

Abstract
A new structurally characterized ferrous corrole [FeII(ttppc)]− (1) binds one equivalent of dioxygen to form [FeIII(O2•−)(ttppc)]− (2). This complex exhibits a 16/18O2-isotope sensitive ν(O–O) stretch at 1128 cm−1 concomitantly with a single ν(Fe–O2) at 555 cm−1, indicating it is an η1-superoxo (“end-on”) iron(III) complex. Complex 2 is the first well characterized Fe-O2 corrole, and mediates the following biologically relevant oxidation reactions: dioxygenation of an indole derivative, and H-atom abstraction from an activated O–H bond.
Keywords: iron, superoxide, hydrogen atom transfer, porphyrinoids, dioxygen activation
Graphical Abstract
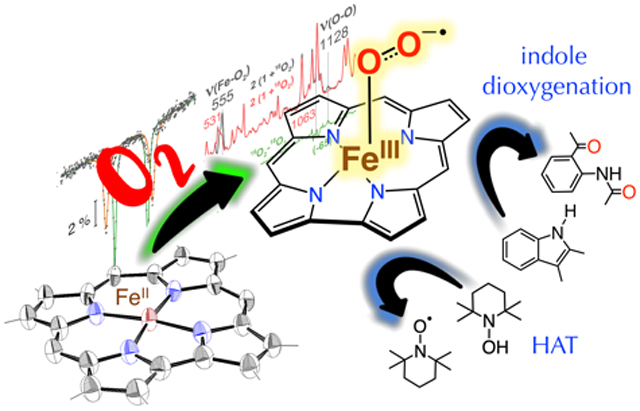
An iron-superoxide corrole has been generated from a ferrous corrole and dioxygen, and was found to be capable of indole dioxygenation and H-atom transfer reactions.
The activation of dioxygen by metalloenzymes usually involves one-electron transfer from a reduced metal center (Mn+) to O2 to give a metal-superoxo (Mn+1(O2−•)) species. A subset of these species involves iron enzymes, and there is significant interest in the biological roles of iron-superoxo intermediates, including their proposed involvement in substrate oxidations.[1] This reactivity has been proposed for heme enzymes such as tryptophan/indoleamine 2,3-dioxygenases (TDO/IDO) (indole dioxygenation),[2] MarE (indole monooxygenation),[3] and nitric oxide synthase (NOS),[4] and nonheme iron enzymes such as isopenicillin-N-synthase (hydrogen atom transfer (HAT)),[5] myo-inositol oxygenase (HAT),[6] and cysteine dioxygenase (S-oxygenation).[1b, 7] Metal-superoxo species are also proposed as key intermediates in a number of synthetic, oxidative catalytic cycles.[8]
The inherent instability of FeIII(O2−•) species makes their preparation and direct characterization a challenge. Iron-superoxo porphyrins derived from O2 have been known for some time,[1a] and recently, analogous nonheme FeIII(O2−•) species have been reported.[9] For the porphyrin complexes, studies describing the oxidative reactivity of the FeO2 adducts are beginning to emerge,[10] but these data remain relatively scarce, and the key factors that influence this reactivity (e.g. bond strengths, spin state, redox potentials, pKa) remain under significant debate.
Corroles are a remarkably versatile family of tetrapyrrolic compounds that have been employed in a wide range of applications, including catalysis, as synthetic models of metalloenzymes,[11] and as artificial cofactors for heme proteins (e.g. myoglobin, peroxidases).[12] Many of these applications involve oxidative transformations, and the reactivity between metallocorroles and oxidants such as O2 or related analogs (e.g. H2O2, ROOH) comprises a critical step in their mechanisms of action. The activation of O2 with biologically relevant iron complexes is anticipated to involve an initial FeO2 adduct. However, essentially nothing is known about FeO2 corroles. We set out to conclusively identify such a species with a sterically bulky corrole ligand, and provide the first measure of its oxidative reactivity.
Herein we report the synthesis and characterization of an iron(III)-superoxo corrole, prepared from a new FeII corrole precursor and O2. The superoxo complex was characterized by UV-vis, Mössbauer, 1H NMR, and resonance Raman spectroscopies, and to our knowledge, is the first example of a well characterized Fe(O2)(corrole) complex.[13] This complex is capable of indole dioxygenation and H-atom abstraction, two fundamental processes proposed to be mediated by biological Fe(O2) species.
Reduction of brown FeIII(ttppc)[11c] (ttppc = 5, 10, 15-tris((2,4,6-triphenyl)phenyl)corrolato(3-)), with sodium/mercury amalgam (1.5% Na in Hg) gives [FeII(ttppc)]− (1). Single crystal X-ray diffraction reveals a 4-coordinate ferrous corrole, [Na(H2O)(2-MeTHF)(1)] (Figure 1). A sodium cation is weakly bound to an Npyrrole atom (Na–Npyrrole = 2.553(5) Å) and the nearest phenyl ring (Na–C28 = 2.861(7) Å). There is one H2O and one 2-MeTHF molecule bound to the Na+ ion. The Fe–N bond distances range between 1.862(4) – 1.893(4) Å, and the iron atom lies along the plane of the macrocycle (Fe-to-mean plane(23 atom core) = 0.044 Å). These metrical parameters are close to those reported for [FeII(tpc)]−, the only other structurally characterized FeII corrole.[13] Complex [Na(H2O)(2-MeTHF)(1)] provides a rare example of a square-planar FeII compound.[14] Magnetic susceptibility measurements for 1 (Evans Method) gave μeff = 3.44 ± 0.2 μB, higher than the theoretical value for an S = 1 ion (μeff(spin-only) = 2.83 μB), but similar to other FeII (S = 1) complexes.[15]
Figure 1.
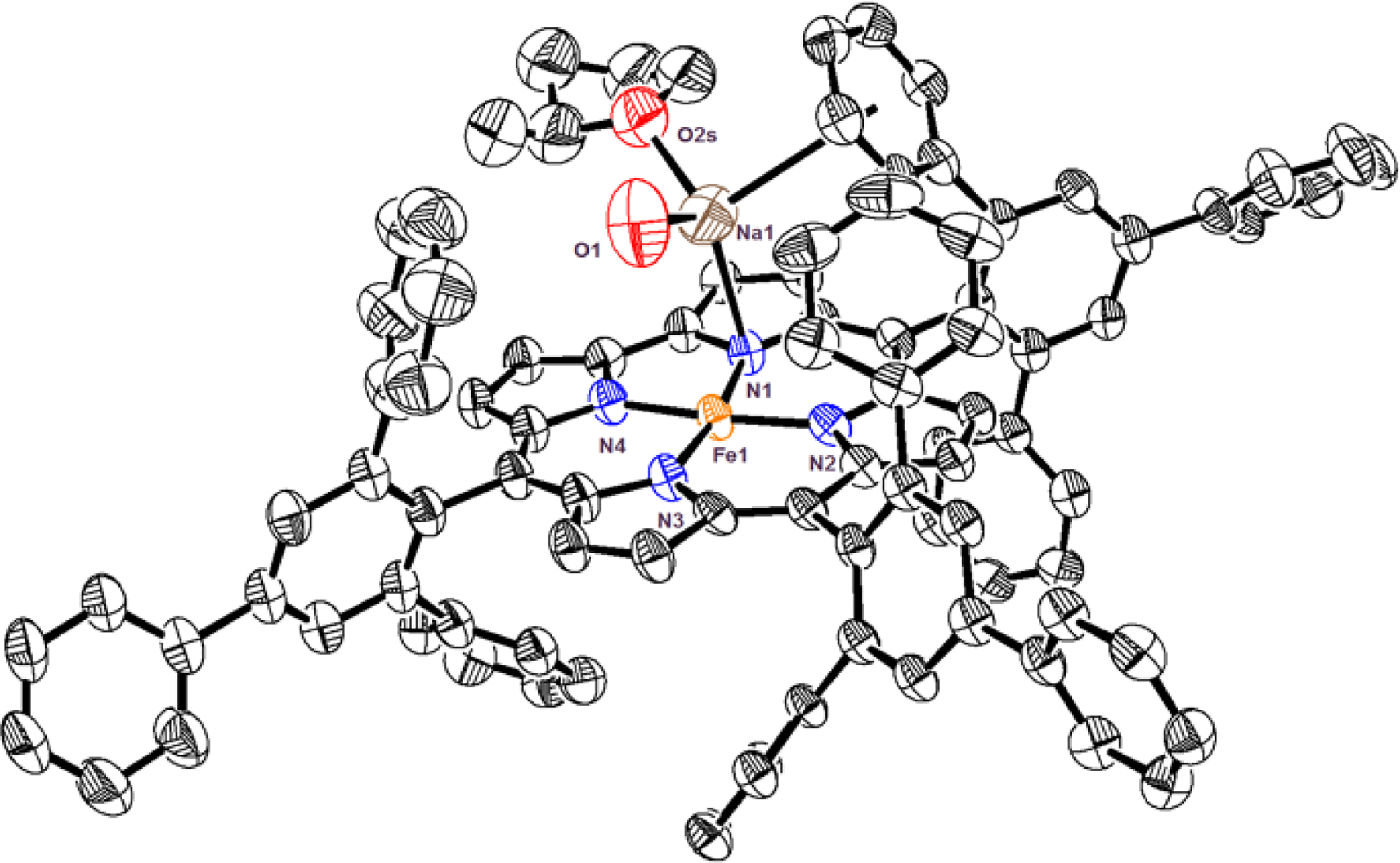
Displacement ellipsoid plot (50% probability level) for [Na(H2O)(2-MeTHF)(1)] at 110(2) K. Hydrogen atoms omitted for clarity.[27]
The UV-vis spectrum of 1 (Figure 2) reveals distinct peaks at λmax (ε in M−1 cm−1) = 465 (56300), 545 (15200), 588 (15700), and 645 (29200) nm, and the 1H-NMR spectrum of 1 in THF-d8 exhibits relatively sharp, paramagnetically-shifted peaks between +16 to −83 ppm (Figure S2). The zero-field Mössbauer spectrum of an 57Fe-enriched sample of 1 reveals a single, sharp quadrupole doublet with δ = 0.25 mm s−1 and |ΔEQ| = 2.45 mm s−1 (Figure 2). There are no reported Mössbauer spectra of other iron(II) corroles for comparison, but the isomer shift of 1 is comparable to other ferrous (S=1) porphyrin/porphyrinoids.[16]
Figure 2.
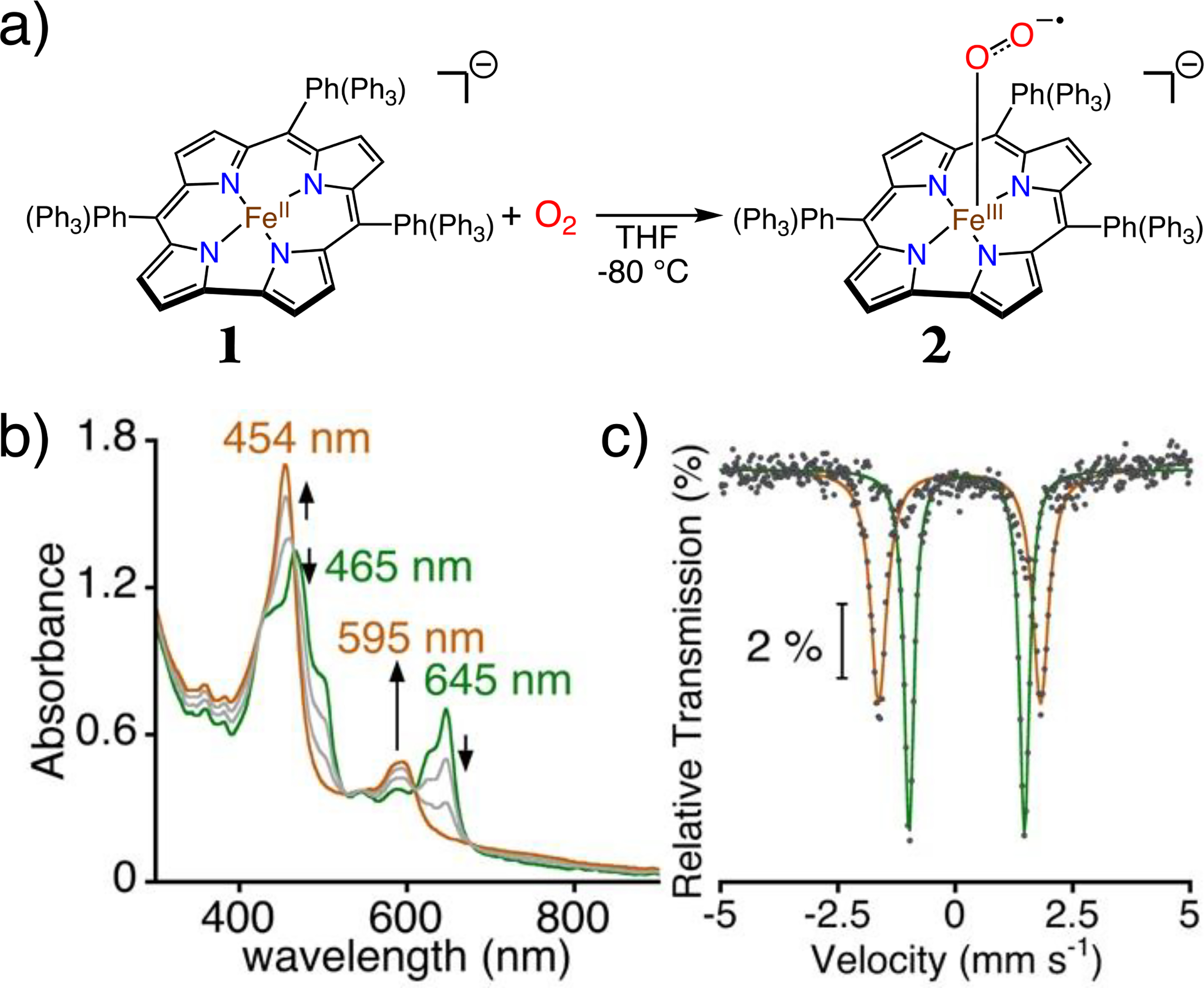
(a) Generation of 2 from 1 + O2 in THF at −80 °C. (b) Titration of 1 (24 μM) with O2 (0 – 1.0 equiv) at −80 °C. (c) Overlay of zero-field 57Fe Mössbauer spectra for 1 and 2 in THF at 80 K. Global fits for 1 (green) and 2 (brown).
Exposure of 1 to air in THF at 23 °C results in immediate oxidation back to FeIII. However, reaction of 1 with excess O2 at −80 °C results in isosbestic absorption changes and new peaks at λmax = 454 nm (70800 M−1 cm−1), 595 nm (20500 M−1 cm−1) assigned to 2. Spectral titrations support a 1:1 Fe:O2 stoichiometry (Figure 2b). The new species exhibits less than 10% decay over 1 hour at −60 °C, but complete decay to FeIII(ttppc) within two hours at −40 °C. These observations suggest an assignment for 2 as a superoxo complex, [FeIII(O2•−)(ttppc)]−.
Zero-field Mössbauer spectroscopy of 2 at 80 K reveals a single, sharp quadrupole doublet with δ = 0.10 mm s−1, |ΔEQ| = 3.45 mm s−1 (88% of the total fit) (Figure 2c). A similar quadrupole doublet was observed upon lowering the temperature to 4 K (Figure S4). The lack of hyperfine splitting at 4 K is consistent with an integer-spin species, as seen for other iron-superoxide porphyrins.[17] 1H NMR (−80 °C) spectra of 2 gives only poorly defined peaks in the diamagnetic region (Figure S5).
The assignment of 2 as an FeIII(O2−•) species was solidified by resonance Raman (RR) spectroscopy. RR spectra of 2 at 110 K were obtained with a 458-nm laser excitation and showed intensely resonance enhanced bands attributable to corrole macrocycle vibrations (Figure 3). Comparing RR spectra of samples prepared with 16O2 and 18O2 identifies a strong band at 555 cm−1 that down shifts to 531 cm−1 with 18O-labeling. These frequencies and the 24 cm−1 downshift are consistent with a ν(Fe–O) stretching mode assignment. Another band observed at 1128 cm−1 in the spectrum of 2 prepared with 16O2 and absent in its 18O2-counterpart falls within the expected range for a ν(O–O) stretching mode of mononuclear η1-superoxo (end-on) metal complexes.[18] The isotope-edited difference spectrum supports this assignment and reveals that the downshifted ν(18O–18O) mode expected around 1064 cm−1 (diatomic harmonic oscillator approximation) affects the intensities of strong corrole vibrations at 1043 and 1069 cm−1. Similar mode mixing and intensity borrowing processes have been observed with porphyrin iron-superoxo complexes.[18c–e] These RR spectra identify 2 as an iron(III)-superoxide species. The high ν(Fe–O2) and low ν(O–O) frequencies sustain the back-bonding correlations previously observed for XO adducts of Fe(II) porphyrins.[19] Moreover, while a side-on superoxo species would be expected to display two Fe–O2 stretches (i.e. symmetric and asymmetric stretches), the concomitant detection of a single ν(Fe–O) at 555 cm−1 with a ν(O–O) at 1128 cm−1, and with intensities and frequencies that are similar to those observed in the RR spectra of end-on iron(III)-superoxo tetrakis(2,6-difluorophenyl)porphyrinate and yeast oxyhemoglobins,[20] support an end-on binding geometry of the superoxo ligand in 2 along with the presence of a weakly coordinating THF as a sixth ligand.
Figure 3.
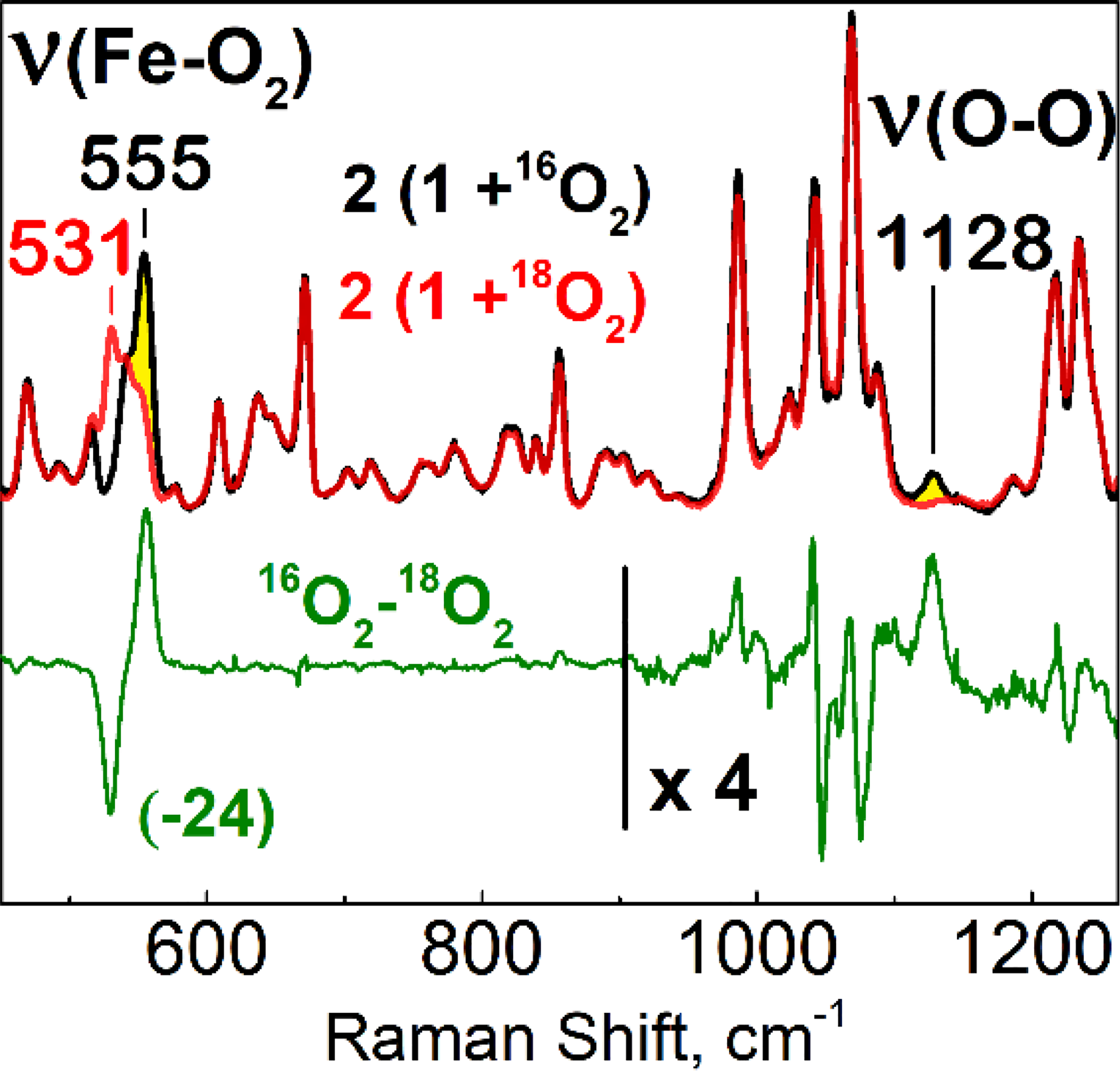
Resonance Raman spectra of 2 prepared with 16O2 (black) and 18O2 (red) in 2-MeTHF at 110 K (λexc = 458 nm); difference spectrum (16O2–18O2) (green) and was multiplied by a factor 4 above 900 cm−1 to better visualize the 1128-cm−1 band and accompanying perturbations near 1064 cm−1 where the ν(18O-18O) is expected to occur.
We set out to establish the oxidative capabilities of 2 in two biologically relevant reactions: indole dioxygenation and H-atom abstraction. The initial step of indoleamine oxidation in TDO/IDO is postulated to involve an FeIII(O2•−)(porphyrin) intermediate, although direct evidence for this step remains scant.[10c, 21] Addition of 2,3-dimethylindole (2,3-DMI) to 2 in THF at −40 °C gives an immediate color change from brown-green to red, and analysis by TLC and 1H NMR spectroscopy identified N-(2-acetyl-phenyl)-acetamide as the major product (86%)(Figure 4). When O2 was bubbled into a solution of 1 preloaded with 2,3-DMI at −40 °C, similar results were obtained. The use of 18O2 leads to a prominent ion at 181 m/z in the electron impact mass spectrum, which corresponds to the incorporation of two 18O atoms in the dioxygenated product (Figure S7). A peak at 179 m/z is also observed, corresponding to the incorporation of a single 18O, suggesting that there is one or more intermediates that can undergo exchange with an 16O source (e.g. trace H2O).
Figure 4.
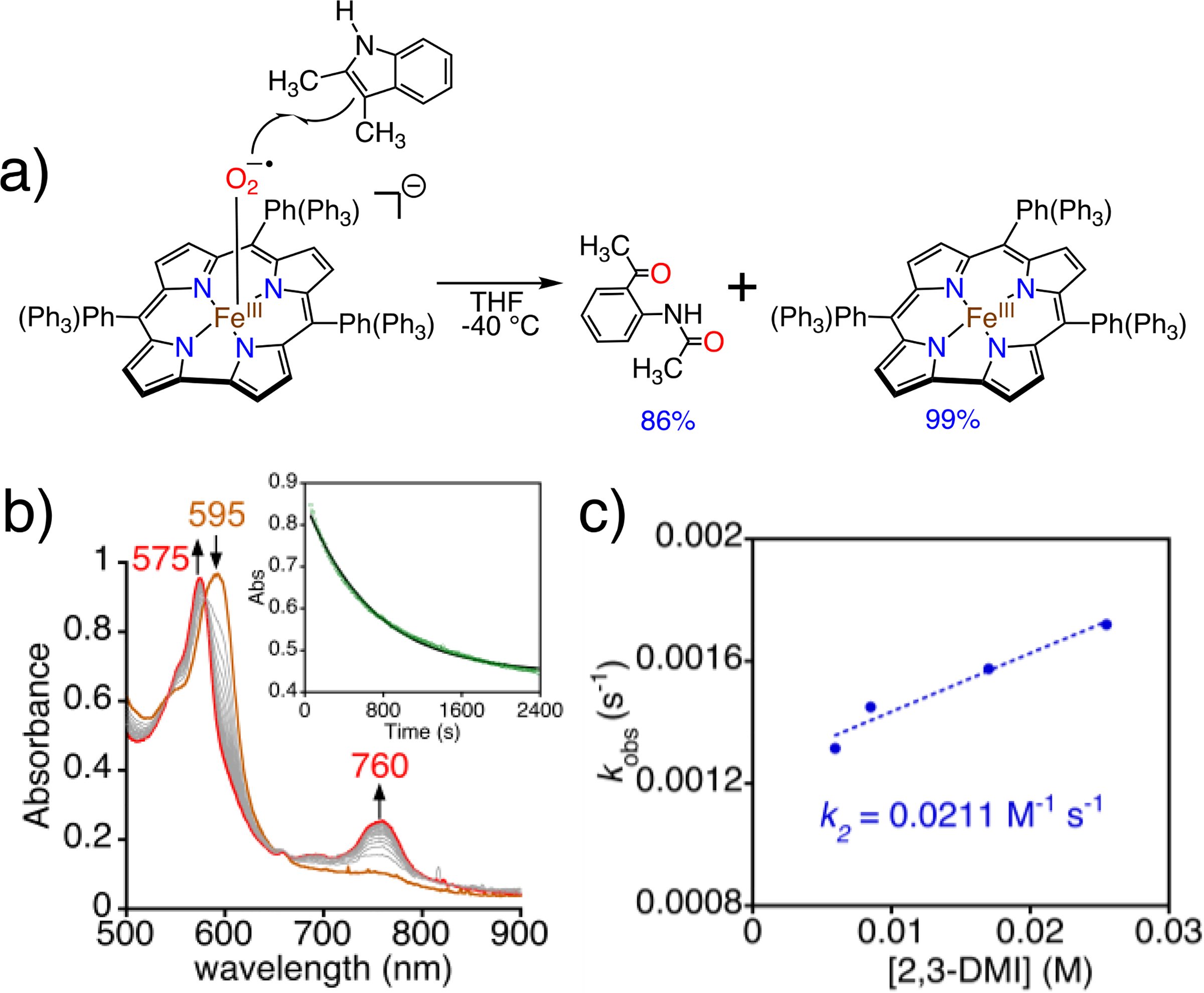
a) Reaction of 2 with 2,3-dimethylindole. b) Time-resolved UV-vis spectra (0 – 45 min) for the reaction between 2 (60 μM) and 2,3-DMI (100 equiv) in THF at −40 °C. Inset: changes in A595 nm over time. c) Plot of pseudo-first-order rate constants (kobs) vs [2,3-DMI] together with best-fit line.
It is known that free superoxide anion reacts with tryptophan derivatives to give ring-opened, dioxygenated products.[22] Given that 2 decays slowly to an FeIII product at −40 °C, we considered the possibility that there may be some free superoxide in solution, or there may be a rapid dynamic equilibrium between bound and free O2•− to give a steady state amount of O2−•, which in turn could react directly with 2,3-DMI. A control reaction was run involving addition of a KO2 mixture to 2,3-DMI under the same conditions as employed for oxidations with 2. No appreciable formation of any oxidation products was observed (Figure S8). This control reaction rules out participation of free O2−• in the oxidation of the indole substrate.
Monitoring the reaction between 2 and 2,3-DMI by UV-vis showed the characteristic peak for 2 (595 nm) converts to a peak at 575 nm assigned to FeIII(ttppc) following first-order kinetics, with isosbestic points at 580 and 664 nm (Figure 4b). Varying the substrate concentration led to a second-order rate constant k2 = 0.02(0) M−1 s−1 (Figure 4c). The lack of an observed intermediate suggests that attack of 2 on 2,3-DMI is the rate-determining step. Subsequent steps may follow those proposed for the enzymes, although the enzymatic mechanisms are still not well understood and remain a topic of significant debate.[23] Proposed mechanisms for certain TDO enzymes also suggest that the initial reaction between the putative FeIII(O2•−) species and the substrate is the rate-determining step.[2c]
Initial efforts to determine the ability of 2 to abstract hydrogen atoms began with relatively weak C-H substrates, e.g. cyclohexadiene (CHD). However, no reaction was observed. Reaction of 2 with the O–H bond donor TEMPOH led to an isosbestic conversion to FeIII(ttppc) (Figure 5), and TEMPO radical (97%), consistent with a hydrogen atom transfer (HAT) reaction. Attempts to detect the H2O2 product via an iodometric method[24] were unsuccessful.
Figure 5.
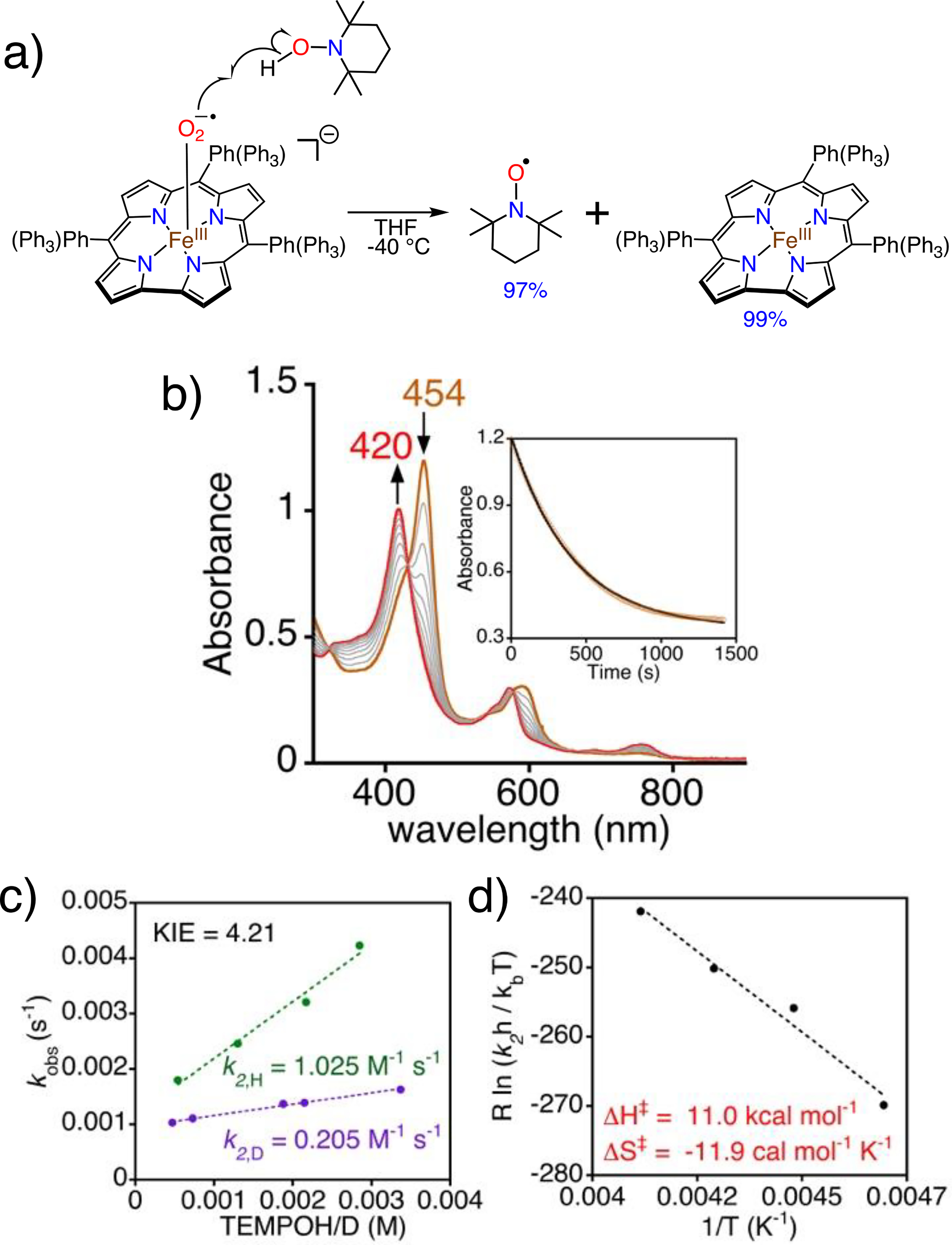
a) Reaction of 2 with TEMPOH. b) UV-vis spectra (0 – 30 min) for 2 (19 μM) and TEMPOH (75 equiv) in THF at −40 °C. Inset: changes in A454 nm over time. c) Plot of pseudo-first-order rate constants (kobs) vs [TEMPOH] (green circles) or [TEMPOD] (purple circles) together with best-fit lines. d) Eyring plot.
Kinetic analysis shows pseudo-first-order behavior with excess TEMPOH, yielding k2,H = 1.03 M−1 s−1 at −40 °C. Deuterated TEMPOD gave k2,D = 0.205 M−1 s−1 and a KIE(kH/kD) = 4.21(Figure 5c). An Eyring analysis yielded activation parameters of ΔH‡ = 11.0 kcal mol−1 and ΔS‡ = −11.9 kcal mol−1 K−1 (Figure 5d). The data clearly point to a bimolecular reaction involving hydrogen atom transfer between TEMPOH and 2.
The extrapolated k2 for 2 at −80 °C is 0.016(0) M−1 s−1, which is ~30 to 130-fold slower than those reported for FeIII(O2•−)(porph) complexes.[10a, 10d] The steric encumbrance of the meso substituents in 2 could be a factor in slowing H-atom abstraction, but previous examination of M(OH)(ttppc) (M = Fe, Mn) showed that these complexes reacted with O–H substrates at rates similar to other less hindered M(OH)/(O) complexes.[11e]
A control reaction with free KO2 was carried out for TEMPOH, and did show production of TEMPO radical (Figure S13). This result was not unexpected given the relative ease of oxidation of TEMPOH. At this time we have no evidence for the presence of free superoxide in these reactions, but we cannot rule out the participation of free O2•− in the oxidation of TEMPOH. We note that the earlier reports on metal-superoxide reactivity with TEMPOH do not appear to have addressed this issue,[10a, 10b, 10d, 25] but the possible involvement of free O2•− in the mechanism of HAT does need to be considered for metal-superoxide species.
In summary, we report the first well-characterized iron(III)-superoxo corrole via dioxygen activation by a ferrous corrole precursor. The ferrous corrole is only the second example to be structurally characterized, and confirms a rare, square-planar iron(II) configuration for this complex. The spectroscopic characterization of 2 provides benchmarks for identifying other FeIII(O2•−) corroles, such as in artificial proteins or oxidation catalysts. Complex 2 was capable of indole dioxygenation, adding to the very few examples of iron-superoxide complexes[10c, 21] shown to carry out the initial step hypothesized for certain TDO/IDO enzymes. These results support the proposed enzyme mechanism, but also indicate that dioxygenation of indole by FeIII(O2•−) is possible in the absence of an optimized enzyme/cofactor active site.[10c, 21] Complex 2 was also competent to oxidize TEMPOH by HAT, although significantly slower than a porphyrin analog. The factors that influence the dramatic differences in HAT reactivity for metal-superoxide species remain poorly understood,[25a, 26] and future studies should help enlighten researchers in this arena.
Supplementary Material
Acknowledgements
The authors acknowledge the support by the NIH (Grant R01GM101153 to D.P.G.). We also thank Dr. D. Cummins and V. Yadav for assistance.
References
- [1].a) Huang X, Groves JT, Chem. Rev. 2018, 118, 2491–2553; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gordon JB, Goldberg DP, in Comprehensive Coordination Chemistry III, 3rd ed. (Eds.: Constable E, Parkin G, Que L), Elsevier, 2021, pp. 333–377; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sahu S, Goldberg DP, J. Am. Chem. Soc. 2016, 138, 11410–11428; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ray K, Pfaff FF, Wang B, Nam W, J. Am. Chem. Soc. 2014, 136, 13942–13958; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) van der Donk WA, Krebs C, Bollinger JM Jr., Curr. Opin. Struct. Biol. 2010, 20, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Lewis-Ballester A, Batabyal D, Egawa T, Lu C, Lin Y, Marti MA, Capece L, Estrin DA, Yeh SR, Proc. Natl. Acad. Sci. 2009, 106, 17371–17376; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Makino R, Obayashi E, Hori H, Iizuka T, Mashima K, Shiro Y, Ishimura Y, Biochemistry 2015, 54, 3604–3616; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Basran J, Booth ES, Lee M, Handa S, Raven EL, Biochemistry 2016, 55, 6743–6750; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lewis-Ballester A, Forouhar F, Kim SM, Lew S, Wang Y, Karkashon S, Seetharaman J, Batabyal D, Chiang BY, Hussain M, Correia MA, Yeh SR, Tong L, Sci. Rep. 2016, 6, 35169; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Geng J, Weitz AC, Dornevil K, Hendrich MP, Liu A, Biochemistry 2020, 59, 2813–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang Y, Zou Y, Brock NL, Huang T, Lan Y, Wang X, Deng Z, Tang Y, Lin S, J. Am. Chem. Soc. 2017, 139, 11887–11894. [DOI] [PubMed] [Google Scholar]
- [4].Zhu Y, Silverman RB, Biochemistry 2008, 47, 2231–2243. [DOI] [PubMed] [Google Scholar]
- [5].Tamanaha E, Zhang B, Guo Y, Chang WC, Barr EW, Xing G, St Clair J, Ye S, Neese F, Bollinger JM Jr., Krebs C, J. Am. Chem. Soc. 2016, 138, 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM Jr., Proc. Natl. Acad. Sci. 2006, 103, 6130–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tchesnokov EP, Faponle AS, Davies CG, Quesne MG, Turner R, Fellner M, Souness RJ, Wilbanks SM, de Visser SP, Jameson GN, Chem. Commun. 2016, 52, 8814–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Pegis ML, Wise CF, Martin DJ, Mayer JM, Chem. Rev. 2018, 118, 2340–2391; [DOI] [PubMed] [Google Scholar]; b) Wang D, Weinstein AB, White PB, Stahl SS, Chem. Rev. 2018, 118, 2636–2679; [DOI] [PubMed] [Google Scholar]; c) Sterckx H, Morel B, Maes BUW, Angew. Chem. Int. Ed. 2019, 58, 7946–7970. [DOI] [PubMed] [Google Scholar]
- [9].a) Fischer AA, Lindeman SV, Fiedler AT, Chem. Commun. 2018, 54, 11344–11347; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blakely MN, Dedushko MA, Yan Poon PC, Villar-Acevedo G, Kovacs JA, J. Am. Chem. Soc. 2019, 141, 1867–1870; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hong S, Lee Y-M, Ray K, Nam W, Coord. Chem. Rev. 2017, 334, 25–42; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Winslow C, Lee HB, Field MJ, Teat SJ, Rittle J, J. Am. Chem. Soc. 2021, 143, 13686–13693; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Pan HR, Chen HJ, Wu ZH, Ge P, Ye S, Lee GH, Hsu HF, JACS Au 2021, 1, 1389–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Kim H, Rogler PJ, Sharma SK, Schaefer AW, Solomon EI, Karlin KD, J. Am. Chem. Soc. 2020, 142, 3104–3116; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim H, Rogler PJ, Sharma SK, Schaefer AW, Solomon EI, Karlin KD, Angew. Chem. Int. Ed. 2021, 60, 5907–5912; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mondal P, Wijeratne GB, J. Am. Chem. Soc. 2020, 142, 1846–1856; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mondal P, Ishigami I, Gérard E, Lim C, Yeh S-R, de Visser S, Wijeratne GB, Chem. Sci. 2021, 12, 8872–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Mondal S, Naik PK, Adha JK, Kar S, Coord. Chem. Rev. 2019, 400, 213043–213068; [Google Scholar]; b) Sacramento JJD, Goldberg DP, Acc. Chem. Res. 2018, 51, 2641–2652;30403479 [Google Scholar]; c) Zaragoza JPT, Yosca TH, Siegler MA, Moenne-Loccoz P, Green MT, Goldberg DP, J. Am. Chem. Soc. 2017, 139, 13640–13643;28930448 [Google Scholar]; d) Zaragoza JPT, Siegler MA, Goldberg DP, J. Am. Chem. Soc. 2018, 140, 4380–4390;29542921 [Google Scholar]; e) Zaragoza JPT, Cummins DC, Mubarak MQE, Siegler MA, de Visser SP, Goldberg DP, Inorg. Chem. 2019, 58, 16761–16770;31804814 [Google Scholar]; f) Cummins DC, Alvarado JG, Zaragoza JPT, Effendy Mubarak MQ, Lin YT, de Visser SP, Goldberg DP, Inorg. Chem. 2020, 59, 16053–16064.33047596 [Google Scholar]
- [12].a) Oohora K, Hayashi T, Meth. Enzymol. 2016, 580, 439–454; [DOI] [PubMed] [Google Scholar]; b) Matsuo T, Hayashi A, Abe M, Matsuda T, Hisaeda Y, Hayashi T, J. Am. Chem. Soc. 2009, 131, 15124–15125; [DOI] [PubMed] [Google Scholar]; c) Lemon CM, Marletta MA, Inorg. Chem. 2021, 60, 2716–2729. [DOI] [PubMed] [Google Scholar]
- [13].The UV-vis spectrum for a postulated FeIII(O2)(corrole) was reported: Caulfield KP, Conradie J, Arman HD, Ghosh A, Tonzetich ZJ, Inorg. Chem. 2019, 58, 15225–15235. [DOI] [PubMed] [Google Scholar]
- [14].Milocco F, de Vries F, Siebe HS, Engbers S, Demeshko S, Meyer F, Otten E, Inorg. Chem. 2021, 60, 2045–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Boyd PDW, Buckingham DA, Mcmeeking RF, Mitra S, Inorg. Chem. 1979, 18, 3585–3591. [Google Scholar]
- [16].a) Lang G, Spartalian K, Reed CA, Collman JP, J. Chem. Phys. 1978, 69, 5424–5427; [Google Scholar]; b) Collman JP, Hoard JL, Kim N, Lang G, Reed CA, J. Am. Chem. Soc. 1975, 97, 2676–2681.166106 [Google Scholar]
- [17].a) Chiang CW, Kleespies ST, Stout HD, Meier KK, Li PY, Bominaar EL, Que L Jr., Munck E, Lee WZ, J. Am. Chem. Soc. 2014, 136, 10846–10849; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Munck E, Lipscomb JD, Proc. Natl. Acad. Sci. 2010, 107, 16788–16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Schatz M, Raab V, Foxon SP, Brehm G, Schneider S, Reiher M, Holthausen MC, Sundermeyer J, Schindler S, Angew. Chem. Int. Ed. 2004, 43, 4360–4363; [DOI] [PubMed] [Google Scholar]; b) Burke JM, Kincaid JR, Peters S, Gagne RR, Collman JP, Spiro TG, J. Am. Chem. Soc. 1978, 100, 6083–6088; [DOI] [PubMed] [Google Scholar]; c) Wagner W-D, Paeng IR, Nakamoto K, J. Am. Chem. Soc. 1988, 110, 5565–5567; [DOI] [PubMed] [Google Scholar]; d) Mizutani Y, Hashimoto S, Tatsuno Y, Kitagawa T, J. Am. Chem. Soc. 1990, 112, 6809–6814; [DOI] [PubMed] [Google Scholar]; e) Proniewicz LM, Paeng IR, Nakamoto K, J. Am. Chem. Soc. 1991, 113, 3294–3303. [DOI] [PubMed] [Google Scholar]
- [19].Vogel KM, Kozlowski PM, Zgierski MZ, Spiro TG, J. Am. Chem. Soc. 1999, 121, 9915–9921. [Google Scholar]
- [20].a) Kim E, Helton ME, Wasser IM, Karlin KD, Lu S, Huang HW, Moënne-Loccoz P, Incarvito CD, Rheingold AL, Honecker M, Kaderli S, Zuberbuhler AD, Proc. Natl. Acad. Sci. 2003, 100, 3623–3628; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Das TK, Couture M, Ouellet Y, Guertin M, Rousseau DL, Proc. Natl. Acad. Sci. 2001, 98, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sacramento JJD, Goldberg DP, Chem. Commun. 2020, 56, 3089–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Itakura K, Uchida K, Kawakishi S, Tetrahedron Lett. 1992, 33, 2567–2570. [Google Scholar]
- [23].Raven EL, J. Biol. Inorg. Chem. 2017, 22, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lee JY, Peterson RL, Ohkubo K, Garcia-Bosch I, Himes RA, Woertink J, Moore CD, Solomon EI, Fukuzumi S, Karlin KD, J. Am. Chem. Soc. 2014, 136, 9925–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Sacramento JJD, Goldberg DP, Chem. Commun. 2019, 55, 913–916; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wind ML, Braun-Cula B, Schax F, Herwig C, Limberg C, Isr. J. Chem. 2019, 60, 1057–1060; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bhadra M, Transue WJ, Lim H, Cowley RE, Lee JYC, Siegler MA, Josephs P, Henkel G, Lerch M, Schindler S, Neuba A, Hodgson KO, Hedman B, Solomon EI, Karlin KD, J. Am. Chem. Soc. 2021, 143, 3707–3713; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bailey WD, Dhar D, Cramblitt AC, Tolman WB, J. Am. Chem. Soc. 2019, 141, 5470–5480; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hong S, Sutherlin KD, Park J, Kwon E, Siegler MA, Solomon EI, Nam W, Nat. Commun. 2014, 5, 5440; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Lin YH, Cramer HH, van Gastel M, Tsai YH, Chu CY, Kuo TS, Lee IR, Ye S, Bill E, Lee WZ, Inorg. Chem. 2019, 58, 9756–9765; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Duan PC, Manz DH, Dechert S, Demeshko S, Meyer F, J. Am. Chem. Soc. 2018, 140, 4929–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Gordon JB, Vilbert AC, Siegler MA, Lancaster KM, Moenne-Loccoz P, Goldberg DP, J. Am. Chem. Soc. 2019, 141, 3641–3653; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kindermann N, Gunes CJ, Dechert S, Meyer F, J. Am. Chem. Soc. 2017, 139, 9831–9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].CCDC 2097620 for 1 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.