

Abstract
Metabolic rewiring is one of the hallmarks of cancer. Altered de novo lipogenesis is one of the pivotal metabolic events deregulated in cancers. Sterol regulatory element-binding transcription factor 1 (SREBP1) controls the transcription of major enzymes involved in de novo lipogenesis, including ACLY, ACACA, FASN, and SCD. Studies have shown the increased de novo lipogenesis in human hepatocellular carcinoma (HCC) samples. Multiple mechanisms, such as activation of the AKT/mechanistic target of rapamycin (mTOR) pathway, lead to high SREBP1 induction and the coordinated enhanced expression of ACLY, ACACA, FASN, and SCD genes. Subsequent functional analyses have unraveled these enzymes’ critical role(s) and the related de novo lipogenesis in hepatocarcinogenesis. Importantly, targeting these molecules might be a promising strategy for HCC treatment. This paper comprehensively summarizes de novo lipogenesis rewiring in HCC and how this pathway might be therapeutically targeted.
Keywords: hepatocellular carcinoma, lipogenesis, targeted therapy
Hepatocellular carcinoma (HCC), a prominent type of primary liver cancer, is the fifth common tumor and the third leading cancer-related death disease worldwide.1 Infection by hepatitis viruses, including hepatitis B and hepatitis C viruses, and alcohol consumption are the main etiological factors for HCC.2 In recent years, fatty liver-related diseases, including nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH), have also become relevant predisposing conditions for the development of this disease.3
For patients with early-stage HCC, liver resection and liver transplant are effective treatment options. However, most HCCs are diagnosed at a late stage, precluding radical therapies to these patients. Traditional chemotherapy is not effective against HCC. Sorafenib, a multikinase inhibitor, has been the first-line treatment drug for advanced HCC for the past decade. However, it has minimal efficacy.4 During the past 2 years, the treatment landscape for HCC has been dramatically expanded with the approval of several multikinase inhibitors, including Regorafenib, Cabozantinib, and Ramucirumab as first or second-line therapies for progressed HCC.5,6 Significantly, recent clinical studies have demonstrated the efficacy of immunotherapy against HCC.7 Specifically, atezolizumab (an anti-PDL1 antibody) combined with bevacizumab (an antivascular endothelial growth factor antibody) led to better overall and progression-free survival than Sorafenib in patients with unresectable HCC.8 Consequently, immunotherapy has become the first-line treatment strategy against advanced liver cancer. However, a significant percentage of patients do not respond to this combination therapy, and reliable biomarkers for patient selection are lacking. Thus, there is a great need to understand the molecular mechanisms underlying hepatocarcinogenesis to develop novel and effective therapies against this malignancy.
Recent genetic studies have uncovered the genomic landscape of HCC, indicating that HCC is a highly heterogeneous disease.9–11 The most common genetic events occurring in HCC include mutations of TP53, CTNNB1, AXIN1, ARID1A, and ARID2 genes, and amplification of c-MYC, CCND1, and FGF19. Unfortunately, except for the FGF19/FGFR4 pathway, none of these genetic alternations appears to be druggable.12
Mounting evidence has demonstrated that deregulated cellular metabolism is a hallmark of various cancer types,13,14 including HCC.15 To maintain rapid cell growth and proliferation, the tumor cells undergo a metabolic adaptation event known as the “Warburg effect,” consisting of the switch of the metabolism from oxidative phosphorylation to aerobic glycolysis.16,17 The aerobic glycolysis provides energy and substrates for biomass production for cancer cells and, consequently, increases pyruvate accumulation. While most pyruvate is transferred to lactate and eliminated, some is converted into acetyl-CoA, the primary substrate for de novo lipogenesis (DNL).18
Increased DNL is one of the most relevant aberrant metabolic events in tumor development and progression.19,20 Studies have demonstrated the pivotal role of DNL in hepatocarcinogenesis, suggesting that targeting DNL might effectively prevent and treat HCC. Here, we provide a review of the genes involved in the DNL pathway, their function(s) in hepatocarcinogenesis, and the therapeutic approaches to target this metabolic pathway for HCC treatment.
Overview of the de Novo Lipogenesis Pathway
DNL is a metabolic process leading to the biosynthesis of fatty acids from carbohydrates.21 When the carbohydrate intake exceeds the body’s storage and oxidation capacities, energy can be stored as fat in the form of triglycerides or other lipid molecules through the DNL pathway. Dysregulation of DNL can lead to various metabolic consequences. For example, studies suggest that elevated DNL is involved in NAFLD pathogenesis,22 and it is linked to insulin resistance.23,24
The biochemical process of DNL is modulated by a series of enzymatic reactions (Fig. 1). As the primary substrate for DNL, glucose from dietary carbohydrates undergoes a series of glycolysis reactions and generates pyruvate. Pyruvate produces citrate via the tricarboxylic acid cycle in the mitochondria. The citrate is transported back to the cytoplasm and converted into acetyl-CoA by the action of ATP-citrate lyase (ACLY). Through Acetyl-CoA carboxylase (ACAC) activity, especially ACACα (ACACA), acetyl-CoA is carboxylated to malonyl-CoA, which is utilized for the production of 16-carbon saturated palmitate. The critical rate-limiting enzyme in this step is fatty acid synthase (FASN). Palmitate can be further elongated into long-chain fatty acids. Palmitate can also be converted into monounsaturated fatty acids (MUFAs), an event catalyzed by stearoyl-CoA desaturase (SCD). All these fatty acids can be incorporated into complex lipids, including triglycerides, phospholipids, and cholesterol.
Fig. 1.
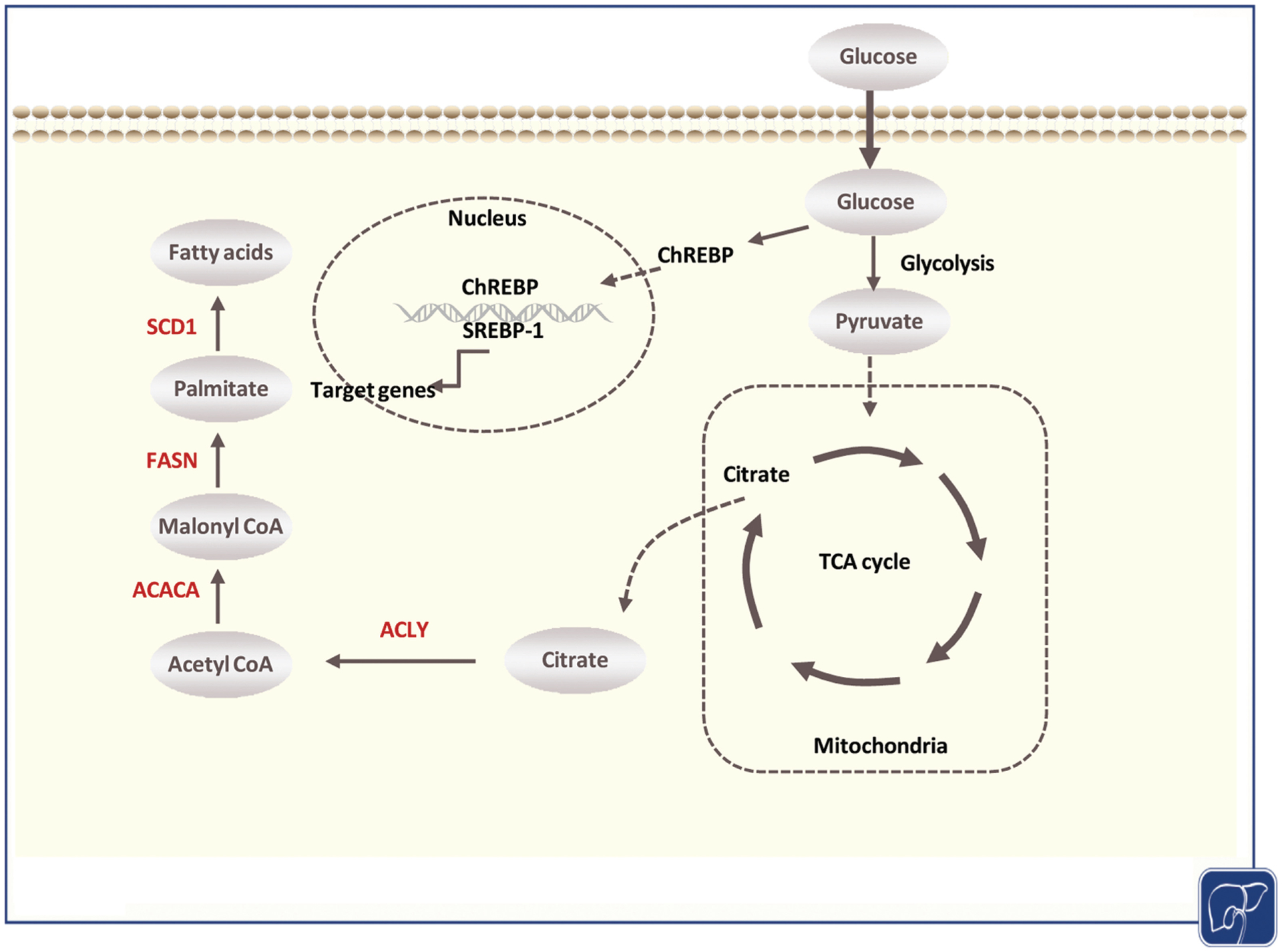
Overview of the de novo lipogenesis pathway. Dietary glucose undergoes a series of reactions of glycolysis to generate pyruvate. Pyruvate enters mitochondria to be used in the tricarboxylic acid (TCA) cycle to produce energy and form citrate. After transported back to the cytoplasm, citrate is converted to acetyl-CoA by the action of ATP-citrate lyase (ACLY). Acetyl-CoA is subsequently converted to malonyl-CoA, which is utilized for the 16-carbon saturated palmitate synthesis by fatty acid synthase (FASN) activity. Stearoyl-CoA desaturase (SCD) converts saturated fatty acids to monounsaturated fatty acids. Regulation of de novo lipogenesis (DNL) is mainly mediated by sterol regulatory element-binding protein-1 (SREBP-1), which activates DNL enzymes’ expression, including ACLY, ACC, FASN, and SCD. Based on the TCGA LIHC database, these enzymes (labeled in red) are significantly upregulated at the transcriptional level in liver cancer compared with matched adjacent nontumorous tissues. TCGA LIHC, The Cancer Genome Atlas Liver Hepatocellular Carcinoma.
The activity of DNL is strongly affected by the nutritional status. High insulin and high glucose concentrations induce the transcription of lipogenic enzymes,25,26 which ultimately triggers fatty acids synthesis. Dietary fructose significantly improves the efficacy of enzymes involved in DNL.27 Regulation of lipogenesis is predominantly at the transcriptional level. The effects of nutrients on lipogenic gene expression are mediated by sterol regulatory element-binding protein-1 (SREBP-1)28 and carbohydrate response element-binding protein.29
In physiological conditions, DNL mainly occurs in metabolic tissues such as liver and adipose tissues,30 and the liver is the major contributor to whole-body lipogenesis. However, in cancer cells, an increased DNL is often detected, providing fatty acids for tumor cell growth, even in the presence of exogenous lipids.31 Indeed, the reprogramming of fatty acid metabolism in cancer cells is pivotal for tumor survival, proliferation, and metastasis. Highly proliferative cancer cells require fatty acids to satisfy their unlimited energy demands for various purposes, such as membrane biogenesis, energy production, and lipid modification of proteins.17 Therefore, deregulated DNL directly leads to cellular fatty-acid accumulation and affects fundamental cellular processes, including signal transduction and gene expression. Several studies have shown that the dysregulation of lipogenic enzymes involved in DNL occurs in multiple cancer types, including HCC.20,31,32
SREBP1
SREBPs are a family of transcriptional regulators that modulate cellular lipid metabolism and homeostasis.33 Mammalian cells express three isoforms of SREBP proteins, SREBP1a, SREBP1c, and SREBP2, encoded by two genes: SREBF1 and SREBF2. SREBP1a and SREBP1c are formed from the same gene (SREBF1) but are transcribed by different promoters.34 SREBP1 transcriptionally activates major DNL enzymes’ expression, including ACLY, ACC, FASN, and SCD. SREBP2, on the other hand, is the central transcription factor regulating cholesterol biosynthesis.
Due to its crucial function in regulating DNL, SREBP1 plays a central role in modulating cancer metabolism.35 Previous studies revealed the activation of SREBP1 in prostate cancer, breast cancer, and glioblastoma.36–38 SREBP1 upregulation has also been observed in human HCC,39 where SREBP1 expression correlates with tumor proliferation rate and dismal prognosis.40 Moreover, the Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA LIHC) database analysis revealed the coordinated upregulation of SREBP1 downstream targets, including ACLY, ACACA, FASN, and SCD.
Multiple mechanisms lead to the increased expression of SREBP1, the master regulator of the DNL pathway, in HCC. Activated AKT kinase is the critical signaling pathway activating SREBP1 via the mTORC1 cascade during hepatocarcinogenesis.20 When an activated form of AKT is expressed in the mouse liver, it upregulates SREBP1 and its downstream effectors, and eventually, the fatty liver phenotype.41 A similar phenotype is observed in liver-specific Pten knockout (KO) mice,42 which depends on AKT2 overactivation.43 Also, it has been shown that activated AKT phosphorylates cytosolic phosphoenolpyruvate carboxykinase 1 (PCK1), INSIG1, and INSIG2, leading to nuclear accumulation of SREBP1.44 Finally, a recent study showed that ZHX2, a tumor suppressor, inhibits SREBP1 and its mediated DNL. As loss of ZHX2 expression often occurs in HCC, this might represent an alternative mechanism of SREBP1-unconstrained activation in this tumor type.45
Once activated, SREBP1 proteins induce the transcription of DNL genes, promoting tumor proliferation, and eventually, tumor development in mice.44 Silencing of SREBP1 suppresses HCC cell growth in culture.20,39 Moreover, the enhanced SREBP1 protein stability promotes proliferation, migration, and invasion of HCC in vitro and xenograft liver cancer models.46 Genetic or pharmacologic inhibition of SREBP1 dramatically reduced diethylnitrosamine (DEN)-induced HCC progression in mice.47 Altogether, these data demonstrate the functional role of SREBP1 in regulating HCC lipogenesis and growth.
ACLY
As a transcriptional target of SREBP1, ACLY catalyzes the conversion of citrate to acetyl-CoA and oxaloacetate,48 providing the essential components for fatty acid biosynthesis (Fig. 1). Overexpression of ACLY correlates with a dismal prognosis, progression, and metastasis of HCC patients.49 The TCGA LIHC dataset also confirmed that high ACLY expression is associated with poor survival. ACLY is involved in several oncogenic pathways. For instance, ACLY can participate in the Wnt/β-catenin cascade in HCC by regulating the stability of β-catenin.49 AKT directly regulates the phosphorylation and activation of ACLY and upregulates its mRNA levels through SREBP1.50 The combination of an ACLY inhibitor and anti-PD-L1 therapy dramatically suppressed DEN-induced hepatocarcinogenesis in mice. The antitumor effect of ACLY suppression was mediated by increased reactive oxygen species production and AMP-activated protein kinase (AMPK) activation/phosphorylation.51 BMS-303141, an ACLY inhibitor, triggers endoplasmic reticulum stress and induces apoptosis of HCC cells. Moreover, inhibition of ACLY synergizes with Sorafenib for improved efficacy against HCC in vivo.52 Also, a recent study suggests that ACLY inhibition is effective in tumors with high aerobic glycolysis rates because they are more dependent on ACLY for acetyl-CoA generation.53
ACAC
ACAC is one of the key regulators of DNL and catalyzes the carboxylation of acetyl-CoA to malonyl-CoA (Fig. 1). There are two isoforms (α and β) of ACAC in mammals, and ACACA is the major isoform involved in DNL. As the first rate-limiting enzyme in lipogenesis, ACACA was hypothesized to play essential functions in regulating cancer metabolism and tumor development. The increased expression of ACACA has been reported in human HCC samples.54 Moreover, bioinformatics analysis revealed that overexpression of ACACA is highly associated with a poor prognosis in liver cancer,55 a finding confirmed by the TCGA LIHC dataset. In vitro, suppression of ACACA in human HCC cell lines reduces cell proliferation and viability.20 In vivo, it has been suggested that ACACA-mediated DNL determines the intracellular lipid content, and overexpression of ACACA promotes HCC growth.56 ACACA has also been revealed to regulate fatty acid oxidation in this tumor type.56 At the molecular level, ACACA is inhibited via phosphorylation by AMPK. Loss-of-function mutations in AMPK phosphorylation sites of ACAC lead to elevated hepatic lipogenesis and fibrosis development in mice.57 In the DEN-induced murine HCC model, ACAC nonphosphorylatable mutant knock-in mice were found to have increased liver tumor lesions compared to wild-type mice,58 indicating that ACAC phosphorylation would inhibit the development of HCC. A potent liver-specific ACAC inhibitor, ND-654, effectively suppressed liver tumor burden, inflammation, and proliferation in the DEN model in rats.58 This evidence suggests a potential therapeutic target role of ACAC in HCC.
FASN
FASN is the key lipogenic enzyme to catalyze fatty acid synthesis. Specifically, FASN catalyzes the synthesis of palmitate from acetyl-CoA and malonyl-CoA in the presence of NADPH (Fig. 1). Since FASN is a critical enzyme during DNL, and no other isoforms of this gene exist, FASN has been extensively studied as a possible target for suppressing DNL in various diseases, including cancer. Numerous studies have reported the increased expression of FASN in multiple tumor types, including HCC.15,32,59,60 While SREBP1 is the pivotal transcription factor inducing FASN overexpression in HCC, FASN has been shown to be also regulated at posttranscriptional levels. For example, it has been demonstrated that the USP2A protein inhibits proteasome-mediated FASN degradation, inducing FASN stabilization and increased activity.61 Besides, ACAT1-induced GNPAT acetylation also promotes FASN stabilization in HCC.62 The functional significance of FASN in hepatocarcinogenesis has been demonstrated both in vitro and in vivo. In vitro, inhibition of FASN via siRNA or small-molecule inhibitors effectively suppresses HCC cell proliferation.63 In genetically engineered mouse models, liver-specific deletion of Fasn completely prevented hepatocarcinogenesis driven by the overexpression of AKT, either alone or in combination with c-Met proto-oncogene.64,65 Knocking down FASN in hepatocytes via AAV-shFasn or treating mice with the FASN inhibitor Orlistat significantly reduced tumor burden in an mTOR-driven HCC mouse model.66 Orlistat treatment also delayed HCC driven by activated AKT and c-Met oncogenes in mice.67 Additional investigations have demonstrated that FASN is required for c-Myc-dependent hepatocarcinogenesis.68 Furthermore, loss of FASN retards HCC development induced by overexpression of c-Met and loss of Pten (c-Met/sgPten) in mice.69 Overall, these data indicate that FASN suppression is a promising therapeutic strategy for HCC treatment.
SCD
SCD is the lipogenic enzyme catalyzing palmitate desaturation and the production of MUFAs (Fig. 1). The conversion of saturated fatty acids into MUFA is tightly linked to the activation of glycolysis and fatty acid synthesis in cancer cells,70 implying that SCD might be relevant in tumorigenesis. In HCC, SCD is upregulated and associated with shorter disease-free survival.71,72 In a combined proteomic and lipidomic profiling study, upregulated hepatic SCD was identified as a reliable marker for HCC diagnosis and progression.73 Mechanistically, SCD expression is induced by the AKT/mTOR pathway, whereas AMPK suppresses its expression.41,70,74,75 SCD overexpression is also observed in human HCC cell lines. At the molecular level, SCD has been found to regulate HCC cell proliferation by inducing MYCN expression.76 Enhanced activity of SCD is also associated with chemotherapy resistance and cell proliferation in vitro through phosphatidylinositol 3 kinase (PI3K)/c-Jun N-terminal kinase activation.71 In a patient-derived HCC xenograft model, SCD inhibitors enhanced sensitivity to Sorafenib via modulation of the ER stress.72 Therefore, targeting SCD might be therapeutically beneficial to HCC patients by hindering fatty acid biosynthesis and alleviating drug resistance.
Molecular Classification of HCC and de Novo Lipogenesis
There have been significant efforts to clarify human HCCs based on molecular and genetic signatures during the past decade. HCCs have been divided into different subgroups in different studies.9,77–80 Notably, it has become clear that HCCs can be classified into two major groups: the proliferation and nonproliferation classes.81 While the expression of lipogenic pathway genes has not been analyzed in detail in these molecular subgroups, it has been found that the HCC proliferation class tends to be p-RPS6(+) and frequently harbors mutations of RPS6KA3 or TSC1/2,81 suggesting the activation of mTOR pathway. As activated mTOR is the principal regulator of lipogenesis in HCC, it is tempting to hypothesize that increased DNL is enriched in the proliferation class of human HCCs. Consistent with the hypothesis, the proliferation class of HCC shows higher AFP expression.81 In a previous study, it has been demonstrated that increased ACACA expression is associated with elevated AFP levels in HCC.58 In addition, HCCs with mutation of CTNNB1 belong to the nonproliferation class. Significantly, DNL is not elevated in CTNNB1 mutant HCC, and mouse HCC lesions induced by activated β-catenin do not require DNL for their growth.15 At the molecular level, it is conceivable that highly proliferative HCC cells require increased fatty acids as building blocks and/or energy sources, thus leading to increased DNL. Obviously, additional detailed analyses of the expression of DNL genes in different HCC molecular subclasses are required to address this issue entirely.
De novo Lipogenesis in HNF-1α-Mutated Hepatocellular Adenoma
Hepatocellular adenoma (HCA) is a rare benign liver tumor that predominantly occurs in young women. Hepatocyte nuclear factor 1α (HNF-1α) mutations are present in 30 to 40% of HCA,82 which show striking tumor steatosis. HNF-1α mutations induce steatosis by promoting DNL activity by increasing lipogenic gene expression and downregulating liver fatty acid-binding protein.83 In addition, HNF-1α has been demonstrated to act as a tumor suppressor gene, and the inactivation of HNF-1α may be an early step in HCC development.84 These findings suggest that signaling pathways responsible for oncogenic transformation may also contribute to the increased DNL in tumor lesions.
Targeting de Novo Lipogenesis for HCC Treatment
As discussed previously, increased DNL in cancer is required for tumor progression, making this metabolic pathway an attractive therapeutic target for cancers, including HCC.19 Targeting DNL for HCC is particularly appealing as NAFLD and NASH are increasingly common as the underlying etiological factors for HCC. DNL inhibitors may be able to suppress both HCC growth and NAFLD/NASH phenotypes. Small molecules targeting various enzymes in the DNL pathway, including SREBP1, ACLY, ACAC, FASN, and SCD, have been developed (Table 1).85,86 Both in vitro and in vivo studies have provided evidence that these molecules may be effective against HCC.
Table 1.
Small-molecule inhibitors targeting the DNL pathway
| Drug/small molecule | Target gene | Current status | Disease | Reference(s) or clinical trial no. |
|---|---|---|---|---|
| Betulin | SREBP1 | Preclinical | Hepatocellular carcinoma | Li et al 201747, Yin et al 201988 |
| Curcumin | SREBP1 | Preclinical | Hepatocellular carcinoma | You et al 201890 |
| Fatostatin | SREBP1 | Preclinical | Hepatocellular carcinoma | Yu et al 202045 |
| Hydroxycitrate | ACLY | Preclinical | Glioblastomas, breast cancer | Beckner et al 2010115, Ismail et al 2020116 |
| ETC-1002 | ACLY | Phase 3 clinical trial | Cardiovascular diseases | NCT02993406 117 |
| BMS-303141 | ACLY | Preclinical | Hepatocellular carcinoma | Zheng et al 202152 |
| TOFA | ACAC | Preclinical | Hepatocellular carcinoma | Calvisi et al 201120 |
| ND-654 | ACAC | Preclinical | Hepatocellular carcinoma | Lally et al 201958 |
| NDI-010976 | ACAC | Phase 1 clinical trial | Overweight and/or obesity | NCT02876796 118 |
| C75 | FASN | Preclinical | Hepatocellular carcinoma | Gao et al 200663 |
| Orlistat | FASN | FDA approved | Obesity | Guri et al 201766, Zhang et al 202067 |
| Fenofibrate | FASN | FDA approved | Hypercholesterolemia | You et al 2019100 |
| TVB-2640 | FASN | Phase 1 clinical trial | Metabolic syndrome | NCT02948569 119 |
| A939572 | SCD | Preclinical | Hepatocellular carcinoma | Ma et al 201772 |
| SSI-4 | SCD | Preclinical | Hepatocellular carcinoma | Ma et al 201772 |
| CAY10566 | SCD | Preclinical | Hepatocellular carcinoma | Huang et al 2015102 |
| MK8245 | SCD | Phase 1 clinical trial | Type 2 diabetes | NCT00790556 120 |
Abbreviation: FDA, Food and Drug Administration.
SREBP1 is the master regulator of DNL in HCC. Betulin is a lupane-type pentacyclic triterpenoid, and it binds directly to SCAP (SREBP cleavage-activating protein), which inhibits the cleavage and activation of SREBP1.87 Betulin has been shown to prevent DEN-induced HCC formation in mice.47 Betulin also enhances Sorafenib antitumor activity in HCC cells and xenograft models.88 Curcumin is a nonspecific SREBP1 inhibitor.89 It has been reported that curcumin could inhibit HCC cell growth via downregulating SREBP1 expression at mRNA and protein levels.90 Fatostatin is another SREBP1 inhibitor, and it suppresses spontaneous HCC formation in the liver-specific Zhx2 KO mice.45
ACLY could be targeted by inhibitors such as hydroxycitrate91 and bempedoic acid (ETC-1002)92 (Table 1). ACLY inhibition might provide a potential advantage over therapeutic strategies targeting other lipogenic enzymes because it lies upstream of the other lipogenic enzymes.93 ETC-1002, also known as bempedoic acid, is a potent ACLY inhibitor,94 and it is currently in phase 3 clinical trial to prevent vascular diseases.95 In the DEN-induced HCC model, combined ETC-1002 and anti-PDL1 antibody treatment dramatically suppressed HCC formation in mice.51 BMS‐303141 is a cell-permeable ACLY inhibitor.96 The combination of BMS‐303141 and Sorafenib hindered HCC cell growth in a xenograft model.52
ACAC has been a significant target of drugs for the treatment of NAFLD and NASH.97 Some of these drugs have been investigated for their efficacy against HCC. For instance, the ACAC inhibitor TOFA can inhibit AKT-dependent HCC cell growth in vitro.20 ND-654 is a liver-specific ACAC inhibitor. Combined administration of ND-654 and Sorafenib suffices to suppress HCC formation in DEN-induced HCC in rats.58
As a central regulator of DNL, FASN is a major focus for cancer therapeutic studies,98,99 including HCC.15 The FASN inhibitor C75 has been shown to hamper HCC cell growth in vitro.42,49 Also, Orlistat can suppress hepatocarcinogenesis in an mTOR-driven mouse model and in a mouse model consisting of the simultaneous activation of AKT and c-Met oncogenes.66,67 Fenofibrate inhibits FASN via binding to its thioesterase domain of FASN, similar to Orlistat. This drug can induce Hep3B cell apoptosis and necroptosis.100
SCD is another DNL enzyme that has been studied for cancer treatment.101 The SCD inhibitor A939572 suppresses the migration and invasion of human HCC cells.72 Also, the SCD inhibitor SSI-4 reduces HCC growth and enhances Sorafenib toxicity in mouse HCC models.72 CAY10566, another SCD inhibitor, activates the AMPK pathway, leading to HCC cell death.102 MK8245, another SCD inhibitor, has been shown to possess antiproliferative effects on zebrafish HCC models and human HCC cells.103
In summary, numerous small molecules against DNL pathway genes have been developed, and the experimental data support the targeting of DNL for HCC treatment. However, most of these small molecules have yet to be investigated in human HCC. One of the most promising drugs is TVB-2640, a FASN inhibitor currently in phase 1 clinical trial for metabolic syndrome. Recent results suggest that TVB-2640 is well tolerated and effectively reduces DNL in patients with metabolic diseases.104 Similar results have been obtained with NDI-010976, an ACAC inhibitor.105 Thus, it would be essential to investigate whether these drugs are effective in inhibiting HCC growth using experimental approaches.
Challenges and Future Direction
An investigation by Calvisi et al published in 2011 is the first comprehensive analysis of the DNL pathway in HCC.20 Since then, multiple studies have been published, validating the major conclusion that DNL is necessary for hepatocarcinogenesis, and targeting DNL may be an effective treatment strategy against this aggressive malignancy. Despite this progress, many challenges remain, and our understanding of this metabolic pathway in HCC is still incomplete.
One key issue to delineate is that most of the investigations focused on HCC cells. The role of DNL in the HCC microenvironment cells, such as immune cells, cancer-associated fibroblasts, and endothelial cells, has not been adequately analyzed. However, previous evidence has demonstrated the importance of DNL in these cells.106 Of particular importance is the study of DNL in immune cells. Evidence suggests that tumor-infiltrating immune cells undergo metabolic changes that influence their phenotype and functions.107 For instance, increased DNL promotes the proliferation and differentiation of effector T cells.108 In TLR (toll-like receptor)-mediated dendritic cells, lipid biosynthesis promotes their activation by affecting the ER and Golgi expansion.109 Inhibition of ACAC prevents the formation of T helper 17 lineage cells and induces the development of Treg cells.110 As tumor cells increase DNL to sustain their growth, this may also lead to high lipid content in the tumor microenvironment. These lipids could be readily taken up by the cells within the tumor microenvironment, such as M2 macrophages. Studies have shown that these macrophages depend on exogenous fatty acids and fatty acid oxidation for their function.111 Therefore, the fatty acids secreted by the tumor cells may be used by these cells for their development and activity. It is important to note that while there are some studies about DNL in immune cells, few such studies have been conducted in HCC. As concerns immunotherapy, especially immune-checkpoint inhibitors, such as anti-PD1 and/or anti-PDL1-based treatment, are becoming the first line therapeutics against HCC. Thus, it would be critical to determine whether these treatments affect DNL in tumor-infiltrating immune cells. The detailed analysis of these metabolic changes within the tumor cells and immune cells will provide clues for possible combination therapies for improved efficacy.
While DNL produces fatty acids required for tumor growth, fatty acids could also be taken up into cells. Therefore, the exogenous fatty acid uptake can compensate for the loss of DNL in HCC cells.112 Additional studies are required to elucidate the molecular crosstalk between DNL and fatty acid uptake in HCC pathogenesis.
As cellular metabolism is an integrated system, deregulation of DNL may lead to other metabolic alterations as compensatory feedback mechanisms. For example, ablation of Fasn and its mediated DNL in a murine HCC model induced by loss of Pten and overexpression of c-Met led to the compensatory upregulation of the cholesterol biosynthesis pathway. The latter cascade can sustain HCC growth in the absence of DNL. Notably, it has been demonstrated that targeting both DNL and cholesterol biosynthesis completely prevents HCC development in mice.69 Comprehensive analysis of the metabolic pathways modulated by inhibition of DNL will therefore be necessary to design effective combination therapies against HCC.
It is worthwhile to mention that many of the genes responsible for DNL have additional functions besides regulating fatty acid synthesis. For example, ACAC is involved in antioxidant defense and hepatocyte survival.113 Thus, it was not surprising to see that DEN-induced hepatocarcinogenesis in liver-specific depletion of Acac mice leads to increased HCC burden than in wild-type mice, suggesting the possible tumor-suppressing role of ACAC in HCC. Therefore, it is necessary to fully illustrate DNL enzymes’ functions to select the most effective and safe molecules for HCC treatment.
Finally, it is also important to note that not all HCCs have elevated DNL or depend on DNL for their growth. Murine HCCs induced by activated mutant of β-catenin and overexpression of c-Met develop at the same latency and efficiency in wild-type or liver-specific Fasn KO mice, suggesting that DNL is dispensable for this murine HCC model.114 Thus, it is critical to identify reliable biomarkers that can predict whether patients will respond or not to anti-DNL therapies. Since the AKT/mTOR pathway is a major regulator of DNL in HCC,20 genetic events leading to the activation of this signaling cascade, for example, TSC1/2 or PIK3CA mutations,9 may be used as biomarkers for patient selection. Clearly, additional studies are required to validate this hypothesis.
In summary, deregulated DNL is one of the pivotal metabolic aberrations in HCC. Targeting DNL has been identified as a possible therapeutics and prevention strategies against HCC. Despite all the progress during the past 10 years, much still need to be learned about this pathway’s enzymes, how they function as an integrated system, how they regulate the HCC microenvironment, and how we can identify patients who will benefit from anti-DNL therapies. Studies focusing on these crucial issues will undoubtedly provide novel insight into the molecular mechanisms leading to hepatocarcinogenesis and help scientists to generate better and safer drugs for HCC prevention and treatment.
Financial Support
This study is supported by NIH grants R01CA190606 and R01CA239251 to XC; P30DK026743 to UCSF Liver Center.
Footnotes
Conflict of Interest
The authors declare no potential conflicts of interest.
References
- 1.Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71(03):209–249 [DOI] [PubMed] [Google Scholar]
- 2.Akinyemiju T, Abera S, Ahmed M, et al. ; Global Burden of Disease Liver Cancer Collaboration. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the Global Burden of Disease Study 2015. JAMA Oncol 2017;3(12):1683–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64(01):73–84 [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Ricci S, Mazzaferro V, et al. ; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359(04):378–390 [DOI] [PubMed] [Google Scholar]
- 5.Huang A, Yang XR, Chung WY, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther 2020;5(01):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu European Association for the Study of the Liver. Management of hepatocellular carcinoma. J Hepatol 2018;69(01):182–23629628281 [Google Scholar]
- 7.Kole C, Charalampakis N, Tsakatikas S, et al. Immunotherapy for hepatocellular carcinoma: a 2021 update. Cancers (Basel) 2020; 12(10):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finn RS, Qin S, Ikeda M, et al. ; IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382(20):1894–1905 [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169:1327–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahn SM, Jang SJ, Shim JH, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 2014;60(06): 1972–1982 [DOI] [PubMed] [Google Scholar]
- 11.Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015;149(05):1226.e4–1239.e4 [DOI] [PubMed] [Google Scholar]
- 12.Raja A, Park I, Haq F, Ahn SM. FGF19-FGFR4 signaling in hepatocellular carcinoma. Cells 2019;8(06):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016;23(01):27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell 2017;168 (04):657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Che L, Paliogiannis P, Cigliano A, Pilo MG, Chen X, Calvisi DF. Pathogenetic, prognostic, and therapeutic role of fatty acid synthase in human hepatocellular carcinoma. Front Oncol 2019;9:1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaupel P, Schmidberger H, Mayer A. The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol 2019;95(07):912–919 [DOI] [PubMed] [Google Scholar]
- 17.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324(5930):1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(05):646–674 [DOI] [PubMed] [Google Scholar]
- 19.Mashima T, Seimiya H, TsuruoT. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer 2009;100(09):1369–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calvisi DF, Wang C, Ho C, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011;140(03): 1071–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 2013;6(06):1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115(05):1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen-Urstad AP, Semenkovich CF. Fatty acid synthase and liver triglyceride metabolism: housekeeper or messenger? Biochim Biophys Acta 2012;1821(05):747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism 2014;63(07): 895–902 [DOI] [PubMed] [Google Scholar]
- 25.Foufelle F, Ferré P New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J 2002;366(Pt 2):377–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dentin R, Girard J, Postic C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005;87(01):81–86 [DOI] [PubMed] [Google Scholar]
- 27.Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci 2016;61 (05):1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kersten S Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep 2001;2(04):282–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iizuka K, Takao K, Yabe D. ChREBP-mediated regulation of lipid metabolism: involvement of the gut microbiota, liver, and adipose tissue. Front Endocrinol (Lausanne) 2020;11:587189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollands MA, Cawthorne MA. Important sites of lipogenesis in the mouse other than liver and white adipose tissue. Biochem J 1981;196(02):645–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016;16(11):732–749 [DOI] [PubMed] [Google Scholar]
- 32.Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition 2000;16(03): 202–208 [DOI] [PubMed] [Google Scholar]
- 33.Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol 2017;13(12):710–730 [DOI] [PubMed] [Google Scholar]
- 34.Xu X, So JS, Park JG, Lee AH. Transcriptional control of hepatic lipid metabolism by SREBP and ChREBP. Semin Liver Dis 2013;33 (04):301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des 2014;20 (15):2619–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ettinger SL, Sobel R, Whitmore TG, et al. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res 2004;64(06):2212–2221 [DOI] [PubMed] [Google Scholar]
- 37.Du T, Sikora MJ, Levine KM, et al. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res 2018;20(01):106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo D, Prins RM, Dang J, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2009;2(101):ra82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C, Yang W, Zhang J, et al. SREBP-1 has a prognostic role and contributes to invasion and metastasis in human hepatocellular carcinoma. Int J Mol Sci 2014;15(05):7124–7138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamashita T, Honda M, Takatori H, et al. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J Hepatol 2009;50(01):100–110 [DOI] [PubMed] [Google Scholar]
- 41.Ho C, Wang C, Mattu S, et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology 2012;55(03):833–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stiles B, Wang Y, Stahl A, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci U S A 2004;101(07):2082–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He L, Hou X, Kanel G, et al. The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol 2010;176(05): 2302–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu D, Wang Z, Xia Y, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature 2020;580 (7804):530–535 [DOI] [PubMed] [Google Scholar]
- 45.Yu X, Lin Q, Wu Z, et al. ZHX2 inhibits SREBP1c-mediated de novo lipogenesis in hepatocellular carcinoma via miR-24–3p. J Pathol 2020;252(04):358–370 [DOI] [PubMed] [Google Scholar]
- 46.Heo MJ, Kang SH, Kim YS, et al. UBC12-mediated SREBP-1 neddylation worsens metastatic tumor prognosis. Int J Cancer 2020;147(09):2550–2563 [DOI] [PubMed] [Google Scholar]
- 47.Li N, Zhou ZS, Shen Y, et al. Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice. Hepatology 2017;65(06):1936–1947 [DOI] [PubMed] [Google Scholar]
- 48.Icard P, Wu Z, Fournel L, Coquerel A, Lincet H, Alifano M. ATP citrate lyase: a central metabolic enzyme in cancer. Cancer Lett 2020;471:125–134 [DOI] [PubMed] [Google Scholar]
- 49.Han Q, Chen CA, Yang W, et al. ATP-citrate lyase regulates stemness and metastasis in hepatocellular carcinoma via the Wnt/β-catenin signaling pathway. Hepatobiliary Pancreat Dis Int 2021;20(03):251–261 [DOI] [PubMed] [Google Scholar]
- 50.Migita T, Narita T, Nomura K, et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res 2008;68(20):8547–8554 [DOI] [PubMed] [Google Scholar]
- 51.Gu L, Zhu Y, Lin X, et al. The IKKβ-USP30-ACLY axis controls lipogenesis and tumorigenesis. Hepatology 2021;73(01): 160–174 [DOI] [PubMed] [Google Scholar]
- 52.Zheng Y, Zhou Q, Zhao C, Li J, Yu Z, Zhu Q. ATP citrate lyase inhibitor triggers endoplasmic reticulum stress to induce hepatocellular carcinoma cell apoptosis via p-eIF2α/ATF4/CHOP axis. J Cell Mol Med 2021;25(03):1468–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hatzivassiliou G, Zhao F, Bauer DE, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005;8 (04):311–321 [DOI] [PubMed] [Google Scholar]
- 54.Yahagi N, Shimano H, Hasegawa K, et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer 2005;41(09):1316–1322 [DOI] [PubMed] [Google Scholar]
- 55.Ye B, Yin L, Wang Q, Xu C. ACC1 is overexpressed in liver cancers and contributes to the proliferation of human hepatoma Hep G2 cells and the rat liver cell line BRL 3A. Mol Med Rep 2019;19(05): 3431–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang MD, Wu H, Fu GB, et al. Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology 2016;63(04):1272–1286 [DOI] [PubMed] [Google Scholar]
- 57.Fullerton MD, Galic S, Marcinko K, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 2013;19(12): 1649–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lally JSV, Ghoshal S, DePeralta DK, et al. Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab 2019;29(01):174.e5–182.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007;7(10): 763–777 [DOI] [PubMed] [Google Scholar]
- 60.Hao Q, Li T, Zhang X, et al. Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol Rep 2014;32(06): 2471–2476 [DOI] [PubMed] [Google Scholar]
- 61.Graner E, Tang D, Rossi S, et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 2004;5(03):253–261 [DOI] [PubMed] [Google Scholar]
- 62.Gu L, Zhu Y, Lin X, Tan X, Lu B, Li Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene 2020;39(11): 2437–2449 [DOI] [PubMed] [Google Scholar]
- 63.Gao Y, Lin LP, Zhu CH, Chen Y, Hou YT, Ding J. Growth arrest induced by C75, A fatty acid synthase inhibitor, was partially modulated by p38 MAPK but not by p53 in human hepatocellular carcinoma. Cancer Biol Ther 2006;5(08):978–985 [DOI] [PubMed] [Google Scholar]
- 64.Li L, Pilo GM, Li X, et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J Hepatol 2016;64(02):333–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu J, Che L, Li L, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep 2016;6:20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guri Y, Colombi M, Dazert E, et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 2017;32(06):807.e12–823.e12 [DOI] [PubMed] [Google Scholar]
- 67.Zhang C, Sheng L, Yuan M, et al. Orlistat delays hepatocarcinogenesis in mice with hepatic co-activation of AKT and c-Met. Toxicol Appl Pharmacol 2020;392:114918. [DOI] [PubMed] [Google Scholar]
- 68.Jia J, Che L, Cigliano A, et al. Pivotal role of fatty acid synthase in c-MYC driven hepatocarcinogenesis. Int J Mol Sci 2020;21(22):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Che L, Chi W, Qiao Y, et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2020;69(01):177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Igal RA. Stearoyl-CoA desaturase-1: a novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010;31(09): 1509–1515 [DOI] [PubMed] [Google Scholar]
- 71.Bansal S, Berk M, Alkhouri N, Partrick DA, Fung JJ, Feldstein A. Stearoyl-CoA desaturase plays an important role in proliferation and chemoresistance in human hepatocellular carcinoma. J Surg Res 2014;186(01):29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ma MKF, Lau EYT, Leung DHW, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol 2017;67(05):979–990 [DOI] [PubMed] [Google Scholar]
- 73.Muir K, Hazim A, He Y, et al. Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma. Cancer Res 2013;73(15):4722–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li L, Wang C, Calvisi DF, et al. SCD1 Expression is dispensable for hepatocarcinogenesis induced by AKT and Ras oncogenes in mice. PLoS One 2013;8(09):e75104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao Y, Li M, Yao X, et al. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep 2020;33(10):108487. [DOI] [PubMed] [Google Scholar]
- 76.Qin XY, Su T, Yu W, Kojima S. Lipid desaturation-associated endoplasmic reticulum stress regulates MYCN gene expression in hepatocellular carcinoma cells. Cell Death Dis 2020;11(01):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chiang DY, Villanueva A, Hoshida Y, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 2008;68(16):6779–6788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee JS, Chu IS, Heo J, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004;40(03):667–676 [DOI] [PubMed] [Google Scholar]
- 79.Boyault S, Rickman DS, de Reyniès A, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007;45(01):42–52 [DOI] [PubMed] [Google Scholar]
- 80.Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 2009;69(18): 7385–7392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rebouissou S, Nault JC. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J Hepatol 2020; 72(02):215–229 [DOI] [PubMed] [Google Scholar]
- 82.European Association for the Study of the Liver (EASL) EASL Clinical Practice Guidelines on the management of benign liver tumours. J Hepatol 2016;65(02):386–398 [DOI] [PubMed] [Google Scholar]
- 83.Rebouissou S, Imbeaud S, Balabaud C, et al. HNF1alpha inactivation promotes lipogenesis in human hepatocellular adenoma independently of SREBP-1 and carbohydrate-response element-binding protein (ChREBP) activation. J Biol Chem 2007;282(19): 14437–14446 [DOI] [PubMed] [Google Scholar]
- 84.Bluteau O, Jeannot E, Bioulac-Sage P, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet 2002;32(02):312–315 [DOI] [PubMed] [Google Scholar]
- 85.Mounier C, Bouraoui L, Rassart E. Lipogenesis in cancer progression (review). Int J Oncol 2014;45(02):485–492 [DOI] [PubMed] [Google Scholar]
- 86.Braig S Chemical genetics in tumor lipogenesis. Biotechnol Adv 2018;36(06):1724–1729 [DOI] [PubMed] [Google Scholar]
- 87.Tang JJ, Li JG, Qi W, et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab 2011;13(01):44–56 [DOI] [PubMed] [Google Scholar]
- 88.Yin F, Feng F, Wang L, Wang X, Li Z, Cao Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis 2019;10(09):672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen Q, Wang T, Li J, et al. Effects of natural products on fructose-induced nonalcoholic fatty liver disease (NAFLD). Nutrients 2017;9(02):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.You ZLi B, Xu J, Chen L, Ye H. Curcumin suppress the growth of hepatocellular carcinoma via down-regulating SREBF1. Oncol Res 2018. (e-pub ahead of print) . Doi: 10.3727/096504018x15219173841078 [DOI] [PubMed] [Google Scholar]
- 91.Vassallo A, Santoro V, Pappalardo I, et al. Liposome-mediated inhibition of inflammation by hydroxycitrate. Nanomaterials (Basel) 2020;10(10):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Burke AC, Telford DE, Huff MW. Bempedoic acid: effects on lipoprotein metabolism and atherosclerosis. Curr Opin Lipidol 2019;30(01):1–9 [DOI] [PubMed] [Google Scholar]
- 93.Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res 2012;72(15):3709–3714 [DOI] [PubMed] [Google Scholar]
- 94.Pinkosky SL, Filippov S, Srivastava RA, et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J Lipid Res 2013;54(01):134–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ruscica M, Banach M, Sahebkar A, Corsini A, Sirtori CR. ETC-1002 (Bempedoic acid) for the management of hyperlipidemia: from preclinical studies to phase 3 trials. Expert Opin Pharmacother 2019;20(07):791–803 [DOI] [PubMed] [Google Scholar]
- 96.Li JJ, Wang H, Tino JA, et al. 2-hydroxy-N-arylbenzenesulfonamides as ATP-citrate lyase inhibitors. Bioorg Med Chem Lett 2007;17(11):3208–3211 [DOI] [PubMed] [Google Scholar]
- 97.Alkhouri N NASH and NAFLD: emerging drugs, therapeutic targets and translational and clinical challenges. Expert Opin Investig Drugs 2020;29(02):87. [DOI] [PubMed] [Google Scholar]
- 98.Fhu CW, Ali A. Fatty acid synthase: an emerging target in cancer. Molecules 2020;25(17):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Menendez JA, Lupu R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin Ther Targets 2017;21(11): 1001–1016 [DOI] [PubMed] [Google Scholar]
- 100.You BJ, Hour MJ, Chen LY, Luo SC, Hsu PH, Lee HZ. Fenofibrate induces human hepatoma Hep3B cells apoptosis and necroptosis through inhibition of thioesterase domain of fatty acid synthase. Sci Rep 2019;9(01):3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of cancer. Cancers (Basel) 2019;11(07):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang GM, Jiang QH, Cai C, Qu M, Shen W. SCD1 negatively regulates autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway. Cancer Lett 2015;358(02):180–190 [DOI] [PubMed] [Google Scholar]
- 103.Yao Y, Sun S, Wang J, et al. Canonical Wnt signaling remodels lipid metabolism in zebrafish hepatocytes following Ras oncogenic insult. Cancer Res 2018;78(19):5548–5560 [DOI] [PubMed] [Google Scholar]
- 104.Syed-Abdul MM, Parks EJ, Gaballah AH, et al. Fatty acid synthase inhibitor TVB-2640 reduces hepatic de novo lipogenesis in males with metabolic abnormalities. Hepatology 2020;72(01): 103–118 [DOI] [PubMed] [Google Scholar]
- 105.Stiede K, Miao W, Blanchette HS, et al. Acetyl-coenzyme A carboxylase inhibition reduces de novo lipogenesis in over-weight male subjects: a randomized, double-blind, crossover study. Hepatology 2017;66(02):324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wei X, Song H, Yin L, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature 2016; 539(7628):294–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM. Lipid metabolism in cancer: new perspectives and emerging mechanisms. Dev Cell 2021;56(10):1363–1393 [DOI] [PubMed] [Google Scholar]
- 108.Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol 2015;36(02): 81–91 [DOI] [PubMed] [Google Scholar]
- 109.Everts B, Amiel E, Huang SC, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKe supports the anabolic demands of dendritic cell activation. Nat Immunol 2014;15(04): 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berod L, Friedrich C, Nandan A, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 2014;20(11):1327–1333 [DOI] [PubMed] [Google Scholar]
- 111.Huang SC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 2014;15(09):846–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cao D, Song X, Che L, et al. Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells. Liver Int 2017;37(01):80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nelson ME, Lahiri S, Chow JD, et al. Inhibition of hepatic lipogenesis enhances liver tumorigenesis by increasing antioxidant defence and promoting cell survival. Nat Commun 2017;8:14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Che L, Pilo MG, Cigliano A, et al. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle 2017;16(06):499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beckner ME, Fellows-Mayle W, Zhang Z, et al. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int J Cancer 2010;126(10):2282–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ismail A, Doghish AS, Elsadek BEM, Salama SA, Mariee AD. Hydroxycitric acid potentiates the cytotoxic effect of tamoxifen in MCF-7 breast cancer cells through inhibition of ATP citrate lyase. Steroids 2020;160:108656. [DOI] [PubMed] [Google Scholar]
- 117.Evaluation of major cardiovascular events in patients with, or at high risk for, cardiovascular disease who are statin intolerant treated with bempedoic acid (ETC-1002) or placebo (CLEAR Outcomes). Accessed June 15, 2021 at: https://ClinicalTrials-gov/show/NCT02993406
- 118.Study to evaluate the pharmacodynamic effects of a single oral dose of GS-0976 (NDI-010976) in healthy adult subjects. Accessed June 15, 2020 at: https://ClinicalTrials.gov/show/NCT02876796
- 119.Evaluation of 3-V Bioscience-2640 to reduce de novo lipogenesis in subjects with characteristics of metabolic syndrome. Accessed June 15, 2021 at: https://ClinicalTrials.gov/show/NCT02948569
- 120.A study to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of MK8245 (8245–004)(COMPLETED). Accessed June 15, 2021 at: https://ClinicalTrials.gov/show/NCT00790556