

Abstract
Since Paul Ehrlich’s introduction of the “magic bullet” concept in 1908, drug developers have been seeking new ways to target drug activity to diseased cells while limiting effects on normal tissues. In recent years, it has been proposed that coupling riboswitches capable of detecting RNA biomarkers to small interfering RNAs (siRNAs) to create siRNA pro-drugs could selectively activate RNA interference (RNAi) activity in specific cells. However, this concept has not been achieved previously. We report here that we have accomplished this goal, validating a simple and programmable new design that functions reliably in mammalian cells. We show that these conditionally activated siRNAs (Cond-siRNAs) can switch RNAi activity against different targets between clearly distinguished OFF and ON states in response to different cellular RNA biomarkers. Notably, in a rat cardiomyocyte cell line (H9C2), one version of our construct demonstrated biologically meaningful inhibition of a heart-disease-related target gene protein phosphatase 3 catalytic subunit alpha (PPP3CA) in response to increased expression of the pathological marker atrial natriuretic peptide (NPPA) messenger RNA (mRNA). Our results demonstrate the ability of synthetic riboswitches to regulate gene expression in mammalian cells, opening a new path for development of programmable siRNA pro-drugs.
Keywords: RNA therapy, conditional siRNA, RNA biomarker, calcineurin, cardiac hypertrophy, nanobiotechnology, bionanotechnology, RNA nanotechnology, DNA nanotechnology
Graphical abstract
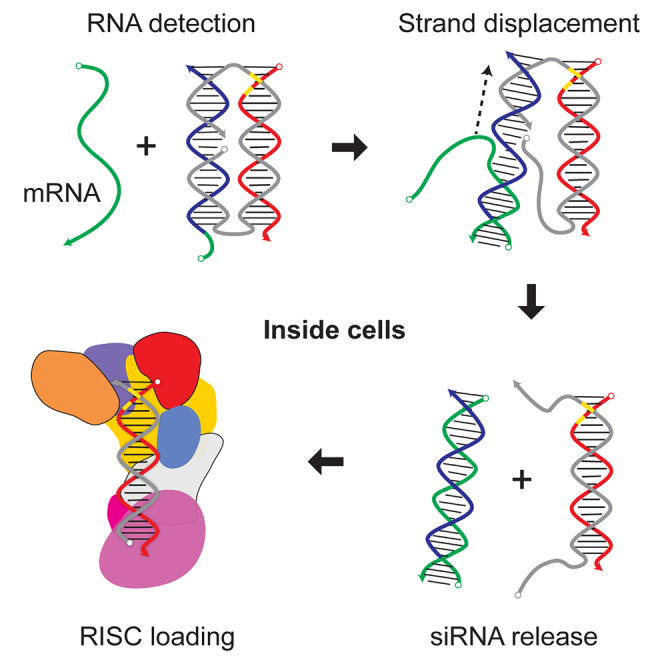
We report development of conditionally activated small interfering RNAs. These programmable oligonucleotide pro-drugs activate RNAi activity against target genes only in response to the presence of specific RNA biomarkers in mammalian cells. This opens new avenues for targeting drug activity to specific cell populations while sparing non-target tissues.
Introduction
The simplest method of disease-selective drug targeting is to target genes that are uniquely important to disease pathways.1 The problem is that in diseases involving dysregulation of human genes, important targets in disease pathways9, 10, 11 can also have critical functions in normal tissues. One way to circumvent this problem is targeted drug delivery to diseased cells using moieties such as antibody drug conjugates12 or nanoparticle drug complexes.13 However, these targeted delivery approaches involve many technical challenges, and diseased cells may lack distinguishing surface markers with properties favorable for efficient drug uptake.
Alternatively, drug developers can use distinctive aspects of cellular biochemistry to activate drugs after delivery. For example, approximately 5%–7% of small-molecule drugs are pro-drugs that are activated by enzymatic processing.14 Although these small pro-drugs can have improved tissue selectivity, their simple activation mechanisms have many limitations regarding the specificity and versatility of targeting that can be achieved.
In contrast, large genetic molecules that interact directly with gene expression and gene regulation pathways can have highly selective and versatile disease-targeting mechanisms. For example, gene therapies can utilize tissue-specific promoters15 or biomolecular logic circuits16,17 to target specific cells, and mRNA therapies can have synthetic miRNA targeting sites that help restrict their activities outside of targeted tissues.18 However, these approaches require delivery of large DNA vectors or mRNAs using viral or nanoparticle transduction, can permanently modify cellular DNA, and can be significantly more expensive and complex to develop and produce than molecular drugs.
In this landscape, modern therapeutic small interfering RNAs (siRNAs) represent a happy medium.19 They are (1) capable of silencing specific RNA transcripts using the endogenous RNAi pathway, (2) much smaller than DNA vectors used in gene therapies, (3) able to act transiently without changing host cell genomes, (4) cost-effective to manufacture via chemical synthesis, and (5) able to incorporate extensive chemical modifications that allow them to be delivered in vivo using simple formulations.20
To give siRNAs enhanced diseased-cell-targeting capabilities, some researchers have sought to create programmable riboswitch-siRNA pro-drugs that would release active siRNA molecules only when their riboswitch portions base pair with disease-indicating RNA biomarkers in the cell.2, 3,4, 5, 6, 7 In theory, such constructs would gain the unique ability to target disease cells according to transcriptome content while maintaining the advantageous properties of siRNAs. In practice, successful implementation of this scheme still faces significant hurdles.
First, existing designs for riboswitch-siRNA combinations cannot reliably suppress RNAi activity in non-targeted mammalian cells or switch ON efficient RNAi activity in targeted mammalian cells. Second, some schemes are not easily programmable for using different biomarkers and target sequences. Third, some designs require an overlap between the base sequences of the biomarker and the target, compromising versatility. Fourth, it has been difficult to integrate all of the desired attributes in an RNA construct that is compact, simple, and stable enough to be manufactured and delivered like conventional siRNAs.
Here we show the feasibility of addressing these concerns using “designer” conditional siRNA (Cond-siRNA) that activate only in the presence of a specific disease biomarker mRNA. Using the test case of cardiac hypertrophy, we demonstrate that, in a rat cardiomyocyte cell line (H9C2)8, our Cond-siRNA constructs can silence calcineurin, a key regulator of hypertrophy, only upon activation with atrial natriuretic peptide (ANP) mRNA (gene natriuretic peptide A, NPPA), a key biomarker for cardiac hypertrophy. Our studies lay the platform for a portfolio of RNA therapeutic agents that can be activated selectively in diseased cells and can target critical disease effector molecules of interest across a wide spectrum of diseases.
Results
Cond-siRNA design
The Cond-siRNA design is shown in Figure 1 (see also Table S1). This Cond-siRNA construct is composed of three strands (sensor, core, and guide) that self-assemble into two associated RNA duplexes via Watson-Crick base-pairing. Because of the routing of the core strand, the duplexes are held in a rigid parallel configuration by crossovers at their two ends.21
-
•
OFF state. In the inactive configuration (as assembled), the sensor duplex and strategically placed chemical modifications block RNAi pathway enzymes19 from binding to and processing the siRNA duplex, keeping RNAi activity switched OFF by steric hindrance (Figure 1A).
-
•
ON state. In cells expressing RNA biomarkers complementary to the sensor strand, the biomarker RNA base-pairs with the sensor strand to disassemble the sensor duplex via toehold-mediated strand displacement (Figures 1B and S1).22 This releases the siRNA duplex, switching the RNAi activity ON. Vestigial overhangs on the siRNA are removed by endogenous nucleases (Figures S2 and S3).
Figure 1.

Design, operating concept, and chemical modifications of Cond-siRNAs
(A) The secondary and tertiary structure of Cond-siRNAs. Docking of the tertiary structure model to the X-ray crystal structure of Giardia Dicer shows that the sensor can sterically block RNAi pathway enzymes from binding to the siRNA. This prevents RNAi processing in the OFF state. (B) RNAi activation via strand displacement. When a complementary input RNA meets the Cond-siRNA (I), the input forms a toe with a 3′ or 5′ single-stranded overhang on the sensor strand (II), leading to strand displacement (III). Displacement results in release of the siRNA (IV). Cellular nucleases remove the core-strand overhang siRNA (V), leaving an active siRNA that is free to enter the RNAi pathway (VI). (C) Chemical modifications that improve OFF/ON RNAi switching in mammalian cells include (1) PS modifications in the sensor overhang, (2) LNA and 2′OMe modifications in the base-paired portion of the sensor strand (3) 2′OMe modifications at the 5′ and-3′ termini of the core strand (highlighted in yellow), (4) PS backbone and base modifications at the ends of the siRNA to block exonucleases, (5) LNA modifications to improve Tm of the siRNA, (6) and base modifications in the siRNA duplex.
Mammalian cells have complex and robust pathways for regulation and degradation of RNA molecules.23, 24, 25 Many past concepts for riboswitch siRNA combinations have not worked correctly in this environment. We find that chemical modifications known to increase resistance to nuclease activity and thermodynamic stability are crucial to achieve correct functioning in cells. These modifications are used extensively on our constructs in six important regions (Figure 1C).
-
(1)
The single-stranded toehold on the sensor strand should be modified with phosphorothioate (PS)26 backbone linkages, locked nucleic acids (LNAs),27 and 2′-O-methyl (2′OMe) bases.
-
(2)
The duplex forming region of the sensor strand should have LNA and 2′OMe bases but no PS linkages.
-
(3)
The 5′ and -3′ termini of the core strand forming the nick in the middle of the sensor duplex should be terminated with 2′OMe bases.
-
(4)
The regions on the core strand adjacent to the 5′ and 3′ overhangs (when the siRNA is released from the sensor) should be modified with base and PS backbone modifications to stop exonucleases from invading the siRNA duplex after degradation of the overhangs on the core strand.
-
(5)
Suppression of OFF-state RNAi activity is enhanced by LNA modification of the ends of the siRNA duplex.
-
(6)
Addition of 2′-OMe modifications to the siRNA duplex further reduces background RNAi activity.
3D structures of Cond-siRNAs
We used atomistic molecular dynamics (MD)28, 29, 30, 31, 32 to predict the 3D structures of two Cond-siRNA constructs incorporating some combinations of these modifications (Figure 2; Table S2). MD shows that the constructs largely conformed to the design parameters. The two duplexes remained stable throughout our simulations. In both constructs, potential cleavage sites on the siRNA duplex for the RNAi pathway enzyme Dicer remained oriented toward the interior of the two parallel duplexes, conferring steric protection. Also, the gap between the 3′ and 5′ termini of the core strand remained oriented toward the center of the construct and did not create any unwanted distortions in the structure.
Figure 2.

3D structures of two different Cond-siRNAs obtained using MD simulations in explicit solvent
Orange and purple arrows denote the directions of strand displacement from the 3’ (orange) and 5’ (purple) ends of the sensor strand. (A) structure I.1 from Table S1. (B) Structure III.1 from Table S1. These simulations included 69,030 and 68,727 atoms per periodic cell, respectively.
We found a key difference in the sensor-unwinding pathway for RNA biomarkers binding to the sensor via a 3′ toehold versus a 5′ toehold. The MD geometry suggested that an RNA biomarker binding first to a 5′ toehold on the sensor strand would encounter steric hindrance as it attempts to unwind the sensor toward the interior of the construct (Figures 1A and 1B, purple arrows). On the other hand, unwinding of the sensor from the 3′ end of the sensor strand (orange arrows) would wind initially toward the exterior of the construct, encountering less hindrance. Indeed, certain aspects of our experimental data were consistent with these expectations.
Functional assessment of Cond-siRNAs
We tested constructs with sensor and guide sequences designed for specific combinations of biomarkers and targets. For each design, we tested different patterns of chemical modifications (Table S1). Experiments were conducted on a human colorectal cancer cell line (see the experimental protocols; Figure 3). HCT116 cells33 were first transfected with two different DNA plasmids:
-
(1)
a PsiCheck-2 dual-luciferase reporter (Promega) carrying the siRNA target in the 3′ UTR of Renilla luciferase, and
-
(2)
a pBlueScript34 vector with a polymerase III (Pol III) promoter expressing short RNA transcripts that had different biomarker sequences35 (Figure S4).
Figure 3.

RNAi activity of Cond-siRNAs in HCT116 cells expressing irrelevant (irrel) or activating (act) RNA biomarkers
Activity was measured by dual-luciferase assay. Cond-siRNAs are designed for different combinations of biomarkers and targets and have various chemical modifications corresponding to features identified in Figure 1C. In each panel, optimized constructs are on the left, followed by versions with less optimal motifs. (A) AML:U5K2 constructs with differing modifications in region 2 (Figure 1C) of the sensor strand (Table S1C, III.5, III.6, and III.7). (B) AML:U5K2 constructs with differing modifications in region 3 (Table S1C, III.1, III.2, and III.3). (C) AML:U5K2 constructs with differing modifications to regions 4 and 5 (Table S1B, II.6, II.2, and II.1). (D) AML:MCL1 construct with and without 2′OMe modifications in region 6. In addition to mismatched (irrel) and fully matched (act) biomarkers, the constructs were also tested against biomarkers with toehold (toe) or duplex region complementarity to the sensor strand (Table S1D, IV.3 and IV.2). Significance was calculated by two-way ANOVA. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001. Error bars denote one standard deviation.
Eight hours later, we transfected Cond-siRNAs into the cells.
48 h after the second transfection, we processed the cells for dual-luciferase readout.
Figures 3 and S5–S7 report our results. We found that optimized versions of Cond-siRNAs (the first cohort in each panel) exhibited dose-dependent RNAi silencing in cells expressing the correct RNA biomarkers (activating [act]) and had significantly reduced RNAi activity in cells expressing incorrect biomarkers (irrelevant [irrel], toehold [toe], and duplex).
In contrast, changing chemical modification patterns away from the ideal configuration shown in Figure 1C leads to decreased control over RNAi activity:
-
•
OFF-state RNAi activity is increased,
-
•
ON-state RNAi activity is decreased,
-
•
or both (Figures S6 and S7).
For example,
-
•
in Figure 3A, removal of LNA and 2′OMe modifications from the sensor strand (Figure 1C, feature 1) leads to complete loss of RNAi switching;
-
•
in Figure 3B, substitution of 2′OMe modifications protecting the 5′ and-3′ ends of the core strand with PS backbone modifications leads to inferior switching,
and removal of all modifications from the 3′ end leads to complete loss of RNAi switching (Figure S6).
In line with expectations from MD simulations, the Cond-siRNAs with 3′ sensor toes (AML:U5K2 and AML:MCL1 in Figures 3A, 3B, and 3D) had better RNAi activation than the Cond-siRNA with the 5′ toe (TAT/REV:U5K2 in Figure 3B). This observation was consistent with results from another construct that had 3′ and 5′ toes (Figure S5). Further experiments are needed to confirm this effect for diverse sequences.
A Cond-siRNA to target pathological cardiac hypertrophy
To test the ability of Cond-siRNAs to detect disease-indicating mRNAs in a more biologically realistic scenario, we tested a Cond-siRNA designed to detect atrial natriuretic peptide, ANP (NPPA) mRNA8 and silence calcineurin (Protein Phosphatase 3 catalytic subunit alpha, PPP3CA) in a rat cardiomyocyte cell line (H9C2) (Figures 4A and 4B; Table S1E). ANP expression is a validated biomarker in H9C2 cells following exposure to phenylephrine (a validated in vitro model for cardiac hypertrophy), and the increase in PPP3CA expression is causally related to development of hypertrophy. In H9C2 cells transfected with self-delivering ANP-calcineurin Cond-siRNA (materials and methods) and exposed to 50 μM phenylephrine, the increased expression of PPP3CA was ameliorated (compared with a scrambled siRNA control) in response to increasing doses of Cond-siRNA. This effect was similar to that observed with a commercial siRNA against PPP3CA. For Cond-siRNA-transfected H9C2 cells exposed to the PBS control (where ANP expression would not be expected to increase), no significant or dose-dependent changes from baseline expression levels of ppp3ca were observed. The observed decrease in PPP3CA mRNA corresponded to a decrease in PPP3CA (calcineurin) protein in rat cardiomyocytes treated with the Cond-siRNA in the presence of phenylephrine (which would be expected to increase the biomarker ANP; Figure 4C).
Figure 4.

RNAi activity of a self-delivering NPPA:PPP3CA Cond-siRNA in a rat cardiomyoblast model
(A) qRT-PCR results show that NPPA mRNA was overexpressed in phenylephrine (PE)-exposed cells but not in PBS-exposed cells. (B) The Cond-siRNA-activated RNAi activity against PPP3CA (calcineurin) in PE-exposed cells and reduced overexpression of PPP3CA in a dose-dependent manner up to 5 nM. In cells exposed to PBS, there was no significant RNAi silencing of PPP3CA. The negative control was scrambled siRNA. The positive control was a commercial PPP3CA siRNA (A and B). The activated Cond-siRNA had similar silencing activity as the commercial siRNA. Significance was calculated by two-way ANOVA. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001. #, p value comparison between PBS and PE; ∗, comparison with the control. Error bars denote one standard deviation. (C) Quantification of protein levels of calcineurin (Cell Signaling Technology, 2614S) in the cytoplasm of NRVMs treated with 1 nM Cond-siRNA constructs or the scrambled control siRNA. 24 h after isolation, primary rat cardiomyocytes were transfected with the Cond-siRNA; after 24 h, cells were treated with 50 μM PE for 48 h, and protein expression was determined 72 h after transfection. Data are shown as fold versus scramble (scrm) PBS. Statistics: unpaired t test; ∗p ≤ 0.05, ∗∗p ≤ 0.01.
Discussion
Design of drug molecules that can specifically target disease-modifying genes in diseased tissues without significant off-target effects or targeting of bystander healthy tissue is a goal that is rarely achieved in the therapeutic arena. Here we demonstrated a strategy to develop Cond-siRNAs that are activated only in the presence of disease biomarkers, conferring cellular and molecular specificity. Our breakthrough technology enables siRNAs to regulate their own RNAi activities in mammalian cells based on whether the transfected cells express specific RNA biomarkers.
-
•
In cells lacking the cognate biomarkers, RNAi activity stays OFF.
-
•
In cells expressing those biomarkers, RNAi activity switches ON.
This makes it possible to silence arbitrary genes in specifically targeted cells that are identified by their internal RNA transcripts, providing a unique new strategy for targeting drug activity to specific cells or tissues in the body.
At the heart of our work is a new type of siRNA pro-drug called Cond-siRNA. Each Cond-siRNA is composed of a sensor domain connected to a siRNA. With the sensor connected, enzymatic processing of the siRNA is prevented by steric hindrance. This inactivates RNAi activity. In cells that are expressing cognate biomarkers, such as messenger RNAs (mRNA) or microRNA (miRNA) associated with particular diseased states, the biomarker RNAs can base-pair with the sensor strands of Cond-siRNAs and release the attached siRNAs via a biophysical mechanism called strand displacement. This removes the steric hindrance, allowing processing of the siRNAs by the RNAi pathway. The sensor is completely sequence independent from the siRNA, allowing the Cond-siRNA to use one gene as a biomarker to activate silencing of an entirely different gene.
The most important application of our technology will be for RNAi therapeutic agents. Cond-siRNAs can potentially target drug activity to specific types or states of cells involved in disease. Many diseases that are difficult to treat today involve dysregulation of important endogenous signaling pathways controlled by master regulators such as calcineurin, transforming growth factor β (TGF-β), nuclear factor κB (NF-κB), BCL-2 family apoptosis inhibitors, and others. Inhibition of these master regulator genes can lead to potent disease treatment effects, but their important functions in many non-diseased tissues create serious safety problems. Targeted treatment of specific diseased cells or tissues could ameliorate safety problems by switching OFF drug activity in healthy tissues. This would allow improved safety for existing treatments and open therapeutic windows for new indications. We demonstrated targeting of calcineurin (PPP3CA), a gene causally involved in development of pathological cardiac hypertrophy in H9C2 cells, only in the presence of the disease biomarker ANP. Our findings offer a broad platform for development of cell- and disease-specific RNA therapies for a wide variety of diseases.
Because the sensor and siRNA duplexes do not overlap, the sensor and guide strands are completely sequence independent and easily programmed, an architecture that makes it easier to keep the OFF state structure thermodynamically stable. The entire construct is only approximately two times the molecular weight of modern siRNAs with similar critical dimensions (e.g., length), which, we found, presents no significant differences in requirements for delivery of Cond-siRNAs versus conventional siRNAs in vitro. Although our studies focused on the design and feasibility aspects of Cond-siRNAs, our pilot experiments in H9C2 cells suggest that self-delivery of Cond-siRNAs to cells is feasible with appropriate modifications of the sensor strands. However, whether this translates into in vivo models and the biodistribution of Cond-siRNAs in animal models are subjects for future investigations.
A missing element awaiting development is a systematic bioinformatics method for designing sensors to detect arbitrary mRNA biomarkers. Such a method would have to select binding sites for the sensor that are specific to the biomarker and accessible for base pairing. The specificity of the binding site can be determined using tools such as NCBI BLAST.36 Accessibility of the binding site is a more complex issue. Pertinent factors include secondary and tertiary structure, protein binding activity, and subcellular localization of the mRNA at different stages of the mRNA’s life cycle. Research into design methods for siRNAs and antisense oligonucleotides19 offers important lessons and suggests that thermodynamics predictions and bioinformatics analyses will need to be combined with empirical screening to identify suitable binding sites.
In addition to being a programmable and reliable siRNA pro-drug, our Cond-siRNA offers some useful lessons for development of riboswitches that function in mammalian cells.
-
•
First, it is critical to use chemical modifications to protect terminal strand regions from exonuclease activity, but familiar motifs from oligonucleotide medicinal chemistry can suffice.
-
•
Second, recent intracellular riboswitches by other authors have used 2′OMe strands to improve intracellular functioning over natural RNA oligonucleotides, demonstrating switching over hours-long periods. Our experimental results during optimization of Cond-siRNAs show that these motifs are still not sufficient to suppress all spurious construct activation (presumably from dissociation or degradation of the sensor duplexes) during operation for extended periods (days). This is especially important with biologically active outputs, such as siRNAs, that can catalytically amplify leakage signals. On the other hand, our combination of LNA and 2′OMe modifications on the signal binding strand (our sensor strand) was necessary and sufficient for reliable activation with low background.
-
•
Third, when evaluating different designs for siRNA pro-drugs, we find that designs that completely release siRNAs from the sensor yield better RNAi activity than designs where the siRNA and sensor remain coupled after activation (Figure S8). This should be important for achieving optimal RNAi switching.
Summary
We have shown successful design of Cond-siRNAs that couple activation in response to disease-specific biomarkers to silence disease-modifying genes. Our Cond-siRNAs, unlike conventional siRNAs, have low activation in the absence of the disease-specific biomarker and have activity comparable with commercial siRNAs when activated. In a cellular model of cardiac hypertrophy, we were able to silence PPP3CA, a gene causally implicated in cardiac hypertrophy only upon activation by the disease-specific biomarker ANP.
Materials and methods
Construct design
Cond-siRNAs were designed for specific pairing of inputs and targets using an iterative protocol.
-
1.
Obtain a suitable 21-nt guide strand sequence for the RNAi domain from previously validated siRNAs, literature sources, or siRNA design tools.
-
2.Create a 23-bp Dicer substrate from the chosen guide strand by adding four GC-rich bases to the 5′ of the guide strand.
- a.
-
3.From the sequence of the input biomarker, generate a list of all possible 31- to 33-nt sensor segments (antisense to the input).
-
a.For the CBFβ-MYH11 fusion sequence, only sensor segments that approximately meet parameters illustrated in Figure 3B were considered.
-
a.
-
4.Rank sensor sequences for uniqueness in the transcriptome of the target animal using NCBI BLAST.36
-
a.For human cancer cell lines, we checked sequences against human transcript and genomic collection using the BLASTn algorithm.
-
b.Where possible, eliminate sensor segments that have more than 17 bases of sequence complementarity and complete overhang complementarity to known or predicted RNA transcripts.
-
a.
-
5.Starting with the most unique sensor segments, construct core-strand sequences in accordance with desired structural parameters for the Cond-siRNA.
-
a.Core strands have sequences of the form 5′-B-C3-P-C3-A-3′, where A and B are complementary to the 5′ and 3′ ends of the sensor strand’s putative duplex domain, P is complementary to the putative guide strand, and C3 and C3’ are linkers.
-
a.
-
6.Use Nupack to rank the thermodynamic stability of the duplexes formed between sensor strand segments and their corresponding 5′ and 3′ core strand overhangs.
-
a.Use RNA strand, Mathews et al.39 parameters, with some dangle treatment.
-
b.Ideally, more than 95% of strands should be base-paired at 1 nM strand concentration.
-
c.Also check that the core strand does not have heavy internal secondary structure.
-
a.
-
7.
Choose the best constructs (guide, core, and sensor sequences) generated in steps 1–6.
-
8.
Add chemical modifications according to patterns described in this manuscript.
-
9.Use Exiqon’s oligonucleotide design tools (https://www.exiqon.com/oligo-tools) to optimize placement of LNA modifications.
-
a.LNA modifications are added to the sensor strand approximately 1 per every 3–4 bases.
-
b.Using the LNA Oligo Optimizer tool, check that the LNA pattern used does not lead to secondary structure or self-pairing interactions with scores higher than 60. Minimize self-complementarity and self-pairing scores as much as possible.
-
a.
Synthesis of strands
Strands with LNA bases were synthesized by Exiqon (now a division of QIAGEN). Strands without LNA were synthesized by GE Life Sciences Dharmacon (now a division of Horizon Discovery Group). All strands were ordered with PAGE or HPLC purification according to recommendations of the manufacturer.
Assembly of Cond-siRNAs
Cond-siRNAs were assembled by thermal annealing in 1× phosphate-buffered saline (PBS). Constructs can be assembled with or without purification. Assembly quality can be assessed using non-denaturing gel electrophoresis on 10%–15% PAGE in 1× Tris-borate-EDTA (TBE) at 4°C.
For assembly without purification, we mixed sensor, core, and guide strands at a 1.1 to 1.00 to 1.1 molar ratio at 50 nM or 100 nM concentration in 1× PBS (pH ∼7.0). Using a slight excess of sensor and guide strands helps to prevent production of constitutively RNAi active guide and core duplexes. Using a PCR thermocycler, we used the following program.
-
•
Heat lid to 105°C.
-
•
Hold at 85°C for 30 s to denature the strand.
-
•
Cool to 50°C at a rate of 0.1°C/s.
-
•
Hold for 45 min at 50°C.
-
•
Cool to 37°C at a rate of 0.02°C/s.
-
•
Cool to 4°C at maximum rate and hold.
For assembly with purification, we mixed and assembled sensor, core, and guide strands at 1 μM nominal concentration in 1× PBS using the annealing protocol described above. Assembled constructs were then loaded on Bio-Rad Mini Protean 10% native PAGE gels in TBE buffer and run at 125 V at 4°C for ∼45 min. The bands corresponding to the Cond-siRNA were visualized and excised under UV lamp illumination.
Excised bands were extracted by electro-elution using a Harvard Apparatus Electroprep system according to the manufacturer’s instructions. Gel pieces were placed in a 0.5-mL chamber sealed by a 100,000 MWCO filter membrane and a 2,000 MWCO filter membrane. Constructs were eluted through the 100,000 MWCO membrane and trapped in an adjacent 0.5-mL chamber formed by the 100,000 MWCO membrane and a second 2,000 MWCO membrane. Elution occurred in 0.1 M Na2HOP4 buffer (∼pH 7.0) at 4°C for ∼45 min. The power supply was set to maintain a constant current of 15 mA with a voltage cutoff of 65 V.
Concentrations of purified constructs were calculated by comparison with Cond-siRNA standards at a known concentration using SYBR Gold staining on non-denaturing PAGE with quantitation using a Bio-Rad ChemiDoc XRS+ Imager.
Assembled constructs are best used immediately after assembly or purification. Constructs can also be stored indefinitely in aliquots at −80°C. However, we found that freeze-thaw cycles compromise construct quality and resulted in construct disassembly. Disassembled constructs can be reassembled by repeating the thermal annealing immediately prior to the assay.
For the data in this paper, we used unpurified constructs because the assembly yield was already excellent, and purification did not consistently improve construct performance.
Strand displacement assay
Pre-assembled constructs were prepared at 50 nM nominal concentration and combined 1:1 with 50 nM oligonucleotide activators (or PBS for control) at 37°C in PBS buffer to obtain mixtures with 25 nM input signals and constructs. Construct-input combinations were then incubated in a PCR thermocycler at a constant 37°C over 4 h. Samples were collected at the indicated time points and immediately frozen at −80°C in 1× native PAGE loading dye. At the end of the experiment, all samples were thawed rapidly and analyzed using non-denaturing PAGE.
Generation of dual-luciferase reporter and activator plasmids
All clones were generated using standard molecular biology protocols by annealing DNA oligos for the specific insert followed by ligation into the indicated sites of the parental vector. The accuracy of all constructs was verified by DNA sequencing.
PsiCHECK dual-luciferase reporters
The DNA oligos below were annealed and ligated into the XhoI and NotI sites of a psiCHECK 2 (Promega) dual-luciferase reporter. Nucleotides in bold font constitute the sense target sequence. Lowercase nucleotides indicate restriction site 5′ overhangs.
HIV U5 region target
5′- tcgaGTCTGGTAACTAGAGATCCCTCAGACCC-3ʹ
5′- ggccGGGTCTGAGGGATCTCTAGTTACCAGAC-3ʹ
MCL-1 target
5′- tcgaGCTGCATCGAACCATTAGCAGAAA-3ʹ
5′- ggccTTTCTGCTAATGGTTCGATGCAGC-3ʹ
RNA input transcripts for signal activation experiments
The activator sequences were expressed as part of a chimeric tRNA transcript. The first part consists of a modified35 tRNALys3 with 3′ terminal CCA, (the mature sequence is shown in its entirety below). The CCA prevents endonucleolytic cleavage by the pre-tRNA processing enzyme tRNAse Z. tRNA Pol III promoters are internal and contained within the coding sequence of the tRNA DNA.
For cloning, we used a parental plasmid containing the first 69 nt of tRNALys3, terminating in an NruI restriction site.35 Digestion of the parental plasmid with NruI generates a blunt end immediately following nucleotide tRNA 69. Annealed overlapping oligos encode the remaining modified tRNA nucleotides, followed by the specified activation sequence. Each activation sequence terminates in a 12-base tetraloop (GGCGCAAGCC) followed by a T6 run encoding the Pol III terminating sequence. U4+RNA transcript sequences are listed below.
For constructs I and II, tRNA Lys3 leader sequence:
5ʹ-GCCCGGAUAGCUCGGUCGGUGGAGCAUCAGACUUUUAAUCUGAGGGUCCAGGGUUCGAGUCCCUGUUCGUGCACCA-3ʹ – activator sequence.
The bolded region is the binding site for the northern blot probe.
Activator sequences for constructs I and II
Regions meant to align with the sensor strand are bold and highlighted in gray. Regions meant to align with the toe are underlined. Segments complementary to the sensor strand are uppercase. Segments mismatched with sensor strand are lowercase.
Activator sequences for constructs III and IV
Regions meant to be aligned with the sensor strand are bold and highlighted in gray. Regions meant to align with the toe are underlined. Segments complementary to the sensor strand are uppercase. Segments mismatched with the sensor strand are lowercase:
DNA probes for northern blots
| tRNA probe | 5ʹ-CTGGACCCTCAGATTAAAAGTC-3ʹ |
| CBFβ probe | 5ʹ-CTCCATTTCCTCCCGATGAGACCTGTC-3ʹ |
| MYH11 probe | 5ʹ-CGCTTGGACTTCTCCAGCTCATGGAC-3ʹ |
| U5 guide strand probe | 5ʹ-AAAGGTAACTAGAGATCCCTCAGA-3ʹ |
Tissue culture
All analyses utilized HCT 116 colorectal carcinoma cells. Cells were maintained using McCoy’s 5A basal medium (Irvine Scientific, USA) supplemented with 10% fetal bovine serum (FBS), 1.5 mM L-glutamine (Irvine Scientific, USA), and 10 mM pyruvate (Irvine Scientific, USA) without antibiotics and kept in a humidified 5% CO2 incubator at 37°C. Conditional siRNAs were also tested in rat cardiomyocyte line H9C2 cells or in neonatal rat ventricular myocytes (NRVMs). Rat myocytes were isolated from postnatal day 1 Wistar rat pups using collagenase II and pancreatin based enzymatic digestion, purified via Percoll gradient, and used for experiments 24h post isolation. NRVMs were cultured in DMEM (Life Technologies, Cat.# 11995073) supplemented with 10% horse serum (Thermo Fisher Scientific, Cat.# 26050-088), 5% FBS (Life Technologies, Cat.# 10437028), 1% Penicillin-Streptomycin (Thermo Fisher Scientific, Cat.# 15140122) and 1% L-Glutamine (Thermo Fisher Scientific, Cat.# 25030-081). H9C2 cells were grown in DMEM supplemented with 10% FBS, 1% Penicillin-Streptomycin and 1% sodium pyruvate (Sigma). All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC).
Northern and western blot analysis
Analysis of activator expression was performed in 6-well plates using 2 μg of plasmid DNA in 250 μL OptiMEM and 250 μL 1:50 diluted Lipofectamine 2000. Liposomes were allowed to form according to the manufacturer’s instructions and added to cells with 2 mL fresh full medium. The medium was replaced at 18 h and, depending on the length of transfection, at least once each subsequent day and 6 h prior to RNA harvest. Analysis of OFF and pre-activated (ON) c-siRNAs were performed similarly; however, the indicated amount of RNAi complex was added to 2 μg of pBluescript plasmid as carrier in 250 μL OptiMEM.
Total RNA was harvested using 1,000 μL RNA Stat-60 (Tel-Test) and processed according to the manufacturer’s instructions with addition of a second organic extraction using 1:1 phenol:chloroform extraction prior to precipitation. RNA pellets were washed twice with 70% ethanol prior to evaporation of excess ethanol and resuspension in RNase-free TE (pH 6.8).
For northern blot analysis, 15 μg of total RNA was run on 8% (for activators) or 12% (for c-siRNAs) urea-PAGE gels (15 cm) with 32P-labeled Ambion Decade markers. Gels were electroblotted to Hybond XL (GE Healthcare Life Sciences), prehybridized and hybridized at 37°C using Sigma Perfect Hyb Plus, and hybridized with 5–10 pmol of P32-5′ end-labeled oligo probe. Blots were washed at 37°C with 4–5 changes of 2× saline sodium citrate (SSC)/1% SDS. With serial hybridizations, the old oligo probe was removed from the membrane according to the manufacturer’s instructions and checked by re-exposure prior to rehybridization unless otherwise indicated. Hybridization of U6 small nuclear RNA (snRNA) was used as a loading control. Cloning procedures and oligos were specified in the section and all probe sequences are listed in tables in the preceeding sections starting from “HIV U5 region target” to “DNA probes for northern blots”.
For western blot analysis, cytoplasmic and nuclear proteins from NRVMs were extracted through the Thermo Scientific NE PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Cat.# 78833). The Pierce™ BCA protein assay (Thermo Fisher Scientific, Cat.# 23227) was performed to quantify lysates’ protein concentration, and 20µg of each sample were used for 4-20% SDS-PAGE electrophoresis. Gels were transferred to PVDF or membranes (BioRad) and blocked with 5%BSA for 1h at RT. Primary antibodies were incubated ON at 4°C rocking at a 1:1000 concentration. The primary antibody used was the following one: calcineurin (Cell Signaling Technology, Cat. #2614S). Secondary HRP-antibodies (Agilent) were incubated for 1h at RT rocking. Blots were developed using the Supersignal Femto developer (Thermo Scientific, Ct. # 34095).
Dual-luciferase assays
Dual luciferase assays were performed using the Promega Dual-Luciferase Reporter Assay System according to the manufacturer’s instructions. The RNAi target sequence was cloned into the 3′ UTR of the Renilla luciferase gene on a psiCHECK-2 (Promega) vector, and firefly luciferase was used as a reference control.
Cells were incubated and transfected in 48-well cluster plates. Cells were seeded 1 day prior to transfection and transfected at 50% confluency. Each experiment was repeated in its entirety at least three times to obtain biological replicates.
We used a single-step transfection protocol for Figures 2B–2H and a two-step transfection protocol for Figures 2I–2K and 3A.
After transfection, at the designated time point for each experiment, 48-well plates were removed. The medium was carefully aspirated from each well. The wells were then washed once with 1× PBS and aspirated dry. 100 μL of 1× Promega passive lysis buffer was added to each well. The plates were then covered in aluminum foil and frozen at −80°C or placed on a shaker for gentle agitation (∼70 rpm) at room temperature for ∼30 min. If frozen, the cells were thawed on a shaker with gentle agitation for at least 30 min prior to the dual-luciferase assay. Before the assay, a visual inspection of the wells was performed to ensure that the cells were well lysed.
Cell lysates were assayed using the Dual-Luciferase Reporter Assay Kit (Promega) according to the manufacturer’s instructions. Renilla luciferase values were normalized to firefly luciferase in each technical replicate (each well).
Technical triplicates within the experiment were averaged to obtain a single biological replicate value. All graphs represent the results of at least three independent biological replicate experiments.
Single-step co-transfection protocol
For each experiment, we created a master mix of the psiCHECK (Promega) reporter plasmid in OptiMEM (Thermo Fisher Scientific). This master mix was separated into aliquots, and the pBluescript (Agilent) control or one of the activator plasmids was added. The new mixtures were then partitioned again for addition of Cond-siRNA complexes at varying concentrations. Finally, a 1:50 dilution of Lipofectamine 2000 (L2K) was added at a 1:1 volume ratio to the plasmid + Cond-siRNA mixtures to achieve the manufacturer’s recommended dilution of 1:100 L2K and incubated at room temperature according to the manufacturer’s recommendation.
For each experimental condition (combination of activator and c-siRNA at a specific concentration), we prepared enough of the mixture (3.3× the amount needed) to transfect 3 separate wells as technical replicates.
Thus, each well in the 48-well plate received 40 μL of the transfection mixture, consisting of
-
•
16 μL psiCHECK and activator plasmids in OptiMEM,
-
•
4 μL 50× Cond-siRNA in 1× PBS buffer, and
-
•
20 μL 1:50 dilution of L2K.
PBS is PBS without calcium or magnesium treated with DEPC (diethyl pyrocarbonate) to remove any RNase activity.
Immediately prior to transfection, we replaced the medium in each well with 160 μL of fresh medium and then added 40 μL of the transfection mixture for a final volume of 200 μL/well with
-
•
40 ng psiCHECK-2 dual-luciferase reporter plasmid,
-
•
120 ng pBluescript or activator-expressing plasmid, and
-
•
Cond-siRNA complexes at the indicated concentrations.
Two-step transfection protocol
This protocol was used to generate data for Figures 2I–2K and 3A.
-
1.Transfection 1 with target and activator plasmids (time −8 h).
-
•For the two-step transfections, we created a master mix of the psiCHECK (Promega) reporter plasmid in OptiMEM (Thermo Fisher Scientific). This master mix was separated into aliquots, and the pBluescript (Agilent) control or one of the activator plasmids was added. A 1:50 dilution of L2K was added at a 1:1 volume ratio to the plasmid mixtures to achieve the manufacturer’s recommended dilution of 1:100 L2K and incubated at room temperature to form lipoplexes according to the manufacturer’s recommendation.
-
•For each experimental condition, we prepared enough mixture to transfect 3 separate wells as technical replicates.
-
•Thus, each well in the 48-well plate received 40 μL of the transfection mixture, consisting of
-
i.20 μL of psiCHECK and activator plasmids in Opti-MEM and
-
ii.20 μL of 1:50 dilution L2K.
-
i.
-
•Immediately prior to transfection, we replaced the medium in each well with 160 μL of fresh medium and then added 40 μL of the transfection mixture for a final volume of 200 μL/well containing
-
i.40 ng psiCHECK-2 dual-luciferase reporter plasmid and
-
ii.120 ng pBluescript or activator-expressing plasmid.
-
i.
-
•The transfection mixture was removed and gently washed with medium after 6 h (−2 h). 160 μL fresh medium was added to each well, and cell incubation continued until the second transfection.
-
•
-
2.Transfection 2 with Cond-siRNA complexes (time 0).
-
•8 h after transfection 1, we transfected Cond-siRNAs at varying concentrations, as specified for the experiment, using RNAiMAX reagent (Thermo Fisher Scientific).
-
•For each experimental condition, we prepared enough of each concentration of Cond-siRNA for technical triplicates of each target/activator combination in PBS. Each Cond-siRNA dilution was mixed with an equal volume of 1:50 RNAiMAX in OptiMEM and incubated at room temperature to form lipoplexes according to the manufacturer’s instructions.
-
•Specifically, each well in the 48-well plate received 40 μL transfection mixture consisting of
-
i.20 μL Cond-siRNA at 10× final concentration (8 μL of PBS +12 μL of OptiMEM) and
-
ii.20 μL 1:50 dilution RNAiMAX.
-
i.
-
•
-
3.Maintenance
-
•Time 0 is marked by addition of the co-transfection mixture to cells for the single-step protocol and as the time of addition of the Cond-siRNA complexes (transfection 2) for the two-step protocol. The medium was replaced 18 h after transfection, at least once each following day, and 6 h before lysate preparation.
-
•
MD simulations
Atomistic models of Cond-siRNAs were built using Nucleic Acid Builder40 and custom scripts and edited with the Accelrys (now BIOVIA, a division of Dassault Systems) Cerius2 package to create appropriate chemical modifications.
A hybrid force field (FF) was created by combining previous Amber FF parameters reported for RNA,41 2ʹOMe,30 LNA,29 and PS42 modifications. Prior reports did not give a parameter set for LNA thymidine. Thus, FF parameters for the LNA sugar ring were derived from the LNA FF, and parameters for the base were derived from the Amber03 FF. Charges were calculated using the RESP ESP charge Derive (RED) server (http://q4md-forcefieldtools.org/REDServer/).
FF parameters for non-DNA components, such as the C3 linker, terminal amine modifications, and terminal PEG linkers, were taken from the GAFF FF.32
All structures were placed in a periodic box with 15-Å spacing on each side and then solvated with TIP3 waters.43 Mg2+ ions were first added to neutralize half of the charge, and then Na+ was added to neutralize the second half. Finally, additional Na+ and Cl− ions were added to 150 mM concentration.
MD simulations were run using the LAMMPS44 GPU-compatible release (December 21, 2016) on NVidia K80 GPUs. Structures were minimized first with the steepest descent option and then with conjugate gradient algorithms for 500 steps and then equilibrated by MD simulations at 310 K using the NVT ensemble over the course of 10 ps using a 1-fs time step. The resulting structures then underwent 10 ps of NPT MD at 310 K and 350 atm to relax the periodic box and ensure positive net pressure. For the NVT simulations, we used a Nosé-Hoover thermostat with a 100-fs time constant. For the NPT simulations, we used a Nosé-Hoover barostat with a 1-ps time constant.
Equilibrated structures then underwent 20 ns of MD at 310°K (NVT ensemble, 1-fs time step).
To obtain the structures presented in Figure 3, the structure with the lowest potential energy from the MD trajectory was extracted, and conjugate gradient energy minimization was applied for 500 steps.
Constructs were visualized using the UCSF Chimera package.45 Helical parameters were calculated from simulation trajectories using the X3DNA.46
Statistical analysis
Data were analyzed using GraphPad Prism 9. Statistical comparisons were made using ordinary two-way ANOVA. p values are shown as ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001.
Acknowledgments
This research was supported by grants from the NSF (NSF EFRI-ODISSEI 1332411 and CMMI-SNM 1120890), the NIH (NIH R01 AI042552, NIH/NCI P30 CA033572 and NIH R35HL150807), and the Margaret E. Early Medical Research Trust and by gifts to the MSC and to the Gehr Family Center for Leukemia Research. We thank Paul Rothemund for useful comments on the manuscript and feedback and advice during the course of the research. We thank Erik Winfree for helpful comments on the manuscript.
Author contributions
S.-p.H., L.S., M.G., A.M.S., M.B.H.S., R.M., S.S., R.H., J.D., Y.-H.K., and G.M. conducted experiments. S.-p.H., L.S., S.D., J.J.R., and W.A.G. designed the experiments. S.-p.H. and S.D. wrote the manuscript. S.D., J.J.R., and W.A.G. provided critical editing and revision of the manuscript. S.D., J.J.R., and W.A.G. were responsible for funding of the project.
Declaration of interests
S.-p.H., L.S., W.A.G., S.D., and J.J.R. hold financial interests in Switch Therapeutics, a company formed to develop therapeutic applications of conditional siRNAs. S.-p.H. and L.S. are now employees of Switch Therapeutics. Switch Therapeutics played no role in the funding, design, or analysis of any of the studies described here. The authors are inventors on granted and pending patents covering novel aspects of conditional RNAi technologies and applications: US 9,029,524 (granted; Caltech; S.-p.H., Robert D. Barish, and W.A.G.; fundamental concepts and designs for Cond-siRNAs), US 9,115,355 (granted; Caltech, City of Hope; S.-p.H., W.A.G., L.S., and J.J.R.; Exonuclease blocking domain for released siRNA), US 9,725,715 (granted; Caltech, City of Hope; S.-p.H., W.A.G., L.S., and J.J.R.; design of Cond-siRNA), PCT/US2019/046075 (pending; Caltech, City of Hope; M.B.H.S., S.-p.H., W.A.G., L.S., and J.J.R.; chemical modifications for Cond-siRNAs), and PCT/US2018/046379 (pending; Caltech, City of Hope, Massachusetts General Hospital; S.D., A.M.S., S.-p.H., L.S., J.D., R.H., S.S., W.A.G., and J.J.R.; Cond-siRNAs for treatment of cardiac hypertrophy).
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.12.039.
Contributor Information
Saumya Das, Email: sdas@mgh.harvard.edu.
John J. Rossi, Email: jrossi@coh.org.
William A. Goddard, III, Email: wagoddard3@gmail.com.
Supplemental information
References
- 1.Strebhardt K., Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat. Rev. Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y.-J., Groves B., Muscat R.A., Seelig G. DNA nanotechnology from the test tube to the cell. Nat. Nano. 2015;10:748–760. doi: 10.1038/nnano.2015.195. [DOI] [PubMed] [Google Scholar]
- 3.Groves B., Chen Y.-J., Zurla C., Pochekailov S., Kirschman J.L., Santangelo P.J., Seelig G. Computing in mammalian cells with nucleic acid strand exchange. Nat. Nano. 2016;11:287–294. doi: 10.1038/nnano.2015.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benenson Y., Gil B., Ben-Dor U., Adar R., Shapiro E. An autonomous molecular computer for logical control of gene expression. Nature. 2004;429:423–429. doi: 10.1038/nature02551. [DOI] [PubMed] [Google Scholar]
- 5.Kumar D., Kim S.H., Yokobayashi Y. Combinatorially inducible RNA interference triggered by chemically modified oligonucleotides. J. Am. Chem. Soc. 2011;133:2783–2788. doi: 10.1021/ja1107436. [DOI] [PubMed] [Google Scholar]
- 6.Hochrein L.M., Ge T.J., Schwarzkopf M., Pierce N.A. Signal transduction in human cell lysate via dynamic RNA nanotechnology. ACS Synth. Biol. 2018;7:2796–2802. doi: 10.1021/acssynbio.8b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bindewald E., Afonin K.A., Viard M., Zakrevsky P., Kim T., Shapiro B.A. Multistrand structure prediction of nucleic acid assemblies and design of RNA switches. Nano Lett. 2016;16:1726–1735. doi: 10.1021/acs.nanolett.5b04651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerjee P., Bandyopadhyay A. Cytosolic dynamics of annexin A6 trigger feedback regulation of hypertrophy via atrial natriuretic peptide in cardiomyocytes. J. Biol. Chem. 2014;289:5371–5385. doi: 10.1074/jbc.M113.514810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkins B.J., Dai Y.-S., Bueno O.F., Parsons S.F., Xu J., Plank D.M., Jones F., Kimball T.R., Molkentin J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 10.Meng X.-m, Nikolic-Paterson D.J., Lan H.Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 11.Youle R.J., Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 12.Zolot R.S., Basu S., Million R.P. Antibody-drug conjugates. Nat. Rev. Drug Discov. 2013;12:259–260. doi: 10.1038/nrd3980. [DOI] [PubMed] [Google Scholar]
- 13.Zuckerman J.E., Davis M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015;14:843. doi: 10.1038/nrd4685. [DOI] [PubMed] [Google Scholar]
- 14.Rautio J., Kumpulainen H., Heimbach T., Oliyai R., Oh D., Järvinen T., Savolainen J. Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 15.Toscano M.G., Romero Z., Muñoz P., Cobo M., Benabdellah K., Martin F. Physiological and tissue-specific vectors for treatment of inherited diseases. Gene Ther. 2011;18:117–127. doi: 10.1038/gt.2010.138. [DOI] [PubMed] [Google Scholar]
- 16.Benenson Y. Biomolecular computing systems: principles, progress and potential. Nat. Rev. Genet. 2012;13:455–468. doi: 10.1038/nrg3197. [DOI] [PubMed] [Google Scholar]
- 17.Gao X.J., Chong L.S., Kim M.S., Elowitz M.B. Programmable protein circuits in living cells. Science. 2018;361:1252–1258. doi: 10.1126/science.aat5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain R., Frederick J.P., Huang E.Y., Burke K.E., Mauger D.M., Andrianova E.A., Farlow S., Siddiqui S., Pimental J., Cheung-Ong K., et al. MicroRNAs enable mRNA therapeutics to selectively program cancer cells to self-destruct. Nucleic Acid Ther. 2018;28:285–296. doi: 10.1089/nat.2018.0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Setten R.L., Rossi J.J., Han S.-p. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019;18:421–446. doi: 10.1038/s41573-019-0017-4. [DOI] [PubMed] [Google Scholar]
- 20.Springer A.D., Dowdy S.F. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018;28:109–118. doi: 10.1089/nat.2018.0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X., Yang X., Qi J., Seeman N.C. Antiparallel DNA double crossover molecules as components for nanoconstruction. J. Am. Chem. Soc. 1996;118:6131–6140. [Google Scholar]
- 22.Srinivas N., Ouldridge T.E., Šulc P., Schaeffer J.M., Yurke B., Louis A.A., Doye J.P.K., Winfree E. On the biophysics and kinetics of toehold-mediated DNA strand displacement. Nucleic Acids Res. 2013;41:10641–10658. doi: 10.1093/nar/gkt801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amaral P.P., Dinger M.E., Mercer T.R., Mattick J.S. The eukaryotic genome as an RNA machine. Science. 2008;319:1787–1789. doi: 10.1126/science.1155472. [DOI] [PubMed] [Google Scholar]
- 24.Valencia-Sanchez M.A., Liu J., Hannon G.J., Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 25.Rajkowitsch L., Chen D., Stampfl S., Semrad K., Waldsich C., Mayer O., Jantsch M.F., Konrat R., Blasi U., Schroeder R. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007;4:118–130. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- 26.Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014;24:374–387. doi: 10.1089/nat.2014.0506. [DOI] [PubMed] [Google Scholar]
- 27.Vester B., Wengel J. LNA (locked nucleic acid):high -affinity targeting of complementary RNA and DNA†. Biochemistry. 2004;43:13233–13241. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 28.Jaramillo-Botero A., Nielsen R., Abrol R., Su J., Pascal T., Mueller J., Goddard W.A. In: Multiscale Molecular Methods in Applied Chemistry. Kirchner B., Vrabec J., editors. Vol. 307. Springer Berlin Heidelberg; 2012. First-Principles-based multiscale, multiparadigm molecular mechanics and dynamics methods for describing complex chemical processes; pp. 1–42. [DOI] [PubMed] [Google Scholar]
- 29.Condon D.E., Yildirim I., Kennedy S.D., Mort B.C., Kierzek R., Turner D.H. Optimization of an AMBER force field for the artificial nucleic acid, LNA, and benchmarking with NMR of L(CAAU) J. Phys. Chem. B. 2014;118:1216–1228. doi: 10.1021/jp408909t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aduri R., Psciuk B.T., Saro P., Taniga H., Schlegel H.B., SantaLucia J. AMBER force field parameters for the naturally occurring modified nucleosides in RNA. J. Chem. Theor. Comput. 2007;3:1464–1475. doi: 10.1021/ct600329w. [DOI] [PubMed] [Google Scholar]
- 31.Pérez A., Marchán I., Svozil D., Sponer J., Cheatham T.E., III, Laughton C.A., Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 33.Rajput A., Dominguez San Martin I., Rose R., Beko A., LeVea C., Sharratt E., Mazurchuk R., Hoffman R.M., Brattain M.G., Wang J. Characterization of HCT116 human colon cancer cells in an orthotopic model. J. Surg. Res. 2008;147:276–281. doi: 10.1016/j.jss.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 34.Alting-Mees M.A., Short J.M. pBluescript II: gene mapping vectors. Nucleic Acids Res. 1989;17:9494. doi: 10.1093/nar/17.22.9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scherer L.J., Frank R., Rossi J.J. Optimization and characterization of tRNA-shRNA expression constructs. Nucleic Acids Res. 2007;35:2620–2628. doi: 10.1093/nar/gkm103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T.L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zadeh J.N., Steenberg C.D., Bois J.S., Wolfe B.R., Pierce M.B., Khan A.R., Dirks R.M., Pierce N.A. NUPACK: analysis and design of nucleic acid systems. J. Comput. Chem. 2011;32:170–173. doi: 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- 38.Fornace M.E., Porubsky N.J., Pierce N.A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synth. Biol. 2020;9:2665–2678. doi: 10.1021/acssynbio.9b00523. [DOI] [PubMed] [Google Scholar]
- 39.Mathews D.H., Sabina J., Zuker M., Turner D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure 11 edited by I. Tinoco. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 40.Macke T.J., Case D.A. Molecular Modeling of Nucleic Acids. Vol. 682. American Chemical Society; 1997. Modeling unusual nucleic acid structures; pp. 379–393. [Google Scholar]
- 41.Duan Y., Wu C., Chowdhury S., Lee M.C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 42.Lind K.E., Sherlin L.D., Mohan V., Griffey R.H., Ferguson D.M. Molecular Modeling of Nucleic Acids. Vol. 682. American Chemical Society; 1997. Parameterization and simulation of the physical properties of phosphorothioate nucleic acids; pp. 41–54. [Google Scholar]
- 43.Mark P., Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A. 2001;105:9954–9960. [Google Scholar]
- 44.Plimpton S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995;117:1–19. [Google Scholar]
- 45.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Colasanti A.V., Lu X.-J., Olson W.K. Analyzing and building nucleic acid structures with 3DNA. J. Vis. Exp. 2013;74:e4401. doi: 10.3791/4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.