

Abstract
Objective:
The forkhead box O1 (FOXO1) transcription factor is a key regulator of autophagy. In chondrocytes, reduced FOXO1 expression with aging causes osteoarthritis due to dysfunction of autophagy, but the mechanisms underlying regulation of FOXO1 expression and the reduction in expression with aging remain unclear. We investigated the mechanism by which transforming growth factor β1 (TGFβ1) signaling regulates the FOXO1–autophagy axis.
Methods:
Expression of FOXO1 was measured in chondrocytes after TGFβ1 treatment. Immunohistochemistry was performed to estimate the levels of activin receptor-like kinase 5 (ALK5) and FOXO1 in the knee joints of young, middle-aged and old mice. The effects of the ALK5 inhibitor and SMAD3 or SMAD2 knockdown on FOXO1 expression were evaluated. The role of TGFβ1 in autophagy after hydrogen peroxide (H2O2) treatment was analyzed. The protective effect of TGFβ1 against H2O2 treatment was assessed by cell viability assay and TUNEL assay.
Results:
TGFβ1 promoted the expression of FOXO1 mRNA and protein. Both ALK5 and FOXO1 expression decreased with aging. ALK5 inhibition and SMAD3 knockdown suppressed induction of FOXO1 expression by TGFβ1, whereas SMAD2 knockdown increased it. TGFβ1 promoted the expression of microtubule-associated proteins 1A/1B light chain 3B (LC3)-I protein via the SMAD3–FOXO1 pathway. Furthermore, under H2O2 treatment, TGFβ1 promoted expression of LC3-II. TGFβ1 pretreatment suppressed cell death of chondrocytes following H2O2 treatment, but this protective effect was abolished by FOXO1 knockdown.
Conclusions:
TGFβ1 protects chondrocytes against oxidative stress via the FOXO1–autophagy axis, and a reduction in ALK5 expression might cause reduced FOXO1 expression with aging.
Keywords: Transforming growth factor β1, Activin receptor-like kinase 5, Forkhead box O1, Autophagy, Oxidative stress
Introduction
Aging is a major risk factor for osteoarthritis (OA), and aged chondrocytes exhibit multiple senescent phenotypes1 2 3 4. In particular, dysfunctional autophagy in aged chondrocytes leads to impaired maintenance of cellular homeostasis under oxidative stress5 6. Autophagy prevents oxidative stress–induced cell death by degradation and recycling damaged organelles and proteins7 8 9. Reactivation of autophagy is a promising therapeutic strategy for OA10 11 12 13 14 15. Therefore, it is necessary to further elucidate the mechanism underlying dysfunctional autophagy in OA progression.
Forkhead box O (FOXO) transcription factors play pivotal roles in autophagy-related gene expression and are intimately involved in aging16 17 18 19 20 21. In human chondrocytes, expression of FOXO1 and FOXO3 decreases with aging, and FOXO1 downregulation is associated with reduced expression of LC3 and BECLIN122 23. Furthermore, cartilage-specific Foxo1-knockout mice develop OA at a young age, concomitant with reduced expression of autophagy-related genes24.
The mechanisms underlying regulation of FOXO1 expression and its reduced expression in aged chondrocytes remain unclear. Transforming growth factor β1 (TGFβ1) is a positive regulator of FOXO1 protein expression in human chondrocytes22. In addition, in chondrogenic progenitor cells undergoing chondrogenic differentiation, TGFβ1 promotes the expression and activity of FOXO1 via SMAD3 signaling25. Intriguingly, expression of activin receptor-like kinase 5 (ALK5), a receptor for TGFβ1, also decreases in aged chondrocytes, resulting in OA6 26 27 28.
Hence, we hypothesized that TGFβ1 promotes the expression of FOXO1 through the ALK5-SMAD3 pathway in chondrocytes, and that the reduction in the level of ALK5 might contribute to the reduction in FOXO1 expression with aging. In this study, we investigated the mechanism by which TGFβ1 signaling promotes FOXO1 expression in chondrocytes and assessed the role of TGFβ1 in autophagy.
Method
Cell isolation from human articular cartilage
Human articular cartilage was obtained from 30 donors with OA undergoing knee replacement surgery and providing informed consent with the approval of the Ethics Committee of Kyushu University Hospital. Cartilage was incubated with collagenase (2 mg/ml) in Dulbecco’s Modified Eagle Medium (DMEM)/Ham’s F12 medium (Gibco, Langley, OK, USA) with 10% fetal bovine serum (FBS) (Gibco) for 12 hours. After digestion, chondrocytes were isolated and cultured in 10-cm dishes in DMEM/Ham’s F12 medium with 10% FBS. The chondrocytes were used in experiments after they reached sub-confluence.
Chemical inhibitor treatment
RepSox (Merck Millipore, Billerica, MA, USA) (1 μM) was used to inhibit the activity of ALK529. The inhibitor was dissolved in dimethyl sulfoxide (DMSO), and control groups were incubated with an equivalent amount of DMSO lacking inhibitor.
Chloroquine (CQ) (MBL, Aichi, Japan) (25 μM) was used to assess autophagy flux.
siRNA transfection
Human chondrocytes were seeded in 12-well plates at a density of 1×105 cells/well. After the cells reached sub-confluence, they were transfected with small interfering RNAs (siRNAs) (5 nM) against FOXO1 (siFOXO1), SMAD3 (siSMAD3) or SMAD2 (siSMAD2) (Santa Cruz Biotechnology, Dallas, TX, USA) in Opti-MEM medium (Gibco) for 6 hours using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, culture medium was changed to DMEM/Ham’s F12 with 10% FBS. The control group was transfected with Control siRNA (siCtrl) (Santa Cruz Biotechnology). After 2 days, the cells were used in experiments. The transfection efficiency was evaluated by qRT-PCR and western blotting.
RNA extraction and quantitative real-time RT-PCR (qRT-PCR)
Total RNA was extracted from human chondrocytes using the TRIzol reagent (Thermo Fisher Scientific). Total RNA (100 ng) from each sample was reverse-transcribed to complementary DNA using the PrimeScript RT reagent kit (Takara Bio, Kusatsu, Japan). qRT-PCR was performed on a Light Cycler 2.0 System (F. Hoffmann-La Roche AG, Basel, Switzerland) using TB Green Premix EX TaqⅡ (Takara Bio). The sequences of the forward and reverse primers were shown in Supplementary Table 1. Data were normalized against the corresponding levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or ribosomal protein S18 (RPS18), house keeping genes.
Western blotting
Whole-cell lysates were extracted from human chondrocytes using the Cell Lytic M (Sigma-Aldrich, Saint Louis, MO, USA) with protease inhibitor (cOMplete™ Mini; Sigma-Aldrich) and phosphatase inhibitor (PhosSTOP; Roche Diagnostics, Mannheim, Germany). Nuclear and cytoplasmic extracts were isolated using nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific). Cell lysates were electrophoresed in 4–12% gradient polyacrylamide gels (Thermo Fisher Scientific), and the resolved proteins were transferred to nitrocellulose membranes (Amersham Biosciences, Arlington Heights, IL, USA). Membranes were blocked with blocking buffer (Takara Bio), washed in Tris-buffered saline with Tween (TBST), and incubated with primary antibodies against FOXO1 (#2880; Cell Signaling Technology, Danvers, MA, USA), pFOXO1 (#9461; Cell Signaling Technology), SMAD2/3 (#8685; Cell Signaling Technology), SMAD3 (#9523; Cell Signaling Technology), SMAD2 (#5339; Cell Signaling Technology), pSMAD3 (#9520; Cell Signaling Technology), pSMAD2 (#3108; Cell Signaling Technology), LC3 (#3868; Cell Signaling Technology), GAPDH (#5174; Cell Signaling Technology) diluted 1:1000, and LAMIN A/C (H-110; Santa Cruz Biotechnology) diluted 1:200 in Can Get Signal Immunoreaction Enhancer Solution 1 (TOYOBO, Osaka, Japan). After washing in TBST, secondary anti–rabbit IgG antibodies (#7074; Cell Signaling Technology) diluted 1:1000 in Can Get Signal Immunoreaction Enhancer Solution 2 (TOYOBO, Osaka, Japan) were added. Immunoreactivity was detected with ECL Prime (Amersham Biosciences) and photographed on an Ez Capture MG (ATTO, Tokyo, Japan). Band densities were calculated using CS Analyzer 3.0 (ATTO).
Mice
C57BL/6J male mice were used in the experiments. Mice were housed in a specific pathogen-free condition with freedom to access to food and water. Young, middle-aged and old knee joints were collected from the hind legs of 3-, 13- and 20-month-old mice, respectively (n=5 per group). All animal experiments were approved by the Animal Experiment Committee of Kyushu University. Experiments were performed under the rules of our institution.
Immunohistochemistry
Knee joints from mice were fixed with 4% paraformaldehyde (PFA) (Wako Pure Chemical Industries) for 2 days, decalcified with K-CX (FALMA, Tokyo, Japan) for 2 days, and embedded in paraffin. Sections were cut into 4 μm–thick sections, deparaffinized, and rehydrated; antigen retrieval was performed by incubation overnight with ethylenediaminetetraacetic acid (EDTA) (1 mM) at pH 8.0. Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxidase (H2O2) in methanol for 30 minutes. After blocking with normal horse serum (Vectastain Universal Elite ABC kit; Vector Laboratories, Burlingame, CA, USA) for 30 minutes, sections were incubated with antibodies against ALK5 (sc-398; Santa Cruz Biotechnology), FOXO1 (#2880; Cell Signaling Technology) and normal rabbit IgG (AB-105-C; R&D Systems, Minneapolis, MN, USA) for 1 hour. Sections were incubated with biotinylated secondary antibodies for 30 minutes, followed by incubation with streptavidin–peroxidase complex (Vectastain Universal Elite ABC kit) for 30 minutes. Antibody complexes were visualized using the diaminobenzidine substrate system (Wako Pure Chemical Industries), and counterstained with hematoxylin. The percentage of cells positive for ALK5 and FOXO1 was determined using the BZ-II Analyzer software (Keyence, Osaka, Japan).
Immunofluorescence
Sections were blocked with 5% goat serum (Wako Pure Chemical Industries) and 0.3% Triton X-100 (Sigma-Aldrich) for 1 hour, and then incubated with antibodies against ALK5 (sc-398; Santa Cruz Biotechnology) and FOXO1 (#2880; Cell Signaling Technology) for 1 hour. Subsequently, the sections were washed three times with PBS and incubated with Alexa Fluor 488 (A11008; Thermo Fisher Scientific) and 568 (A11004; Thermo Fisher Scientific) for 1 hour. SlowFade diamond antifade mountant with DAPI (Thermo Fisher Scientific) was used for nuclear staining. Immunostaining was visualized by fluorescence microscopy (BZ-X700; Keyence).
Immunocytochemistry
Human chondrocytes were seeded in 8-well chamber slides at a density of 2×104 cells/well. The cells were fixed with 4% PFA for 10 min at room temperature for antibodies against FOXO1 (#14952; Cell Signaling Technology), SMAD2/3 (#8685; Cell Signaling Technology), SMAD3 (#9523; Cell Signaling Technology), SMAD2 (#5339; Cell Signaling Technology) or ice-cold 100% methanol for 15 min at −20°C for antibody against LC3 (#3868; Cell Signaling Technology), and then blocked with 5% goat serum and 0.3% Triton X-100 for 1 hour. Subsequently, the cells were incubated with each primary antibody for 1 hour. The cells were washed three times with PBS and incubated with Alexa Fluor 488 (A11008; Thermo Fisher Scientific) and 568 (A11004; Thermo Fisher Scientific) for 1 hour.
MTT assay
Human chondrocytes were seeded in 96-well plates at a density of 0.6×104 cells/well and transfected with siCtrl or siFOXO1. After incubation for 3 hours with recombinant human TGFβ1 (R&D Systems, Minneapolis, MN, USA), H2O2 was added to each well to a final concentration of 0, 100, or 250 μM. After 48 hours, 25 μl of MTT (5mg/ml) was added to each well. Three hours later, supernatant was aspirated, and 200 μl of MTT solvent (4 mM HCl in isopropanol) was added. Optical densities were measured at 595 nm using an iMark microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). Data are expressed as the percent absorbance of cells in each group relative to cells transfected with siCtrl, and not treated with TGFβ1 and H2O2.
ATP assay
Human chondrocytes were seeded in 96-well plates at a density of 0.6×104 cells/well and transfected with siCtrl or siFOXO1. After incubation for 3 hours with TGFβ1, H2O2 was added to each well to a final concentration of 0, 100, or 250 μM. After 48 hours, ATP production was evaluated using CellTiter-Glo (Promega). Data are expressed as the percent luminescence of cells in each group relative to cells transfected with siCtrl, and not treated with TGFβ1 and H2O2.
TUNEL assay
TUNEL assay was performed using the in situ Apoptosis Detection kit (Takara Bio). Human chondrocytes were seeded in 8-well chamber slides at a density of 4×104 cells/well and transfected with siCtrl or siFOXO1. After incubation for 3 hours with TGFβ1, H2O2 (0 or 250 μM) was added to each well. After 48 hours, the cells were fixed with 4% PFA for 15 min at room temperature, and then incubated with terminal deoxynucleotidyl transferase buffer for 1hour at 37°C. The percentage of apoptotic cells was determined using the BZ-II Analyzer software (Keyence).
Statistical analysis
All experiments were repeated with at least five samples from independent donors. Data are presented as dot plots with means ± S.D. In each experiment, the effect size and the power were calculated. For in vivo experiment, a sample size n=5 per group provided 82% power, given an α level of 0.05, to detect the difference in means of the percentage of ALK5 positive cells between 48.3 ± 9.5% in 13-month-old group and 31.1 ± 6.9% in 20-month-old group. Similarly, all experiments provided >80% power. Wilcoxon’s rank–sum test was used for two-group comparisons. After normality was confirmed by the Shapiro-Wilk test, multiple comparisons were evaluated by one-way repeated measures ANOVA with the Tukey–Kramer post hoc test. All data analyses were performed using the JMP 14 statistical software (SAS Institute, Cary, NC, USA). P<0.05 was considered statistically significant.
Results
TGFβ1 promotes FOXO1 expression in human chondrocytes
First, we investigated whether TGFβ1 treatment promotes the expression of FOXO1, FOXO3, and FOXO4 in human chondrocytes. Among the three genes, only FOXO1 expression was promoted by TGFβ1 treatment for 24 hours (5.22-fold increase). Conversely, FOXO3 expression was reduced after TGFβ1 treatment (1.82-fold decrease). FOXO4 expression was unchanged (Fig. 1A). The reproducibility of the result in Fig. 1A was confirmed by RPS18, another housekeeping gene (Fig. 1B). Western blotting also revealed that expression of total FOXO1 protein significantly increased after TGFβ1 treatment for more than 8 hours (Fig. 1C). The activity of FOXO1 is negatively regulated by phosphorylation at Ser-25630. For example, FOXO1 is phosphorylated by nutrient load (Fig. S1). By contrast, the level of phosphorylated FOXO1 (pFOXO1) was not changed after incubation with TGFβ1 (Fig. 1C). In addition, we performed immunocytochemistry to confirm the localization of FOXO1. After incubation with TGFβ1 for 24 hours, the level of FOXO1 protein significantly increased in the nucleus (Fig. 1D).
Fig. 1. TGFβ1 promotes FOXO1 expression in human chondrocytes.
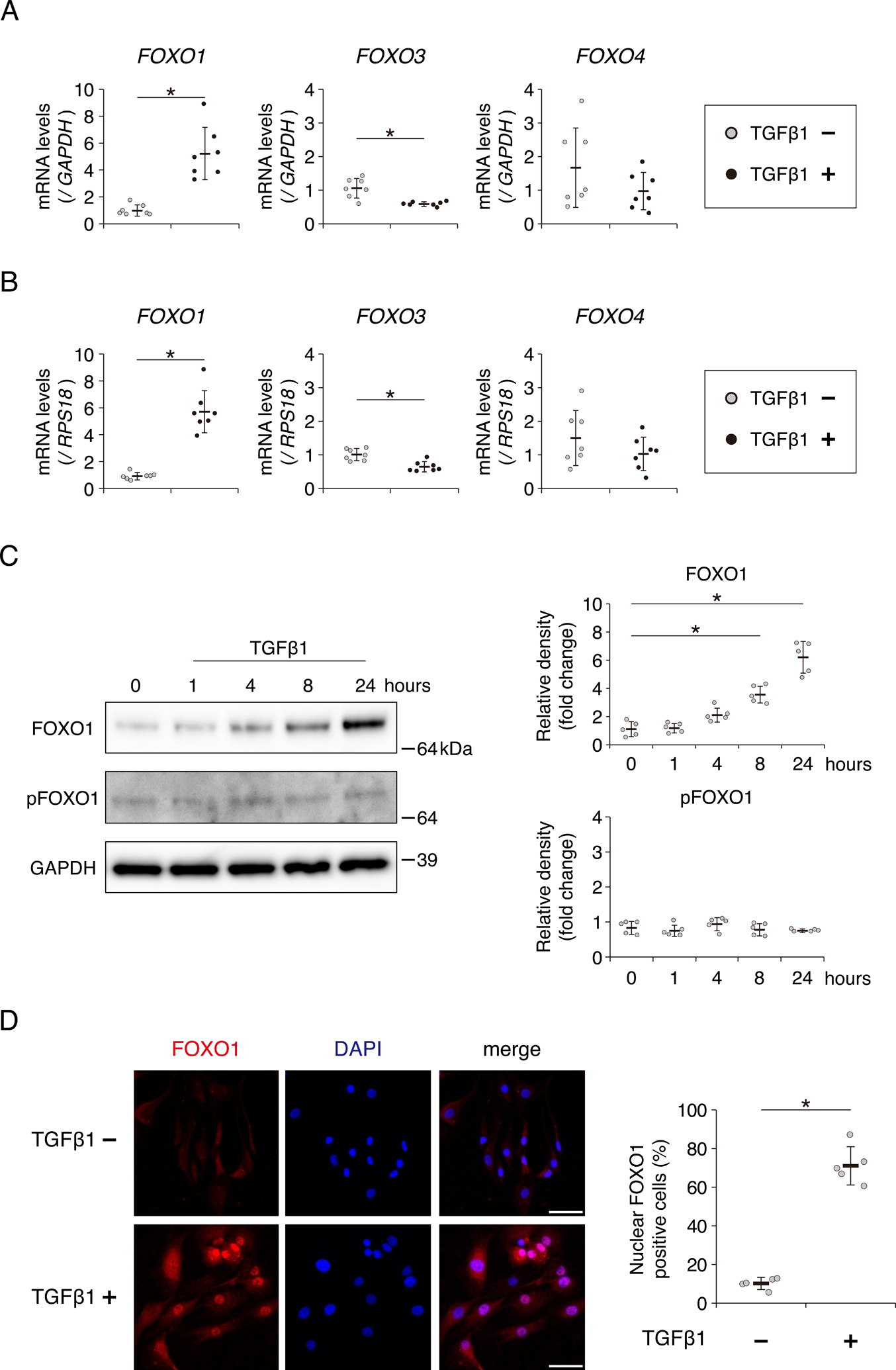
Relative mRNA levels of FOXO1, FOXO3, and FOXO4 after incubation with TGFβ1 (10 ng/ml) for 24 hours, as determined by qRT-PCR using the primer of GAPDH (A) or RPS18 (B), house keeping genes. Gene expression is shown relative to the level in cells incubated without TGFβ1; n = 7 independent experiments. (C) Levels of total FOXO1 and phosphorylated FOXO1 (pFOXO1) at Ser-256 after incubation with TGFβ1 (10 ng/ml) for 1, 4, 8, or 24 hours, as determined by western blotting. GAPDH was used as a loading control. Graphs show levels of total FOXO1 and pFOXO1, relative to the corresponding levels in cells incubated without TGFβ1; n = 5 independent experiments. (D) FOXO1 localization after incubation with TGFβ1 (10 ng/ml) for 24 hours, visualized by immunocytochemistry using anti-FOXO1 antibody (red staining). Nuclei were detected with DAPI (blue staining). Bars, 50 μm. Graph shows the percentage of nuclear FOXO1 positive cells; n = 5 independent experiments. Data are presented as dots showing individual values and as means ± S.D. Statistical analysis in (A), (B) and (D) was performed using Wilcoxon’s rank–sum test. Statistical analysis in (C) was performed using one-way repeated measures ANOVA with the Tukey–Kramer post hoc test. *P < 0.05.
ALK5 and FOXO1 levels decrease in aged mouse joints
To assess whether both ALK5 and FOXO1 expression decrease with aging, we investigated the levels of ALK5 and FOXO1 in young (3 months old, 3M), middle-aged (13 months old, 13M), and old (20 months old, 20M) mouse knee joints. ALK5 was strongly expressed in chondrocytes of young joints at 3M, whereas its expression was significantly reduced with aging (Fig. 2A and B). Similarly, levels of FOXO decreased with aging (Fig. 2A and B). In 13M mouse cartilage, the distribution pattern of FOXO1 expression was similar to ALK5 expression (Fig. 2C).
Fig. 2. Expression of ALK5 and FOXO1 decrease with aging in mouse cartilage.
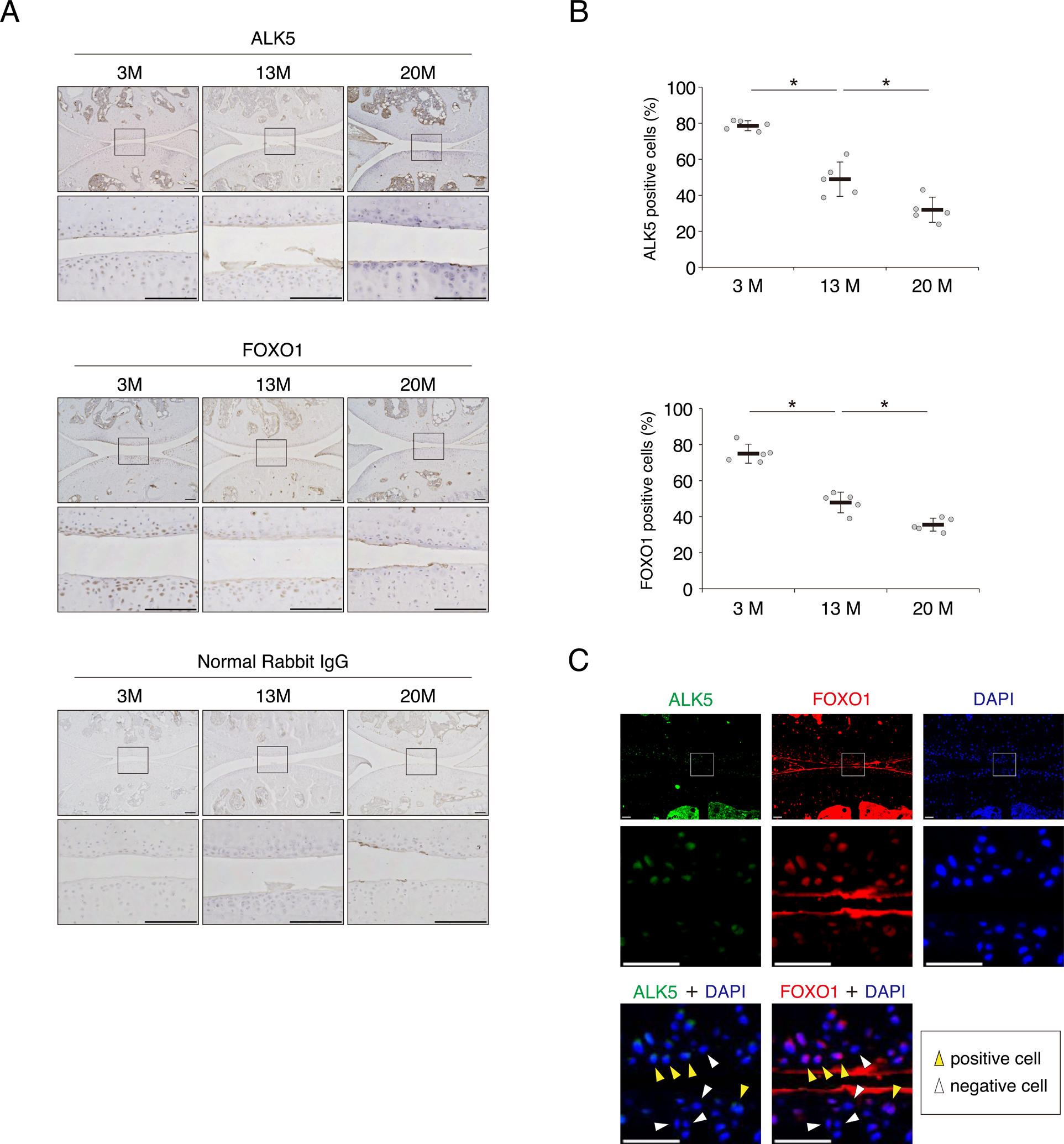
Knee joints were collected from the hind legs of 3-month-old (3M) young mice, 13-month-old (13M) middle-aged mice or 20-month-old (20M) old mice. (A) Representative images of ALK5 and FOXO1, detected by immunohistochemistry. Sections stained with normal rabbit IgG are shown as negative controls. Bottom images in each group show higher-magnification views corresponding to the boxed areas in the top images. Bars, 100 μm. (B) Quantification of ALK5 and FOXO1 positive cells; n = 5 mice per group. (C) Co-localization of ALK5 and FOXO1 in 13M mouse knee joint were visualized by immunofluorescence. ALK5 is shown in green, and FOXO1 is shown in red. Nuclei were detected with DAPI (blue staining). Middle and bottom images show higher-magnification views corresponding to the boxed areas in the top images. Yellow arrows indicate both ALK5 and FOXO1 positive cells, and white arrows indicate both ALK5 and FOXO1 negative cells. Bars, 100 μm. Data are presented as dots showing individual values and as means ± S.D. Statistical analysis was performed using one-way repeated measures ANOVA with the Tukey–Kramer post hoc test. *P < 0.05.
Inhibition of ALK5 suppresses FOXO1 expression induced by TGFβ1
To clarify the effect of ALK5 inhibition on the induction of FOXO1 expression by TGFβ1 in vitro, we incubated human chondrocytes with RepSox, a specific inhibitor of ALK5. Inhibition of ALK5 significantly suppressed the expression of FOXO1 after TGFβ1 treatment (Fig. 3A). Western blotting revealed that induction of FOXO1 protein by TGFβ1 was suppressed by RepSox; however, the level of pFOXO1 was not affected (Fig. 3B). FOXO1 protein both in the cytoplasm and nucleus significantly increased upon incubation with TGFβ1. The induction of FOXO1 in the cytoplasm and nucleus by TGFβ1 was completely suppressed by RepSox (Fig. 3C). Inhibition of ALK5 suppressed the phosphorylation of SMAD2 and SMAD3 after TGFβ1 treatment for 30 min (Fig. 3D). Consistently, nuclear translocation of SMAD2 and SMAD3 was induced after TGFβ1 treatment, but inhibited by RepSox (Fig. 3E). In immunocytochemistry, the localization of both SMAD2/3 and FOXO1 protein to the nucleus following TGFβ1 treatment were suppressed by RepSox (Fig. 3F).
Fig. 3. ALK5 inhibition suppresses induction of FOXO1 expression by TGFβ1 in human chondrocytes.
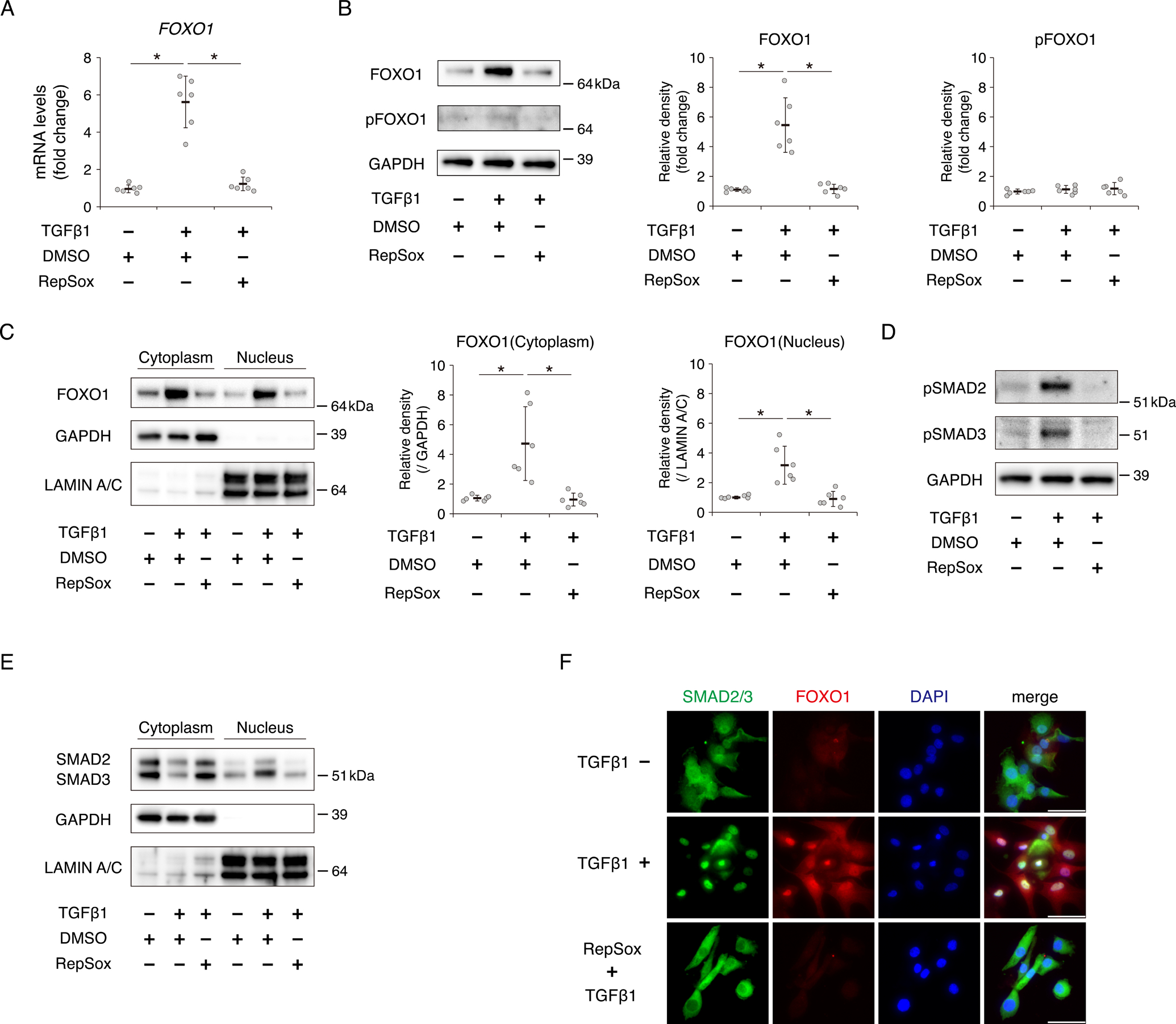
Chondrocytes were incubated with or without TGFβ1 (10 ng/ml) for 24 hours after pretreatment with or without RepSox (1 μM) for 3 hours. (A) Relative mRNA levels of FOXO1 were measured by qRT-PCR. Gene expression at each stage is expressed relative to the level in cells incubated without TGFβ1 and RepSox; n = 6 independent experiments. (B) Levels of total FOXO1 and pFOXO1 were analyzed by western blotting. GAPDH was used as a loading control. Graphs show total FOXO1 protein level and pFOXO1 protein level, relative to the corresponding levels in cells incubated without TGFβ1 and RepSox; n = 6 independent experiments. (C) Levels of cytoplasmic FOXO1 and nuclear FOXO1 were analyzed by western blotting. GAPDH was used as a loading control for cytoplasmic extracts. LAMIN A/C was used as a loading control for nuclear extracts. Graphs show cytoplasmic FOXO1 protein level and nuclear FOXO1 protein level, relative to the corresponding levels in cells incubated without TGFβ1 and RepSox; n = 6 independent experiments. (D) Levels of phosphorylated SMAD2 (pSMAD2) at Ser-465/467 and phosphorylated SMAD3 (pSMAD3) at Ser-423/425 in chondrocytes incubated with or without TGFβ1 (10 ng/ml) for 30 min after pretreatment with or without RepSox (1 μM) for 3 hours, as determined by western blotting. (E) Levels of cytoplasmic SMAD2/3 and nuclear SMAD2/3 in chondrocytes incubated with or without TGFβ1 (10 ng/ml) for 30 min after pretreatment with or without RepSox (1 μM) for 3 hours, as determined by western blotting. (F) SMAD2/3 and FOXO1 localizations in chondrocytes incubated with or without TGFβ1 (10 ng/ml) for 24 hours after pretreatment with or without RepSox (1 μM) for 3 hours were visualized by immunocytochemistry. SMAD2/3 is shown in green, and FOXO1 is shown in red. Nuclei were detected with DAPI (blue staining). Bars, 50 μm. Data are presented as dots showing individual values and as means ± S.D. Statistical analysis was performed using one-way repeated measures ANOVA with the Tukey–Kramer post hoc test. *P < 0.05.
FOXO1 expression via TGFβ1 is mediated by SMAD3 and repressed by SMAD2
To verify the discrete effect of SMAD3 and SMAD2 on the expression of FOXO1 by TGFβ1, we transfected human chondrocytes with siSMAD3 or siSMAD2. Gene and protein expression of SMAD3 significantly decreased after TGFβ1 treatment for 24 hours, and were further suppressed by siSMAD3 (Fig. 4A and C). SMAD3 knockdown decreased expression of FOXO1 at mRNA and protein level after TGFβ1 treatment (Fig. 4B and C). In immunocytochemistry, SMAD3 knockdown in the nucleus with siSMAD3 suppressed the induction of FOXO1 protein by TGFβ1 (Fig. 4D). These findings were consistent with the effect of ALK5 inhibition by RepSox.
Fig. 4. FOXO1 expression via TGFβ1 is mediated by SMAD3 and repressed by SMAD2.
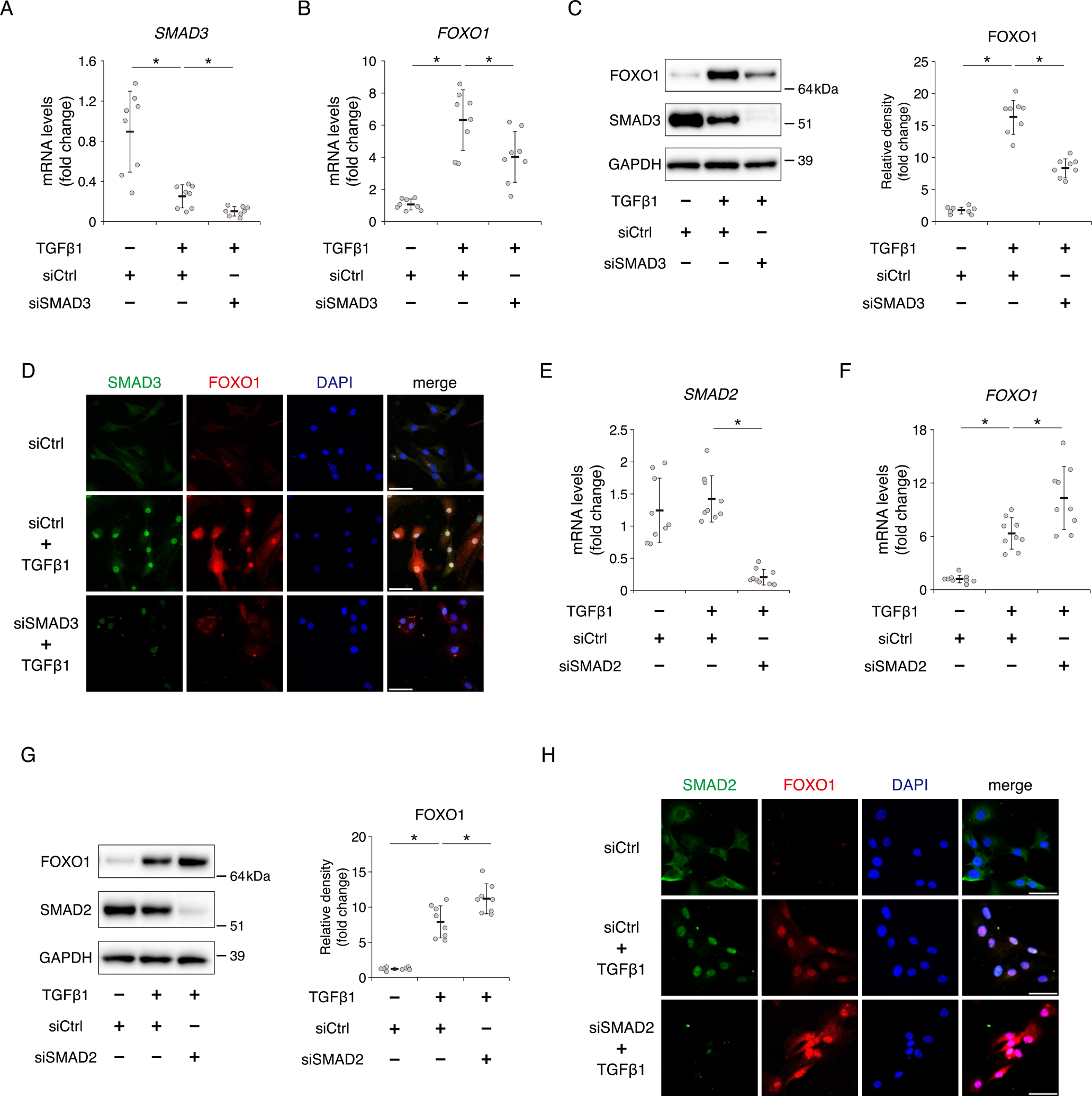
Chondrocytes were incubated with or without TGFβ1 (10 ng/ml) for 24 hours after transfection with siCtrl or siSMAD3. Relative SMAD3 (A) and FOXO1 (B) mRNA levels were measured by qRT-PCR. Gene expression at each stage is expressed relative to the level in cells transfected with siCtrl and incubated without TGFβ1; n = 8 independent experiments. (C) Levels of total FOXO1 and SMAD3 were analyzed by western blotting. GAPDH was used as a loading control. Graphs show total levels of FOXO1, relative to the corresponding levels in cells transfected with siCtrl and incubated without TGFβ1; n = 8 independent experiments. (D) SMAD3 and FOXO1 proteins were visualized by immunocytochemistry. SMAD3 is shown in green, and FOXO1 is shown in red. Nuclei were detected with DAPI (blue staining). Bars, 50 μm. Chondrocytes were incubated with or without TGFβ1 (10 ng/ml) for 24 hours after transfection with siCtrl or siSMAD2. Relative SMAD2 (E) and FOXO1 (F) mRNA levels were measured by qRT-PCR. Gene expression at each stage is expressed relative to the level in cells transfected with siCtrl and incubated without TGFβ1; n = 9 independent experiments. (G) Levels of total FOXO1 and SMAD2 were analyzed by western blotting. Graphs show total levels of FOXO1, relative to the corresponding levels in cells transfected with siCtrl and incubated without TGFβ1; n = 8 independent experiments. (H) SMAD2 and FOXO1 proteins were visualized by immunocytochemistry. SMAD2 is shown in green, and FOXO1 is shown in red. Bars, 50 μm. Data are presented as dots showing individual values and as means ± S.D. Statistical analysis was performed using one-way repeated measures ANOVA with the Tukey–Kramer post hoc test. *P < 0.05.
On the other hand, TGFβ1 treatment did not affect the gene and protein expression of SMAD2 (Fig. 4E and G). In contrast to SMAD3, SMAD2 knockdown increased expression of FOXO1 at mRNA and protein level after TGFβ1 treatment (Fig. 4F and G). In immunocytochemistry, SMAD2 knockdown in the nucleus with siSMAD2 increased the induction of FOXO1 protein by TGFβ1 (Fig. 4H).
TGFβ1 promotes autophagy under oxidative stress through the SMAD3-FOXO1 pathway
We next investigated whether TGFβ1 can activate autophagy in human chondrocytes. First, we evaluated the effect of TGFβ1 on the expression of autophagy-related genes. Expression of autophagy related 3 (ATG3) and microtubule associated protein 1 light chain 3 beta (MAP1LC3B) increased after incubation with TGFβ1 (Fig. 5A). MAP1LC3B encodes LC3, which has two forms: LC3-I and LC3-II. When autophagy is activated, LC3-I is converted to LC3-II, a phosphatidylethanolamine (PE)-conjugated form31. In western blotting, TGFβ1 increased the expression of LC3-I, but had no effect on LC3-II (Fig. 5B). To further evaluate autophagy flux after TGFβ1 treatment, chondrocytes were incubated with chloroquine (CQ) for 2 hours after TGFβ1 pretreatment for 22 hours. In this condition, TGFβ1 also increased the expression of LC3-I, but did not affect LC3-II (Fig. 5C). Next, we assessed the effect of TGFβ1 on the activity of autophagy under oxidative stress. H2O2 treatment increased the expression of LC3-II relative to cells not exposed H2O2 (Fig. 5B and D). Furthermore, expression of LC3-II was higher in chondrocytes pretreated with TGFβ1 than in cells not pretreated with TGFβ1 as well as LC3-I (Fig. 5D). In chondrocytes incubated with H2O2 and CQ, TGFβ1 pretreatment also increased the expression of LC3-I and LC3-II (Fig. 5E). By immunocytochemistry, we assessed the level of LC3 puncta (dots) after incubation with H2O2. The number of LC3 dots increased following exposure to H2O2, and TGFβ1 pretreatment increased the number of LC3 dots under oxidative stress relative to cells not pretreated with TGFβ1 (Fig. 5F). To confirm that the effect of TGFβ1 on the regulation of LC3 protein expression was mediated by the SMAD3–FOXO1 pathway, we transfected chondrocytes with siSMAD3 or siFOXO1. FOXO1 knockdown decreased the induction of LC3-I protein by TGFβ1 (Fig. 5G). SMAD3 knockdown also decreased the induction of FOXO1 and LC-3I protein by TGFβ1 (Fig. 5G). Furthermore, following exposure to H2O2, knockdown of either FOXO1 or SMAD3 suppressed the induction of LC3-I and LC3-II by TGFβ1 pretreatment (Fig. 5H).
Fig. 5. TGFβ1 promotes autophagic activity under oxidative stress through the SMAD3–FOXO1 pathway.
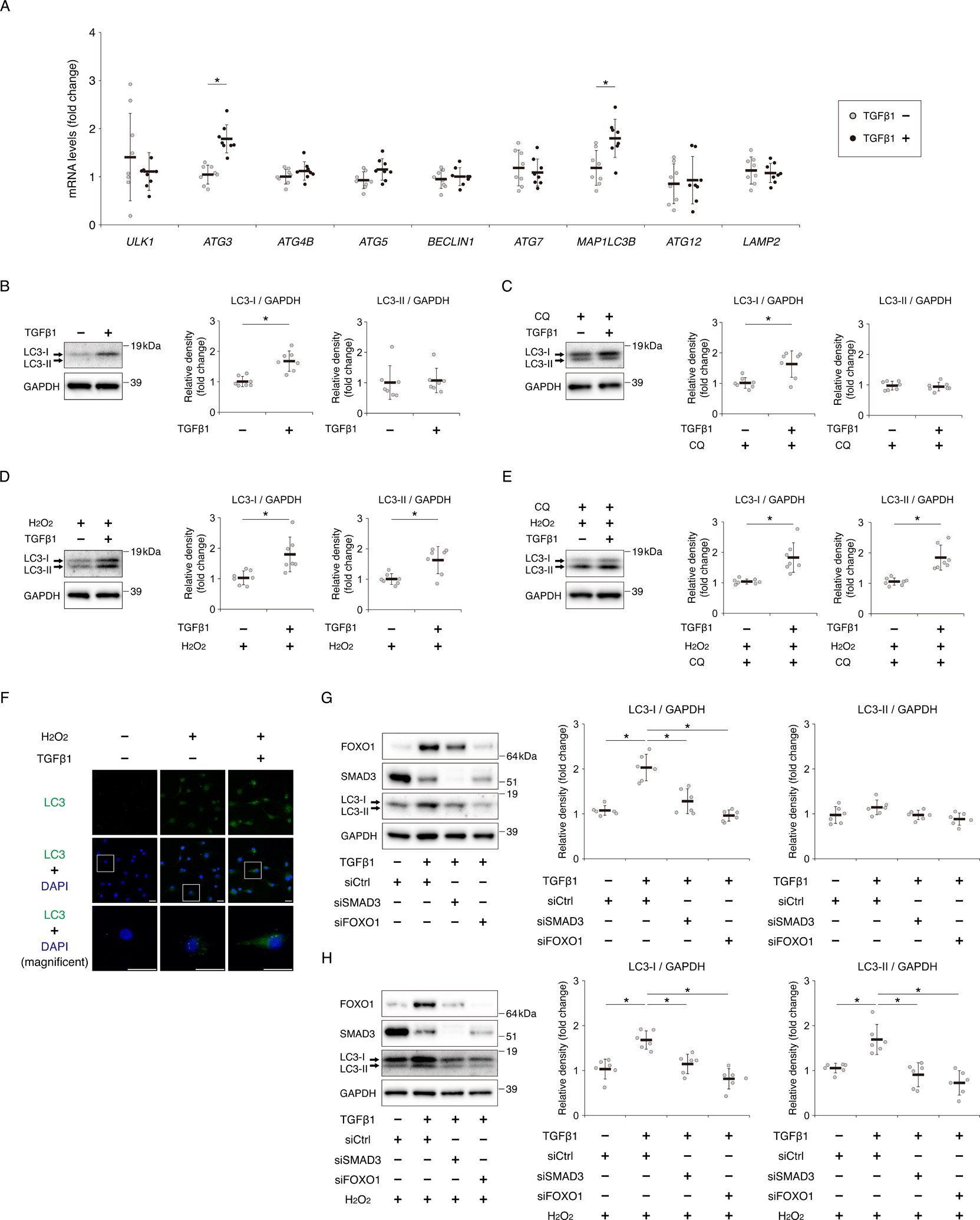
(A) Relative mRNA levels of ULK1, ATG3, ATG4B, ATG5, BECLIN1, ATG7, MAP1LC3B, ATG12, and LAMP2 in human chondrocytes after incubation with TGFβ1 (10 ng/ml) for 24 hours, as determined by qRT-PCR. Gene expression is shown relative to the level in cells incubated without TGFβ1; n = 8 independent experiments. (B) Levels of LC3-I and LC3-II after incubation with TGFβ1 (10 ng/ml) for 24 hours, as determined by western blotting. GAPDH was used as a loading control. Graphs show levels of LC3-I and LC3-II, relative to the corresponding levels in cells incubated without TGFβ1; n = 7 independent experiments. (C) Levels of LC3-I and LC3-II in chondrocytes incubated with chloroquine (CQ) (25 μM) for 2 hours after pretreatment with or without TGFβ1(10 ng/ml) for 22 hours, as determined by western blotting. Graphs show levels of LC3-I and LC3-II, relative to the corresponding levels in cells incubated without TGFβ1; n = 7 independent experiments. (D) Levels of LC3-I and LC3-II in chondrocytes exposed to H2O2 (500 μM) for 2 hours after pretreatment with or without TGFβ1(10 ng/ml) for 22 hours, as determined by western blotting. Graphs show levels of LC3-I and LC3-II, relative to the corresponding levels in cells pretreated without TGFβ1; n = 7 independent experiments. (E) Levels of LC3-I and LC3-II in chondrocytes incubated with CQ (25 μM) and H2O2 (500 μM) for 2 hours after pretreatment with or without TGFβ1(10 ng/ml) for 22 hours, as determined by western blotting. Graphs show levels of LC3-I and LC3-II, relative to the corresponding levels in cells incubated without TGFβ1; n = 7 independent experiments. (F) LC3 proteins in chondrocytes incubated with or without H2O2 (500 μM) for 2 hours after pretreatment with or without TGFβ1 (10 ng/ml) for 22 hours were visualized by immunocytochemistry. LC3 is shown as green dots. Nuclei were detected with DAPI (blue staining). Bottom images show higher-magnification views corresponding to the boxed areas in the middle images. Bars, 30 μm. (G) Levels of FOXO1, SMAD3, LC3-I, and LC3-II, as determined by western blotting, in chondrocytes transfected with siCtrl, siSMAD3, or siFOXO1 after incubation with or without TGFβ1 (10 ng/ml) for 24 hours. Graphs show total levels of LC3-I and LC3-II, relative to the corresponding levels in cells transfected with siCtrl and incubated without TGFβ1; n = 6 independent experiments. (H) Levels of FOXO1, SMAD3, LC3-I, and LC3-II, as determined by western blotting, in chondrocytes transfected with siCtrl, siSMAD3, or siFOXO1 following exposure to H2O2 (500 μM) for 2 hours after pretreatment with or without TGFβ1 (10 ng/ml) for 22 hours. Graphs show total levels of LC3-I and LC3-II, relative to the corresponding levels in cells transfected with siCtrl and incubated without TGFβ1; n = 6 independent experiments. Data are presented as dots showing individual values and as means ± S.D. Statistical analysis in (A), (B), (C), (D) and (E) was performed using Wilcoxon’s rank–sum test. Statistical analysis in (G) and (H) was performed using one-way repeated measures ANOVA with the Tukey–Kramer post hoc test. *P < 0.05.
TGFβ1 protects chondrocytes against oxidative stress–induced cell death via FOXO1
To investigate the protective effect of TGFβ1 against cell death induced by oxidative stress via FOXO1, we assessed the cell viability after exposure to H2O2. After pretreatment with or without TGFβ1 in chondrocytes transfected with siCtrl or siFOXO1, the cells were exposed to H2O2. In MTT assay, exposure to H2O2 decreased cell viability in a dose-dependent manner relative to cells not exposed to H2O2, and in chondrocytes transfected with siCtrl, TGFβ1 pretreatment suppressed the decrease in cell viability following exposure to H2O2 (Fig. 6A). On the other hand, in chondrocytes transfected with siFOXO1, the protective effect of TGFβ1 against exposure to H2O2 was abolished (Fig. 6A). ATP assay revealed that ATP production was decreased by FOXO1 knockdown (Fig. 6B). TGFβ1 pretreatment suppressed the decrease of ATP production following exposure to H2O2, whereas in cells transfected with siFOXO1, the decrease of ATP production was not suppressed by TGFβ1 pretreatment (Fig. 6B). Consistently, in TUNEL assay, TGFβ1 suppressed cell apoptosis against exposure to H2O2 via FOXO1 (Fig. 6C).
Fig. 6. TGFβ1 protects chondrocytes against oxidative stress–induced cell death via FOXO1.
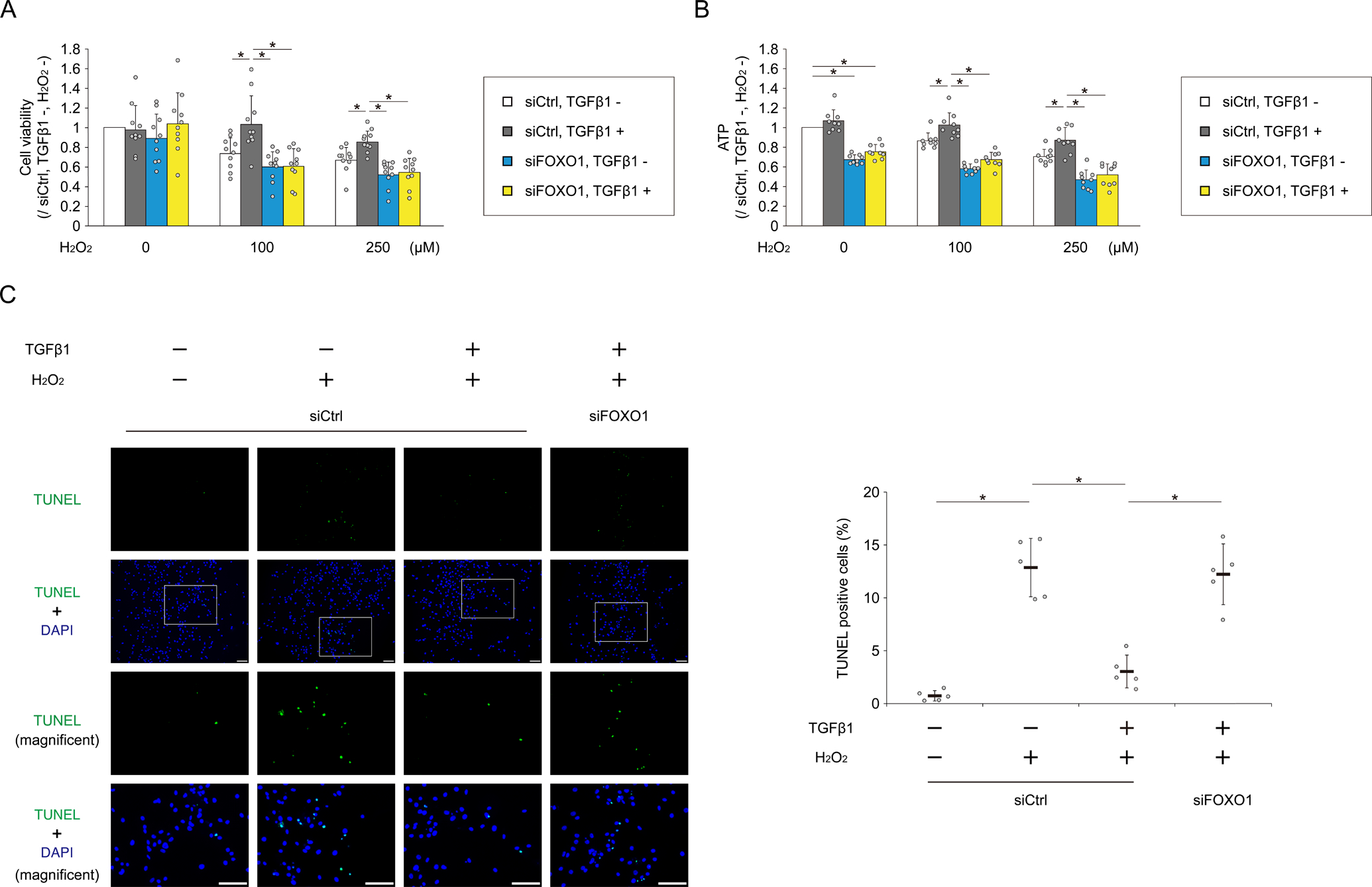
Human chondrocytes were transfected with siCtrl or siFOXO1. After incubation with or without TGFβ1 for 3 hours, H2O2 was added to each well to a final concentration of 0, 100, or 250 μM for 48 hours. (A) Cell viability was analyzed by MTT assay; n = 10 independent experiments. (B) ATP production was analyzed using the CellTiter-Glo assay; n = 9 independent experiments. Data are expressed as the percent absorbance (A) or luminescence (B) of cells in each group relative to cells transfected with siCtrl, and not treated with TGFβ1 and H2O2. Human chondrocytes were transfected with siCtrl or siFOXO1. After incubation with or without TGFβ1 for 3 hours, H2O2 (0 or 250 μM) was added to each well. After 48 hours, cell apoptosis was analyzed by TUNEL assay (C). Higher-magnification views are corresponding to the white boxed areas in each group. Graph shows the percentage of TUNEL positive cells; n = 5 independent experiments. Data are presented as dots showing individual values and as means ± S.D. Statistical analysis was performed using one-way repeated measures ANOVA with the Tukey–Kramer post hoc test. *P < 0.05.
Discussion
In this study, we clarified the mechanism by which TGFβ1 signaling promotes FOXO1 expression in chondrocytes. It is well known that the activity of FOXO proteins is strictly regulated by post-translational modifications such as phosphorylation and acetylation32 33. In particular, insulin and insulin-like growth factor I (IGF-I) are the main negative regulators of FOXO proteins, and promote phosphorylation of FOXO protein through the phosphatidylinositol 3-kinase (PI3K)/AKT pathway34. Phosphorylated FOXO proteins are exported from the nucleus to the cytoplasm, followed by cytoplasmic retention mediated by binding to 14-3-3 proteins or ubiquitination-mediated degradation35 36. However, a previous report revealed that in chondrocytes, IGF-I did not affect either the expression or the phosphorylation of FOXO proteins22. In addition, that report demonstrated that TGFβ1 promoted expression of the FOXO1 protein. These previous findings motivated us to investigate the mechanistic details of FOXO1 regulation by TGFβ1 signaling in chondrocytes.
We first showed that only FOXO1 mRNA expression was promoted by TGFβ1 treatment, whereas FOXO3 and FOXO4 were not promoted. In addition, we showed that TGFβ1 promoted the expression of the FOXO1 protein without affecting phosphorylation at Ser-256, a key site regulated by the PI3K/AKT pathway30. Considering the result that TGFβ1 promoted the expression of cytoplasmic FOXO1 protein, our findings indicate that TGFβ1 promotes the expression of FOXO1 at a transcriptional level, rather than via post-translational modifications. Little is known about the regulation of FOXO at the transcriptional level, although a few studies have described the effects of transcription factors such as E2F1 and Forkhead box C1 on the expression of FOXO genes37 38. Furthermore, although previous studies described the regulation of FOXOs through TGFβ1 signaling in some cell types, they reported that TGFβ1 promoted the expression of FOXO proteins by inhibiting cytoplasmic translocation and did not refer to the expression of FOXO genes39 40. Our findings demonstrate the potential of TGFβ1 to promote the expression of FOXO1 genes in chondrocytes. In addition, we revealed that TGFβ1 promotes the localization of FOXO1 to the nucleus. This indicates that TGFβ1 promotes not only the expression of FOXO1, but also activation of FOXO1 by nuclear translocation.
Next, we confirmed the decreases in ALK5 and FOXO1 levels in aged chondrocytes in vivo. In addition, we showed that inhibition of ALK5 suppressed the induction of FOXO1 expression by TGFβ1 in vitro. In the canonical pathway, after binding with TGFβ1, ALK5 phosphorylates SMAD2 and SMAD3, which form a complex with SMAD4. The complex translocates to the nuclei and regulates their target genes41. In our results, although the nuclear translocations of SMAD3 and SMAD2 were induced via the TGFβ1-ALK5 pathway, the inverse impact of siRNA between SMAD3 and SMAD2 on the expression of FOXO1 was observed. SMAD3 knockdown inhibited the induction of FOXO1 expression by TGFβ1, whereas SMAD2 knockdown promoted TGFβ1-induced FOXO1 expression. Recent studies showed that SMAD3 and SMAD2 have distinct roles. In dendritic cells, the TGFβ1-SMAD3 pathway positively regulates TGFβ1 autoinduction, whereas SMAD2 negatively regulates TGFβ1 expression42. In pancreatic ductal adenocarcinoma cells, SMAD3 mediates the inhibition of cell growth and migratory response by TGFβ1, whereas SMAD2 induces cell growth and migration43. The regulatory mechanism of FOXO1 expression by TGFβ1 in chondrocytes is similar to the results of these reports. Our findings suggest that TGFβ1 promotes the expression of FOXO1 through the ALK5-SMAD3 pathway, whereas SMAD2 negatively regulate FOXO1 expression (Fig. 7A), and a decrease in ALK5 expression suppresses the activity of SMAD3 signaling, resulting in the reduction of FOXO1 expression in aged chondrocytes (Fig. 7B). Further studies will provide more information on the regulatory mechanism of FOXO1 expression by TGFβ1 signaling. For example, the observation of mice younger than 13 months old will determine the detailed time point at which the expression of ALK5 and FOXO1 begins to decline.
Fig. 7. Schematic model of the regulation of the FOXO1–autophagy axis by TGFβ1 signaling.
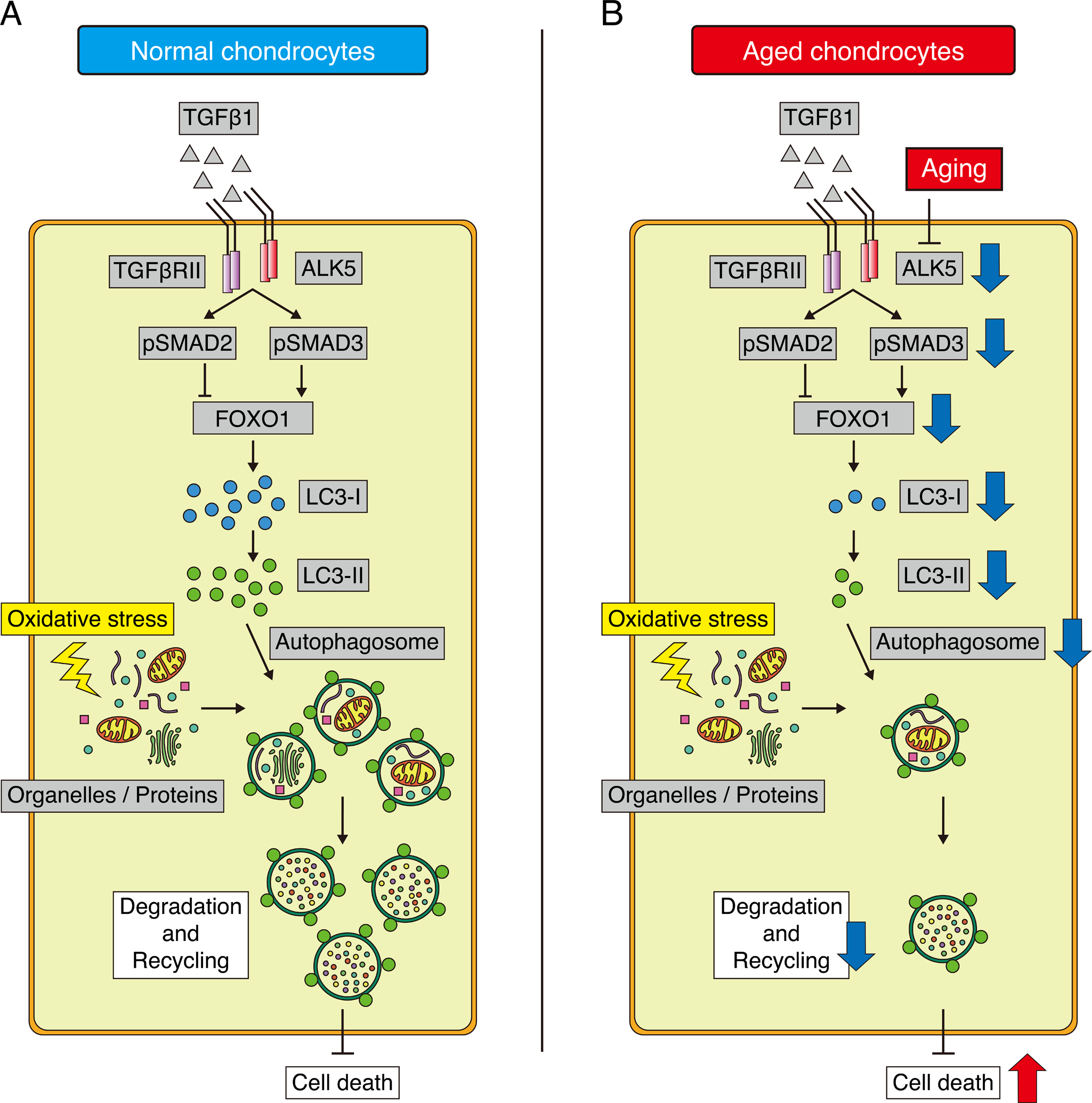
(A) In young normal chondrocytes, TGFβ1 promotes FOXO1 expression via the ALK5-SMAD3 pathway, whereas SMAD2 negatively regulate the expression of FOXO1. SMAD3-FOXO1 axis maintains the expression of LC3-I. Organelles and proteins damaged by oxidative stress are degraded and recycled by autophagosomes, and chondrocytes are protected against cell death. (B) In aged chondrocytes, the reduction in ALK5 expression suppresses SMAD3 activity and FOXO1 expression. Under these conditions, organelles and proteins damaged by oxidative stress are not degraded effectively because of a dysfunction of autophagy caused by reductions in LC3-I and LC3-II expression, resulting in induction of cell death.
TGFβ1–ALK5–SMAD3 signaling is an important pathway for the maintenance of the extracellular matrix in articular cartilage44 45 46 47. On the other hand, little has been reported about the role of TGFβ1 signaling in autophagy in chondrocytes, although TGFβ1 has been reported to promote the activity of autophagy in other cell types48 49. In this study, we demonstrated that TGFβ1 signaling activates autophagy via FOXO1. We first showed that expression of MAP1LC3B increased after TGFβ1 treatment. Consistent with this, overexpression of FOXO1 in chondrocytes promotes the expression of MAP1LC3B24. Next, we examined the expression of LC3 proteins in order to assess autophagic flux. When cells are exposed to stressful conditions, autophagy is activated7 8. When this happens, LC3-I is conjugated to phosphatidylethanolamine and forms LC3-II, which is involved in formation of the autophagosome membrane. Therefore, elevated expression of LC3-II indicates activation of autophagic flux31. Importantly, TGFβ1 treatment promoted only LC3-I expression, whereas the level of LC3-II did not change. This observation indicates TGFβ1 treatment is not stressful for chondrocytes, and TGFβ1 alone does not induce autophagic flux. In addition, we investigated LC3-II expression under oxidative stress because autophagic flux is induced by oxidative stress50. TGFβ1 promoted oxidative stress–induced expression of LC3-II. This result was consistent with the observation in the cells incubated with CQ. On the other hand, SMAD3 and FOXO1 knockdown suppressed induction of LC3-I expression by TGFβ1 and subsequently decreased oxidative stress–induced expression of LC3-II. These findings suggest that TGFβ1 plays an essential role in the expression of LC3-I by promoting the expression of MAP1LC3B via the SMAD3-FOXO1 pathway, and that under oxidative stress, TGFβ1 signaling makes autophagy more effective (Fig. 7A). Conversely, reduced TGFβ1 signaling causes a reduction in FOXO1 expression, which in turn decreases expression of LC3-I. This results in dysfunction of autophagy under oxidative stress because autophagosomes are not formed effectively due to insufficient expression of LC3-II (Fig. 7B). To clarify further detailed regulatory mechanism of autophagy by TGFβ1 signaling, we have to do some additional experiments in the future, for instance, the observation of autophagosomes by electron microscopy.
FOXO1 is important for protecting cells against oxidative stress19. Because TGFβ1 promotes autophagy under oxidative stress via FOXO1, we speculated that TGFβ1 signaling could protect chondrocytes against oxidative stress. Indeed, TGFβ1 suppressed cell death following oxidative stress in a FOXO1-dependent manner. This is the first evidence that TGFβ1 signaling protects chondrocytes against oxidative stress.
In summary, our findings indicate that TGFβ1 promotes the expression of FOXO1 via the ALK5–SMAD3 pathway, maintains the expression of LC3-I, and protects chondrocytes against oxidative stress–induced cell death by activating autophagy (Fig. 7A). Conversely, in aged chondrocytes, the reduction in ALK5 expression may cause a reduction in the level of FOXO1 and subsequent defects in autophagy. Consequently, because damaged organelles and proteins are not effectively degraded and recycled, chondrocytes cannot maintain homeostasis and undergo cell death (Fig. 7B). Our findings reveal a novel function of TGFβ1 signaling in the maintenance of cartilage homeostasis under oxidative stress, mediated by the FOXO1–autophagy axis, and emphasize the important role of TGFβ1 signaling in preventing OA.
Supplementary Material
Acknowledgments
We thank Hitomi Kimura for the supporting histological preparing.
Funding sources
This work was supported by a Grant-in-Aid for Young Scientists (A) 17H05097 from the Japan Society for the Promotion of Science and a grant from Takeda Science Foundation.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol 2016;12:412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone 2012;51:241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCulloch K, Litherland GJ, Rai TS. Cellular senescence in osteoarthritis pathology. Aging Cell 2017;16:210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 2017;23:775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum 2010;62:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hui W, Young DA, Rowan AD, Xu X, Cawston TE, Proctor CJ. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann Rheum Dis 2016;75:449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008;451:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol 2015;11:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ansari MY, Khan NM, Ahmad I, Haqqi TM. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthritis Cartilage 2018;26:1087–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen T, Alvarez-Garcia O, Li Y, Olmer M, Lotz MK. Suppression of Sestrins in aging and osteoarthritic cartilage: dysfunction of an important stress defense mechanism. Osteoarthritis Cartilage 2017;25:287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Q, Zheng G, Feng Z, Chen Y, Lou Y, Wang C, et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis 2017;8:e3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis 2012;71:575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouderlique T, Vuppalapati KK, Newton PT, Li L, Barenius B, Chagin AS. Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann Rheum Dis 2016;75:627–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasheghani F, Zhang Y, Li YH, Blati M, Fahmi H, Lussier B, et al. PPARgamma deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann Rheum Dis 2015;74:569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature 1993;366:461–4. [DOI] [PubMed] [Google Scholar]
- 17.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Sci STKE 2003;2003:Re5. [DOI] [PubMed] [Google Scholar]
- 18.Coffer P OutFOXing the grim reaper: novel mechanisms regulating longevity by forkhead transcription factors. Sci STKE 2003;2003:Pe39. [DOI] [PubMed] [Google Scholar]
- 19.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 2013;14:83–97. [DOI] [PubMed] [Google Scholar]
- 20.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 2008;20:126–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fullgrabe J, Ghislat G, Cho DH, Rubinsztein DC. Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci 2016;129:3059–66. [DOI] [PubMed] [Google Scholar]
- 22.Akasaki Y, Hasegawa A, Saito M, Asahara H, Iwamoto Y, Lotz MK. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthritis Cartilage 2014;22:162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akasaki Y, Alvarez-Garcia O, Saito M, Carames B, Iwamoto Y, Lotz MK. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol 2014;66:3349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuzaki T, Alvarez-Garcia O, Mokuda S, Nagira K, Olmer M, Gamini R, et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci Transl Med 2018;10:eaan0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurakazu I, Akasaki Y, Hayashida M, Tsushima H, Goto N, Sueishi T, et al. FOXO1 transcription factor regulates chondrogenic differentiation through transforming growth factor β1 signaling. J Biol Chem 2019;294:17555–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaney Davidson EN, Remst DF, Vitters EL, van Beuningen HM, Blom AB, Goumans MJ, et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J Immunol 2009;182:7937–45. [DOI] [PubMed] [Google Scholar]
- 27.van Caam A, Madej W, Thijssen E, Garcia de Vinuesa A, van den Berg W, Goumans MJ, et al. Expression of TGFβ-family signalling components in ageing cartilage: age-related loss of TGFβ and BMP receptors. Osteoarthritis Cartilage 2016;24:1235–45. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Tan QY, Xu W, Qi HB, Chen D, Zhou S, et al. Cartilage-specific deletion of Alk5 gene results in a progressive osteoarthritis-like phenotype in mice. Osteoarthritis Cartilage 2017;25:1868–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gellibert F, Woolven Fau - Fouchet M-H, Fouchet Mh Fau - Mathews N, Mathews N Fau - Goodland H, Goodland H Fau - Lovegrove V, Lovegrove V Fau - Laroze A, et al. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-β type I receptor inhibitors. J Med Chem 2004;47:4494–506. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, et al. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J Biol Chem 2002;277:45276–84. [DOI] [PubMed] [Google Scholar]
- 31.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obsil T, Obsilova V. Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene 2008;27:2263–75. [DOI] [PubMed] [Google Scholar]
- 33.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci U S A 2005;102:11278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004;117:421–6. [DOI] [PubMed] [Google Scholar]
- 35.Bridges D, Moorhead GB. 14-3-3 proteins: a number of functions for a numbered protein. Sci STKE 2005;2005:re10. [DOI] [PubMed] [Google Scholar]
- 36.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A 2003;100:11285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nowak K, Killmer K, Gessner C, Lutz W. E2F-1 regulates expression of FOXO1 and FOXO3a. Biochim Biophys Acta 2007;1769:244–52. [DOI] [PubMed] [Google Scholar]
- 38.Berry FB, Skarie JM, Mirzayans F, Fortin Y, Hudson TJ, Raymond V, et al. FOXC1 is required for cell viability and resistance to oxidative stress in the eye through the transcriptional regulation of FOXO1A. Hum Mol Genet 2008;17:490–505. [DOI] [PubMed] [Google Scholar]
- 39.Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, et al. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010;463:676–80. [DOI] [PubMed] [Google Scholar]
- 40.Vivar R, Humeres C, Munoz C, Boza P, Bolivar S, Tapia F, et al. FoxO1 mediates TGF-beta1-dependent cardiac myofibroblast differentiation. Biochim Biophys Acta 2016;1863:128–38. [DOI] [PubMed] [Google Scholar]
- 41.Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signal 2019;12:eaav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kashiwagi I, Morita R, Schichita T, Komai K, Saeki K, Matsumoto M, et al. Smad2 and Smad3 Inversely Regulate TGF-beta Autoinduction in Clostridium butyricum-Activated Dendritic Cells. Immunity 2015;43:65–79. [DOI] [PubMed] [Google Scholar]
- 43.Ungefroren H, Groth S, Sebens S, Lehnert H, Gieseler F, Fandrich F. Differential roles of Smad2 and Smad3 in the regulation of TGF-β1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: control by Rac1. Mol Cancer 2011;10:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blaney Davidson EN, van der Kraan PM, van den Berg WB. TGF-β and osteoarthritis. Osteoarthritis Cartilage 2007;15:597–604. [DOI] [PubMed] [Google Scholar]
- 45.van der Kraan PM, Goumans M-J, Blaney Davidson E, ten Dijke P. Age-dependent alteration of TGF-β signalling in osteoarthritis. Cell and Tissue Research 2011;347:257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niikura T, Reddi AH. Differential regulation of lubricin/superficial zone protein by transforming growth factor β/bone morphogenetic protein superfamily members in articular chondrocytes and synoviocytes. Arthritis Rheum 2007;56:2312–21. [DOI] [PubMed] [Google Scholar]
- 47.Wang G, Chen S, Xie Z, Shen S, Xu W, Chen W, et al. TGFβ attenuates cartilage extracellular matrix degradation via enhancing FBXO6-mediated MMP14 ubiquitination. Ann Rheum Dis 2020;79:1111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, et al. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 2009;69:8844–52. [DOI] [PubMed] [Google Scholar]
- 49.Ding Y, Kim JK, Kim SI, Na HJ, Jun SY, Lee SJ, et al. TGF-β1 protects against mesangial cell apoptosis via induction of autophagy. J Biol Chem 2010;285:37909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 2010;6:838–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.