not pass the polyphyletic SNP distribution. The remaining three genes corresponded to tp0897, tp1031, and tp0857, discussed above (supplementary table 7, Supplementary Material online).
Selection Analyses
The effects of selection on the 12 recombinant genes as identified by PIM were estimated by the nonsynonymous to synonymous substitution ratios (ω). We found diverse patterns across the genes (table 3).
Table 3.
Recombinant Genes with Their Synonymous and Nonsynonymous Sites and Changes and Estimates of ω=dN/dS for the Recombinant and Nonrecombinant Regions of Each Gene.
| Nonrecombinant Regions |
Recombinant Regions |
ω
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Size (nt) | Syn Sites | Syn Changes | Nonsyn Sites | Nonsyn Changes | Size (nt) | Syn Sites | Syn Changes | Nonsyn Sites | Nonsyn Changes | Nonrecombinant Regions | Recombinant Regions |
| tp0136 | 1,286 | 432 | 18 | 801 | 84 | 202 | 64 | 6 | 128 | 25 | 2.52 | 2.08 |
| tp0164 | 687 | 238 | 0 | 446 | 0 | 114 | 40 | 2 | 72 | 2 | NC | 0.56 |
| tp0179 | 1,495 | 547 | 1 | 947 | 1 | 389 | 137 | 2 | 245 | 7 | 0.58 | 1.96 |
| tp0326 | 1,632 | 567 | 2 | 1,060 | 3 | 930 | 331 | 3 | 580 | 29 | 0.80 | 5.51 |
| tp0462 | 345 | 119 | 2 | 219 | 8 | 834 | 271 | 2 | 545 | 41 | 2.17 | 10.19 |
| tp0488 | 1,898 | 719 | 0 | 1,169 | 18 | 640 | 206 | 2 | 406 | 39 | NC | 9.89 |
| tp0515 | 1,076 | 386 | 1 | 688 | 0 | 1,900 | 647 | 0 | 1,239 | 17 | 0.00 | NC |
| tp0548 | 652 | 211 | 7 | 423 | 27 | 653 | 204 | 5 | 420 | 40 | 1.92 | 3.89 |
| tp0558 | 488 | 170 | 3 | 316 | 1 | 421 | 152 | 4 | 266 | 0 | 0.18 | 0.00 |
| tp0865 | 652 | 211 | 3 | 430 | 10 | 788 | 277 | 14 | 497 | 17 | 1.64 | 0.68 |
| tp0967 | 508 | 173 | 0 | 330 | 1 | 1,046 | 371 | 6 | 666 | 9 | NC | 0.84 |
| tp0968 | 419 | 152 | 1 | 264 | 1 | 1,204 | 421 | 5 | 775 | 17 | 0.58 | 1.85 |
Note.—NC, noncomputable.
Both positive and purifying selections were observed in four genes, tp0179, tp0865, tp0968, and tp0326, with clear differences between recombinant and nonrecombinant regions of these two genes. In tp0179 and tp0968, purifying selection was observed in the nonrecombinant region (ω = 0.58 in both genes) and positive selection in the recombinant region (ω = 1.96 and ω = 1.85, respectively), with fewer changes in the nonrecombinant compared with the recombinant regions (table 3). In contrast, three nonrecombinant and two recombinant regions were observed in tp0865, with indications for positive selection in the former (ω = 1.64) and purifying selection (ω = 0.68) in the latter. Interestingly, a more detailed analysis of ω in the different subregions revealed some additional differences (supplementary table 8, Supplementary Material online). Values of ω were similar in both recombinant regions, with estimates lower than 1 but not indicative of strong purifying selection (0.84 and 0.58, respectively). The three nonrecombinant portions of this gene were not homogeneous: two of them, NR1 and NR3, were very constrained to changes, with only one synonymous mutation in NR3 and one nonsynonymous change in NR1. The central, nonrecombinant portion of tp0865 (NR2) accumulated more nonsynonymous than synonymous changes, leading to ω = 2.25.
For tp0326, a weak signal of purifying selection was detected in the nonrecombinant regions, with two synonymous substitutions and three nonsynonymous substitutions detected. However, the recombinant portion of this gene seems to have evolved under strong positive selection (ω = 5.51, with 29 nonsynonymous changes and three synonymous ones).
Three genes (tp0164, tp0558, and tp0967) showed negative or purifying selection (ω < 1) in both types of regions. For tp0164, we only found SNPs within the recombinant region, associated with the recombination events (table 3). In contrast, tp0558 and tp0967 showed some SNPs in both types of regions, but with fewer nonsynonymous than synonymous changes in the tp0558 gene, and more nonsynonymous changes than synonymous changes in the tp0967 gene (table 3).
Positive selection in both regions was the dominant form of selection in four of the remaining recombinant loci. The tp0136 gene had similar ω values (>1) in the nonrecombinant and recombinant regions indicating pervasive positive selection in this gene (table 3). In tp0462, tp0488, and tp0548, both recombinant and nonrecombinant regions showed a strong signal of positive selection. Finally, tp0515 also presented strong positive selection in the recombinant region, with no synonymous substitutions and 17 nonsynonymous changes, but its nonrecombinant region is highly constrained, with only one synonymous substitution found (table 3).
In addition to examining the role of natural selection in the putative recombinant genes, we also investigated its role in the rest of the genome using SNPeff and Codeml. Among the 965 remaining genes (without considering the final 12 recombinant genes and the tp0897 gene), only 14 yielded estimates of ω > 1 (supplementary table 9, Supplementary Material online), with estimates ranging from 1.11 to 2.61. Among them, only two had estimates of ω > 2, containing eight or fewer SNPs. Thus, the amount of variation contributed by these genes was quite low.
Although some of the genes identified as recombinant and with signs of positive selection had a large number of SNPs, especially for genomes with such a low genetic variability as those of T. pallidum, not all the genes with a relatively high proportion of SNPs have been under the action of adaptive selection. We detected 23 genes (supplementary table 10, Supplementary Material online) in which the number of observed SNPs exceeded twice the number of expected ones, but whose estimates for ω were <1.
Globally, there were only eight genes in the T. pallidum genome with estimates for ω > 2. Six of those corresponded to genes involved in recombination events (tp0136, tp0326, tp0462, tp0488, tp0515, and tp0548), whereas tp0639 and tp0969 had the lowest ω values among the genes in this group and were not involved in recombination. The distribution of ω values along the T. pallidum genome is shown in figure 3 (detailed results of ω values per gene are available in supplementary table 11, Supplementary Material online).
Fig. 3.

Distribution of ω values along the Treponema pallidum genome (detailed results of ω value per gene are in supplementary table 11, Supplementary Material online). Genes with ω > 2 are indicated. This parameter cannot be estimated for tp0515 because it does not present synonymous substitutions. Genes marked with asterisk were detected as recombinant by PIM.
In addition to the computation of dN/dS, we also checked for the action of natural selection on T. pallidum genes using Hyphy. We analyzed the 12 recombinant genes, the 14 nonrecombinant genes but with ω values >1 and the 23 nonrecombinant genes without signal of natural selection but with a large number of SNPs. Of these 49 genes, five genes could not be tested due to the presence of indels in their sequences. The BUSTED test implemented in Hyphy provided evidence for positive selection for 18 genes, comprising nine recombinant and nine nonrecombinant genes (P < 0.05) (table 4 and supplementary table 11, Supplementary Material online). Among the nonrecombinant genes, one was found to display evidence of positive selection (supplementary table 9, Supplementary Material online), and eight had a high number of SNPs (supplementary table 10, Supplementary Material online). The two genes (tp0639 and tp0969) with ω > 2 (supplementary table 9, Supplementary Material online) were not detected by Hyphy to be under positive selection. Hyphy detected no genes as evolving under relaxed selection using the RELAX test.
Table 4.
Genes Tested with Hyphy for Positive Selection (Busted Test) with Their Corresponding P Values.
| Gene | P Value | Gene | P Value |
|---|---|---|---|
| tp0110 | 1.00 | tp0618 | 1.00 |
| tp0133 | 0.00 | tp0620 | 0.00 |
| *tp0136 | 0.00 | *tp0639 | 0.97 |
| tp0164 | 0.98 | tp0640 | 1.00 |
| tp0179 | 1.00 | tp0687 | 0.79 |
| tp0304 | 0.71 | tp0691 | 1.00 |
| tp0313 | 0.00 | tp0705 | 0.24 |
| *tp0326 | 0.00 | tp0729 | 0.09 |
| tp0346 | 0.57 | tp0733 | 0.06 |
| tp0369 | 0.95 | tp0746 | 0.27 |
| tp0433 | 0.00 | tp0856 | 0.01 |
| *tp0462 | 0.00 | tp0858 | 0.02 |
| tp0464 | 1.00 | tp0859 | 0.03 |
| tp0483 | 0.00 | tp0861 | 1.00 |
| tp0484 | 0.98 | tp0865 | 0.00 |
| *tp0488 | 0.00 | tp0896 | 0.45 |
| *tp0515 | 0.01 | tp0898 | 1.00 |
| *tp0548 | 0.00 | tp0966 | 0.16 |
| tp0558 | 1.00 | tp0967 | 0.00 |
| tp0564 | 0.97 | tp0968 | 0.00 |
| tp0577 | 1.00 | *tp0969 | 0.82 |
| tp0617 | 0.50 | tp1031 | 0.00 |
Note.—Genes with a significant P value are underlined and those with dN/dS>2 are marked with an asterisk.
Red, recombinant genes with positive selection (table 1); green, nonrecombinant genes with ω > 1 evaluated by codeml + SNPeff (supplementary table 9, Supplementary Material online); light blue, nonrecombinant genes with excess of SNPs (supplementary table 10, Supplementary Material online).
Discussion
In this study, we have examined a large set of nearly complete T. pallidum genomes to comprehensively evaluate recombination and selection in a pathogen displaying high clonality. We have followed a rigorous approach by analyzing the results using three different genome references for mapping, thus avoiding problems arising in differential variant calls (Valiente-Mullor et al. 2021) and three different methods for the detection of recombination: PIM developed in our group, Gubbins (Croucher et al. 2015), and ClonalFrameML (Didelot and Wilson 2015). Using PIM, we were able to identify 12 recombinant genes comprising 19 recombinant regions within them and involving at least 21 different recombination events. The estimates for the detection of recombination by Gubbins and ClonalFrameML were considerably larger. These differences are most likely because Gubbins evaluates the density of SNPs while concurrently constructing a phylogeny based on the putative point mutations outside of these regions. ClonalFrameML uses ML inference to simultaneously detect recombination and account for it in phylogenetic reconstruction. In contrast, PIM requires loci to fulfill three selection criteria to be classified as recombinant. First, the gene is required to have phylogenetic signal, which necessitates not only polymorphisms but specifically those that allow for phylogenetic resolution. Secondly, with PIM, we check for phylogenetic congruence of the individual gene tree topologies with a reference genome tree. Finally, the last requirement is the presence of a minimum number of homoplasic SNPs in the putative recombination events. In this particular case, we found that most genes detected as involved in recombinant events by Gubbins or ClonalFrameML failed the second condition, that is, the reciprocal incongruence between the topologies of the gene and the reference trees when tested with the corresponding multiple alignments. As a result, PIM is more conservative in the identification of recombination events and regions than Gubbins and ClonalFrame.
Few studies have focused on the identification of recombination in T.pallidum subspecies in detail because of the monomorphic nature of the organism, and because no mechanisms for recombination have been identified to date. Nonetheless, these studies have reported examples of recombination in T. pallidum and hypothesized that these events may have played a significant role in the evolution of this species (Gray et al. 2006; Harper et al. 2008; Pětrošová et al. 2012; Čejková et al. 2013; Staudová et al. 2014; Arora et al. 2016; Tong et al. 2017; Marks et al. 2018; Pospíšilová et al. 2018; Schuenemann et al. 2018; Strouhal et al. 2018; Beale et al. 2019; Grillová et al. 2019; Beale and Lukehart 2020; Majander et al. 2020). Although the methods used to detect recombination are different from our workflow, most of the results previously reported were confirmed by our findings. For instance, recombination in tp0136, tp0326, tp0462, tp0488, tp0548, and tp0865, has also been detected in other studies (Gray et al. 2006; Harper et al. 2008; Pětrošová et al. 2012; Čejková et al. 2013; Staudová et al. 2014; Arora et al. 2016; Mikalová et al. 2017, 2020; Tong et al. 2017; Kumar, Caimano, et al. 2018; Marks et al. 2018; Pospíšilová et al. 2018; Schuenemann et al. 2018; Strouhal et al. 2018; Grillová et al. 2019; Majander et al. 2020) but we have identified additional genes (tp0164, tp0179, tp0515, tp0558, tp0967, and tp0968) not previously reported as recombinant, probably as a result of the systematic analysis of 75 genomes. On the contrary, loci tp0117/tp0131, tp0119, tp0317, tp0621, tp0856, and tp0858, and the spacers of rRNA operons, which were detected in previous studies (Brinkman et al. 2008; Kumar, Caimano, et al. 2018; Marks et al. 2018; Strouhal et al. 2018; Grillová et al. 2019) were not detected as recombinant in our analyses because of the large number of missing positions resulting from mapping of short reads with stringent conditions applied to paralogous/duplicated genes.
Remarkably, we observed only one recombination event (tp0136_2) between TPA strains, from one strain of the Nichols clade (Seattle 81-4) to the SS14 clade (SW6). All the other recombination events detected correspond to intersubspecies transfers (TPE/TEN to TPA). Currently, it is not possible to discern whether this pattern is due to the difficulties in detecting intrasubspecific transfers, in light of the low levels of genetic variation in this species, or to the result of mechanisms that favor the transfer and incorporation of foreign material from a different subspecies. This might be the case for those genes in which only the recombinant regions have evolved under positive selection, such as tp0179, and tp0326 and tp0968, but not all the recombinant regions do so.
For the two tpr genes that could be analyzed, a donor for the variable regions could not be identified. None of the 19 recombinant regions (table 1) was identified as donor for the variable regions in the tp0897 gene (table 2). Variation in this gene accrues much more rapidly than in any other portion of the T. pallidum genome (Centurion-Lara et al. 2004; Pinto et al. 2016). Hence, it is likely that gene conversion, the mechanism generating variation in tp0897 is not the mechanism involved in the recombination events in the rest of the genome. In this regard, it is relevant to remark that all but one recombination event corresponds to intersubspecies transfers, likely occurring much earlier than the continuously generated variability in tp0897. We have also identified a putative recombination event in tp1031 involving a donor sequence of unknown origin, but very likely from the Treponema genus, as revealed by the identity in the constant positions (109/128) in the recombinant region (supplementary fig. 14, Supplementary Material online). The contribution of other donors to the genetic variation within and between TPA lineages remains to be explored.
Intersubspecies transfers are particularly striking given that TPE and TEN are, to date, geographically restricted to specific regions. Furthermore, no evidence for coinfection has been found in humans so far. One possibility is that recombination occurred in a host other than the human species, for example, in nonhuman primates. However, the observation of at least one recombination event within the TPA clades Nichols and SS14 suggests that coinfection in humans is possible. These two clades are estimated to have shared their most recent common ancestor in the 18th century (Arora et al. 2016), and all strains so far identified were found in humans. Interestingly, in a recent investigation of medieval skeletons, Majander et al. (2020) revealed the existence of a previously unknown T. pallidum lineage present in Europe probably until medieval times or even later. The historical geographical distribution of T. pallidum subspecies is not known but we cannot discard their possible coexistence, thus providing ecological and temporal opportunities for coinfection and recombination in human hosts.
The absence of recent recombination events in T. pallidum has important implications for the design and use of MLST schemes in these subspecies. If there is only marginal or no recombination, then it is possible to use the genes involved in recombination events in an MLST scheme because most alleles will result from new mutations. The genes tp0136 and tp0548, detected as recombinant in this work, are included in the recently proposed schemes for TPA (Grillová et al. 2018; Pospíšilová et al. 2018) and TPE (Godornes et al. 2017). The former scheme also includes genes tp0462 and tp0865, whereas tp0326 is incorporated in the scheme for TPE. Clearly, more research on the ecology and natural history of current and past T.pallidum strains is necessary to answer these questions.
We have observed a close relationship between recombination and selection. All the recombinant genes identified here display strong signals of either positive or purifying selection. Of the seven genes with evidence of positive selection, six had ω values above 2. Only two of the 14 positively selected, nonrecombinant genes in the rest of the genome had also ω larger than 2, although Hyphy did not identify them to evolve under positive selection. Notably, a strong purifying selection is apparently acting on the recombinant portion of tp0558 (table 3), similar to the nonrecombinant portion of tp0164 and many other genes in the T. pallidum genome.
The genes for which we found evidence of recombination as well as selection are functionally important. Most of these genes encode proteins that reside at the host–pathogen interface (table 1 and supplementary tables 9 and 10, Supplementary Material online). This result is congruent with the results obtained in previous studies (Arora et al. 2016; Kumar, Caimano, et al. 2018; Maděránková et al. 2019) in that the variation present in these genes maintained by the selective forces that act on them contributes significantly to the evolution of T. pallidum. Although lack of a culture system for T. pallidum has prevented experimental confirmation of most inferred protein functions for this species, these genes have been suggested as potentially involved in virulence, with an important role in the defense of the pathogen against the host and the evasion of the immune system (Kumar, Caimano, et al. 2018). These findings suggest that human hosts’ selective pressures drive the diversity of TPA integral outer membrane proteins (OMP). The genes coding for OMPs of T. pallidum detected under positive selection are pivotal in ensuring and maintaining pathogen fitness and pathogenicity for humans and have important implications for the selection of vaccine candidates and the design of a broadly protective syphilis vaccine (supplementary material, note 1, Supplementary Material online).
Our study shows the critical role that recombination and selection play in generating diversity in those genes most critical in the host–pathogen interactions. These processes are known to play a key role in the emergence and adaptive evolution of many pathogens. Recent analyses of complete genome sequences of monomorphic bacteria have also revealed that these processes have been important in the initial stages of speciation, usually along with adaptation to a new niche, as it is the case of Mycobacterium tuberculosis (Chiner-Oms et al. 2019), or Vibrio cholerae (Shapiro et al. 2017). These are examples of the clonal expansion model from a panmictic pool (Shapiro 2016), in which adaptation to a new niche, resulting in a successful spread, might be mediated by the introduction of variation through recombination followed by the action of natural selection which contributes to the maintenance of the changes and the fixation of the carrier alleles in recombinant genes. Our results indicate that this might have been also the case in the early evolution of TPA and its more recent epidemic spread.
Our results point to a significant role of recombination and selection in the evolution and emergence of the syphilis agent as a human pathogen. However, several questions remain to be answered, including, which molecular processes are responsible for recombination, what is the relevance of intraspecies recombination, and, if this is an important phenomenon driving the evolution of this pathogen, how to improve the current methods available for its detection. Additional questions concern the frequency of coinfections, where they occur, and which subspecies/lineages are usually involved. The constantly increasing availability of complete genome sequences along with advances in the in vitro culturing and genetic manipulation (Romeis et al. 2021) of T. pallidum will likely help in shedding light on all these pivotal questions.
Materials and Methods
Read Processing and Data Set Generation
We compiled a set of 75 T. pallidum genomes (67 TPA, seven TPE, and one TEN) from previous studies and public databases. The genomes were selected in order to obtain the best possible representation of the four groups identified in phylogenetic trees at the date (March 2017) when the analyses were started. Short read sequencing data were retrieved for 64 strains from three previous studies (Arora et al. 2016, Pinto et al. 2016, Sun et al. 2016). Complete genome sequences for the remaining 11 strains were downloaded from GenBank (supplementary table 12, Supplementary Material online).
To reconstruct the individual genomes from the raw short read data, we applied EAGER (Peltzer et al. 2016), a pipeline including read preprocessing, mapping, deduplication, indel realignment, and variant identification. This pipeline has been applied in previous studies of TPA (Arora et al. 2016; Majander et al. 2020). The individual steps are briefly described next. After adapter clipping, merging, and quality trimming, the resulting reads for each sample were mapped to the Nichols genome (NC_021490.2) using the BWA-MEM algorithm (Li 2014) with default parameters. PCR duplicates were removed with DeDUP (Peltzer et al. 2016). Coverage breadth of the reference genome and coverage depth were calculated using QualiMap (version 2.17) (Okonechnikov et al. 2016). Indel realignments were performed using GATK (version 3.6) (McKenna et al. 2010). SNVs (Single Nucleotide Variants) for the resulting mappings were called using GATK UnifiedHaplotyper. Sequenced samples were required to cover at least 80% of the Nichols genome by at least three reads to be included in further analyses (Arora et al. 2016). We applied MUSIAL (https://github.com/Integrative-Transcriptomics/MUSIAL) to obtain a multiple genome alignment (MSA) from the resulting VCF files.
To test the effects of the choice of reference genome, the EAGER-based procedure described above was also applied employing two other reference genomes, the dominant TPA strain in current infections, SS14 (NC_010741.1), and a well-studied TPE strain, CDC-2 (NC_016848.1). In total, this yielded three different data sets, each comprising a total of 75 genomes, and each with a different reference genome (Nichols, SS14, or CDC-2). These are referred to as the NIC-mapped, SS14-mapped, and CDC-2-mapped data sets, respectively. In the following, gene positions and annotations, unless otherwise stated, refer to the Nichols strain. A coverage threshold of 3 and a minimum homozygous SNP allele frequency of 0.9 were required to assign a variant against the nucleotide in each reference genome.
In the following, an overview of the methods used for the analyses of recombination and selection events in T. pallidum genomes is given (see also fig. 4 for a summary of the steps).
Fig. 4.

Analysis workflow for the study of recombination and selection in Treponema pallidum genomes for the NIC-mapped data set. The same pipeline was applied to the SS14-mapped and CDC2-mapped data sets.
Recombination Detection: PIM
To infer the presence of recombination in the complete genomes of T. pallidum, we have used PIM, which was developed in our group (Sánchez-Busó et al. 2014; Arora et al. 2016; Beamud et al. 2019). Putative recombination events are identified on a “per gene” basis with further verification of events spanning more than one gene as well as a detailed analysis of the intragenic portions actually involved in those events. This method was applied to each of the three data sets: the NIC-mapped data set, SS14-mapped data set, and the CDC2-mapped data set.
Phylogenetic Signal Test
The initial step consisted of an assessment of the phylogenetic information in each of the protein-coding genes annotated in the reference genome (978 for the NIC-mapped data set, 975 for the SS14-mapped data set, and 1,067 for the CDC2-mapped data set), using the likelihood mapping test in IQ-TREE 1.5.0 (Nguyen et al. 2015). Prior to the test, individual sequences in each of these protein-coding genes were assigned to four groups, corresponding to the three different subspecies of T. pallidum, with TPA sequences further divided into Nichols and SS14 clades (TPE, TEN, TPA-NIC, or TPA-SS14) (Arora et al. 2016). For the test, we obtained 10,000 random quartets comprising one sequence from each group. For each of these draws, the likelihoods of the three possible unrooted trees for the four groups described above were compared. The genes that showed some phylogenetic signal, evaluated as likelihoods falling outside the central region in the LM triangle (Strimmer and von Haeseler 1997), were retained for the ensuing analyses. The tp0897 gene, also known as tprK, was not included in the next recombination analyses because its hypervariable regions undergo intrastrain gene conversion and have been studied in detail elsewhere (Pinto et al. 2016) but was examined in detail in additional analyses explained below.
Phylogenetic Congruence Tests
A topology test was conducted as the second step of the PIM. For this, we constructed ML trees with IQ-TREE for each of the genes showing some phylogenetic signal in the likelihood mapping analysis. We also obtained the ML tree of the whole genome alignment of the 75 genomes, which we used as the reference genome-wide data tree. We used the GTR+G4+I as the evolutionary model in all the phylogenetic reconstructions. Next, we carried out topology tests for each gene, again with IQ-TREE, using two different methods: Shimodaira–Hasegawa (SH, Shimodaira and Hasegawa 1999) and Expected Likelihood Weights (ELW, Strimmer and Rambaut 2002) tests. Each topology test involves two comparisons. First, we compared the likelihood of each individual gene tree and the reference genome-wide data tree using the corresponding gene alignment. Secondly, we compared the same likelihoods using the complete genome alignment. A reciprocal incongruence is called when both tests reject the topology not derived from the corresponding alignment (individual gene in the first comparison, the complete genome in the second). This procedure was performed for the three data sets. Genes for which the two tests rejected the reference tree topology with the gene alignment adopting a conservative approach (P < 0.2, weight value close to 0, for SH and ELW tests, respectively) and the complete genome alignment rejected the topology of the tree built using the gene alignment (reciprocal incongruence, P < 0.2 and weight value close to 0) in at least one of them were selected and examined more closely in the next step.
Polyphyletic SNP Distribution
The selected genes that showed reciprocal incongruence were further analyzed with MEGAX (Kumar, Stecher, et al. 2018) in order to evaluate and define putative recombination events. To retain a gene as recombinant, we required it to contain at least three neighboring homoplasic SNPs, that is, SNPs shared among the different groups (TPE, TEN, TPA-NIC, or TPA-SS14) yielding a polyphyletic distribution. Recombinant regions were delimited by the homoplasic SNPs detected in the gene alignment. Hence, the distance between the flanking SNPs represents the minimum size of the recombination event, but it might extend further into the nonvariable positions upstream or downstream of the homoplasic SNPs given that the exact size of the recombining fragment cannot be estimated. The putative donor and recipient clade/strain of each recombination event were inferred applying a parsimony criterion to the distribution of alternative states of the homoplasic SNPs.
Among the genes selected in the topology tests were some pertaining to the tpr family, which comprises groups of paralogous genes. Due to the repetitive nature of the DNA sequences of these genes, and the challenges of the mapping stage, a large proportion of sites had missing data. When possible we examined these genes manually. In addition, to test for intrastrain recombination in the seven variable regions of the tp0897 (tprK) gene (Centurion-Lara et al. 2004), we generated a BLAST database with the 75 complete T. pallidum genomes described above and used the set of unique variable motifs found in the variable regions as query for BlastN searches.
Recombination Detection with Alternative Tools
We used Gubbins 2.2.0-1 (Croucher et al. 2015) and ClonalFrameML 1.1 (Didelot and Wilson 2015), two widely used programs for the detection of recombination based on genome-wide data, as alternative tools to the PIM method. To reduce the computational load of these programs, they were run for a subset of the NIC-mapped data set selected to obtain a balanced representation of the four groups identified in the phylogenetic trees (TPE, TEN, TPA-NIC, TPA-SS14). To obtain this representative subset, we first concatenated the putative recombinant loci detected by PIM and generated a multiple sequence alignment. For each of the 27 unique haplotypes, the most complete genome was selected as representative. eulerAPE v.3.0 (Micallef and Rodgers 2014) was used to represent common and different genes found to be recombinant using the three different methods.
Selection Analyses
Selection analyses were conducted only with the NIC-mapped data set. For each putative recombinant gene selected in our analyses, we extracted the SNPs, determined whether they were synonymous or nonsynonymous, and computed the nonsynonymous-to-synonymous substitution ratio ω = dN/dS. This ratio was conducted for the entire gene, as well as for the recombinant and nonrecombinant regions separately. In addition, we also estimated ω for all nonrecombinant genes. A ratio above 1 is indicative of positive selection, whereas a ratio below 1 points to purifying selection. Additionally, for the nonrecombinant genes, we used Codeml in the PAML 4.9 package (Yang 2007) to estimate the total number of synonymous and nonsynonymous sites per region/gene, as well as SNPeff (Cingolani et al. 2012) to evaluate the number of synonymous and nonsynonymous changes in each gene. Additional analyses to detect positive selection were performed with BUSTED (Murrell et al. 2015), a gene-based method, and RELAX (Wertheim et al. 2015), a codon-based method, both of which are implemented in HyPhy (Hypothesis Testing Using Phylogenies) (Pond et al. 2005).
Details on the programs, commands, and options used in the analyses described above are publicly available at DOI: 10.5281/zenodo.5528940.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
F.G.-C. and M.P.-D. are supported by grant numbers BFU2017-89594R from Ministerio de Investigación y Ciencia (Spanish Government) and PROMETEO2016-122 from Generalitat Valenciana. M.P.-D. is recipient of a predoctoral contract FPU17/02367 from Ministerio de Educación, Cultura y Deporte (Spanish Government). V.J.S. is supported by the Swiss National Science Foundation: grant number 188963—“Towards the origins of syphilis” and the University of Zurich’s University Research Priority Program “Evolution in Action: From Genomes to Ecosystems.” D.S. is supported by the Grant Agency of the Czech Republic (GA17-25455S; GC18-23521J; gacr.cz). L.S.-B. is funded by Plan GenT (CDEI-06/20-B), Conselleria de Sanitat Universal i Salut Pública, Generalitat Valenciana (Valencia, Spain). L.G. is supported by the NIH U19AI144133 “Syphilis Vaccine to Protect against Local and Disseminated T. pallidum Infection” grant. We would also like to thank two anonymous reviewers for their valuable comments and suggestions for improving the original manuscript.
Author Contributions
Conceptualization: F.G.-C., M.P.-D., and N.A. Methodology: F.G.-C., M.P.-D., N.A., and L.S.-B. Software: M.P.-D., K.N., V.J.S., and L.S.-B. Formal analysis: M.P.-D. and F.G.-C. Resources: M.P.-D., D.S., H.C.B., N.A., V.J.S., and P.P.B. Data curation: M.P.-D. and F.G.-C. Visualization: M.P.-D. and L.G. Supervision: F.G.-C. Drafting: M.P.-D., F.G.-C., N.A., L.S.-B., and L.G. Writing final: All the authors.
References
- Achtman M. 2008. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol. 62:53–70. [DOI] [PubMed] [Google Scholar]
- Achtman M. 2012. Insights from genomic comparisons of genetically monomorphic bacterial pathogens. Philos Trans R Soc Lond B Biol Sci. 367(1590):860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Nielsen R, Yang Z.. 2003. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164(3):1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora N, Schuenemann VJ, Jäger G, Peltzer A, Seitz A, Herbig A, Strouhal M, Grillová L, Sánchez-Busó L, Kühnert D, et al. 2016. Origin of modern syphilis and emergence of a pandemic Treponema pallidum cluster. Nat Microbiol. 2:16245. [DOI] [PubMed] [Google Scholar]
- Awadalla P. 2003. The evolutionary genomics of pathogen recombination. Nat Rev Genet. 4(1):50–60. [DOI] [PubMed] [Google Scholar]
- Beale MA, Lukehart SA.. 2020. Archaeogenetics: what can ancient genomes tell us about the origin of syphilis? Curr Biol. 30(19):R1092–R1095. [DOI] [PubMed] [Google Scholar]
- Beale MA, Marks M, Sahi SK, Tantalo LC, Nori AV, French P, Lukehart SA, Marra CM, Thomson NR.. 2019. Genomic epidemiology of syphilis reveals independent emergence of macrolide resistance across multiple circulating lineages. Nat Commun. 10(1):3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beamud B, Bracho MA, González-Candelas F.. 2019. Characterization of new recombinant forms of HIV-1 from the Comunitat Valenciana (Spain) by phylogenetic incongruence. Front Microbiol. 10:1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman MB, McGill MA, Pettersson J, Rogers A, Matejková P, Šmajs D, Weinstock GM, Norris SJ, Palzkill T.. 2008. A novel Treponema pallidum antigen, TP0136, is an outer membrane protein that binds human fibronectin. Infect Immun. 76(5):1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocchieri L. 2001. Phylogenetic inferences from molecular sequences: review and critique. Theor Popul Biol. 59(1):27–40. [DOI] [PubMed] [Google Scholar]
- Čejková D, Zobaníková M, Chen L, Pospíšilová P, Strouhal M, Qin X, Mikalová L, Norris SJ, Muzny DM, Gibbs RA, et al. 2012. Whole genome sequences of three Treponema pallidum ssp. pertenue strains: yaws and syphilis treponemes differ in less than 0.2% of the genome sequence. PLoS Negl Trop Dis. 6(1):e1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čejková D, Zobaníková M, Pospíšilová P, Strouhal M, Mikalová L, Weinstock GM, Šmajs D.. 2013. Structure of rrn operons in pathogenic non-cultivable treponemes: sequence but not genomic position of intergenic spacers correlates with classification of Treponema pallidum and Treponema paraluiscuniculi strains. J Med Microbiol. 62(Pt 2):196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centurion-Lara A, LaFond RE, Hevner K, Godornes C, Molini BJ, Van Voorhis WC, Lukehart SA.. 2004. Gene conversion: a mechanism for generation of heterogeneity in the tprK gene of Treponema pallidum during infection. Mol Microbiol. 52(6):1579–1596. [DOI] [PubMed] [Google Scholar]
- Chiner-Oms Á, Sánchez-Busó L, Corander J, Gagneux S, Harris SR, Young D, González-Candelas F, Comas I.. 2019. Genomic determinants of speciation and spread of the MTB complex. Sci Adv. 5:eaaw3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM.. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6(2):80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR.. 2015. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43(3):e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Davies D.. 2010. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 74(3):417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan JH, DeGiorgio M, Bryant D, Rosenberg NA.. 2009. Properties of consensus methods for inferring species trees from gene trees. Syst Biol. 58(1):35–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Maiden MCJ.. 2010. Impact of recombination on bacterial evolution. Trends Microbiol. 18(7):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Wilson DJ.. 2015. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol. 11(2):e1004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francés-Cuesta C, Sánchez-Hellín V, Gomila B, González-Candelas F.. 2021. Is there a widespread clone of Serratia marcescens producing outbreaks worldwide? J Hosp Infect. 108:7–14. [DOI] [PubMed] [Google Scholar]
- Gagneux S. 2018. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol. 16(4):202–213. [DOI] [PubMed] [Google Scholar]
- Giacani L, Brandt SL, Puray-Chavez M, Reid TB, Godornes C, Molini BJ, Benzler M, Hartig JS, Lukehart SA, Centurion-Lara A.. 2012. Comparative investigation of the genomic regions involved in antigenic variation of the TprK antigen among treponemal species, subspecies, and strains. J Bacteriol. 194(16):4208–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacani L, Chattopadhyay S, Centurion-Lara A, Jeffrey BM, Le HT, Molini BJ, Lukehart SA, Sokurenko EV, Rockey DD.. 2012. Footprint of positive selection in Treponema pallidum subsp. pallidum genome sequences suggests adaptive microevolution of the syphilis pathogen. PLoS Negl Trop Dis. 6(6):e1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godornes C, Giacani L, Barry AE, Mitja O, Lukehart SA.. 2017. Development of a Multilocus Sequence Typing (MLST) scheme for Treponema pallidum subsp. pertenue: application to yaws in Lihir Island, Papua New Guinea. PLoS Negl Trop Dis. 11(12):e0006113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray RR, Mulligan CJ, Molini BJ, Sun ES, Giacani L, Godornes C, Kitchen A, Lukehart SA, Centurion-Lara A.. 2006. Molecular evolution of the tprC, D, I, K, G, and J genes in the pathogenic genus Treponema. Mol Biol Evol. 23(11):2220–2233. [DOI] [PubMed] [Google Scholar]
- Grillová L, Bawa T, Mikalová L, Gayet-Ageron A, Nieselt K, Strouhal M, Sednaoui P, Ferry T, Cavassini M, Lautenschlager S, et al. 2018. Molecular characterization of Treponema pallidum subsp. pallidum in Switzerland and France with a new multilocus sequence typing scheme. PLoS One 13(7):e0200773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillová L, Oppelt J, Mikalová L, Nováková M, Giacani L, Niesnerová A, Noda AA, Mechaly AE, Pospíšilová P, Čejková D, et al. 2019. Directly sequenced genomes of contemporary strains of syphilis reveal recombination-driven diversity in genes encoding predicted surface-exposed antigens. Front Microbiol. 10:1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper KN, Ocampo PS, Steiner BM, George RW, Silverman MS, Bolotin S, Pillay A, Saunders NJ, Armelagos GJ.. 2008. On the origin of the treponematoses: a phylogenetic approach. PLoS Negl Trop Dis. 2(1):e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SJ, Didelot X, Gandhi K, Dean D, Read TD.. 2011. Interplay of recombination and selection in the genomes of Chlamydia trachomatis. Biol Direct. 6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Caimano MJ, Anand A, Dey A, Hawley KL, LeDoyt ME, La Vake CJ, Cruz AR, Ramirez LG, Paštěková L, et al. 2018. Sequence variation of rare outer membrane protein β-barrel domains in clinical strains provides insights into the evolution of Treponema pallidum subsp. pallidum, the syphilis spirochete. mBio 9(3):e01006–e01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K.. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefébure T, Stanhope MJ.. 2007. Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 8(5):R71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. 2014. A Burrows-Wheeler transform based method for DNA sequence comparison. Comput Biol Bioinformatics. 2(3):33. [Google Scholar]
- Liu X, Gutacker MM, Musser JM, Fu Y-X.. 2006. Evidence for recombination in Mycobacterium tuberculosis. J Bacteriol. 188(23):8169–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison WP, Knowles LL.. 2006. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 55(1):21–30. [DOI] [PubMed] [Google Scholar]
- Maděránková D, Mikalová L, Strouhal M, Vadják Š, Kuklová I, Pospíšilová P, Krbková L, Koščová P, Provazník I, Šmajs D.. 2019. Identification of positively selected genes in human pathogenic treponemes: syphilis-, yaws-, and bejel-causing strains differ in sets of genes showing adaptive evolution. PLoS Negl Trop Dis. 13(6):e0007463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majander K, Pfrengle S, Kocher A, Neukamm J, du Plessis L, Pla-Díaz M, Arora N, Akgül G, Salo K, Schats R, et al. 2020. Ancient bacterial genomes reveal a high diversity of Treponema pallidum strains in early Modern Europe. Curr Biol. 30(19):3788–3803.e10. [DOI] [PubMed] [Google Scholar]
- Marks M, Fookes M, Wagner J, Butcher R, Ghinai R, Sokana O, Sarkodie Y-A, Lukehart SA, Solomon AW, Mabey DCW, et al. 2018. Diagnostics for yaws eradication: insights from direct next-generation sequencing of cutaneous strains of Treponema pallidum. Clin Infect Dis. 66(6):818–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micallef L, Rodgers P.. 2014. euler APE: drawing area-proportional 3-Venn diagrams using ellipses. PLoS One 9(7):e101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikalová L, Janečková K, Nováková M, Strouhal M, Čejková D, Harper KN, Šmajs D.. 2020. Whole genome sequence of the Treponema pallidum subsp. endemicum strain Iraq B: a subpopulation of bejel treponemes contains full-length tprF and tprG genes similar to those present in T. p. subsp. pertenue strains. PLoS One 15(4):e0230926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikalová L, Strouhal M, Oppelt J, Grange PA, Janier M, Benhaddou N, Dupin N, Šmajs D.. 2017. Human Treponema pallidum 11q/j isolate belongs to subsp. endemicum but contains two loci with a sequence in TP0548 and TP0488 similar to subsp. pertenue and subsp. pallidum, respectively. PLoS Negl Trop Dis. 11(3):e0005434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, et al. 2015. Gene-wide identification of episodic selection. Mol Biol Evol. 32(5):1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K, Conesa A, García-Alcalde F.. 2016. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32(2):292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltzer A, Jäger G, Herbig A, Seitz A, Kniep C, Krause J, Nieselt K.. 2016. EAGER: efficient ancient genome reconstruction. Genome Biol. 17:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pětrošová H, Zobaníková M, Čejková D, Mikalová L, Pospíšilová P, Strouhal M, Chen L, Qin X, Muzny DM, Weinstock GM, et al. 2012. Whole genome sequence of Treponema pallidum ssp. pallidum, strain Mexico A, suggests recombination between yaws and syphilis strains. PLoS Negl Trop Dis. 6(9):e1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto M, Borges V, Antelo M, Pinheiro M, Nunes A, Azevedo J, Borrego MJ, Mendonça J, Carpinteiro D, Vieira L, et al. 2016. Genome-scale analysis of the non-cultivable Treponema pallidum reveals extensive within-patient genetic variation. Nat Microbiol. 2:16190. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW, Muse SV.. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21(5):676–679. [DOI] [PubMed] [Google Scholar]
- Pospíšilová P, Grange PA, Grillová L, Mikalová L, Martinet P, Janier M, Vermersch A, Benhaddou N, Del Giudice P, Alcaraz I, et al. 2018. Multi-locus sequence typing of Treponema pallidum subsp. pallidum present in clinical samples from France: infecting treponemes are genetically diverse and belong to 18 allelic profiles. PLoS One 13(7):e0201068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeis E, Tantalo L, Lieberman N, Phung Q, Greninger A, Giacani L.. 2021. Genetic Engineering of Treponema pallidum subsp. pallidum, the Syphilis Spirochete. PLoS Pathog. 17(7):e1009612. [DOI] [PMC free article] [PubMed]
- Sánchez-Busó L, Comas I, Jorques G, González-Candelas F.. 2014. Recombination drives genome evolution in outbreak-related Legionella pneumophila isolates. Nat Genet. 46(11):1205–1211. [DOI] [PubMed] [Google Scholar]
- Schuenemann VJ, Kumar Lankapalli A, Barquera R, Nelson EA, Iraíz Hernández D, Acuña Alonzo V, Bos KI, Márquez Morfín L, Herbig A, Krause J.. 2018. Historic Treponema pallidum genomes from Colonial Mexico retrieved from archaeological remains. PLoS Negl Trop Dis. 12(6):e0006447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro BJ. 2016. How clonal are bacteria over time? Curr Opin Microbiol. 31:116–123. [DOI] [PubMed] [Google Scholar]
- Shapiro BJ, Jesse Shapiro B, Levade I, Kovacikova G, Taylor RK, Almagro-Moreno S.. 2017. Origins of pandemic Vibrio cholerae from environmental gene pools. Nat Microbiol. 2:16240. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M.. 1999. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 16(8):1114–1114. [Google Scholar]
- Singh AE, Romanowski B.. 1999. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 12(2):187–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šmajs D, Norris SJ, Weinstock GM.. 2012. Genetic diversity in Treponema pallidum: implications for pathogenesis, evolution and molecular diagnostics of syphilis and yaws. Infect Genet Evol. 12(2):191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudová B, Strouhal M, Zobaníková M, Cejková D, Fulton LL, Chen L, Giacani L, Centurion-Lara A, Bruisten SM, Sodergren E, et al. 2014. Whole genome sequence of the Treponema pallidum subsp. endemicum strain Bosnia A: the genome is related to yaws treponemes but contains few loci similar to syphilis treponemes. PLoS Negl Trop Dis. 8(11):e3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K, Rambaut A.. 2002. Inferring confidence sets of possibly misspecified gene trees. Proc Biol Sci. 269(1487):137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K, von Haeseler A.. 1997. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc Natl Acad Sci U S A. 94(13):6815–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strouhal M, Mikalová L, Haviernik J, Knauf S, Bruisten S, Noordhoek GT, Oppelt J, Čejková D, Šmajs D.. 2018. Complete genome sequences of two strains of Treponema pallidum subsp. pertenue from Indonesia: modular structure of several treponemal genes. PLoS Negl Trop Dis. 12(10):e0006867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Meng Z, Wu K, Liu B, Zhang S, Liu Y, Wang Y, Zheng H, Huang J, Zhou P.. 2016. Tracing the origin of Treponema pallidum in China using next-generation sequencing. Oncotarget 7(28):42904–42918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M-L, Zhao Q, Liu L-L, Zhu X-Z, Gao K, Zhang H-L, Lin L-R, Niu J-J, Ji Z-L, Yang T-C.. 2017. Whole genome sequence of the Treponema pallidum subsp. pallidum strain Amoy: an Asian isolate highly similar to SS14. PLoS One 12(8):e0182768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente-Mullor C, Beamud B, Ansari I, Francés-Cuesta C, García-González N, Mejía L, Ruiz-Hueso P, González-Candelas F.. 2021. One is not enough: on the effects of reference genome for the mapping and subsequent analyses of short-reads. PLoS Comput Biol. 17(1):e1008678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K.. 2015. RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol. 32(3):820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.