

Abstract

A catalytic electron donor–acceptor (EDA) complex for the visible-light-driven annulation reaction between activated alkenes and N,N-substituted dialkyl anilines is reported. The key photoactive complex is formed in situ between dialkylated anilines as donors and 1,2-dibenzoylethylene as a catalytic acceptor. The catalytic acceptor is regenerated by aerobic oxidation. Investigations into the mechanism are provided, revealing a rare example of a catalytic acceptor in photoactive EDA complexes that can give access to selective functionalization of aromatic amines under mild photochemical conditions.
Introduction
An electron donor–acceptor (EDA) complex is a weak molecular aggregation between an electron-rich donor and an electron-poor acceptor.1 A characteristic of EDA complexes is their associated charge-transfer (CT) band in the electromagnetic spectrum. Excitation by light within this CT band results in a single electron transfer (SET) from the donor to the acceptor. The resulting radical species can subsequently undergo a range of different processes and take part in radical reactions. Due to the red-shifted absorption of the CT band, compared to the individual components of the EDA complex, visible light can often be used to induce reactivity in compounds otherwise not absorbing in this spectral region.2
Over the last decade, a wide range of different approaches to utilize EDA complexes in synthetic organic chemistry have been developed.1−4 Examples include arylations,5 stereoselective alkylations,6 oxidative annulations,7 and acylations.8 The prototypical examples are coupling reactions between a donor and an acceptor, driven by the formation of a stoichiometric EDA complex,7,9 although strategies to harvest the synthetic potential of EDA complexes have rapidly been expanded to include catalytic complexes (Scheme 1).2,10−12
Scheme 1. Examples of Catalytic EDA Complexes in Organic Synthesis.

(a) Working principle of a system with catalytic donors in combination with redox auxiliaries (RA). (b) Working principle of a system using Lewis acids as electron acceptors (LAA). (c) EDA catalysis for visible light-mediated synthesis of tetrahydroquinolines.
Within the catalytic regime, different approaches can be exploited depending on how the EDA complex is formed. The EDA donor or acceptor can be formed in situ from a pre-catalyst, generating highly polarized species from non-reactive substrates. Examples include in situ-formed enamines and enolates as donors, or iminium ions as acceptors, that take part in the formation of photoactive EDA complexes.6,13,14
Alternatively, an external donor can be present in catalytic amount that associates with a redox-active reactant to form an EDA complex (Scheme 1a). After a SET to the reactant, the donor can re-form to close the catalytic cycle. Examples of these systems include the use of electron-rich aromatics and amines.10,11,15,16 Examples of systems using catalytic acceptors in the same manner, however, still remain rare in the literature and are limited to strong Lewis acids (Scheme 1b).17,18 Identification of milder suitable acceptors that could act as catalysts for photo-mediated synthesis would constitute a significant contribution to the field of EDA complex-driven transformations. Inspired by this opportunity, we set out to investigate this methodology in the generation of α-amino alkyl radicals. Herein, we present a protocol using visible light to furnish a range of annulation products from aromatic amines and activated alkenes, driven by the formation of a photoactive organocatalytic acceptor–EDA complex (Scheme 1c).
Results and Discussion
As a model system for this study, 1,2-dibenzoyl ethylene 4a as a catalyst in combination with N,N-dimethylaniline (DMA) 1a was chosen because the EDA complex of the two species absorbs strongly in the visible region (Figure 1).19 It was thought that upon photoexcitation of this complex, a SET from the amine to 4a would occur, resulting in the formation of an α-amino alkyl radical (Scheme 1). This radical could rapidly react with a suitable reaction partner. With a suitable oxidant present, the enol radical formed could be regenerated to 4a, closing the catalytic cycle. Due to the nucleophilic nature of α-amino alkyl radicals, the proposed methodology could be utilized for a broad scope of electrophiles as reaction partners.
Figure 1.

(A) UV–vis spectra of 1a (0.4 M), 2a (73 mM), 4a (6 mM), and their EDA complexes in 1,4-dioxane; (B) pictures of 1a, 2a, 4a, and their EDA complexes; and (C) structures of 1a, 2a, and 4a.
As a starting point for the study, N-substituted maleimides were chosen as reaction partners due to their well-known reactivity with aromatic α-amino alkyl radicals, forming tetrahydroquinolines under a range of reaction conditions.7,20−46 The photochemically driven annulation reaction between DMA 1a and N-phenyl maleimide (2a) to furnish tetrahydroquinoline (3a) proceeded smoothly without an external photocatalyst under UV irradiation.7 However, the initial screening of reaction conditions, presented in Table 1, shows that visible light irradiation (with a white compact fluorescent lamp) in the absence of a catalyst resulted in a low yield. This slow background reactivity prompted us to use this reaction as a model for our system. Notably, the addition of only 1 mol % dibenzoyl ethylene (4a) resulted in a significantly increased yield (Table 1, entry 2), supporting the feasibility of the outlined approach.
Table 1. Optimization of the Reaction Conditions.

| entrya | catalyst (mol %) | light source | eq. 1a | yield 3ab |
|---|---|---|---|---|
| 1 | CFL | 7 | 7c | |
| 2 | 4a (1) | CFL | 7 | 53 |
| 3 | 4a (5) | CFL | 7 | 80 |
| 4 | 4b (5) | CFL | 7 | 26 |
| 5 | 4c (5) | CFL | 7 | 80 |
| 6 | 4d (5) | CFL | 7 | 26 |
| 7 | 4a (5) | CFL | 1 | 40 |
| 8 | 4a (5) | CFL | 2 | 42 |
| 9 | 4a (5) | CFL | 5 | 82 |
| 10 | 4a (5) | 7 | 12d | |
| 11 | 4a (5) | green LED | 7 | 50e |
| 12 | 4a (5) | blue LED | 7 | 85f (80)f,g |
| 13 | 4a (5) | blue LED | 7 | 3f,h |
Conditions: 2a (0.2 mmol), 1a (1–7 equiv), and the catalyst in 3 mL of 1,4-dioxane were irradiated for 6 h under an ambient atmosphere.
Determined by GC-FID using n-dodecane as an internal standard.
Reaction performed in the absence of the catalyst.
Reaction performed with protection from light at 100 °C.
Reaction time, 30 h.
Reaction time, 7 h.
Isolated yield.
Reaction carried out under an atmosphere of nitrogen.
A higher loading of 5 mol % increased the yield further (Table 1, entry 3). The impact of aryl substitutions on the dibenzoyl ethylene catalyst, as a way to tune the catalytic properties, was briefly investigated (Table 1, entries 4–6). Electron-withdrawing groups in the p-position resulted in lower yields, while the introduction of an electron-donating group did not affect the catalytic action significantly. Initially, using the amine in excess (7 equiv), we investigated the influence of decreasing amounts and found that with 5 equiv of amine, the reaction proceeded smoothly to yield the desired product at 82% yield (Table 1, entry 9). In order to investigate the effect of the excitation wavelength on the reaction outcome, green and blue light-emitting diodes (LEDs) were used as light sources (Table 1, entries 11 and 12). Green light (525 nm) could promote the transformation with 50% yield using longer irradiation times. A blue LED (465 nm), on the other hand, proved to be a suitable light source, driving the reaction yield to 85%. This result reflects the stronger absorption of the EDA complex in the blue region of the spectrum.
With our optimized reaction conditions being found, the substrate scope of the reaction was then investigated (Schemes 2 and 3). Due to the known EDA complexes between anilines and maleimides that potentially could drive the reactions to the desired product in the absence of any catalyst, a control experiment without catalyst 4a was performed for each entry (Supporting Information, Scheme S1).
Scheme 2. Scope of Activated Alkenes.

Conditions: alkene (0.3 mmol), amine (7 equiv), and 4a (5 mol %) in 3 mL of 1,4-dioxane were irradiated for 6–72 h under an ambient atmosphere. Yields reported are isolated. a Diastereomeric ratio was determined by 1H NMR. b Reaction time, 18 h. c Reaction time, 72 h.
Scheme 3. Scope of Amines.

Conditions: alkene (0.3 mmol), amine (7 equiv), and 4a (5 mol %) in 3 mL of 1,4-dioxane were irradiated for 6–18 h under an ambient atmosphere. Yields reported are isolated. a Reaction time 18 h. b No product was observed in the crude reaction mixture by 1H NMR analysis.
First, DMA and different N-aryl-substituted maleimides were subjected to the reaction conditions to give products 3b–3f (Scheme 2). Electron-rich p-OMe-substituted maleimide gave product 3b in a low yield, whereas the slightly electron-withdrawing groups chloro and fluoro provided the desired products 3c and 3d in higher yields of 62 and 74%, respectively. The sterically demanding o-substituted tert-butyl group was tolerated in the reaction, giving product 3e in 64% yield as a single diastereomer. On the other hand, compound 3f bearing the smaller methyl group was obtained as a mixture of diastereomers (2:1 dr) in 66% yield. Next, a set of N-alkyl substituted maleimides were tested as Michael acceptors in the radical addition reaction. N-Methyl maleimide provided a significantly slower reaction rate with a yield of 31% after 7 h and 44% yield after 18 h. More bulky substituents, such as propyl and tert-butyl, gave slightly increased yields of 48 and 54% for 3i and 3k, respectively. Non-substituted maleimide provided the desired product 3n in 30% yield; however, longer reaction times promoted the reaction to proceed to give 66% yield. Michael acceptors other than maleimides resulted in sluggish reactions under the current protocol, giving products 5–7 in 17–29% yield.
The effect of substitution on the aniline reaction partner was then explored, giving products 3o–aa in 25–84% yield (Scheme 3). The reaction was found to be highly sensitive to the electronic nature of the donor amine. Electron-deficient amines, such as p-aceto, p-carboxylate, and p-CN, resulted in lower reaction rates, giving products 3s–u in 47, 30, and 25% yields, respectively, after 7 h of reaction time. This is presumably due to the higher oxidation potential of these amines.47 Complete selectivity toward addition of methyl radicals to the Michael acceptors was observed when different N-Me-N-alkyl anilines were subjected to the reaction conditions, providing compounds 3x and 3y, consistent with the literature.20,48 The sterically hindered o-methyl-DMA also resulted in lower reactivity (Scheme 3, entry 3v). When N,N-diethyl aniline was subjected to the reaction conditions, no annulation product could be observed (Scheme 3, entry 3z). When N,N,3-trimethylaniline was used as a starting material, the corresponding tetrahydroquinoline 3aa/3aa′ could be isolated as a mixture of regioisomers in (1.8:1) 42% yield.
To showcase the scalability of the developed protocol, a gram-scale reaction was carried out (Scheme 4). The desired product 3a could efficiently be isolated in 78% yield after simple extraction and washing steps.
Scheme 4. Gram-Scale Synthesis of 3a.

To gain an insight into the reaction mechanism, several control experiments were carried out. First, when the reaction was carried out in the absence of light at 100 °C for 7 h (Table 1, entry 10), a yield of 12% was obtained. This result shows that although a thermal background reaction can be active at elevated temperatures, light irradiation is needed to successfully drive the reaction. From the background reaction without 4a as an additive being investigated for almost all substrates (Supporting Information, Scheme S1), it is evident that the catalyst is crucial for promoting the reaction using blue light. Although amines and maleimides formed EDA complexes with absorption that tailed into the visible region (Figure 1), the excitation of these complexes with visible light alone did not seem to result in the efficient formation of reactive radical species. Even with an LED with increased power (40 W) and an emission maximum of 440 nm, where the complex between 1a and 2a absorbed significantly, the background reaction proceeded slowly (Supporting Information, Figure S3).
When the reaction was carried out in the presence of the radical scavenger butylated hydroxytoluene, a significant decrease in the yield was observed, suggesting that the reaction proceeded via a radical pathway (Scheme 5a). To investigate if the reaction involved a singlet oxygen species,49 a singlet oxygen scavenger, histidine, was investigated, and it was found to have a little impact on the reaction outcome (Scheme 5a).43
Scheme 5. Control Experiments.

Next, kinetic isotope effect (KIE) experiments were carried out (Scheme 5b,c). The intramolecular KIE was found to be 5.8 (Scheme 5b), whereas an intermolecular KIE of 3.8 was observed when running two parallel reactions with DMA-d6 and DMA or as a competition experiment (Scheme 5c). The observed difference suggests that the mechanism would not involve a single-step C–H cleavage, such as hydrogen atom transfer, to a significant degree.50,51 Instead, the formation of an α-amino alkyl radical from 3a likely involves two consecutive steps: oxidation and C–H bond cleavage through proton transfer. The oxidation step is an equilibrium between electron transfer and back electron transfer (BET), common to many EDA complexes.1,2 In such a system, the intramolecular KIE will be higher than the intermolecular KIE with a difference determined by the ratio between the rate of C–H cleavage and the rate of BET.50,52 To investigate the electronic effects of the amine donor, a series of competition experiments were carried out (Figure 2). A significant dependence of the natural logarithms of the relative rates on the Hammett parameter σp was observed with a slope of −3.2, suggesting that a SET is involved to a significant degree in the mechanism.51,53
Figure 2.
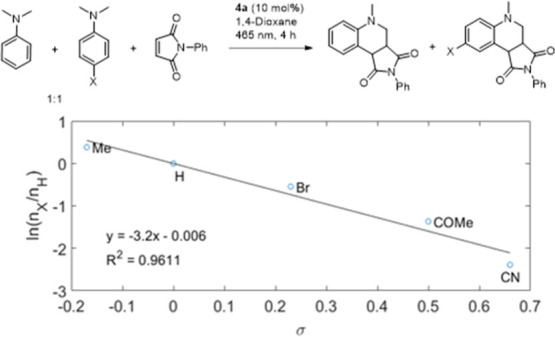
Competition experiment between para-substituted DMA and the plot of the natural logarithms of the relative product ratios, as determined by 1H NMR analysis, versus the Hammett parameter σp.
To investigate the role of air in the reaction, a control experiment under a nitrogen atmosphere was performed which resulted in a product yield of 3% (Table 1, entry 13). The consumption of oxygen was then measured as a function of product formation, and it was observed that a slight excess of oxygen was needed to drive the reaction to completion (Supporting Information, Figure S6).
The quantum yield of the reaction at 450 nm was investigated using standard potassium ferrioxalate actinometry (Supporting Information), and it was found to be 0.07. The low quantum yield can be attributed to the large concentration and absorption by other species such as maleimide and the EDA complex formed between DMA and maleimide, leading to non-productive absorption of photons. Furthermore, given the difference in intra- and intermolecular KIE for the present system, a significant rate of BET can be expected, leading to unproductive absorption of photons.
Based on the control experiments, a possible reaction mechanism can be postulated. The crucial role of 4a in the reaction outcome can be rationalized according to two likely scenarios. In the first scenario (Scheme 6, cycle A), 4a is locally excited and promoted to its excited state 4a*, which can oxidize amine 1via SET to form III, in line with a typical photo-redox catalytic cycle. In the second scenario, however, the key photoactive species is the ground-state EDA complex between 4a and amine 1 (Scheme 6, cycle B). Excitation of this complex results in the formation of radical anion I and the radical cation of the amine via a SET within the complex. Subsequently, I, or its protonated form II, can be oxidized by ground-state molecular oxygen54 (rather than by a singlet oxygen species, see Scheme 5a) to yield a superoxide anion or a hydroperoxyl radical.55 The deprotonation of the amine radical cation III with a pKa of 956 can be achieved by proton transfer to either 1, the radical anion I, or a superoxide radical, and yields an α-amino alkyl radical (IV) that readily reacts with a Michael acceptor to form V, which after aromatization forms the final product 3. The regenerated 4a can associate with an amine, forming another EDA complex to close the catalytic cycle.
Scheme 6. Proposed Mechanism.

To completely distinguish between the two scenarios under the developed conditions is, however, challenging. Due to the overlap in absorption of 4a and its EDA complex with amines (Figure 1), it cannot be ruled out that 4a is locally excited and acts as a sensitizer. However, the observation that the reaction proceeds with excitation by green light (525 nm) suggests that the EDA complex between 4a and the amine is a key intermediate in the reaction (Table 1, entry 11). At this wavelength, 4a has a very low absorption; however, once the EDA complex between 1a and 4a has formed, the absorbance is significantly increased. The observation that maleimide reacts with the formed α-amino alkyl radical much faster than the available Michael acceptor 4a can be rationalized in terms of electrophilicity and the stoichiometry of the reaction conditions. In the absence of light irradiation, a slow background reaction was observed at elevated temperature (Table 1, entry 10). The mechanism for this reaction was not further investigated, although it could be possible that the EDA complex between 1 and 4 or 2 could be activated thermally to some degree, as has been reported for other systems.57,58
In conclusion, a protocol for the visible light-induced aerobic oxidative annulation reaction between dialkylanilines and activated alkenes has been disclosed. The reaction is postulated to proceed via the excitation of an EDA complex formed between the catalyst 1,2-dibenzoyl ethylene and the amine reaction partner. The simple and available 1,2-dibenzoyl ethylene makes the developed protocol attractive as an alternative to the use of complex photoredox catalysts. Furthermore, this example of a catalytic external acceptor could stimulate the field of EDA complex chemistry to pursue novel photoreactions.
Experimental Section
General Information
All reagents and solvents were purchased from Sigma-Aldrich and Alfa Aesar and used without any further purification unless specified. Purification was performed in an automated column chromatograph Biotage Isolera Spektra One with a Biotage SNAP-10 g KP-silica column, together with a 1 g Samplet cartridge using n-heptane or petroleum ether (40–60 °C)/ethyl acetate as solvent mixture unless otherwise noted. 1H (400 MHz) and 13C (101 MHz) NMR spectra were acquired on an Agilent NMR machine at 25 °C. The chemical shifts for 1H and 13C NMR spectra are reported in parts per million (ppm) relative to the residual peak from the solvent CDCl3 as the internal standard: 1H NMR at δ 7.26 ppm and 13C NMR at δ 77.16 ppm for CDCl3. All coupling constants (J) are reported in hertz, and multiplicities are indicated by s (singlet), d (doublet), dd (doublet of doublet), td (triplet of doublet), ddd (doublet of doublets of doublets), (t) triplett, dt (doublet of triplet), and m (multiplet). Infrared (IR) spectra were recorded on a PerkinElmer series ATR FTIR spectrometer and are reported in wavenumber (cm–1).
High-resolution mass spectrometry measurements were performed by CMSI service at Chalmers University of Technology. An Agilent 6520 equipped with an electrospray interface was operated in the positive ionization mode.
UV–vis absorption spectra were recorded on a Cary 4000 UV–vis spectrometer using 1 × 1 cm quartz cuvettes. All light-promoted reactions were carried out in Biotage microwave vials (2–5 mL) under irradiation with a commercial LED strip (Ledsavers, 5 W, λmax 465 nm), a commercial compact fluorescent lamp (Narva Scandinavia, UV light bulb, 15 W, λmax 365 nm), a Kessil PR160L-440 LED lamp, or a Kessil PR160L-525 LED lamp. Gas chromatography studies were performed using an Agilent 7820A equipped with a flame ionization detector and an Agilent HP-5 19091J-413 column. Emission spectra of light sources were measured using an AvaSpec-2048.
Synthesis of Starting Materials
Maleimides 2b, 2d, 2e, 2f, and 2l were synthesized following a modified reported method.59 The appropriate amine (10 mmol) dissolved in diethyl ether (5 mL) was added to a stirred solution of maleic anhydride (10 mmol, 1 g) in diethyl ether (10 mL). The solution was stirred at room temperature for 4–18 h, and the resulting suspension was filtered. The collected solids were washed with diethyl ether and then dried in vacuo and directly added to acetic anhydride (6 mL), followed by sodium acetate (1 equiv). The suspension was heated to 120 °C until full conversion as followed by TLC (1–4 h reaction time). The solvent was then removed under reduced pressure, and the residue was taken up with ethyl acetate and washed with saturated sodium bicarbonate solution and brine. The organic phase was then dried over anhydrous sodium sulfate and was concentrated under reduced pressure. The solid residue was recrystallized from ethanol or ethyl acetate to afford the desired maleimide as a crystalline solid or purified by column chromatography for oils.
1-(4-Methoxyphenyl)-1H-pyrrole-2,5-dione (2b)60
A yellow solid afforded after recrystallization from ethanol, 1.2 g (58%); spectroscopic data were in accordance with the literature;601H NMR (400 MHz, chloroform-d): δ 7.25–7.19 (m, 2H), 7.02–6.94 (m, 2H), 6.84 (s, 2H), 3.83 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 169.9, 159.3, 134.2, 127.7, 123.8, 114.6, 55.6 ppm.
1-(4-Fluorophenyl)-1H-pyrrole-2,5-dione (2d)61
A yellow crystalline solid obtained after recrystallization from ethyl acetate/heptane, 937 mg (50%); spectroscopic data were in accordance with the literature;611H NMR (400 MHz, chloroform-d): δ 7.38–7.28 (m, 2H), 7.21–7.12 (m, 2H), 6.86 (d, J = 0.8 Hz, 2H); 13C{1H} NMR (101 MHz, chloroform-d): δ 169.5, 162.0 (d, J = 248 Hz), 134.4, 128.0 (d, J = 8.7 Hz), 127.2 (d, J = 3.3 Hz), 116.3 (d, J = 22.9 Hz) ppm.
1-(2-(tert-Butyl)phenyl)-1H-pyrrole-2,5-dione (2e)62
An off-white solid obtained after recrystallization thrice from ethanol (500 mg, 36%). Spectroscopic data were in accordance with the literature;621H NMR (400 MHz, chloroform-d): δ 7.59 (dd, J = 8.2, 1.5 Hz, 1H), 7.41 (tdd, J = 7.4, 1.6, 0.8 Hz, 1H), 7.31–7.27 (m, 1H), 6.93–6.87 (m, 3H), 1.30 (s, 9H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 171.0, 149.7, 135.1, 131.5, 130.0, 129.3, 128.8, 127.5, 35.6, 31.7 ppm.
1-(2,4-Dimethylphenyl)-1H-pyrrole-2,5-dione (2f)63
A yellow oil obtained after column chromatography (silica, 10% ethyl acetate in petroleum spirit), 1.47 g (34%); spectroscopic data were in accordance with the literature;631H NMR (400 MHz, chloroform-d): δ 7.14 (dq, J = 2.0, 0.7 Hz, 1H), 7.13–7.08 (m, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.86 (s, 2H), 2.35 (s, 3H), 2.11 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 167.0, 139.7, 136.3, 134.5, 132.0, 128.6, 127.8, 127.3, 21.3, 17.9 ppm.
1-Phenethyl-1H-pyrrole-2,5-dione (2l)64
Pink flakes isolated after recrystallization from ethanol twice, 894 mg (44%); spectroscopic data were in accordance with the literature;641H NMR (400 MHz, chloroform-d): δ 7.33–7.26 (m, 2H), 7.25–7.17 (m, 3H), 6.65 (s, 2H), 3.82–3.72 (m, 2H), 2.95–2.83 (m, 2H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 170.7, 138.0, 134.2, 129.0, 128.7, 126.8, 39.3, 34.7 ppm.
General Procedure for the Visible-Light-Driven Aerobic Oxidative Annulation Reaction
To a 2–5 mL Biotage microwave vial were added N-substituted maleimide (0.3 mmol, 1 equiv), the appropriate amine (1.75 mmol, 7 equiv), and 1,2-dibenzoyl ethylene (0.013 mmol, 0.05 equiv). 1,4-Dioxane (3 mL) was then added, and the solution was irradiated using a 465 nm LED stripe with a distance of 2 cm to the irradiation source for 6–18 h, after which the solvent was removed under reduced pressure and the crude residue was loaded on a silica column and eluted with a mixture of ethyl acetate in petroleum ethers to afford the desired tetrahydroquinoline products 3a–aa and 5–7.
5-Methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3a)7
Starting with 43 mg of N-phenyl maleimide following the general procedure, 3a was obtained as a yellow solid (58.3 mg, 80%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;71H NMR (400 MHz, chloroform-d): δ 7.54 (dd, J = 7.6, 1.4 Hz, 1H), 7.47–7.40 (m, 2H), 7.39–7.32 (m, 1H), 7.29–7.21 (m, 3H), 6.92 (tt, J = 7.5, 1.2 Hz, 1H), 6.76 (d, J = 8.2 Hz, 1H), 4.17 (d, J = 9.6 Hz, 1H), 3.66–3.59 (m, 1H), 3.59–3.51 (m, 1H), 3.14 (ddd, J = 11.4, 4.4, 1.0 Hz, 1H), 2.85 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.8, 175.9, 148.5, 132.1, 130.5, 129.1, 128.8, 128.6, 126.5, 119.9, 118.7, 112.8, 50.8, 43.7, 42.3, 39.6 ppm.
For the gram-scale synthesis version of 3a, to a 100 mL round-bottom flask were added N-phenylmaleimide (762 mg, 4.4 mmol, 1 equiv), 1,2-dibenzoyl ethylene (52 mg, 0.22 mmol, 0.05 equiv), DMA (3.7 g, 30.5 mmol, 6.9 equiv), and 1,4-dioxane (50 mL). The reaction mixture was then placed 5 cm from a 40 W blue LED lamp and irradiated for 7 h under vigorous stirring. The round-bottom flask was kept open during the reaction to facilitate easy diffusion of oxygen to drive the reaction. After full conversion, as judged by TLC (20% ethyl acetate in n-heptane), the solvent was removed under reduced pressure and the residue was taken up in ethyl acetate. Excess aniline was removed by washing with HCl (3 × 10 mL, 1 M). The organic layer was then washed with brine and was dried over sodium sulfate. The solvent was then removed under reduced pressure to yield a brown solid that was suspended in ethyl acetate/heptane and was collected by filtration. The solids were washed with a small amount of ethanol and then excessive amounts of n-heptane. After drying in vacuo, 3a was obtained as an off-white powder (1.0 g, 3.4 mmol, 78%).
2-(4-Methoxyphenyl)-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3b)35
Starting with 49.9 mg of N-(4-methoxyphenyl)maleimide following the general procedure provided 3b as a yellow solid (27.4 mg, 36%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;351H NMR (400 MHz, chloroform-d): δ 7.53 (ddd, J = 7.5, 1.6, 0.8 Hz, 1H), 7.27–7.21 (m, 1H), 7.21–7.14 (m, 2H), 6.98–6.90 (m, 3H), 6.75 (dd, J = 8.2, 1.1 Hz, 1H), 4.14 (d, J = 9.5 Hz, 1H), 3.80 (s, 3H), 3.60 (dd, J = 11.4, 2.8 Hz, 1H), 3.52 (ddd, J = 9.6, 4.4, 2.7 Hz, 1H), 3.12 (dd, J = 11.5, 4.4 Hz, 1H), 2.84 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.1, 176.1, 159.5, 148.6, 130.5, 128.8, 127.7, 124.7, 119.8, 118.8, 114.4, 112.7, 55.6, 50.8, 43.6, 42.2, 39.6 ppm.
2-(4-Chlorophenyl)-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3c)7
Starting with 52.6 mg of N-(4-chlorophenyl)maleimide following the general procedure provided a white solid (51 mg, 62%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;71H NMR (400 MHz, chloroform-d): δ 7.51 (ddd, J = 7.5, 1.7, 0.8 Hz, 1H), 7.45–7.34 (m, 2H), 7.29–7.20 (m, 4H), 6.92 (td, J = 7.5, 1.1 Hz, 1H), 6.75 (dd, J = 8.3, 1.1 Hz, 1H), 4.15 (d, J = 9.6 Hz, 1H), 3.60 (dd, J = 11.5, 2.7 Hz, 1H), 3.52 (ddd, J = 9.6, 4.4, 2.7 Hz, 1H), 3.11 (dd, J = 11.5, 4.4 Hz, 1H), 2.84 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.5, 175.6, 148.6, 134.3, 130.5, 130.4, 129.3, 128.9, 127.7, 119.9, 118.5, 112.7, 50.7, 43.7, 42.2, 39.6 ppm.
2-(4-Fluorophenyl)-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3d)22
Starting with 48.3 mg of N-(4-fluorophenyl)maleimide following the general procedure provided a white solid (57.5 mg, 74%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;221H NMR (400 MHz, chloroform-d): δ 7.52 (ddd, J = 7.6, 1.6, 0.8 Hz, 1H), 7.29–7.20 (m, 3H), 7.15–7.07 (m, 2H), 6.92 (td, J = 7.5, 1.2 Hz, 1H), 6.75 (dd, J = 8.3, 1.1 Hz, 1H), 4.15 (d, J = 9.6 Hz, 1H), 3.60 (dd, J = 11.5, 2.7 Hz, 1H), 3.52 (ddd, J = 9.6, 4.4, 2.6 Hz, 1H), 3.11 (dd, J = 11.5, 4.4 Hz, 1H), 2.84 (s, 3H); 13C{1H} NMR (101 MHz, chloroform-d): δ 177.8, 175.8, 162.2 (d, 1JC–F = 248.4 Hz), 148.6, 130.4, 128.8, 128.3 (d, 3JC–F = 8.7 Hz), 128.0 (d, 4JC–F = 3.3 Hz), 119.8, 118.5, 116.1 (d, 2JC–F = 22.9 Hz), 112.7, 50.7, 43.7, 42.2, 39.5 ppm.
2-(2-(tert-Butyl)phenyl)-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3e)
Starting with 58.9 mg of N-(2-tertbutylphenyl)maleimide following the general procedure with an irradiation time of 18 h provided 3e as an orange oil (56.9 mg, 64%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). 1H NMR (400 MHz, chloroform-d): δ 7.54 (dddd, J = 22.0, 7.5, 1.5, 0.7 Hz, 2H), 7.39–7.32 (m, 1H), 7.29–7.23 (m, 1H), 7.22–7.16 (m, 1H), 6.92 (tt, J = 7.5, 0.9 Hz, 1H), 6.78 (dd, J = 8.2, 1.1 Hz, 1H), 6.68 (dd, J = 7.8, 1.5 Hz, 1H), 4.16 (d, J = 9.6 Hz, 1H), 3.61 (ddd, J = 11.4, 2.8, 0.7 Hz, 1H), 3.58–3.48 (m, 1H), 3.11 (ddd, J = 11.4, 4.6, 0.8 Hz, 1H), 2.86 (d, J = 0.7 Hz, 3H), 1.36 (t, J = 0.6 Hz, 9H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 179.1, 176.9, 162.5, 148.9, 147.8, 131.0, 130.6, 129.8, 128.8, 128.7, 127.4, 119.9, 119.1, 112.6, 51.1, 44.0, 42.6, 39.6, 35.8, 31.8 ppm; FTIR ν: 2958, 1709, 1495, 1375, 1316, 1195, 1181 cm–1; HRMS (ESI) m/z: calcd C22H24N2O2 [M + H]+, 349.1916; found, 349.1917.
2-(2-(tert-Butyl)phenyl)-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3e′)
Diastereomer 3e′ was furnished from 3e as follows: to a 2–5 mL Biotage microwave vial was added 3e (20 mg, 0.057 mmol). The vial was then capped and heated to 160 °C using a metal heating block. After 1 h of heating, the vial was cooled to room temperature and the crude mixture was purified using silica flash chromatography (10% ethyl acetate in petroleum ether) to afford 3e (8.4 mg, 42%) and 3e′ (6.9 mg, 35%) as white solids. mp 195.4–196.7 °C; 1H NMR (400 MHz, chloroform-d): δ 7.54–7.46 (m, 2H), 7.36 (ddd, J = 8.1, 7.3, 1.6 Hz, 1H), 7.29 (dd, J = 7.5, 1.5 Hz, 1H), 7.25–7.19 (m, 1H), 6.92–6.85 (m, 2H), 6.73 (dd, J = 8.3, 1.1 Hz, 1H), 4.18 (d, J = 9.5 Hz, 1H), 3.70 (dd, J = 11.5, 2.2 Hz, 1H), 3.54 (ddd, J = 9.6, 4.0, 2.2 Hz, 1H), 3.16 (dd, J = 11.5, 4.0 Hz, 1H), 2.84 (s, 3H), 1.00 (s, 9H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 179.1, 177.0, 148.8, 148.6, 131.4, 130.9, 130.3, 129.8, 128.8, 128.4, 127.5, 119.8, 118.1, 112.4, 50.4, 45.0, 43.1, 39.1, 35.1, 30.7 ppm; FTIR ν: 2966, 1710, 1497, 1371, 1265, 1180 cm–1; HRMS (ESI) m/z: calcd C22H24N2O2 [M + H]+, 349.1916; found, 349.1921.
2-(2,4-Dimethylphenyl)-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3f)
Starting with 53.0 mg of N-(2,4-dimethylphenyl)maleimide following the general procedure provided 3f as a 2:1 mixture of diastereomers as an orange oil (55.3 mg, 66%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). 1H NMR (400 MHz, chloroform-d): δ 7.56–7.48 (m, 3H), 7.26–7.22 (m, 3H), 7.15–6.98 (m, 8H), 6.95–6.88 (m, 3H), 6.86–6.72 (m, 4H), 4.23–4.12 (m, 3H), 3.66–3.50 (m, 6H), 3.15–3.06 (m, 3H), 2.86 (s, 3H), 2.85 (s, 6H), 2.35–2.28 (m, 9H), 2.16 (s, 3H), 1.80 (s, 6H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.1, 177.9, 176.1, 175.9, 148.7 (2C), 139.6, 139.4, 135.6, 135.0, 131.9 (2C), 130.5, 130.4, 128.7, 128.6, 128.0, 127.6 (3C), 119.8 (2C), 119.2, 118.8, 112.6, 112.4, 51.2, 50.9, 44.4, 43.7, 42.9, 42.4, 39.6, 39.3, 21.3 (2C), 17.8, 17.0 ppm; FTIR ν: 2924, 1706, 1498, 1386, 1197, 1180 cm–1; HRMS (ESI) m/z: calcd C20H20N2O2 [M + H]+, 321.1603; found, 321.1612.
5-Methyl-2-(1-phenylethyl)-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3g)65
Starting with 54.4 mg of 1-(1-phenylethyl)-1H-pyrrole-2,5-dione following the general procedure provided 3g as a 1:1 mixture of diastereomers as a brown oil (54.6 mg, 63%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature.651H NMR (400 MHz, chloroform-d): δ 7.48–7.43 (m, 2H), 7.35 (ddtd, J = 6.1, 4.6, 1.6, 0.7 Hz, 4H), 7.32–7.18 (m, 8H), 6.89 (dtd, J = 8.7, 7.5, 1.2 Hz, 2H), 6.72 (ddd, J = 8.2, 2.2, 1.1 Hz, 2H), 5.47–5.31 (m, 2H), 3.98–3.87 (m, 2H), 3.44 (ddd, J = 11.4, 9.2, 3.0 Hz, 2H), 3.37–3.24 (m, 2H), 3.04 (ddd, J = 11.3, 6.4, 4.6 Hz, 2H), 2.82 (s, 3H), 2.78 (s, 3H), 1.79 (d, J = 7.3 Hz, 3H), 1.75 (d, J = 7.3 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.6, 178.5, 176.7, 176.5, 148.6 (2C), 139.5 (2C), 130.3 (2C), 128.6 (2C), 128.5, 128.4, 127.7 (2C), 127.2 (2C), 119.8, 119.7, 119.3, 119.0, 112.5 (2C), 51.2, 51.0, 50.7, 50.6, 43.5, 43.4, 42.1, 42.0, 39.5, 39.4, 16.9, 16.8 ppm.
2,5-Dimethyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3h)35
Starting from 27.8 mg of N-methylmaleimide following the general procedure, 3h as a white solid (18.0 mg, 31% with a 6 h reaction time or 26 mg, 44% with an 18 h reaction time) was isolated after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether); spectroscopic data were in accordance with the literature;351H NMR (400 MHz, chloroform-d): δ 7.47 (ddd, J = 7.4, 1.7, 0.8 Hz, 1H), 7.24–7.14 (m, 1H), 6.89 (td, J = 7.5, 1.1 Hz, 1H), 6.69 (dd, J = 8.3, 1.1 Hz, 1H), 3.99 (d, J = 9.4 Hz, 1H), 3.53 (dd, J = 11.5, 2.5 Hz, 1H), 3.36 (ddd, J = 9.4, 4.4, 2.4 Hz, 1H), 3.03 (dd, J = 11.5, 4.4 Hz, 1H), 2.98 (s, 3H), 2.79 (s, 3H); 13C{1H} NMR (101 MHz, chloroform-d): δ 178.9, 176.9, 148.5, 130.3, 128.7, 119.8, 118.9, 112.7, 50.6, 43.7, 42.1, 39.6, 25.5 ppm.
5-Methyl-2-propyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3i)7
Starting from 34.1 mg of N-propylmaleimide following the general procedure, 3i as a white solid (30.2 mg, 48%) was isolated after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;71H NMR (400 MHz, chloroform-d): δ 7.48–7.42 (m, 1H), 7.22–7.14 (m, 1H), 6.87 (td, J = 7.4, 1.2 Hz, 1H), 6.69 (dd, J = 8.2, 1.3 Hz, 1H), 3.95 (d, J = 9.3 Hz, 1H), 3.53–3.40 (m, 3H), 3.33 (ddd, J = 9.0, 4.3, 2.4 Hz, 1H), 3.01 (dd, J = 11.5, 4.5 Hz, 1H), 2.78 (s, 3H), 1.66–1.47 (m, 2H), 0.81 (td, J = 7.4, 1.7 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.9, 176.9, 148.4, 130.3, 128.6, 119.8, 119.2, 112.6, 50.6, 43.6, 42.1, 41.0, 39.5, 21.0, 11.1 ppm.
2-Cyclohexyl-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3j)7
Starting from 47.4 mg of N-cyclohexylmaleimide following the general procedure, 3j as a yellow oil (49.9 mg, 64%) was isolated after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;71H NMR (400 MHz, chloroform-d): δ 7.46 (ddd, J = 7.5, 1.7, 0.7 Hz, 1H), 7.23–7.14 (m, 1H), 6.88 (td, J = 7.5, 1.1 Hz, 1H), 6.69 (dd, J = 8.2, 1.1 Hz, 1H), 4.00–3.85 (m, 2H), 3.45 (dd, J = 11.4, 3.0 Hz, 1H), 3.28 (ddd, J = 9.5, 4.6, 3.0 Hz, 1H), 3.02 (dd, J = 11.4, 4.6 Hz, 1H), 2.79 (s, 3H), 2.21–1.96 (m, 2H), 1.84–1.72 (m, 2H), 1.66–1.58 (m, 1H), 1.58–1.49 (m, 2H), 1.37–1.09 (m, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.8, 176.9, 148.5, 130.3, 128.5, 119.5, 119.1, 112.4, 52.2, 50.9, 43.1, 41.8, 39.5, 28.9, 28.8, 25.9, 25.8, 25.1 ppm.
2-(tert-Butyl)-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3k)7
37.8 mg of N-tBu-maleimide provided an orange oil, 37.3 mg (54%), after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;71H NMR (400 MHz, chloroform-d): δ 7.45 (dddd, J = 7.5, 1.7, 0.9, 0.4 Hz, 1H), 7.25–7.18 (m, 1H), 6.88 (td, J = 7.5, 1.2 Hz, 1H), 6.72 (dd, J = 8.2, 1.1 Hz, 1H), 3.83 (d, J = 9.7 Hz, 1H), 3.44 (dd, J = 11.4, 3.1 Hz, 1H), 3.21 (ddd, J = 9.7, 4.6, 3.1 Hz, 1H), 3.01 (dd, J = 11.4, 4.6 Hz, 1H), 2.81 (s, 3H), 1.54 (s, 9H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 179.6, 177.9, 148.2, 130.4, 128.6, 119.7, 119.4, 112.7, 58.9, 51.0, 43.1, 42.2, 39.7, 28.5 ppm.
5-Methyl-2-phenethyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3l)
Starting with 52.9 mg of N-phenethylmaleimide following the general procedure provided 3l as an orange oil (39.4 mg, 47%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). 1H NMR (400 MHz, chloroform-d): δ 7.45 (ddd, J = 7.6, 1.6, 0.8 Hz, 1H), 7.25–7.13 (m, 4H), 7.10 (dd, J = 7.7, 1.9 Hz, 2H), 6.89 (td, J = 7.4, 1.1 Hz, 1H), 6.71 (dd, J = 8.3, 1.1 Hz, 1H), 3.93 (d, J = 9.4 Hz, 1H), 3.74 (t, J = 7.4 Hz, 2H), 3.47 (dd, J = 11.5, 2.6 Hz, 1H), 3.28 (ddd, J = 9.4, 4.4, 2.6 Hz, 1H), 3.01 (dd, J = 11.5, 4.4 Hz, 1H), 2.85 (td, J = 7.2, 1.4 Hz, 2H), 2.78 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.5, 176.6, 148.4, 137.8, 130.2, 128.9, 128.6, 128.5, 126.6, 119.6, 118.8, 112.5, 50.4, 43.5, 42.0, 40.5, 39.5, 33.5 ppm; ATR-FTIR ν: 2949, 1698, 1499, 1401, 1354, 1159 cm–1; HRMS (ESI) m/z: calcd C20H20N2O2 [M + H]+, 321.1603; found, 321.1610.
2-Benzyl-5-methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3m)7
Starting with 46.5 mg of N-benzylmaleimide following the general procedure provided a white solid (45.3 mg, 60%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;71H NMR (400 MHz, chloroform-d): δ 7.51–7.41 (m, 1H), 7.38–7.13 (m, 6H), 6.90 (td, J = 7.5, 1.2 Hz, 1H), 6.71 (dd, J = 8.2, 1.1 Hz, 1H), 4.76–4.51 (m, 2H), 3.99 (d, J = 9.4 Hz, 1H), 3.50 (dd, J = 11.5, 2.7 Hz, 1H), 3.36 (ddd, J = 9.4, 4.6, 2.8 Hz, 1H), 3.05 (dd, J = 11.5, 4.6 Hz, 1H), 2.80 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.4, 176.5, 148.5, 135.7, 130.3, 128.7, 128.7, 128.4, 127.9, 119.8, 119.0, 112.6, 50.8, 43.7, 42.9, 42.2, 39.5 ppm.
5-Methyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3n)66
Starting with 25.9 mg of maleimide following the general procedure provided 3n as an off-white solid after column chromatography using 0–4% methanol in dichloromethane (17.2 mg, 30%). Spectroscopic data were in accordance with the literature;661H NMR (400 MHz, chloroform-d): δ 8.56 (s, 1H), 7.50–7.36 (m, 1H), 7.25–7.17 (m, 1H), 6.89 (td, J = 7.4, 1.1 Hz, 1H), 6.72 (dd, J = 8.3, 1.1 Hz, 1H), 4.02 (d, J = 9.5 Hz, 1H), 3.50 (dd, J = 11.5, 2.6 Hz, 1H), 3.41 (ddd, J = 9.5, 4.5, 2.6 Hz, 1H), 3.02 (ddd, J = 11.5, 4.4, 0.8 Hz, 1H), 2.81 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 179.0, 177.0, 148.5, 130.2, 128.8, 119.9, 118.5, 112.7, 50.5, 44.9, 43.4, 39.6 ppm.
5,8-Dimethyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3o)7
Starting with 43.9 mg of N-phenylmaleimide following the general procedure provided a white solid (65.0 mg, 84%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;71H NMR (400 MHz, chloroform-d): δ 7.46–7.38 (m, 2H), 7.37–7.30 (m, 2H), 7.29–7.23 (m, 2H), 7.07–6.98 (m, 1H), 6.65 (d, J = 8.3 Hz, 1H), 4.11 (d, J = 9.5 Hz, 1H), 3.58 (dd, J = 11.4, 2.7 Hz, 1H), 3.50 (ddd, J = 9.6, 4.4, 2.7 Hz, 1H), 3.05 (dd, J = 11.4, 4.4 Hz, 1H), 2.79 (s, 3H), 2.29 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.9, 176.0, 146.4, 132.1, 131.0, 129.4, 129.2, 129.1, 128.6, 126.5, 118.7, 112.7, 51.1, 43.7, 42.3, 39.8, 20.6 ppm.
5,7,9-Trimethyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3p)22
Starting with 42.4 mg of N-phenylmaleimide following the general procedure provided 3p as a yellow solid (41.8 mg, 53%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;221H NMR (400 MHz, chloroform-d): δ 7.44–7.37 (m, 2H), 7.36–7.29 (m, 1H), 7.28–7.22 (m, 2H), 6.71–6.36 (m, 2H), 4.45 (d, J = 9.8 Hz, 1H), 3.54 (dd, J = 11.3, 1.6 Hz, 1H), 3.48 (ddd, J = 9.8, 4.8, 1.7 Hz, 1H), 2.91 (dd, J = 11.3, 4.8 Hz, 1H), 2.76 (s, 3H), 2.53 (s, 3H), 2.27 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.7, 176.0, 150.1, 138.4, 137.9, 132.2, 129.1, 128.6, 126.6, 123.7, 116.6, 111.6, 52.7, 44.8, 40.0, 39.4, 21.7, 20.4 ppm.
8-Bromo-5-methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3q)43
Starting with 46.3 mg of N-phenylmaleimide following the general provided 3q as a yellow solid (69.3 mg, 70%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;431H NMR (400 MHz, chloroform-d): δ 7.64 (m, 1H), 7.48–7.39 (m, 2H), 7.40–7.33 (m, 1H), 7.31 (m, 1H), 7.29–7.21 (m, 2H), 6.60 (dd, J = 8.8, 1.9 Hz, 1H), 4.10 (dd, J = 9.6, 1.8 Hz, 1H), 3.60 (m, 1H), 3.56–3.45 (m, 1H), 3.11 (m, 1H), 2.82 (d, J = 1.9 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.4, 175.2, 147.6, 132.8, 131.9, 131.6, 129.2, 128.8, 126.4, 120.5, 114.4, 111.8, 50.5, 43.4, 41.9, 39.6 ppm.
5-Methyl-2-phenyl-8-(pyridin-2-yl)-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3r)
Starting with 18.1 mg of N-phenylmaleimide following the general procedure using an irradiation time of 18 h provided 3r as a yellow foam (26.4 mg, 68%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). mp 183.5–184.6 °C; 1H NMR (400 MHz, chloroform-d): δ 8.16 (p, J = 1.0 Hz, 1H), 7.90 (dt, J = 8.7, 1.7 Hz, 1H), 7.69 (dt, J = 4.9, 1.4 Hz, 2H), 7.40 (ddd, J = 7.6, 6.6, 1.4 Hz, 2H), 7.37–7.29 (m, 1H), 7.26 (dt, J = 8.3, 1.4 Hz, 3H), 7.14 (qd, J = 4.6, 1.3 Hz, 1H), 6.82 (dd, J = 8.6, 1.3 Hz, 1H), 4.28–4.18 (m, 1H), 3.64 (ddd, J = 11.5, 2.9, 1.3 Hz, 1H), 3.56 (dddd, J = 8.7, 4.3, 2.8, 1.3 Hz, 1H), 3.18 (ddd, J = 11.5, 4.4, 1.3 Hz, 1H), 2.89 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.6, 175.8, 157.0, 149.4, 149.2, 137.0, 132.1, 130.6, 129.1, 129.0, 128.7, 127.5, 126.5, 121.5, 120.0, 118.5, 113.0, 50.4, 43.5, 42.2, 39.6 ppm; FTIR ν: 1704, 1568, 1469, 1370, 1152, 776, 761, 691, 576 cm–1; HRMS (ESI) m/z: calcd C23H19N3O2 [M + H]+, 370.1556; found, 370.1556.
8-Acetyl-5-methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3s)21
Starting with 43.5 mg of N-phenylmaleimide following the general procedure, 3s was obtained as an off-white solid (39.5 mg, 47%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;211H NMR (400 MHz, chloroform-d): δ 8.13 (dd, J = 2.2, 0.9 Hz, 1H), 7.85 (dd, J = 8.7, 2.1 Hz, 1H), 7.47–7.39 (m, 2H), 7.39–7.32 (m, 1H), 7.28–7.20 (m, 2H), 6.74 (d, J = 8.7 Hz, 1H), 4.19 (dd, J = 9.5, 0.9 Hz, 1H), 3.67 (dd, J = 11.7, 3.0 Hz, 1H), 3.58 (ddd, J = 9.6, 4.5, 2.9 Hz, 1H), 3.25 (dd, J = 11.7, 4.5 Hz, 1H), 2.93 (s, 3H), 2.54 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 196.5, 177.0, 175.4, 151.8, 131.9, 131.3, 129.6, 129.1, 128.7, 128.6, 126.3, 117.2, 112.1, 49.6, 43.0, 41.7, 39.5, 26.3 ppm.
Ethyl-5-methyl-1,3-dioxo-2-phenyl-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,4-c]quinoline-8-carboxylate (3t)
Starting with 43.3 mg of N-phenylmaleimide following the general procedure, the product was obtained as an off-white solid after silica column chromatography using 0–10% EtOAc in petroleum ether. Subsequent recrystallization from hot ethyl acetate afforded compound 3t as a yellow crystalline solid (26.9 mg, 30%). mp 156.5–157.8 °C; 1H NMR (400 MHz, chloroform-d): δ 8.21 (d, J = 2.0 Hz, 1H), 7.91 (dd, J = 8.7, 2.0 Hz, 1H), 7.42 (dd, J = 8.3, 6.6 Hz, 2H), 7.38–7.32 (m, 1H), 7.25 (dd, J = 7.4, 1.6 Hz, 2H), 6.73 (d, J = 8.7 Hz, 1H), 4.42–4.27 (m, 2H), 4.19 (d, J = 9.4 Hz, 1H), 3.67 (dd, J = 11.6, 2.5 Hz, 1H), 3.62–3.51 (m, 1H), 3.22 (dd, J = 11.7, 4.2 Hz, 1H), 2.92 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.2, 175.4, 166.5, 151.7, 131.9, 131.9, 130.7, 129.1, 128.7, 126.4, 121.3, 117.4, 112.1, 60.7, 49.9, 43.2, 41.8, 39.5, 14.6 ppm; ATR-FTIR ν: 1709, 1700, 1606, 1517, 1500, 1373, 1272, 1178, 1152, 759 cm–1; HRMS (ESI+) m/z: calcd C21H20N2O4 [M + H]+, 365.1501; found, 365.1501.
5-Methyl-1,3-dioxo-2-phenyl-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,4-c]quinoline-8-carbonitrile (3u)
Starting with 39.7 mg of N-phenylmaleimide following the general procedure provided 3u as a yellow solid (18 mg, 25%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). mp 194.0–194.9 °C; 1H NMR (400 MHz, chloroform-d): δ 7.82 (dd, J = 2.1, 0.8 Hz, 1H), 7.51–7.41 (m, 3H), 7.41–7.35 (m, 1H), 7.29–7.20 (m, 2H), 6.74 (d, J = 8.6 Hz, 1H), 4.16 (dd, J = 9.6, 0.8 Hz, 1H), 3.69 (dd, J = 11.8, 3.1 Hz, 1H), 3.59 (ddd, J = 9.6, 4.5, 3.1 Hz, 1H), 3.29 (dd, J = 11.8, 4.5 Hz, 1H), 2.94 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 176.7, 174.8, 151.2, 134.2, 133.1, 131.7, 129.3, 128.9, 126.3, 119.6, 118.3, 112.8, 101.8, 49.4, 42.8, 41.4, 39.5 ppm; FTIR ν: 3066, 2931, 2217, 1710, 1604, 1514, 1497, 1374 cm–1; HRMS (ESI) m/z: calcd C19H15N3O2 [M + H]+, 318.1243; found, 318.1253.
5,6-Dimethyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3v)35
Yellow oil (32.9 mg, 43%) obtained after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;351H NMR (400 MHz, chloroform-d): δ 7.57 (ddt, J = 7.6, 1.6, 0.7 Hz, 1H), 7.49–7.41 (m, 2H), 7.41–7.34 (m, 1H), 7.29–7.23 (m, 2H), 7.14 (ddt, J = 7.4, 1.6, 0.7 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 4.16 (d, J = 8.9 Hz, 1H), 3.60–3.45 (m, 2H), 3.45–3.32 (m, 1H), 2.74 (s, 3H), 2.30 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.9, 175.9, 146.8, 132.9, 132.1, 130.6, 129.3, 129.1, 128.8, 128.6, 126.4, 124.0, 123.1, 51.5, 42.2, 41.7, 39.0, 17.8 ppm.
8-Ethynyl-5-methyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3w)21
Starting with 41.3 mg of N-phenylmaleimide following the general procedure provided 3w as a yellow solid (19 mg, 25%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;211H NMR (400 MHz, chloroform-d): δ 7.68 (dd, J = 2.0, 0.9 Hz, 1H), 7.49–7.40 (m, 2H), 7.40–7.31 (m, 2H), 7.30–7.21 (m, 3H), 6.67 (d, J = 8.5 Hz, 1H), 4.12 (dt, J = 9.6, 0.6 Hz, 1H), 3.63 (dd, J = 11.6, 2.8 Hz, 1H), 3.54 (ddd, J = 9.6, 4.4, 2.8 Hz, 1H), 3.17 (dd, J = 11.6, 4.4 Hz, 1H), 3.00 (s, 1H), 2.86 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.4, 175.4, 148.7, 134.2, 132.8, 132.0, 129.2, 128.7, 126.4, 118.2, 112.8, 112.6, 83.9, 76.1, 50.2, 43.3, 41.8, 39.5 ppm.
5-Benzyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3x)20
Starting with 40.5 mg of N-phenylmaleimide following the general procedure provided 3x as a yellow oil (36.6 mg, 42%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;201H NMR (400 MHz, chloroform-d): δ 7.60–7.52 (m, 1H), 7.47 (dd, J = 8.3, 6.8 Hz, 2H), 7.43–7.36 (m, 1H), 7.34–7.22 (m, 7H), 7.19–7.11 (m, 1H), 6.90 (td, J = 7.5, 1.1 Hz, 1H), 6.76 (d, J = 1.0 Hz, 1H), 4.55–4.25 (m, 2H), 4.19 (d, J = 9.5 Hz, 1H), 3.70 (dd, J = 11.7, 2.9 Hz, 1H), 3.55 (ddd, J = 9.5, 4.4, 2.8 Hz, 1H), 3.28 (dd, J = 11.7, 4.4 Hz, 1H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.6, 175.9, 147.6, 137.8, 132.2, 130.7, 130.3, 129.2, 128.7, 128.7, 128.6, 127.5, 127.4, 126.5, 119.9, 119.0, 113.6, 55.5, 49.2, 44.3, 42.6 ppm.
5-Allyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3y)20
Starting with 41.6 mg of N-phenylmaleimide following the general procedure provided 3y as a yellow oil (44.1 mg, 58%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;201H NMR (400 MHz, chloroform-d): δ 7.54 (ddd, J = 7.6, 1.7, 0.8 Hz, 1H), 7.47–7.40 (m, 2H), 7.40–7.33 (m, 1H), 7.30–7.23 (m, 2H), 7.24–7.16 (m, 1H), 6.89 (td, J = 7.5, 1.1 Hz, 1H), 6.78 (dd, J = 8.3, 1.1 Hz, 1H), 5.87 (ddt, J = 17.3, 10.3, 5.8 Hz, 1H), 5.34–5.21 (m, 2H), 4.15 (d, J = 9.5 Hz, 1H), 3.96–3.74 (m, 2H), 3.68 (dd, J = 11.8, 2.9 Hz, 1H), 3.54 (ddd, J = 9.5, 4.3, 2.8 Hz, 1H), 3.15 (dd, J = 11.8, 4.3 Hz, 1H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 177.7, 175.9, 147.4, 133.3, 132.1, 130.7, 129.1, 128.6, 128.6, 126.4, 119.6, 118.9, 118.3, 113.3, 53.6, 47.8, 44.1, 42.5 ppm.
5,9-Dimethyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3aa) and 5,7-Dimethyl-2-phenyl-3a,4,5,9b-tetrahydro-1H-pyrrolo[3,4-c]quinoline-1,3(2H)-dione (3aa′)22
Starting with 41.6 mg of N-phenylmaleimide following the general procedure provided a mixture of regioisomers 3aa and 3aa′ as an off-white solid (31.8 mg, 42%) after purification using column chromatography (SiO2, 0–7% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;221H NMR (400 MHz, chloroform-d): δ 7.46–7.39 (m, 4H), 7.38–7.32 (m, 1.6H), 7.32–7.26 (m, 3H), 7.13 (t, J = 7.9 Hz, 1H), 6.82 (d, J = 7.5 Hz, 1H), 6.77–6.70 (m, 0.6H), 6.64 (d, J = 8.2 Hz, 1H), 6.56 (d, J = 1.5 Hz, 0.6H), 4.53 (d, J = 9.8 Hz, 1H), 4.13 (d, J = 9.6 Hz, 0.6H), 3.63–3.57 (m, 1.8H), 3.56–3.49 (m, 1.6H), 3.11 (dd, J = 11.5, 4.4 Hz, 1H), 2.96 (dd, J = 11.3, 4.8 Hz, 1H), 2.83 (s, 1.6H), 2.79 (s, 3H), 2.59 (s, 3H), 2.33 (s, 1.6H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 178.7, 178.0, 176.1, 175.8, 150.2, 148.5, 138.74, 138.69, 132.2, 132.1, 130.3, 129.2, 129.1, 128.7, 128.6, 128.2, 126.6, 126.5, 122.8, 120.7, 119.6, 115.7, 113.4, 110.8, 52.7, 50.8, 45.0, 43.7, 42.0, 40.0, 39.62, 39.60, 21.8, 20.5 ppm.
1-Methyl-1,2,3,4-tetrahydroquinoline-3,4-dicarbonitrile (5)
Starting with fumaronitrile (18.7 mg) following the general procedure, 5 was afforded as a white solid (8.9 mg, 19%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). 1H NMR (400 MHz, chloroform-d): δ 7.33–7.14 (m, 3H), 6.81 (td, J = 7.5, 1.1 Hz, 1H), 6.72 (dd, J = 8.3, 1.0 Hz, 1H), 4.25 (d, J = 5.7 Hz, 1H), 3.69 (dd, J = 12.0, 3.1 Hz, 1H), 3.59–3.41 (m, 2H), 3.01 (s, 3H); 13C{1H} NMR (101 MHz, chloroform-d): δ 144.4, 130.7, 129.5, 118.5, 118.2, 117.4, 112.8, 111.6, 50.1, 39.2, 32.9, 28.8 ppm; FTIR ν: 2843, 2247, 1601, 1507, 1349, 1310, 1223, 740 cm–1; HRMS (ESI) m/z: calcd C12H11N3 [M + H]+, 198.1031; found, 198.1031.
1-Methyl-3-phenyl-2,3-dihydroquinoline-4,4(1H)-dicarbonitrile (6)65
Starting with benzylidenemalonitrile (34 mg), 6 was afforded as a white solid (9.9 mg, 17%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;651H NMR (400 MHz, chloroform-d): δ 7.91 (ddd, J = 8.5, 1.3, 0.6 Hz, 1H), 7.51–7.42 (m, 5H), 7.39–7.32 (m, 1H), 6.82 (ddd, J = 7.8, 7.3, 1.1 Hz, 1H), 6.75 (dd, J = 8.4, 1.1 Hz, 1H), 3.97 (dd, J = 12.4, 11.4 Hz, 1H), 3.61 (dd, J = 11.4, 3.9 Hz, 1H), 3.52 (dd, J = 12.4, 3.9 Hz, 1H), 3.03 (s, 3H); 13C{1H} NMR (101 MHz, chloroform-d): δ 144.1, 134.9, 131.9, 129.6, 129.3, 129.0, 128.6, 117.8, 115.3, 114.2, 112.9, 112.7, 51.5, 45.7, 42.6, 38.9 ppm.
Diethyl (3R,4R)-1-Methyl-1,2,3,4-tetrahydroquinoline-3,4-dicarboxylate (7)28
Starting with diethyl fumarate (25.8 mg), following the general procedure, 7 was obtained as a colorless oil (13 mg, 29%) after purification using column chromatography (SiO2, 0–10% ethyl acetate in petroleum ether). Spectroscopic data were in accordance with the literature;281H NMR (400 MHz, chloroform-d): δ 7.22 (dt, J = 7.7, 1.4 Hz, 1H), 7.14 (dddd, J = 8.1, 7.3, 1.7, 0.7 Hz, 1H), 6.81–6.58 (m, 2H), 4.30–4.07 (m, 5H), 3.59–3.49 (m, 1H), 3.46–3.34 (m, 2H), 2.92 (s, 3H), 1.29 (t, J = 7.1 Hz, 3H), 1.23 (t, J = 7.1 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d): δ 173.3, 172.2, 145.8, 129.2, 128.5, 117.9, 117.3, 112.0, 61.4, 61.2, 50.4, 44.8, 41.3, 39.5, 14.3 (2C) ppm.
Acknowledgments
This work was supported by grants from the Swedish Research Council FORMAS (2019-00699) and from Olle Engkvists Stiftelse.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c02776.
UV–vis studies, emission spectrum of the irradiation source, mechanistic investigations, and NMR spectra (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Lima C. G. S.; de M. Lima T.; Duarte M.; Jurberg I. D.; Paixão M. W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. 10.1021/acscatal.5b02386. [DOI] [Google Scholar]
- Crisenza G. E. M.; Mazzarella D.; Melchiorre P. Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.-q.; Majumder S.; Yang M.-h.; Guo S.-r. Recent Advances in Catalyst-Free Photochemical Reactions via Electron-Donor-Acceptor (EDA) Complex Process. Tetrahedron Lett. 2020, 61, 151506. 10.1016/j.tetlet.2019.151506. [DOI] [Google Scholar]
- Postigo A. Electron Donor-Acceptor Complexes in Perfluoroalkylation Reactions. Eur. J. Org. Chem. 2018, 2018, 6391–6404. 10.1002/ejoc.201801079. [DOI] [Google Scholar]
- Tobisu M.; Furukawa T.; Chatani N. Visible Light-Mediated Direct Arylation of Arenes and Heteroarenes Using Diaryliodonium Salts in the Presence and Absence of a Photocatalyst. Chem. Lett. 2013, 42, 1203–1205. 10.1246/cl.130547. [DOI] [Google Scholar]
- Arceo E.; Jurberg I. D.; Álvarez-Fernández A.; Melchiorre P. Photochemical Activity of a Key Donor–Acceptor Complex Can Drive Stereoselective Catalytic α-Alkylation of Aldehydes. Nat. Chem. 2013, 5, 750–756. 10.1038/nchem.1727. [DOI] [PubMed] [Google Scholar]
- Hsu C.-W.; Sundén H. α-Aminoalkyl Radical Addition to Maleimides via Electron Donor–Acceptor Complexes. Org. Lett. 2018, 20, 2051–2054. 10.1021/acs.orglett.8b00597. [DOI] [PubMed] [Google Scholar]
- Guillemard L.; Colobert F.; Wencel-Delord J. Visible-Light-Triggered, Metal- and Photocatalyst-Free Acylation of N-Heterocycles. Adv. Synth. Catal. 2018, 360, 4184–4190. 10.1002/adsc.201800692. [DOI] [Google Scholar]
- Kandukuri S. R.; Bahamonde A.; Chatterjee I.; Jurberg I. D.; Escudero-Adán E. C.; Melchiorre P. X-Ray Characterization of an Electron Donor–Acceptor Complex That Drives the Photochemical Alkylation of Indoles. Angew. Chem., Int. Ed. 2015, 54, 1485–1489. 10.1002/anie.201409529. [DOI] [PubMed] [Google Scholar]
- McClain E. J.; Monos T. M.; Mori M.; Beatty J. W.; Stephenson C. R. J. Design and Implementation of a Catalytic Electron Donor–Acceptor Complex Platform for Radical Trifluoromethylation and Alkylation. ACS Catal. 2020, 10, 12636–12641. 10.1021/acscatal.0c03837. [DOI] [Google Scholar]
- Bosque I.; Bach T. 3-Acetoxyquinuclidine as Catalyst in Electron Donor–Acceptor Complex-Mediated Reactions Triggered by Visible Light. ACS Catal. 2019, 9, 9103–9109. 10.1021/acscatal.9b01039. [DOI] [Google Scholar]
- de Pedro Beato E.; Spinnato D.; Zhou W.; Melchiorre P. A General Organocatalytic System for Electron Donor–Acceptor Complex Photoactivation and Its Use in Radical Processes. J. Am. Chem. Soc. 2021, 143, 12304–12314. 10.1021/jacs.1c05607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woźniak Ł.; Murphy J. J.; Melchiorre P. Photo-Organocatalytic Enantioselective Perfluoroalkylation of β-Ketoesters. J. Am. Chem. Soc. 2015, 137, 5678–5681. 10.1021/jacs.5b03243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morack T.; Mück-Lichtenfeld C.; Gilmour R. Bioinspired Radical Stetter Reaction: Radical Umpolung Enabled by Ion-Pair Photocatalysis. Angew. Chem., Int. Ed. 2019, 58, 1208–1212. 10.1002/anie.201809601. [DOI] [PubMed] [Google Scholar]
- Fu M.-C.; Shang R.; Zhao B.; Wang B.; Fu Y. Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide. Science 2019, 363, 1429–1434. 10.1126/science.aav3200. [DOI] [PubMed] [Google Scholar]
- Quint V.; Morlet-Savary F.; Lohier J.-F.; Lalevée J.; Gaumont A.-C.; Lakhdar S. Metal-Free, Visible Light-Photocatalyzed Synthesis of Benzo[b]Phosphole Oxides: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2016, 138, 7436–7441. 10.1021/jacs.6b04069. [DOI] [PubMed] [Google Scholar]
- Aramaki Y.; Imaizumi N.; Hotta M.; Kumagai J.; Ooi T. Exploiting Single-Electron Transfer in Lewis Pairs for Catalytic Bond-Forming Reactions. Chem. Sci. 2020, 11, 4305–4311. 10.1039/D0SC01159B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W.; Huang J.; Xu X.; Wang L.; Tang X.-Y. B(C6F5)3-Catalyzed Electron Donor–Acceptor Complex-Mediated Aerobic Sulfenylation of Indoles under Visible-Light Conditions. Org. Lett. 2021, 23, 7139–7143. 10.1021/acs.orglett.1c02553. [DOI] [PubMed] [Google Scholar]
- Runemark A.; Zacharias S. C.; Sundén H. Visible-Light-Driven Stereoselective Annulation of Alkyl Anilines and Dibenzoylethylenes via Electron Donor–Acceptor Complexes. J. Org. Chem. 2021, 86, 1901–1910. 10.1021/acs.joc.0c02819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perumal G.; Kandasamy M.; Ganesan B.; Govindan K.; Sathya H.; Hung M.-Y.; Chandru Senadi G.; Wu Y.-C.; Lin W.-Y. Visible Light-Induced N-Methyl Activation of Unsymmetric Tertiary Amines. Tetrahedron 2021, 80, 131891. 10.1016/j.tet.2020.131891. [DOI] [Google Scholar]
- Sharma K.; Das B.; Gogoi P. Synthesis of Pyrrolo[3,4-c]Quinoline-1,3-Diones: A Sequential Oxidative Annulation Followed by Dehydrogenation and N-Demethylation Strategy. New J. Chem. 2018, 42, 18894–18905. 10.1039/c8nj04443k. [DOI] [Google Scholar]
- Yang X.-L.; Guo J.-D.; Lei T.; Chen B.; Tung C.-H.; Wu L.-Z. Oxidative Cyclization Synthesis of Tetrahydroquinolines and Reductive Hydrogenation of Maleimides under Redox-Neutral Conditions. Org. Lett. 2018, 20, 2916–2920. 10.1021/acs.orglett.8b00977. [DOI] [PubMed] [Google Scholar]
- Ranieri A. M.; Burt L. K.; Stagni S.; Zacchini S.; Skelton B. W.; Ogden M. I.; Bissember A. C.; Massi M. Anionic Cyclometallated Platinum(II) Tetrazolato Complexes as Viable Photoredox Catalysts. Organometallics 2019, 38, 1108–1117. 10.1021/acs.organomet.8b00913. [DOI] [Google Scholar]
- Firoozi S.; Hosseini-Sarvari M.; Koohgard M. Solvent-Free and Room Temperature Visible Light-Induced C–H Activation: CdS as a Highly Efficient Photo-Induced Reusable Nano-Catalyst for the C–H Functionalization Cyclization of t-Amines and C–C Double and Triple Bonds. Green Chem. 2018, 20, 5540–5549. 10.1039/C8GC03297A. [DOI] [Google Scholar]
- Hosseini-Sarvari M.; Koohgard M.; Firoozi S.; Mohajeri A.; Tavakolian H. Alizarin Red S–TiO2-Catalyzed Cascade C(Sp3)–H to C(Sp2)–H Bond Formation/Cyclization Reactions toward Tetrahydroquinoline Derivatives under Visible Light Irradiation. New J. Chem. 2018, 42, 6880–6888. 10.1039/C8NJ00476E. [DOI] [Google Scholar]
- Yadav A. K.; Yadav L. D. S. Visible Light Photoredox Catalysis with N-Hydroxyphthalimide for [4+2] Cyclization between N-Methylanilines and Maleimides. Tetrahedron Lett. 2017, 58, 552–555. 10.1016/j.tetlet.2016.12.077. [DOI] [Google Scholar]
- Guo J.-T.; Yang D.-C.; Guan Z.; He Y.-H. Chlorophyll-Catalyzed Visible-Light-Mediated Synthesis of Tetrahydroquinolines from N,N-Dimethylanilines and Maleimides. J. Org. Chem. 2017, 82, 1888–1894. 10.1021/acs.joc.6b03034. [DOI] [PubMed] [Google Scholar]
- Song Z.; Antonchick A. P. Catching α-Aminoalkyl Radicals: Cyclization between Tertiary Alkylanilines and Alkenes. Tetrahedron 2016, 72, 7715–7721. 10.1016/j.tet.2016.04.052. [DOI] [Google Scholar]
- Nicholls T. P.; Constable G. E.; Robertson J. C.; Gardiner M. G.; Bissember A. C. Brønsted Acid Cocatalysis in Copper(I)-Photocatalyzed α-Amino C–H Bond Functionalization. ACS Catal. 2016, 6, 451–457. 10.1021/acscatal.5b02014. [DOI] [Google Scholar]
- Yadav A. K.; Yadav L. D. S. Intermolecular Cyclization of N-Methylanilines and Maleimides to Tetrahydroquinolines via K2S2O8 Promoted C(Sp3)–H Activation. Tetrahedron Lett. 2016, 57, 1489–1491. 10.1016/j.tetlet.2016.02.078. [DOI] [Google Scholar]
- Nikitas N. F.; Theodoropoulou M. A.; Kokotos C. G. Photochemical Reaction of N,N-Dimethylanilines with N-Substituted Maleimides Utilizing Benzaldehyde as the Photoinitiator. Eur. J. Org. Chem. 2021, 2021, 1168–1173. 10.1002/ejoc.202001593. [DOI] [Google Scholar]
- Huo C.; Chen F.; Quan Z.; Dong J.; Wang Y. Cobalt-Catalyzed Aerobic Oxidative Povarov Reaction of Tertiary Anilines with Dihydrofuran for the Synthesis of Hexahydrofuroquinolines. Tetrahedron Lett. 2016, 57, 5127–5131. 10.1016/j.tetlet.2016.10.031. [DOI] [Google Scholar]
- Wang Z. J.; Ghasimi S.; Landfester K.; Zhang K. A. I. Bandgap Engineering of Conjugated Nanoporous Poly-Benzobisthiadiazoles via Copolymerization for Enhanced Photocatalytic 1,2,3,4-Tetrahydroquinoline Synthesis under Visible Light. Adv. Synth. Catal. 2016, 358, 2576–2582. 10.1002/adsc.201600125. [DOI] [Google Scholar]
- Sakai N.; Matsumoto S.; Ogiwara Y. Cobalt-Catalyzed Oxidative Annulation of Aromatic Tertiary Amines with Electron-Deficient Maleimides Leading to Tetrahydroquinoline Derivatives. Tetrahedron Lett. 2016, 57, 5449–5452. 10.1016/j.tetlet.2016.10.071. [DOI] [Google Scholar]
- Tang J.; Grampp G.; Liu Y.; Wang B.-X.; Tao F.-F.; Wang L.-J.; Liang X.-Z.; Xiao H.-Q.; Shen Y.-M. Visible Light Mediated Cyclization of Tertiary Anilines with Maleimides Using Nickel(II) Oxide Surface-Modified Titanium Dioxide Catalyst. J. Org. Chem. 2015, 80, 2724–2732. 10.1021/jo502901h. [DOI] [PubMed] [Google Scholar]
- Liang Z.; Xu S.; Tian W.; Zhang R. Eosin Y-Catalyzed Visible-Light-Mediated Aerobic Oxidative Cyclization of N,N-Dimethylanilines with Maleimides. Beilstein J. Org. Chem. 2015, 11, 425–430. 10.3762/bjoc.11.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju X.; Li D.; Li W.; Yu W.; Bian F. The Reaction of Tertiary Anilines with Maleimides under Visible Light Redox Catalysis. Adv. Synth. Catal. 2012, 354, 3561–3567. 10.1002/adsc.201200608. [DOI] [Google Scholar]
- Nishino M.; Hirano K.; Satoh T.; Miura M. Copper-Catalyzed Oxidative Direct Cyclization of N-Methylanilines with Electron-Deficient Alkenes Using Molecular Oxygen. J. Org. Chem. 2011, 76, 6447–6451. 10.1021/jo2011329. [DOI] [PubMed] [Google Scholar]
- Roy R. B.; Swan G. A. A Novel Formation of Quinoline Derivatives. Chem. Commun. 1968, 1446–1447. 10.1039/C19680001446. [DOI] [Google Scholar]
- Almansaf Z.; Hu J.; Zanca F.; Shahsavari H. R.; Kampmeyer B.; Tsuji M.; Maity K.; Lomonte V.; Ha Y.; Mastrorilli P.; Todisco S.; Benamara M.; Oktavian R.; Mirjafari A.; Moghadam P. Z.; Khosropour A. R.; Beyzavi H. Pt(II)-Decorated Covalent Organic Framework for Photocatalytic Difluoroalkylation and Oxidative Cyclization Reactions. ACS Appl. Mater. Interfaces 2021, 13, 6349–6358. 10.1021/acsami.0c21370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z.; Li F.; Niu L.; Li H.; Zheng J.; Han R.; Ju Z.; Li S.; Li D. CuBr/NHPI Co-Catalyzed Aerobic Oxidative [3 + 2] Cycloaddition-Aromatization to Access 5,6-Dihydro-Pyrrolo[2,1-a]Isoquinolines. Org. Biomol. Chem. 2020, 18, 6889–6898. 10.1039/D0OB01403F. [DOI] [PubMed] [Google Scholar]
- Hloušková Z.; Klikar M.; Pytela O.; Almonasy N.; Růžička A.; Jandová V.; Bureš F. Structural Elaboration of Dicyanopyrazine: Towards Push–Pull Molecules with Tailored Photoredox Activity. RSC Adv. 2019, 9, 23797–23809. 10.1039/C9RA04731J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Bao W.; Zhang Y.; Rao Y. Cercosporin-Photocatalyzed Sp3 (C–H) Activation for the Synthesis of Pyrrolo[3,4-c]Quinolones. Org. Biomol. Chem. 2019, 17, 8958–8962. 10.1039/C9OB01946D. [DOI] [PubMed] [Google Scholar]
- Yang X.; Liang T.; Sun J.; Zaworotko M. J.; Chen Y.; Cheng P.; Zhang Z. Template-Directed Synthesis of Photocatalyst-Encapsulating Metal–Organic Frameworks with Boosted Photocatalytic Activity. ACS Catal. 2019, 9, 7486–7493. 10.1021/acscatal.9b01783. [DOI] [Google Scholar]
- Mandal T.; Das S.; De Sarkar S. Nickel(II) Tetraphenylporphyrin as an Efficient Photocatalyst Featuring Visible Light Promoted Dual Redox Activities. Adv. Synth. Catal. 2019, 361, 3200–3209. 10.1002/adsc.201801737. [DOI] [Google Scholar]
- Nicholls T. P.; Burt L. K.; Simpson P. V.; Massi M.; Bissember A. C. Tricarbonyl Rhenium(i) Tetrazolato and N-Heterocyclic Carbene Complexes: Versatile Visible-Light-Mediated Photoredox Catalysts. Dalton Trans. 2019, 48, 12749–12754. 10.1039/C9DT02533B. [DOI] [PubMed] [Google Scholar]
- Macdonald T. L.; Gutheim W. G.; Martin R. B.; Guengerich F. P. Oxidation of Substituted N,N-Dimethylanilines by Cytochrome P-450: Estimation of the Effective Oxidation-Reduction Potential of Cytochrome P-450. Biochemistry 1989, 28, 2071–2077. 10.1021/bi00431a016. [DOI] [PubMed] [Google Scholar]
- Leng L.; Fu Y.; Liu P.; Ready J. M. Regioselective, Photocatalytic α-Functionalization of Amines. J. Am. Chem. Soc. 2020, 142, 11972–11977. 10.1021/jacs.0c03758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitas N. F.; Tzaras D. I.; Triandafillidi I.; Kokotos C. G. Photochemical Oxidation of Benzylic Primary and Secondary Alcohols Utilizing Air as the Oxidant. Green Chem. 2020, 22, 471–477. 10.1039/C9GC03000J. [DOI] [Google Scholar]
- Baciocchi E.; Lanzalunga O.; Lapi A.; Manduchi L. Kinetic Deuterium Isotope Effect Profiles and Substituent Effects in the Oxidative N-Demethylation of N,N-Dimethylanilines Catalyzed by Tetrakis(Pentafluorophenyl)Porphyrin Iron(III) Chloride. J. Am. Chem. Soc. 1998, 120, 5783–5787. 10.1021/ja980187i. [DOI] [Google Scholar]
- Baciocchi E.; Bietti M.; Gerini M. F.; Lanzalunga O. Electron-Transfer Mechanism in the N-Demethylation of N,N-Dimethylanilines by the Phthalimide-N-Oxyl Radical. J. Org. Chem. 2005, 70, 5144–5149. 10.1021/jo0503916. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Watanabe Y.; Fukuzumi S.; Jones J. P.; Dinnocenzo J. P. Mechanisms of N-Demethylations Catalyzed by High-Valent Species of Heme Enzymes:Novel Use of Isotope Effects and Direct Observation of Intermediates. J. Am. Chem. Soc. 1998, 120, 10762–10763. 10.1021/ja981357u. [DOI] [Google Scholar]
- Ratnikov M. O.; Doyle M. P. Mechanistic Investigation of Oxidative Mannich Reaction with Tert-Butyl Hydroperoxide. The Role of Transition Metal Salt. J. Am. Chem. Soc. 2013, 135, 1549–1557. 10.1021/ja3113559. [DOI] [PubMed] [Google Scholar]
- Skolia E.; Apostolopoulou M. K.; Nikitas N. F.; Kokotos C. G. Photochemical Synthesis of Benzimidazoles from Diamines and Aldehydes. Eur. J. Org. Chem. 2021, 2021, 422–428. 10.1002/ejoc.202001357. [DOI] [Google Scholar]
- Hayyan M.; Hashim M. A.; AlNashef I. M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. 10.1021/acs.chemrev.5b00407. [DOI] [PubMed] [Google Scholar]
- Karki S. B.; Dinnocenzo J. P.; Jones J. P.; Korzekwa K. R. Mechanism of Oxidative Amine Dealkylation of Substituted N,N-Dimethylanilines by Cytochrome P-450: Application of Isotope Effect Profiles. J. Am. Chem. Soc. 1995, 117, 3657–3664. 10.1021/ja00118a001. [DOI] [Google Scholar]
- Saritha R.; Annes S. B.; Saravanan S.; Ramesh S. Carbazole Based Electron Donor Acceptor (EDA) Catalysis for the Synthesis of Biaryl and Aryl-Heteroaryl Compounds. Org. Biomol. Chem. 2020, 18, 2510–2515. 10.1039/d0ob00282h. [DOI] [PubMed] [Google Scholar]
- Chen L.; Jin S.; Gao J.; Liu T.; Shao Y.; Feng J.; Wang K.; Lu T.; Du D. N-Heterocyclic Carbene/Magnesium Cocatalyzed Radical Relay Assembly of Aliphatic Keto Nitriles. Org. Lett. 2021, 23, 394–399. 10.1021/acs.orglett.0c03883. [DOI] [PubMed] [Google Scholar]
- Xu W. L.; Tang L.; Ge C. Y.; Chen J.; Zhou L. Synthesis of Tetrahydroisoindolinones via a Metal-Free Dehydrogenative Diels-Alder Reaction. Adv. Synth. Catal. 2019, 361, 2268–2273. 10.1002/adsc.201801436. [DOI] [Google Scholar]
- Lu C.-D.; Chen Z.-Y.; Liu H.; Hu W.-H.; Mi A.-Q.; Doyle M. P. A Facile Three-Component One-Pot Synthesis of Structurally Constrained Tetrahydrofurans That Are t-RNA Synthetase Inhibitor Analogues. J. Org. Chem. 2004, 69, 4856–4859. 10.1021/jo0497508. [DOI] [PubMed] [Google Scholar]
- Matuszak N.; Muccioli G. G.; Labar G.; Lambert D. M. Synthesis and in Vitro Evaluation of N-Substituted Maleimide Derivatives as Selective Monoglyceride Lipase Inhibitors. J. Med. Chem. 2009, 52, 7410–7420. 10.1021/jm900461w. [DOI] [PubMed] [Google Scholar]
- Salewska N.; Boros-Majewska J.; Łącka I.; Chylińska K.; Sabisz M.; Milewski S.; Milewska M. J. Chemical Reactivity and Antimicrobial Activity of N-Substituted Maleimides. J. Enzyme Inhib. Med. Chem. 2012, 27, 117–124. 10.3109/14756366.2011.580455. [DOI] [PubMed] [Google Scholar]
- Samgina T. Y.; Gorshkov V. A.; Vorontsov E. A.; Bagrov V. V.; Nifant’ev I. E.; Lebedev A. T. New Cysteine-Modifying Reagents: Efficiency of Derivatization and Influence on the Signals of the Protonated Molecules of Disulfide-Containing Peptides in Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. J. Anal. Chem. 2010, 65, 1320–1327. 10.1134/S1061934810130034. [DOI] [Google Scholar]
- Jiang S.; Tala S. R.; Lu H.; Zou P.; Avan I.; Ibrahim T. S.; Abo-Dya N. E.; Abdelmajeid A.; Debnath A. K.; Katritzky A. R. Design, Synthesis, and Biological Activity of a Novel Series of 2,5-Disubstituted Furans/Pyrroles as HIV-1 Fusion Inhibitors Targeting Gp41. Bioorg. Med. Chem. Lett. 2011, 21, 6895–6898. 10.1016/j.bmcl.2011.08.081. [DOI] [PubMed] [Google Scholar]
- Nicholls T. P.; Constable G. E.; Robertson J. C.; Gardiner M. G.; Bissember A. C. Bronsted Acid Cocatalysis in Copper(I)-Photocatalyzed α-Amino C-H Bond Functionalization. ACS Catal. 2016, 6, 451–457. 10.1021/acscatal.5b02014. [DOI] [Google Scholar]
- Huang P.; Wang P.; Wang S.; Tang S.; Lei A. Electrochemical Oxidative [4 + 2] Annulation of Tertiary Anilines and Alkenes for the Synthesis of Tetrahydroquinolines. Green Chem. 2018, 20, 4870–4874. 10.1039/c8gc02463d. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.