

Abstract
Treating solid malignancies with chimeric antigen receptor (CAR) T cells typically results in poor responses. Immunomodulatory biologics delivered systemically can augment the cells’ activity, but off-target toxicity narrows the therapeutic window. Here we show that the activity of intratumoural CAR T cells can be controlled photothermally via synthetic gene switches that trigger the expression of transgenes in response to mild temperature elevations (to 40–42 °C). In vitro, heating engineered primary human T cells for 15–30 min led to over 60-fold-higher expression of a reporter transgene without affecting the cells’ proliferation, migration and cytotoxicity. In mice, CAR T cells photothermally heated via gold nanorods produced a transgene only within the tumours. In mouse models of adoptive transfer, the systemic delivery of CAR T cells followed by intratumoural production, under photothermal control, of an interleukin-15 superagonist or a bispecific T cell engager bearing an NKG2D receptor redirecting T cells against NKG2D ligands enhanced antitumour activity and mitigated antigen escape. Localized photothermal control of the activity of engineered T cells may enhance their safety and efficacy.
Engineered T cell therapies such as chimeric antigen receptor (CAR) T cells are transforming clinical care for haematological malignancies, spurring numerous efforts to expand their use for different cancer types and applications. However, this success has not reliably translated to solid tumours1. The factors that contribute to low response rates are multifaceted and include the paucity of tumour-specific antigens, inefficient persistence and expansion of adoptively transferred T cells, and immunosuppression by the tumour microenvironment2,3. Promising approaches to improve antitumour activity of engineered T cells include systemic administration of potent immunostimulatory agents such as cytokines, checkpoint blockade inhibitor antibodies and bispecific T cell engagers (BiTEs)4–7. However, these biologics lack specificity, activate both engineered and endogenous immune cells, and exhibit toxicity in healthy tissue that limits maximum tolerable doses and narrows their therapeutic windows8–11. Thus, expanding current abilities to target and locally augment CAR T cell functions at tumour and disease sites, such as draining lymph nodes, could improve the safety and efficacy of cell-based therapies.
Emerging strategies to control engineered T cells and augment their antitumour activity include the use of biomaterials to co-deliver adjuvants to the tumour microenvironment as well as genetic constructs for autonomous expression of immunostimulatory genes. For example, implantation of biopolymer scaffolds loaded with tumour-specific T cells and immunostimulatory adjuvants at the surgical site improved postoperative responses following primary-tumour resection in mouse models12,13. To provide a localized source of adjuvants, T cells tethered on their cell surface to nanoparticle ‘backpacks’ allowed infiltrating T cells to carry cargo14 and release a one-time dose of drug within tumours15. Increasingly sophisticated genetic circuitry has also allowed T cells to locally produce biologics to overcome immunosuppression or target antigens after tumour infiltration. For example, ‘armoured CARs’ leverage constitutive expression of biologics such as interleukin (IL)-1216, anti-programmed cell death protein 1 single chain variable fragments (αPD-1 scFvs)17 and BiTEs18 to improve antitumour activity. T cells have also been engineered with sense-and-respond biocircuits that conditionally activate in the presence of specific input signals such as hypoxia19,20. These strategies include nuclear factor of activated T cells (NFAT)-inducible cassettes that upregulate expression of cytokines following T cell recognition of a tumour-associated antigen21–23. To further increase specificity, T cells have been engineered to target unique combinations of epitopes expressed in the tumour microenvironment to allow discrimination from healthy cells expressing a single epitope24. Such approaches based on Boolean logic require a specific combination of various target antigens for T cell activation to occur and have demonstrated efficacy in multiple models of focal tumours25–27. Collectively, these approaches illustrate the need to develop strategies to control and improve intratumoural T cell activity.
In this Article, we develop a technology for photothermal control of T cell therapies. Heat treatments are used clinically to sensitize cancer cells to chemotherapy, ablate isolated metastatic nodules and enhance diffusion of small-molecule drugs into tumours28. Both superficial and deep-seeded tumours can be targeted for thermal treatment by technologies including high-intensity focused ultrasound (HIFU)29, laser interstitial thermal therapy (LITT)30 and electromagnetic heating31. To engineer T cells with the ability to respond to heat, we constructed and screened panels of synthetic thermal gene switches containing combinations of heat-shock elements (HSEs) and core promoters to identify an architecture that responds to mild hyperthermia while remaining non-responsive to orthogonal cell stresses. We designed thermal constructs to control broad classes of immunostimulatory genes including BiTEs and a cytokine superagonist to enhance key T cell functions including proliferation and T cell targeting. In adoptive transfer models of cancer, photothermal control of an IL-15 superagonist or a natural killer group 2D ligand (NKG2DL) BiTE reduced tumour burden, improved survival and mitigated antigen escape. Spatial control of T cells by thermal targeting could improve the precision of cellular therapies by enabling site-specific control of antitumour responses.
Results
Engineering thermal-specific gene switches.
The cellular response to hyperthermia is mediated by trimerization of the temperature-sensitive transcription factor heat shock factor 1 (HSF1) and its subsequent binding to HSEs. HSEs comprise multiple inverted repeats of the consensus sequence 5′-nGAAn-3′ (refs.32,33) and are arrayed upstream of the transcription start site of heat-shock proteins (HSPs) to enable their upregulation following thermal stress34. The response of endogenous HSP genes is selective but not specific, for heat as their promoters contains additional regulatory elements (for example, hypoxia response elements35 or metal-responsive elements36) that mediate transcription following exposure to a diverse set of cues including hypoxia37, heavy metals38 and mechanical force39. Moreover, differences in the core promoter (for example, initiator elements such as the TATA box) influence the composition of the pre-initiation complex and its interactions with transcriptional enhancers including HSF1, leading to different thermal responses across tissues and types of cells40,41. Due to this complexity and cross-activation by non-thermal response pathways, we sought to build synthetic gene switches that are activated by heat but not by other sources of stress.
We cloned six candidate constructs consisting of two to seven repeats of the HSE motif 5′-nGAAnnTTCnnGAAn-3′ upstream of the HSPB1 core promoter into Jurkat T cells (labelled 2H-B1 to 7H-B1, Fig. 1a). We initially selected the HSPB1 core promoter as its parent gene was one of two that were upregulated by more than 20-fold at 42 °C in primary murine T cells, in contrast to more than 80 HSP and HSP-related genes that did not respond to heat treatment (Supplementary Fig. 1). We reasoned that selecting a core promoter from an endogenous gene with high thermal response would facilitate transcriptional activity when integrated with HSE repeats. To quantify responses of our thermal switches, transduced Jurkat T cells were transiently heated to 3–5 °C above body temperature (that is, 40–42 °C), which is a mild temperature range in contrast to that used for ablative therapies (>50 °C)29. Compared with control samples at 37 °C, we observed increased expression of the reporter Gaussia luciferase (Gluc) as the temperature and number of HSEs increased (Fig. 1b). Constructs containing five to seven HSE repeats (5H-B1 to 7H-B1) resulted in substantially higher thermal responses compared with those with two to four HSEs (2H-B1 to 4H-B1) (Fig. 1c).
Fig. 1 |. Constructing thermal-specific gene switches.
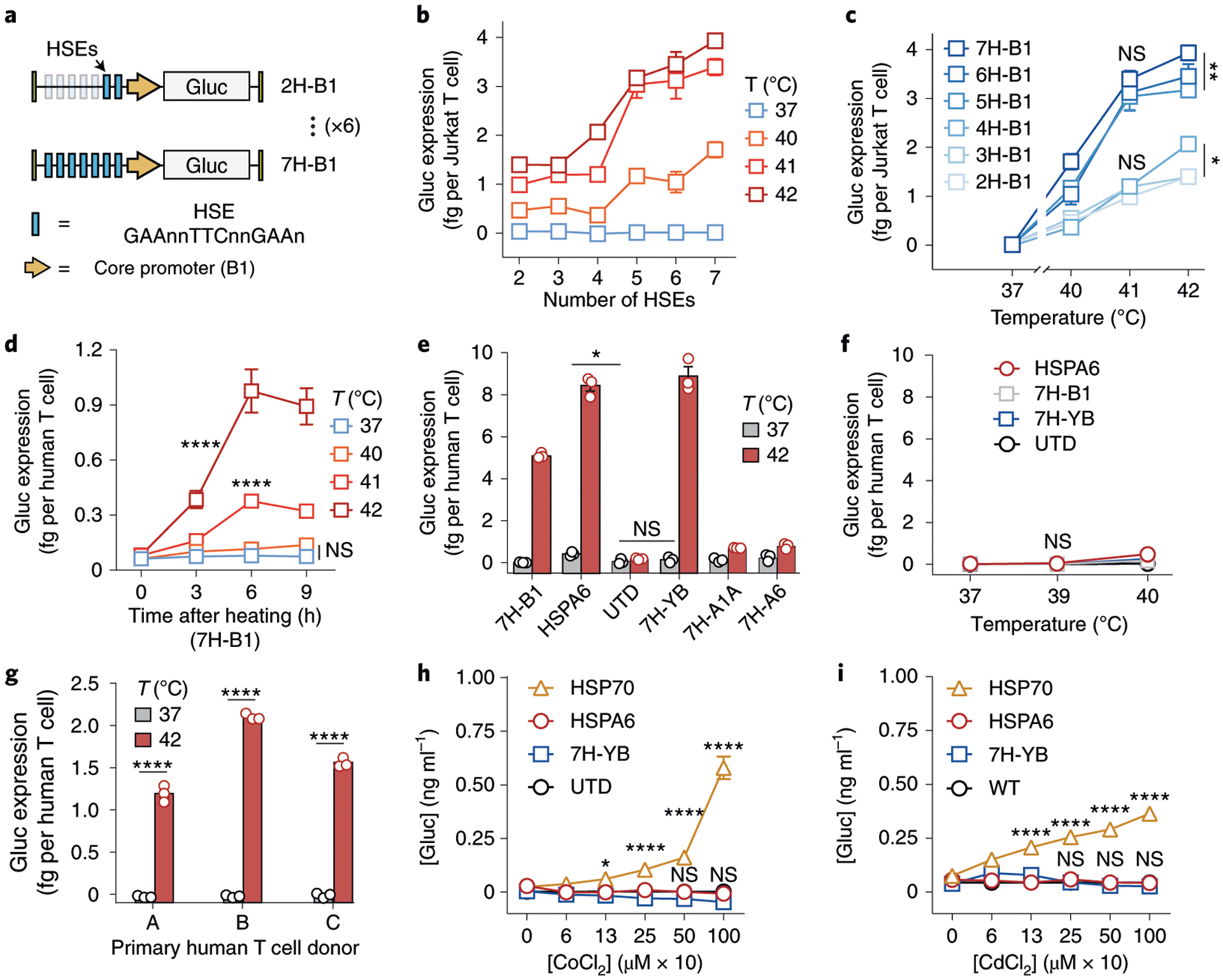
a, Schematic of a panel of six thermal gene-switch constructs consisting of two to seven HSEs upstream of the HSPB1 core promoter (labelled 2H-B1 to 7H-B1). Uppercase base pairs within HSE were conserved while base pairs indicated as n were randomized. b,c Gluc reporter expression by Jurkat T cells following heating as a function of HSE number (b) or temperature (c). NS, not significant; *P = 0.0248 between 2H-B1 and 4H-B1 at 42 °C; **P = 0.0037 between 5H-B1 and 7H-B1 at 42 °C; two-way analysis of variance (ANOVA) and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells. Three independent experiments were performed with similar results. d, Kinetics of Gluc reporter expression by primary human T cells following heat treatments at indicated temperatures. ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells. Two independent experiments were performed with similar results. e, Activity of thermal gene switches containing different core-promoter constructs following heat treatments in primary human T cells. *P = 0.0140 between HSPA6 and untransduced (UTD) groups at 37 °C; one-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells. Two independent experiments were performed with similar results. f, Gluc expression in primary human T cells that were incubated 24 h at the displayed temperatures. Two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells. g, Activity of 7H-YB in primary human T cells from multiple human donors following 30 min heat treatment at the indicated temperatures. ****P < 0.0001; two-tailed t-test; mean ± s.e.m. is depicted; n = 3 biologically independent wells. h, Activity of 7H-YB compared with endogenous HSP70 and HSPA6 promoters in primary human T cells following exposure to CoCl2 to mimic hypoxia or i, to CdCl2 to model heavy-metal toxicity. *P = 0.011; ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells.
To test thermal response in primary human T cells, we transduced T cells with the 7H-B1 construct and observed peak thermal activity approximately 6 h after heating at temperatures above 40 °C (Fig. 1d). Because the HSPB1 core promoter was initially selected from a screen of murine T cells, we reasoned that thermal responses in primary human T cells may further depend on the core-promoter sequence. Thus, we compared core promoters identified in the quantitative PCR (qPCR) screen (HSPA1A, HSPH1, HSPB1) (Supplementary Fig. 1), from the human HSPA6 gene based on our past work42, and a synthetic core promoter (YB) described previously43,44. Among the panel, the 7H-YB construct resulted in the highest increase in Gluc reporter levels after 30 min at 42 °C, corresponding to a ~60-fold increase in activity (Fig. 1e). Basal activity at 37 °C remained low relative to untransduced controls, and negligible activation was observed at temperatures of 37–40 °C for 24 h, which correspond to fever range (Fig. 1f). We further verified 7H-YB thermal activation in T cells derived from three separate donors to confirm lack of donor-dependency (Fig. 1g and Supplementary Fig. 2). On the basis of these data, we selected 7H-YB for subsequent experiments.
We tested thermal specificity using hypoxia and heavy-metal toxicity as two representative non-thermal stresses45–47. As a benchmark, we compared activity against endogenous HSPA6 or HSP70 promoters, which are highly stress-inducible48 and have been previously used for thermal control of gene expression49–52. We tested the gene switches by incubating transduced primary human T cells and Jurkat T cells with the hypoxia-mimetic agent CoCl2, a stabilizer of the hypoxia response’s master regulator HIF-1α53, as well as the heavy-metal complex cadmium chloride (CdCl2), which accumulates in the body by diet or environmental exposure54–56. While the HSP70 or HSPA6 promoter showed dose-dependent activation by hypoxia and cadmium toxicity in primary human T cells or Jurkat T cells respectively (Fig. 1h,i and Supplementary Fig. 3), 7H-YB was not activated and remained similar to untransduced controls up to concentrations above the ranges commonly used to test cellular responses to hypoxia and cadmium53,54,57,58 (1,000 mM CoCl2 and 1,000 μM CdCl2). These results show that our constructs have increased thermal specificity when exposed to non-thermal stresses compared with endogenous HSPs.
Primary T cells maintain key functions after thermal treatment.
We next identified thermal delivery profiles that would be well-tolerated by primary T cells without affecting key functions including proliferation, migration and cytotoxicity. In thermal medicine, heating target sites to temperatures greater than 50 °C is used to locally ablate tissue by inducing tumour-cell apoptosis and coagulative necrosis29. By contrast, mild hyperthermia therapy (40–42 °C) is used to enhance transport of small molecules such as in hyperthermic intraperitoneal chemotherapy (HIPEC) where abdominal infusions of heated chemotherapy serve as an adjuvant treatment following surgical debulking in advanced ovarian cancer patients28,59,60. At temperatures below 45 °C, transient exposure to mild hyperthermia is well-tolerated by cells and tissues due to induction of stress-response pathways including HSPs61. In addition, we also considered T cell responses to continuous and fractionated heat treatments. Dose fractionation is commonly used in radiotherapy to reduce damage to normal tissues61 while maximizing the effect of radiation on cancer. On the basis of our previous observations that thermal pulse trains increased Jurkat T cell tolerance compared with continuous heat treatments with an identical treatment area under the curve42, we sought to additionally probe the effect of thermal dose fractionation on primary T cells.
We first compared pulsed heat treatments at 67% duty cycles consisting of three discrete thermal pulses (5 or 10 min each) separated by intervening rest periods at 37 °C (2.5 or 5 min each) to their unfractionated counterparts (15 or 30 min continuous heating) (Fig. 2a). In primary human T cells transduced with the 7H-YB Gluc vector, pulsed heat treatments resulted in up to 87% higher reporter expression as compared to continuous treatments with the same 30 min area under the curve (Fig. 2b). To assess T cell viability, we quantified death (propidium iodide, PI) and apoptotic (annexin V, AnV) markers and observed significant improvements for primary T cells that received pulsed treatments at a 67% duty cycle for durations of 30–60 min (Fig. 2c). By contrast, as high as ~33% reduction in T cell viability was observed in samples that received continuous heat treatments for greater than 40 min. Similar trends were observed in T cell proliferation assays by CellTrace Violet dilution where the percentage of proliferated T cells following incubation with CD3/28 beads was unaffected by both continuous and pulsed heating for 30 min at 42 °C or 43 °C, while samples that were heated for 60 min resulted in reduced T cell proliferation (Fig. 2d and Supplementary Fig. 4).
Fig. 2 |. Thermal treatments are well-tolerated by primary human T cells.
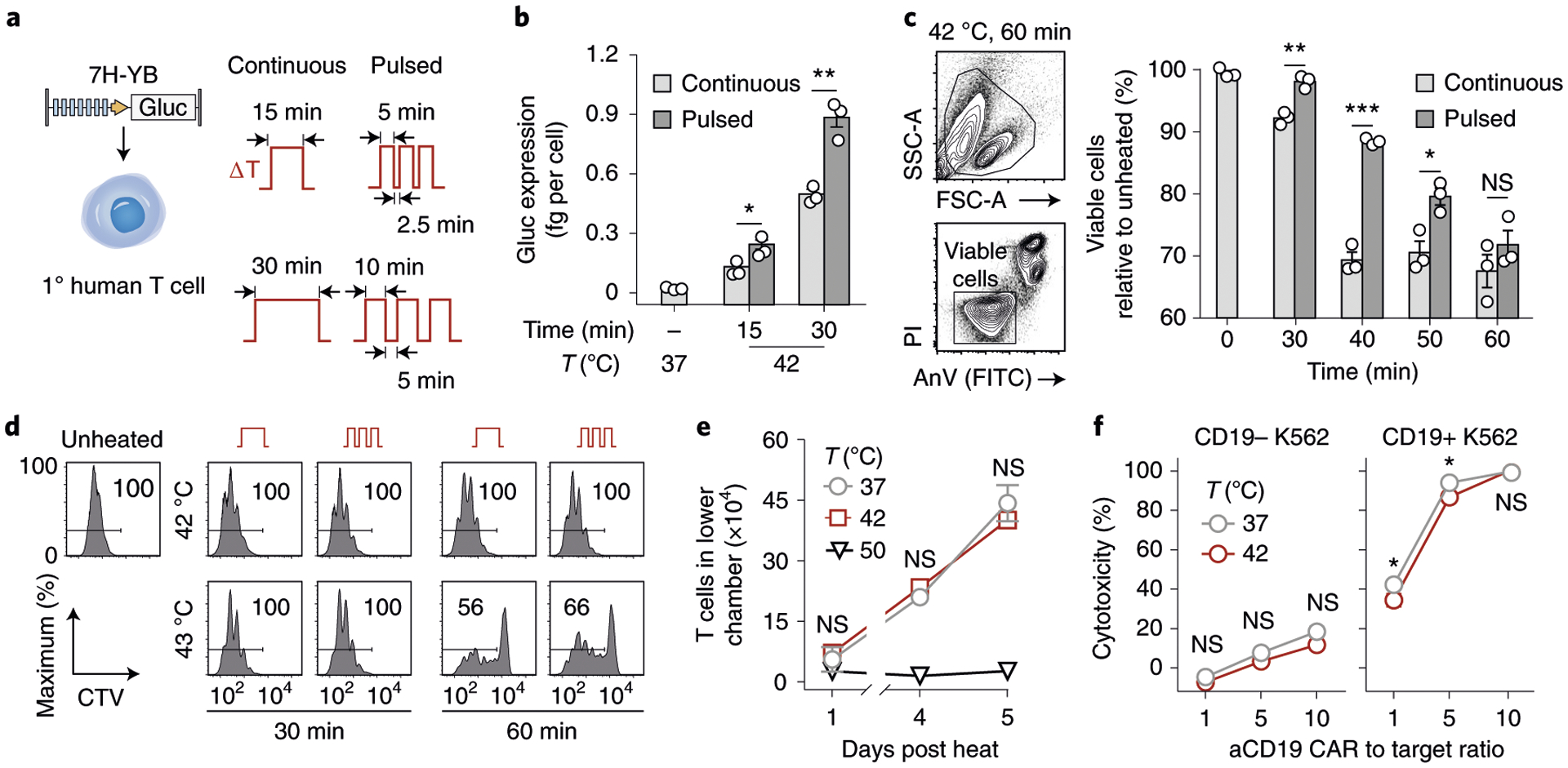
a, Schematic representation of transduced primary human T cells and thermal delivery profiles. b, Gluc activity of the 7H-YB thermal switch in primary human T cells after continuous (light grey) and pulsed (dark grey) heat treatments with temperatures, total durations and heating profiles as indicated. *P = 0.033; **P = 0.0021; two-tailed t-test; mean ± s.e.m. is depicted; n = 3 biologically independent wells. c, PI and AnV flow staining of CD3+ T cells. Bars represent viable populations (PI−AnV−) normalized to unheated samples. *P = 0.0455; **P = 0.0034; ****P < 0.0001; two-way ANOVA and Šidák post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells; FSC-A, forward scatter area; SSC-A, side scatter area. Two independent experiments were performed with similar results. d, CellTrace Violet (CTV) flow histograms of T cells after heat treatments and incubation with CD3/28 beads at a 3:1 bead to T cell ratio. Values in histogram boxes represent percentage in indicated gate. Two independent experiments were performed with similar results. e, Number of cells in lower well of a transwell plate containing CXCL12. T cells were heated and loaded into the top well before sampling at indicated timepoints. Two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells. Two independent experiments were performed with similar results. f, Percent cytotoxicity observed in CD19− or CD19+ luciferized K562 cells after incubation with T cells constitutively expressing CARs after heating, with effector to target ratios as indicated. *P = 0.0166 at a 1:1 CAR T cell to target cell ratio; *P = 0.0251 at a 5:1 CAR T cell to target cell ratio; two-way ANOVA and Šidák post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells. Two independent experiments were performed with similar results.
To probe T cell migration by chemotaxis under mild hyperthermia, we used transwell assays and observed that heat treatments (42 °C for 30 min) did not significantly affect T cell migration into lower wells containing the chemokine CXCL12, whereas T cells heated to 50 °C were affected (Fig. 2e). To test longitudinal activation of the thermal switch, we re-heated T cells over the course of 8 d and observed similar increases in green fluorescent protein (GFP) median fluorescent intensity, as well as in GFP activation and decay half-lives (t1/2 ≈ 0.5 and 1 d, respectively), indicating that the magnitude and kinetics of T cell responses are unaffected by multiple heat treatments (Supplementary Fig. 5). To quantify the effect of heat on T cell cytotoxicity, we incubated primary human T cells expressing an αCD19 CAR under a constitutive EF1α promoter with either CD19+ or CD19− K562 tumour cells containing a luciferase reporter to allow quantification of cell death by loss of luminescence (Fig. 2e). At all effector to target cell ratios tested (1:1, 5:1, 10:1), heated T cells maintained greater than 90% of the cytotoxicity observed in unheated samples, while no significant difference in cytotoxicity was observed in samples containing CD19− K562 target cells (Fig. 2f). Similarly, no statistical difference was observed with longitudinal heat treatments where αCD19 CAR T cells were heated four times over the course of 8 d before co-incubation with CD19+ K562 cells (Supplementary Fig. 6). Collectively, these data demonstrate that primary human T cells maintain the ability to proliferate, migrate and kill target cells following short heat treatments delivered in continuous or pulsed wave forms for less than 30 min duration.
Photothermal activation of T cells in vivo.
We next sought to demonstrate spatially targeted activation of adoptively transferred T cells by photothermal heating. To locally heat tumours, we used plasmonic gold nanorods (AuNRs) as antennas to convert incident near-infrared (NIR) light (~650–900 nm) into heat62. Polyethylene glycol (PEG)-coated AuNRs are well-studied nanomaterials with long circulation times that passively accumulate in tumours following intravenous (i.v.) administration63,64. To confirm photothermal heating and thermal switch activation, primary T cells transduced with 7H-YB driving firefly luciferase (TS-Fluc) and constitutively expressing αCD19 CAR were co-incubated with AuNRs in 96-well plates and irradiated with 808 nm laser light. In wells that reached 40–45 °C as monitored by a thermal camera, we observed a marked increase in luminescent signals after 6 h when TS-Fluc αCD19 CAR T cells were present but not in wells containing untransduced controls (Fig. 3a), confirming plasmonic photothermal control of engineered T cells.
Fig. 3 |. Photothermal activation of engineered T cells in vivo.
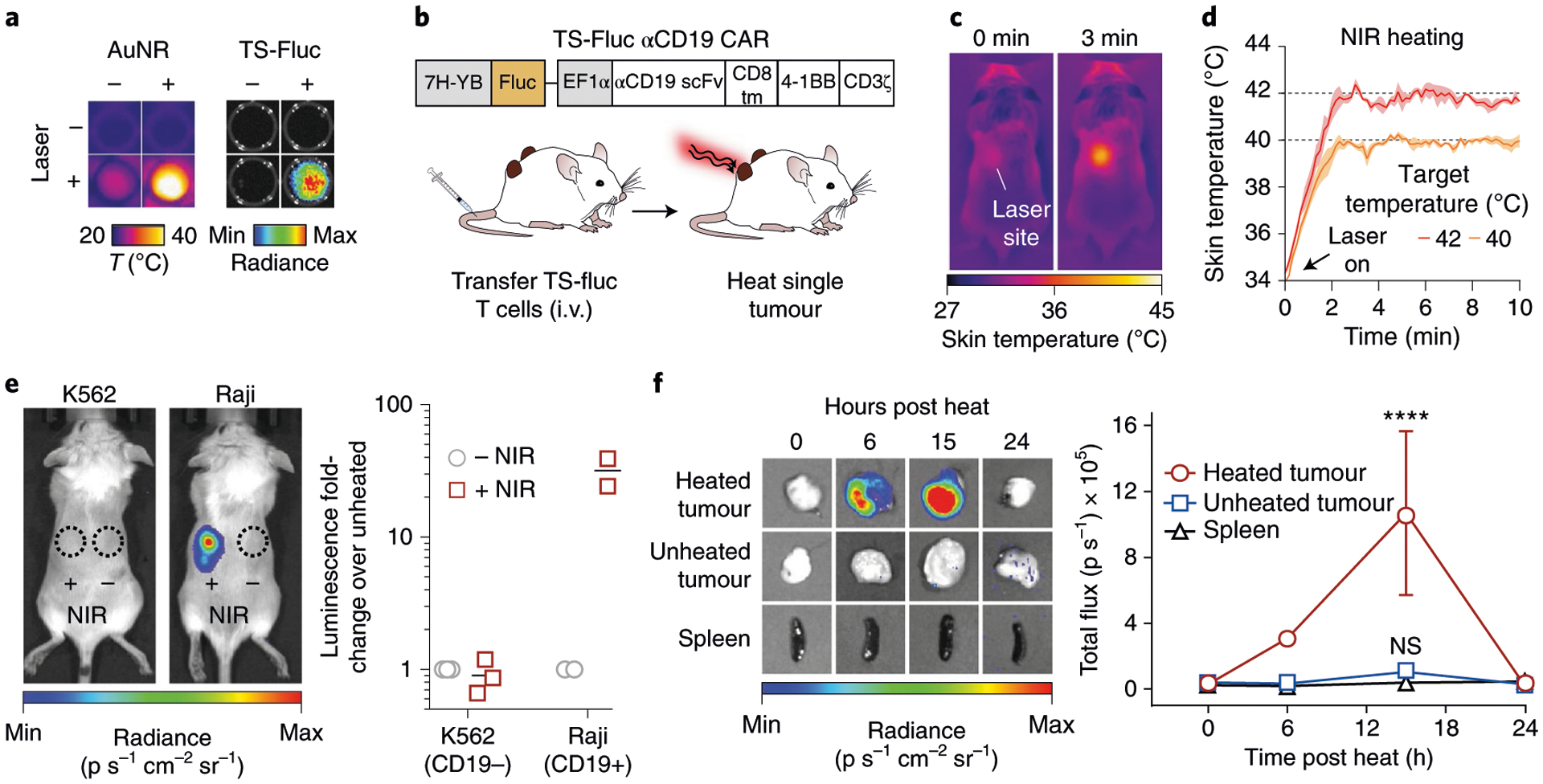
a, Thermal and luminescence images of wells containing TS-Fluc T cells after irradiation with NIR laser light. Thermal images (left) were acquired using a FLIR thermal camera while luminescence images (right) were acquired using an IVIS Spectrum CT system 6 h after heating. b, Schematic representation of TS-Fluc αCD19 CAR construct transduced into primary human T cells before transfer into NSG mice with two flank (K562 or Raji) tumours followed by photothermal heating of single tumour. c, Thermal images of mouse during laser irradiation of tumour site at 0 and 3 min. d, Kinetic traces (coloured lines) showing average skin temperature of a 3 × 3 pixel region of interest (ROI) centred on laser site. Shaded regions show s.d. of three heating runs. e, Left: luminescence images of heated mice bearing either K562 (CD19−) or Raji (CD19+) tumours. Signal indicates luciferase activity by transferred TS-Fluc T cells. Dotted circles represent ROIs. Right: luminescence of each tumour site relative to the luminescence from the unheated tumour in the same animal. A separate experiment with repeated heating of Raji tumours was conducted to confirm reproducibility of experimental results (Supplementary Fig. 8). f, Mice bearing two Raji (CD19+) tumours with one site heated. Left: luminescence images of excised tumours (heated and unheated) and spleen. Right: quantification of luminescence following heat treatments (0, 6, 12, 18, 24 h). ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 4 to 5 biologically independent mice.
To implement photothermal targeting in vivo, we inoculated NSG mice with bilateral flank tumours, with one cohort receiving CD19− K562 cells and a separate cohort receiving CD19+ Raji cells to model CAR antigen-negative and -positive tumours, respectively (Supplementary Fig. 7). Following intravenous injection of AuNRs and adoptive transfer of TS-Fluc αCD19 CAR T cells (Fig. 3b), we irradiated tumours with NIR laser light under the guidance of a thermal camera (Fig. 3c) to maintain target skin temperatures (Fig. 3d). After 20 min heat treatments, luminescence increased by more than 30-fold in Raji tumours that received NIR light compared with unheated tumours in the same animal (Fig. 3e) and similar to in vitro experiments, thermal activation repeated twice over the course of 4 d did not result in loss of luminescent signals (Supplementary Fig. 8). By contrast, we did not observe increased luminescence in CD19− K562 tumours that were treated with or without NIR light following infusion of TS-Fluc αCD19 CAR T cells (Fig. 3e). We attributed this lack of heat-induced activity to the absence of CD19 CAR antigen, which resulted in a 20-fold lower density of intratumoural αCD19 CAR T cells in resected K562 tumours compared with CD19+ Raji tumours (Supplementary Fig. 9). Although heat-triggered expression of transgenes is spatially controlled by photothermal targeting, heat activated T cells could migrate out of tumours and therefore result in off-target expression of transgenes. Therefore, in mice bearing bilateral CD19+ Raji flank tumours, we heated a single tumour site and quantified Fluc activity in the distal tumour and the spleen. Whereas luminescence in heated tumours increased by approximately 40-fold within 15 h after heating, unheated tumours and spleens remained at baseline levels, indicating that transgene expression in TS-Fluc αCD19 CAR T cells was spatially confined to the heated site (Fig. 3f). Collectively, these data demonstrate photothermal control of intratumoural T cells engineered with thermal gene switches.
Photothermal control of IL-15 SA enhances adoptive T cell transfer.
We next investigated whether thermal control could enhance the effectiveness of adoptive T cell therapies in vivo. To do this, we cloned a single-chain IL-15 superagonist (IL-15 SA) consisting of the cytokine tethered to the sushi domain of the IL-15Rα subunit65 under the control of our thermal switch (TS-IL15). IL-15 SA is a potent stimulant of CD8 T cells and natural killer (NK) cells and a clinical candidate, ALT-803, is currently under investigation for a wide range of cancers66,67. To test whether heat-induced IL-15 SA was functionally active, we developed a T cell proliferation assay using carboxyfluorescein succinimidyl ester (CFSE)-labelled wild-type T cells incubated with CD3/28 beads at a 1:10 ratio without supplemental cytokines. We found that this condition was insufficient to induce T cell proliferation compared with conditions when cytokines such as IL-2 were present in media (Supplementary Fig. 10). Therefore, to test thermal control of IL-15 SA, we added heated or unheated TS-IL-15 αCD19 CAR T cells to samples containing CFSE-labelled wild-type T cells with CD3/28 beads at a 10:1 T cell to bead ratio (Fig. 4a and Supplementary Fig. 11). Compared with unheated controls, we found that CFSE-labelled T cells in heated samples expanded with significantly higher proliferation and division indices (Fig. 4b), demonstrating that TS-IL-15 αCD19 T cells can produce physiologically active levels of IL-15 SA following a single thermal treatment. To further characterize the thermal effect of heat-triggered secretion of IL-15 SA, we analysed conditioned media by ELISA and found that IL-15 SA levels increased with the duration and temperature of thermal treatment (Fig. 4c).
Fig. 4 |. Photothermal control of IL-15 SA enhances adoptive T cell transfer and overall survival in mice.
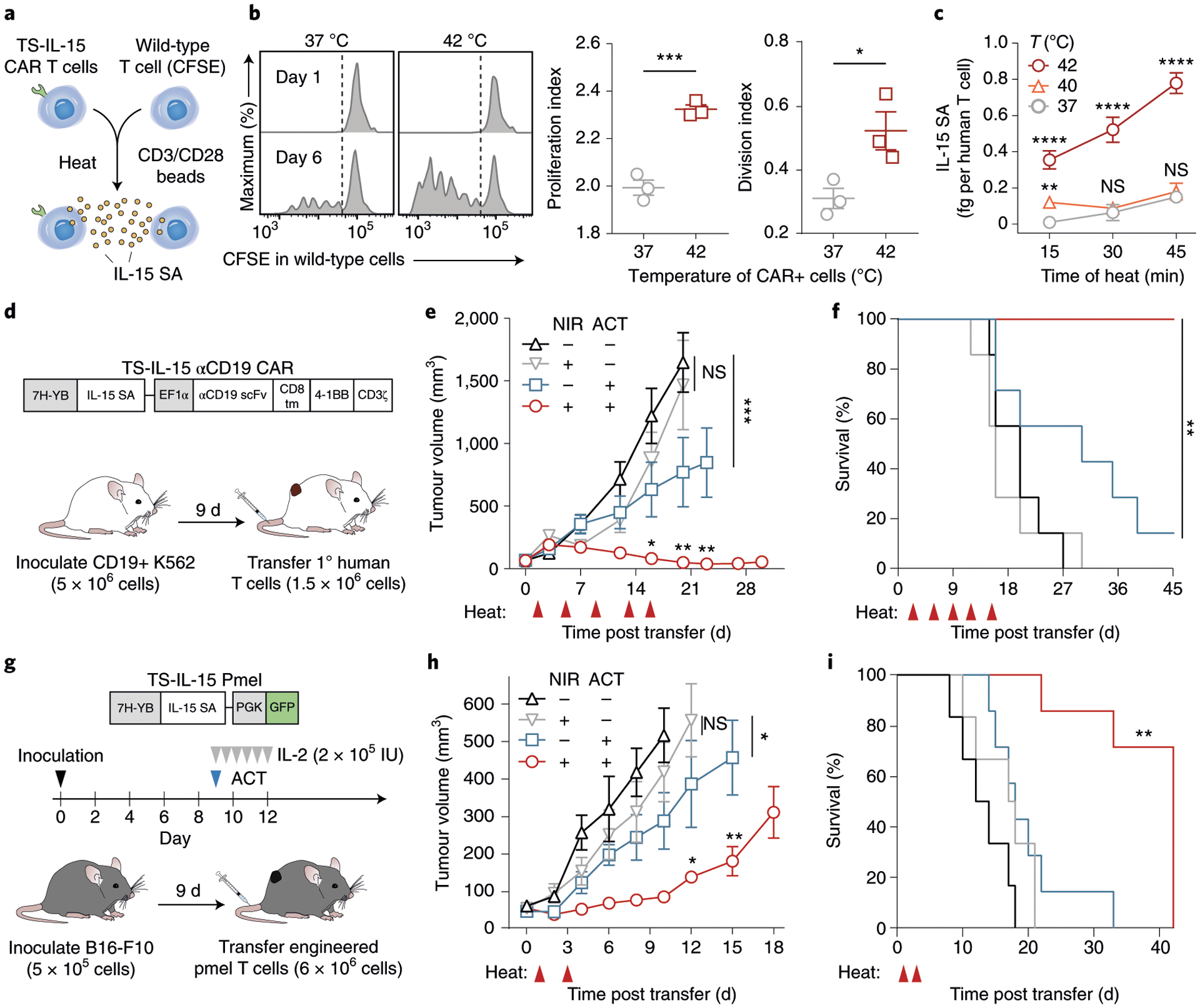
a, Schematic of co-culture assay of heated TS-IL-15 αCD19 CAR T cells and CFSE-labelled wild-type cells. CD3/28 beads were added at 1:10 bead to T cell ratio. b, Representative flow histograms (left), quantified proliferation (middle) and division indices (right) of the CFSE-labelled wild-type T cell population, as calculated by FlowJo proliferation tool, after a 30 min thermal treatment at indicated temperatures. ***P = 0.0009; *P = 0.0352; two-tailed t-test; mean ± s.e.m. is depicted; n = 3 biologically independent wells. c, IL-15 superagonist concentrations in supernatant of TS-IL-15 αCD19 CAR T cells following heat treatments. Temperature and duration of treatments are as indicated. **P = 0.0032, ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 3 biologically independent wells, comparisons displayed between sample and unheated control. Two independent experiments were performed with similar results. d, Schematic of TS-IL-15 αCD19 CAR vector used to transduce primary human T cells before transfer into tumour-bearing NSG mice. e, Tumour growth curves of CD19+ K562s following transfer of TS-IL-15 αCD19 CAR T cells on day 0 and heat treatments on days 2, 6, 9, 13 and 16. ***P = 0.0004 between cells only and untreated cohort on day 23; *P = 0.0157 between cohorts receiving cells and heat versus cells only on day 20; **P = 0.0047 between cohorts receiving cells and heat versus cells only on day 23; **P = 0.0012 between cohorts receiving cells and heat versus cells only on day 27; two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 7 biologically independent mice. Three independent experiments were performed with similar results. f, Survival curves of tumour-bearing mice in d and e following transfer of TS-IL-15 αCD19 CAR T cells and heat treatments. **P = 0.000022, log-rank (Mantel–Cox) test with correction for six multiple comparisons, n = 7 biologically independent mice. g, Schematic of TS-IL-15 vector transduced into primary murine Pmel-1 T cells transferred into tumour-bearing C57BL/6 J mice. h, Tumour growth curves following inoculation of B16-F10 after transfer of TS-IL-15 Pmel-1 T cells on day 0 and heat treatments on days 1 and 3. *P = 0.0438 between untreated and heat-only cohorts on day 10; *P = 0.0166 between cohorts receiving cells and heat versus cells only on day 12; **P = 0.0058 between cohorts receiving cells and heat versus cells only on day 15; two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 6 to 7 biologically independent mice. i, Survival curves of tumour-bearing mice in g and h following transfer of TS-IL-15 Pmel-1 T cells and heat treatments. **P = 0.000043, log-rank (Mantel–Cox) test with correction for six multiple comparisons, n = 6 to 7 biologically independent mice.
To explore the therapeutic effect of thermal targeting, we adoptively transferred TS-IL-15 αCD19 CAR T cells into NSG mice bearing CD19+ K562 tumours when tumours averaged 70 mm3 in volume (Fig. 4d). Photothermal heating of tumours was then carried out every 3–4 d after adoptive cell transfer (ACT) (days 2, 6, 9, 13 and 16) for a total of five treatments. Compared with control mice that did not receive CAR T cells or heat treatments (black), thermal treatment of tumour sites alone did not lead to reduction in tumour burden or improvement in survival (grey) (Fig. 4e,f). Transfer of TS-IL-15 αCD19 CAR T cells alone significantly reduced tumour burden (blue), yet greater than 85% (6/7) of animals reached euthanasia criteria within 39 d of ACT. By contrast, ACT of TS-IL-15 αCD19 CAR T cells combined with NIR treatments markedly reduced tumour burden and no animals reached euthanasia criteria within the time window of the study.
As NSG mice lack an intact immune system, we further tested our technology in immunocompetent C57BL/6 J mice bearing syngeneic B16-F10 melanoma tumours with transgenic T cell receptor (TCR) Pmel-1 T cells that recognize the melanoma self-antigen gp100 (Fig. 4g). Following peptide activation of Pmel-1 splenocytes with hGP10025–33, we verified CD8+ purity, transduction efficiency and thermal production of IL-15 SA (Supplementary Fig. 12). TS-IL-15 Pmel-1 T cells were adoptively transferred on day 9 after lymphodepletion (~52 mm3 average tumour volume) and 2 × 105 IU of IL-2 was given twice a day for 3 d to expand transferred cells5,68,69. Under these conditions, we observed that two cycles of photothermal treatment (day 1 and 3 post ACT) led to significantly enhanced control of tumour growth (red) compared with cohorts that received TS-IL-15 Pmel-1 + IL-2 but without heat treatment (blue), heat treatments only (grey) or untreated animals (Fig. 4h). Whereas all control mice reached euthanasia criteria within 33 d after ACT, photothermal treatment of TS-IL-15 Pmel-1 T cells resulted in significantly extended survival to day 42 (Fig. 4i). We repeated these experiments with well-established and vascularized B16-F10 tumours (~120 mm3 average tumour volume) and likewise observed significant improvements in tumour control in heat-treated mice (Extended Data Fig. 1). Our results are consistent with previous studies that showed IL-2 and IL-15 in combination improves antitumour activity compared with treatment with IL-2 alone70. Together, our data indicate that photothermal control of IL-15 SA production by CAR or TCR engineered T cells significantly improves tumour control.
TS-BiTE αHER2 CAR T cells mitigate antigen escape.
Heterogenous expression of antigens can lead to tumour escape from CAR T cells that are directed against a single antigen18,71. We therefore sought to explore whether heat-triggered expression of a BiTE targeting NKG2D ligands—which are upregulated on a wide range of cancers as well as suppressor cells72–75—could mitigate antigen escape. We cloned a previously described NKG2DL-BiTE containing CD3-recognition domains from the OKT3 antibody linked to the extracellular domain of the human NKG2D receptor73. This vector (TS-BiTE) included an Igκ leader sequence for BiTE secretion, a HisTag reporter and a constitutive αCD19 CAR (Fig. 5a). After heat treatment, we observed that TS-BiTE T cells were stained positively by anti-HisTag antibodies compared with TS-Fluc control cells (Fig. 5b). We therefore postulated that TS-BiTE T cells would undergo autocrine activation before BiTEs would engage bystander T cells for paracrine activation. To test this, we heated a mixture of TS-BiTE Jurkat T cells with untransduced cells as bystanders before co-incubation with NKG2DL + CD19 − K562 target cells (Fig. 5c–e) to isolate T cell activation by BiTE engagement without confounding factors due to CD19 CAR binding. We found that expression of the early activation marker CD69 on TS-BiTE Jurkat T cells was significantly upregulated compared with bystander cells as heating durations were extended (red versus black) (Fig. 5f,g). By contrast, CD69 was minimally upregulated on bystander cells compared with untransduced Jurkat T cells that were incubated with K562 cells and heated in separate wells as controls (black versus grey). These data provide support that TS-BiTE T cells are primarily activated in an autocrine path. To quantify cytotoxicity from heat-triggered expression of BiTEs, we co-incubated primary human TS-BiTE αCD19 CAR T cells with NKG2DL + CD19 − K562 cells. In contrast to untransduced or TS-Fluc αCD19 CAR controls, TS-BiTE αCD19 CAR T cells secreted increasing levels of Th1 cytokines interferon (IFN)-γ and tumor necrosis factor (TNF)-α as temperatures were raised from 37 °C to 42 °C (Fig. 5h). We also observed temperature-dependent increases in K562 cytotoxicity compared with untransduced controls but not at 37 °C, demonstrating lack of BiTE-induced killing at basal temperatures (Fig. 5i). Our data show that TS-BiTE αCD19 CAR T cells can be redirected to target antigen-negative tumour cells that express NKG2DL by thermal control.
Fig. 5 |. Expanding CAR T cell targeting via heat-triggered BiTEs.
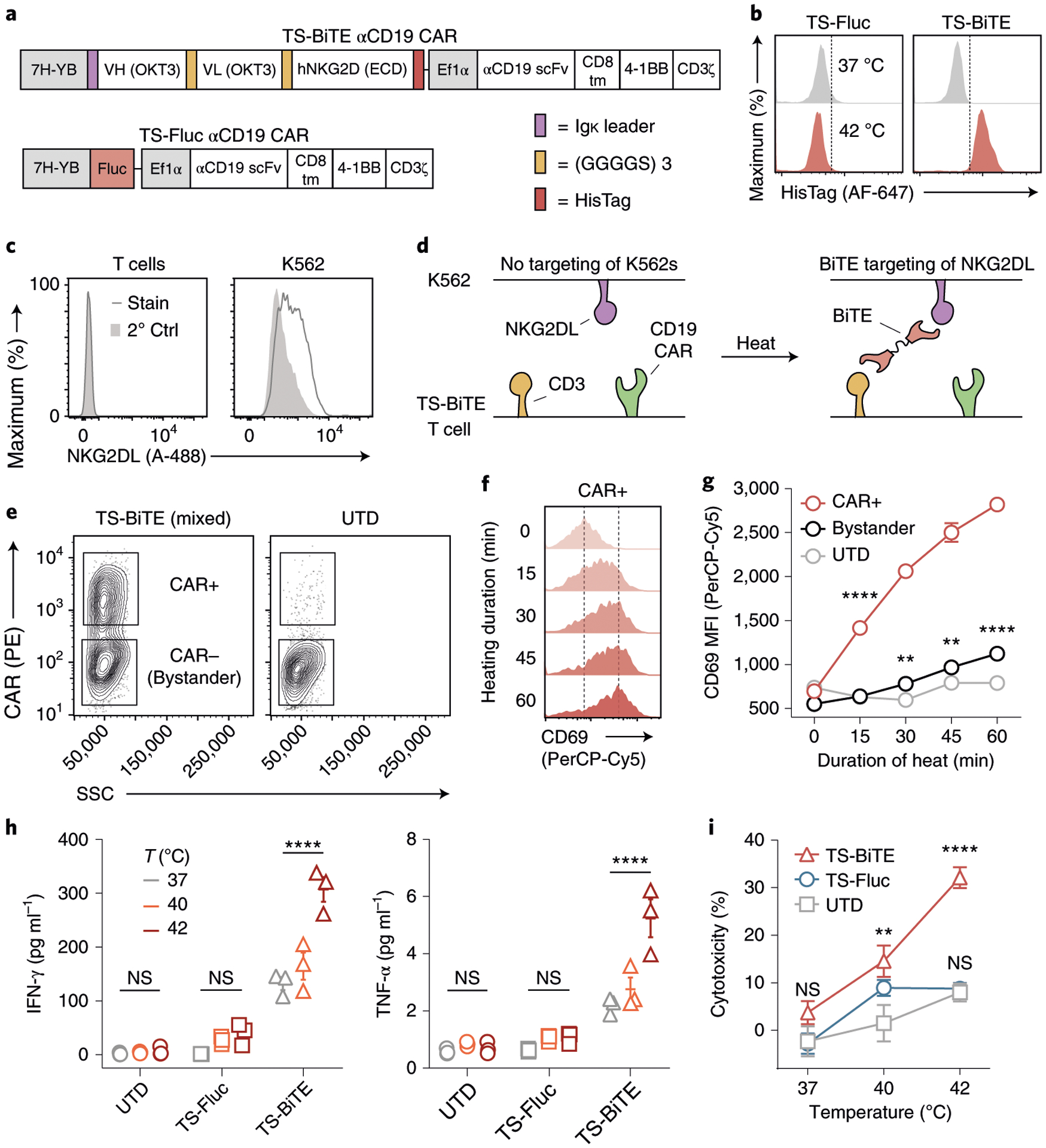
a, Schematic of TS-BiTE and TS-Fluc thermal switches containing heat-triggered BiTE or Fluc reporters. Both constructs contained constitutive αCD19 CARs. b, Histograms for HisTag flow staining in TS-BiTE and TS-Fluc primary T cells following heating. c, NKG2DL flow staining on primary human T cells and K562s using an NKG2D-Fc chimera and an αFc-A488 secondary antibody. Stain, full staining; 2° Ctrl, secondary antibody only. d, Schematic depicting BiTE-mediated targeting of K562 target cells lacking the CAR target antigen via BiTE binding to NKG2DL and CD3. e, Flow-gating strategy for defining bystander cells on the basis of CD19 CAR expression using phycoerythrin (PE) in Jurkat co-culture assays with K562s. UTD controls were gated on the lower (CAR−) population for graphing in g. f,g Flow staining of CD69 on Jurkat T cells following heating and incubation with K562s. TS-BiTE CAR+ histograms (f) and summary data of indicated populations (g) are graphed. Statistics show comparison to UTD; **P = 0.0025 between bystander and UTD populations after 30 min heating; **P = 0.0044 between bystander and UTD populations after 45 min heating; ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; mean + s.e.m. is depicted; n = 3 biologically independent wells. Two independent experiments were performed with similar results. MFI, median fluorescent intensity. h, Cytokine concentrations in supernatant of primary human T cells after heat treatments and incubation with K562 cells. T cells were either UTD or transduced with the indicated thermal switches. ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; mean + s.e.m. is depicted; n = 3 biologically independent wells. i, Cytotoxicity against K562s as quantified by luciferase assay after incubation with primary human T cells. T cells were either UTD or transduced with the indicated thermal switches. **P = 0.0043 between UTD and TS-BiTE cells; ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; comparisons are to UTD control; mean + s.e.m. is depicted; n = 3 biologically independent wells.
To test mitigation of antigen escape in vivo, we developed a heterogenous model of breast cancer consisting of a mixture of HER2+ and HER2− MDA-MB-468 tumour cells. We verified endogenous expression of NKG2DL in wild-type cells and transduced them with either HER2 (Supplementary Fig. 13a) or Fluc (Supplementary Fig. 13b) to allow luminescent quantification of antigen-negative cells in vivo. We also confirmed that both TS-BiTE or TS-Rluc αHER2 CAR T cells selectively target and kill HER2+ MDA-MB-468 cells (Fig. 6a and Supplementary Fig. 13c). By contrast, targeting of HER2− cells required thermal treatment of TS-BiTE αHER2 CAR T cells as confirmed by temperature-dependent elevation of the activation markers CD69, PD-1 and CD107a (Fig. 6b and Supplementary Fig. 14). To test whether thermal control of NKG2DL BiTE could treat tumours with heterogenous antigen expression, we inoculated NSG mice with HER2+ and HER2− MDA-MB-468 cells at a 3:1 ratio and transferred TS-BiTE or TS-Rluc αHER2 CAR T cells on day 44 when tumours were well-established and vascularized (~110 mm3 average volume) (Fig. 6c). For approximately 40 d following ACT and with longitudinal heating (days 45, 47, 52, 59, 66 and 72), we observed significant tumour regression in mice treated with either TS-BiTE or TS-Rluc αHER2 CAR T cells, the latter of which we attributed to killing of the HER2+ fraction of the tumours. However, by day 74, tumours from mice treated with TS-Rluc αHER2 CAR T cells began to relapse relative to TS-BiTE-treated cohorts, resulting in tumours that were ~12 times larger in volume on average by day 100. Tumours from four out of six TS-BiTE mice and one out of six TS-Rluc mice were undetectable by caliper measurements and palpation (Fig. 6d). To further corroborate our findings and determine whether relapse was attributable to outgrowth of antigen-negative cells, we quantified tumour luminescence from HER2− cells and observed significant signal reduction in TS-BiTE compared with TS-Rluc groups by day 58 before tumour volumes began to diverge (Fig. 6e). We also observed evidence of residual disease in one mouse from both the TS-BiTE and TS-Rluc groups that initially appeared to be a complete responder by caliper measurements but had luminescent signals that were above background, which we defined as two standard deviations above the average. We therefore considered three of six TS-BiTE mice that had unpalpable tumours and luminescence within background levels for over ~45 d to be complete responders (Fig. 6f). Our data indicate that thermal control of NKG2DL BiTE has the potential to mitigate antigen escape in tumours with heterogenous antigen expression.
Fig. 6 |. Photothermal control of TS-BiTE αHER2 CAR T cells mitigates outgrowth of antigen-negative tumours.
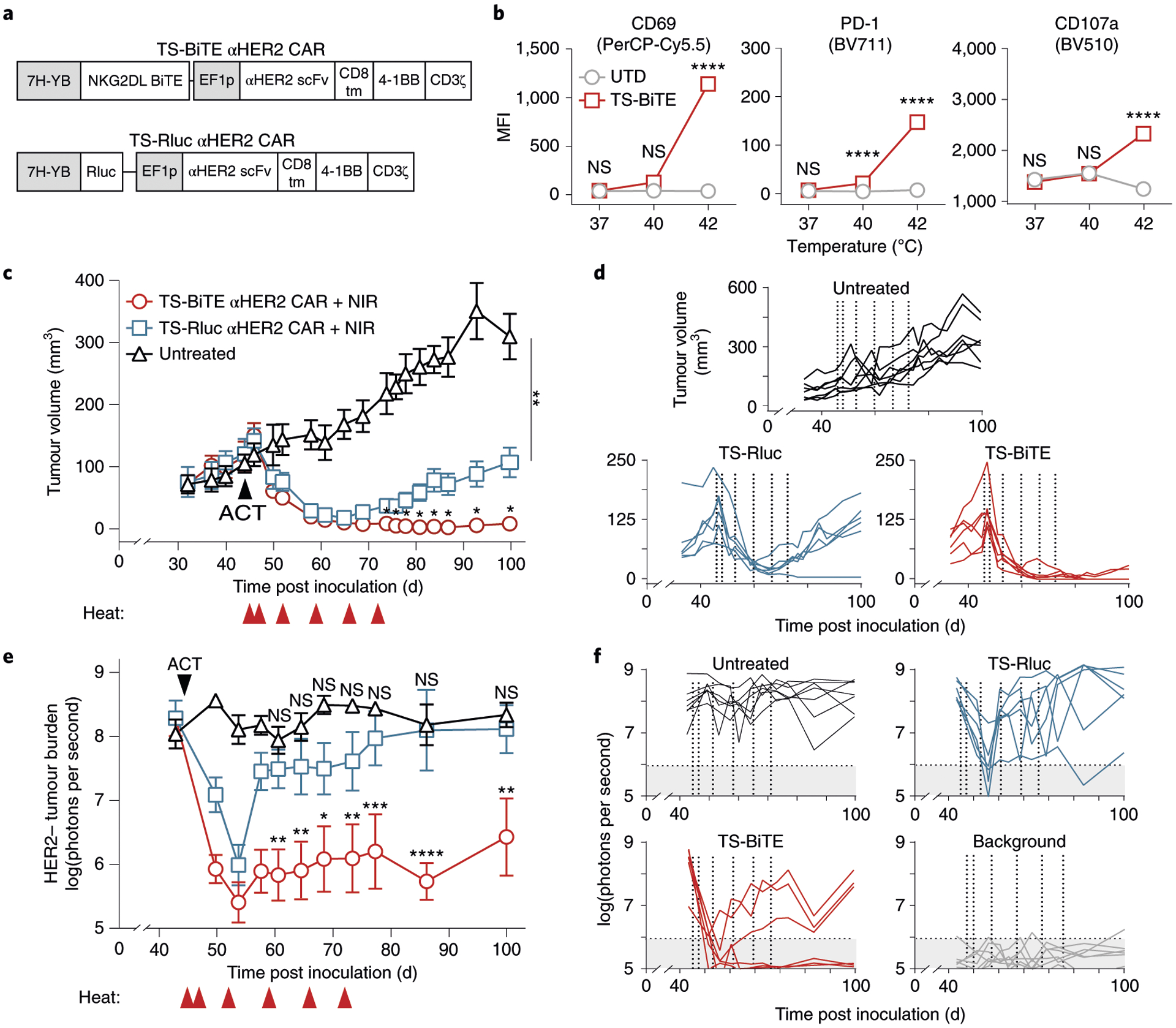
a, Schematic of TS-BiTE and TS-Rluc αHER2 CAR vectors. b, Flow cytometry quantification of activation markers CD69, PD-1 and CD107a by TS-BiTE αHER2 CAR T cells after 30 min heating and co-culture with HER2−MDA-MB-468 target cells. ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; error bars show s.e.m.; n = 4 biologically independent wells). c, Tumour growth curves of MDA-MB-468 tumours inoculated at a 3:1 HER2+ to HER2− ratio and treated with TS-BiTE or TS-Rluc αHER2 CAR T cells. Heat treatments were carried out on days 45, 47, 52, 59, 66 and 72. *P = 0.0351 between TS-Rluc and TS-BiTE cohorts on day 74; *P = 0.0206 between TS-Rluc and TS-BiTE cohorts on day 76; *P = 0.0340 between TS-Rluc and TS-BiTE cohorts on day 78; *P = 0.0141 between TS-Rluc and TS-BiTE cohorts on day 81; *P = 0.0158 between TS-Rluc and TS-BiTE cohorts on day 84; *P = 0.0170 between TS-Rluc and TS-BiTE cohorts on day 87; *P = 0.0172 between TS-Rluc and TS-BiTE cohorts on day 93; *P = 0.0147 between TS-Rluc and TS-BiTE cohorts on day 100; **P = 0.0017 between TS-Rluc and untreated cohorts on day 100; two-way ANOVA and Dunnett post-test and correction; mean ± s.e.m. is depicted; n = 6 to 7 biologically independent mice. d, Spider plots of individual tumours, with vertical dotted lines indicating heat treatments. e, In vivo luminescence imaging time course and f, individual spider plots acquired by an IVIS Spectrum CT system representative of the HER2−/Fluc+ cell population in the mixed MDA-MB-468 tumour model. Transfer of engineered human T cells occurred on day 44 and heat treatments on days 45, 47, 52, 59, 66 and 72. Grey background represents mean ± 2 s.d. above and below background measurements (n = 6) taken throughout the time course of the experiment. **P = 0.0022 between TS-Rluc and TS-BiTE cohorts on day 58; ***P = 0.0009 between TS-Rluc and TS-BiTE cohorts on day 61; **P = 0.0013 between TS-Rluc and TS-BiTE cohorts on day 65; **P = 0.0068 between TS-Rluc and TS-BiTE cohorts on day 69; **P = 0.0029 between TS-Rluc and TS-BiTE cohorts on day 74; ***P = 0.0003 between TS-Rluc and TS-BiTE cohorts on day 78; ***P = 0.0007 between TS-Rluc and TS-BiTE cohorts on day 101; ****P < 0.0001; two-way ANOVA and Tukey post-test and correction; mean ± s.e.m. is depicted; n = 6 to 7 biologically independent mice.
Discussion
The ability to better control engineered T cell activity within tumour sites has the potential to improve therapy against solid tumours. Here we developed a technology for photothermal control of T cell activity. To provide T cells with the capacity to respond to heat, we designed synthetic thermal gene switches consisting of arrays of HSEs upstream of a core promoter. This architecture eliminated sensitivity to non-thermal stresses such as hypoxia and its thermal response was tunable on the basis of the number of HSEs or different core promoters. While we tested constructs containing up to seven HSEs paired with four core promoters, future work could explore a larger library of building parts including temperature-sensitive transcription factors such as HSF1 to tune response to heat. Importantly, we observed negligible activation of our thermal gene switches at temperatures ≤40 °C when T cells were incubated for over 24 h, providing support that the temperature threshold for activation is higher than the range of typical fevers (~38–40 °C)76–78 in patients with cytokine release syndrome, which would prevent T cell activation without a targeted thermal input. In the future, we envision that designing thermal gene switches with lower temperature activation thresholds may be useful as a sense-and-respond circuit to autonomously detect fever temperatures and trigger expression of therapeutic agents such as tocilizumab to attenuate cytokine release syndrome.
We demonstrated that thermal control of T cell activity with an IL-15 superagonist and a NKG2DL BiTE enhanced antitumour responses. Engineered T cells that constitutively express similar classes of molecules have demonstrated strong antitumour efficacy, but their therapeutic applications are limited by off-tumour effects and toxicities in healthy tissues1,2. Thus, targeted expression of these genes within tumours could potentially contain potent T cell activity and improve therapeutic outcomes. We showed that thermal induction of transgenes is transient and reversible, and found that thermally activated T cells remained localized to the heated site while the transgene was expressed, reducing the potential for off-target expression of transgenes. In K562 and syngeneic B16-F10 tumours, we found that photothermal control of IL-15 SA expression by either αCD19 CAR or TCR-transgenic Pmel-1 T cells resulted in enhanced antitumour activity compared with adoptive transfer of T cells alone. We further demonstrated photothermal control of a NKG2DL BiTE to mitigate antigen escape by allowing CAR T cells to target antigen-negative tumour cells that express NKG2D ligands. In a mixed model of HER2+ and HER2− breast cancer, treatment with TS-BiTE αHER2 CAR T cells led to elimination of well-established tumours without detectable residual disease in three of six mice, or significantly delayed relapse compared with treatment with αHER2 CAR T cells that targeted a single antigen. In light of our results, we expect that a wide range of biologics are amenable to thermal control without potential loss of function due to protein misfolding or aggregation in T cells by heat stress.
Looking forward, we envision that additional advances are required to address the limitations of the current study. Photothermal targeting by NIR light is limited by penetration depth to a few centimetres and by the need for a plasmonic transducer79–81; therefore, alternative heating modalities29 capable of image-guided, deep-tissue activation without transducers are required, such as focused ultrasound, which was recently demonstrated for the control of T cell gene expression82,83 or for the thermal control of bacteria engineered with temperature-sensitive repressors84. Whether local thermal treatments would lead to responses in distal tumours (that is, the abscopal effect) remains to be studied; therefore, the advances of this current study will probably not be applicable for the treatment of broadly disseminated tumours, because targeting individual metastases would preclude therapy. Rather, we envision potential use cases for localized primary tumours such as glioblastoma or head-and-neck cancers. Another limitation of our study is the need for the repeat application of heat; however, it should be noted that adoptive cell therapies engineered to constitutively express immunostimulatory transgenes have been associated with severe adverse toxicities21,23,85, which has spurred numerous inducible strategies to control T cell activity86–88. We envision that the thermally induced production of biologics may enhance therapeutic outcomes; nevertheless, a direct comparison with systemic administration of constitutively expressed transgenes is necessary before clinical application. In thermal oncology, repeated heat applications can be performed using technologies such as LITT due to the minimally invasive nature of the procedure, which provides clinical precedent89–91. Lastly, some of the conclusions from our studies are context specific. For example, our in vitro experiments showing that BiTE activation occurs primarily by an autocrine mechanism can be affected by secretion rates, diffusion and effector-to-bystander ratios, as these parameters are tunable. Taken together, our results support the photothermal targeting of engineered T cell therapies as a strategy for the improvement of responses against solid tumours.
Methods
Plasmid construction.
Synthetic thermal switches were produced as gene blocks by Integrated DNA Technologies (IDT) and cloned into the Lego-C (Addgene plasmid 27348) or pMKO.1 (Imgenex) backbones. The core promoters were truncated immediately upstream of their previously described TATA boxes at their 5′-termini and at their translational start site on their 3′-termini92–94. The genomic HSPA6 promoter was amplified from genomic DNA using PCR primers listed in a previous publication42. The NKG2DL BiTE sequence (US20120294857A1) was described previously75 and modified to include an Igκ leader sequence to facilitate secretion from T cells as well as a HisTag for construct detection. This combined sequence was synthesized (ATUM) and cloned downstream of synthetic thermal gene switches. The IL-15 superagonist sequence was described previously66 and synthesized by Atum without modification. The constitutive αCD19 CAR (US9499629B2) was kindly provided by Dr Krishnendu Roy (Georgia Institute of Technology. The αHER2 CAR (US20180326032A1) was described previously93. All unique materials can be made available by the corresponding author on reasonable request.
Culture of primary human T cells and cell lines.
CD19+ K562 and wild-type K562s were cultured in Isocove’s modified Dulbecco’s medium (ThermoFisher 12440053) supplemented with 10% FBS (Fisher 16140071) and 10 U ml−1 penicillin–streptomycin (Life Technologies 15140-122). Raji cells were cultured in RPMI-1640 media supplemented with 10% FBS. MDA-MB-468 (ATCC, HTB-132) and B16-F10 (ATCC, CRL-6475) cells were cultured in Dulbecco’s modified Eagle medium (Gibco 11995073) supplemented with 10% FBS (Fisher 16140071) and 10 U ml−1 penicillin–streptomycin (Life Technologies 15140-122). Human cell lines were authenticated using short tandem repeat (STR) analysis (Labcorp) (Supplementary Table 1). Primary human CD3+ cells were obtained from an anonymous donor blood after apheresis (AllCells) and were cryopreserved in 90% FBS and 10% dimethylsulfoxide until subsequent use. After thawing, cells were cultured in human T cell media consisting of X-VIVO 10 (Lonza 04-380Q), 5% human AB serum (Valley Biomedical HP1022), 10 mM N-acetyl l-cysteine (Sigma A9165) and 55 μM 2-mercaptoethanol (Sigma M3148-100ML) supplemented with 50 units per ml human IL-2 (Sigma 11147528001). Seven total donors were utilized for experimentation. Figure 1 used donors 1, 2, 6 and 7; Fig. 2 used donors 2 and 3; Fig. 3 used donor 2; Fig. 4 used donor 4; Figs. 5 and 6 used donor 2.
Isolation and expansion of Pmel-1 T cells.
Splenocytes from Pmel-1 transgenic mice were depleted of red blood cells using RBC lysis buffer (Biolegend 420302) and cultured in complete medium with 100 units ml−1 recombinant human IL-2 (Sigma 11147528001) in the presence of 1 μg ml−1 hgp10025–33 peptide for 2 d (Tufts University Core Facility). Splenocytes were resuspended at 10 × 106 cells per ml in a media spiked with retrovirus at a multiplicity of infection of ten and centrifuged for 90 min at 2,000 × g. Cells were cultured at a concentration of 1 × 106 cells per ml in complete media supplemented with 100 units per ml IL-2 before intravenous transfer into C57BL/6 J mice 6 d after isolation.
Viral production and primary human T cell transduction.
VSV-G pseudotyped lentivirus was produced via transfection of HEK293 T cells (ATCC, CRL-3216) using psPAX2 (Addgene 12260) and pMD2.G (Addgene 12259); viral supernatant was concentrated using PEG-it virus precipitation solution (System Biosciences LV825A-1) according to the manufacturer’s instructions. Retrovirus was produced via transfection of HEK293 T cells (ATCC, CRL-3216) using pCL-Eco and pMKO.1 vector (Imgenex) encoding for the thermal switch circuit (TS-IL-15); after 48 h, viral supernatant was concentrated using Retro-Concentin retroviral concentration reagent (System Biosciences RV100A-1) according to the manufacturer’s instructions and frozen at −80 °C. For viral transductions of primary human T cells, cells were thawed, incubated for 24 h and activated with human T-Activator Dynabeads (Life Technologies 11131D) at a 3:1 bead:cell ratio for 24 h. To transduce the activated T cells, concentrated lentivirus was added to non-tissue culture treated 6-well plates that were coated with retronectin (Takara T100B) according to the manufacturer’s instructions and spun at 1,200 × g for 90 min at room temperature. Following centrifugation, viral solution was aspirated and 2 ml of human T cells (2.5 × 105 cells per ml) in human T cell media containing 100 units ml−1 hIL-2 was added to the wells, spun at 1,200 × g for 60 min at 37 °C and moved to an incubator. Cells were incubated on a virus-coated plate for 24 h before expansion, and Dynabeads were removed 7 d after T cell activation. For cells flow-sorted before adoptive cell transfer, Dynabeads were added immediately after sorting at 3:1 ratios for 48 h.
Staining and flow cytometry.
To detect CD19 CAR expression, biotinylated CD19 (10 μg ml−1; Acro Biosystems CD9-H8259) and streptavidin-APC (ThermoFisher S868) were used according to the manufacturer’s instructions. NKG2DL expression was assessed by staining with NKG2D-Fc chimera (10 μg ml−1; Fisher 1299NK050) followed by an αFc secondary stain (Invitrogen A-10631). NIR Live/Dead (ThermoFisher L34976), CFSE (LifeTech C34554) and CellTrace Violet (LifeTech C34557) were used according to the manufacturer’s instructions. Human Fc block (BD 564220) was used before staining with any antibodies. Antibodies for CD69 (FN50; BD, RRID: AB_1727510), CD4 (RPA-T4; BioLegend, RRID: AB_314076); hCD8 (RPA-T8; BioLegend, RRID: AB_10669564), CD3 (UCHT1; BD, RRID: AB_2744387), CD45 (HI30; BD, RRID: AB_395875), CD19 (HIB19; BioLegend, RRID: AB_395813), PD-1 (EH12.2H7; Biolegend, RRID: AB_10900818), CD107a (H4A3; Biolegend, RRID: AB_2562647), mCD8 (53-6.7; BioLegend, RRID: AB_10897101), HER2 (24D2; Biolegend, RRID: AB_756121), and HisTags (4E3D10H2/E3; ThermoFisher, RRID: AB_2610637) and human Fc (HP6069; ThermoFisher, RRID: AB_2534050) were all used at 1:100 dilutions.
In vitro luciferase and thermotolerance assays.
Primary human T cells were heated in a thermal cycler and transferred to culture plates for incubation at 37 °C. Unless otherwise noted, cellular supernatant was sampled for luciferase activity 24 h after conclusion of thermal treatment. Non-thermal treatments were conducted by incubating engineered cells at indicated concentrations of CoCl2 (Sigma 232696-5 G) or CdCl2 (Sigma 202908). When indicated, luminescence was compared to a ladder of recombinant Gaussia luciferase (NanoLight 321-500) quantified using a Gaussia Luciferase Glow Assay kit (ThermoFisher 16161) according to the manufacturer’s instructions. For viability and proliferation studies, primary human T cells were heated in the thermal cycler before assaying with an apoptosis detection kit (BD 556547) or CellTrace Violet (Fisher C34571). Viability was assessed 24 h after heating and gating strategies are depicted in Supplementary Fig. 4. For migration studies, wild-type cells were added to the top insert of a transwell plate (Sigma CLS3421), while CXCL12 (50 ng ml−1, Peprotech 300-28 A) was added to the lower chamber. Cells in the lower chamber were counted using a hemocytometer at indicated times.
Cytotoxicity and T cell activation assays.
For cytometric analysis, T cells were heated in a thermal cycler and co-incubated with K562 target cells at a 10:1 effector cell to target cell ratio for 24 h before staining as described above. For luciferase-based assays, K562s were luciferized with either firefly luciferase (CD19+) or Renilla luciferase (CD19−) and incubated with effector cells after heating. Unless otherwise noted, a 10:1 effector to target ratio was used. After incubation, either d-luciferin (Fisher LUCK-2G; 150 μg ml−1 read concentration) or Rluc substrate (VWR PAP1232; 17 μM read concentration) was added to the sample. Maximum cytotoxicity was defined as luminescent signal from wells containing only media, while no cytotoxicity was defined by wells containing only target cells. Supernatant was collected after incubation and assayed for cytokines using the human Th1/Th2/Th17 CBA kit (BD 560484). IL-15 superagonist was quantified using the human IL-15/IL-15R alpha complex DuoSet ELISA (R&D Systems DY6924). For BiTE experiments with primary human T cells, two heat treatments (42 °C, 30 min) separated by 6 h were applied to T cells before incubation with target cells. The ability of engineered T cells to kill tumour target cells was also measured by lactate dehydrogenase (LDH) release assay. Briefly, engineered T cells were co-cultured with target cells at a 2:1 effector to target ratio for 24 h in a 96-well plate. Then LDH release was measured by the LDH-Cytotoxicity Assay kit (Fluorometric) (Abcam 197004) according to the manufacturer’s instructions.
IL-15 superagonist Dynabead experiment.
Wild-type primary human T cells were labelled with CFSE and incubated with either heated or unheated TS-IL-15 cells. Beads were added at a 10:1 T cell to bead ratio that was determined not to induce strong proliferation in untransduced T cells without cytokine support (Supplementary Fig. 10). CFSE labelling allowed discrimination from TS-IL-15 cells (Supplementary Fig. 11) and proliferation and division indices were calculated in FlowJo using the Proliferation tool.
Animals.
NSG mice were bred and housed in the Georgia Tech Physiological Research Laboratory (GT PRL) before use at an age of 8–16 weeks. C57BL/6 mice and transgenic Pmel-1 mice (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J) were purchased from Jackson Laboratories. Six- to eight-week-old C57BL/6 and Pmel-1 mice were used at the outset of experiments. All animal protocols were approved by Georgia Tech Institutional Animal Care and Use Committee (protocols no. A100190 and A100191). All authors complied with relevant ethical regulations while conducting this study.
Photothermal heating and in vivo bioluminescence imaging.
AuNRs were purchased from Nanopartz (A12-10-808-CTAB-500) and PEGylated (Laysam Bio MPEG-SH-5000-5g) to replace the CTAB coating. These AuNRs were intravenously injected into tumour-bearing mice (10 mg kg−1) ~24–48 h before adoptive transfer of T cells. Mice were anaesthetized with isoflurane gas, and target sites were irradiated using an 808 nm laser (Coherent) under the guidance of a thermal camera (FLIR model 450sc). Fluc activity was measured using an IVIS Spectrum CT (PerkinElmer) ~5 min after intravenous injections or 20 min after intraperitoneal injection of d-luciferin (Fisher LUCK-2G). The detection limit was identified by calculating the mean ± 2 s.d. of background measurements.
ACT experiments.
At GT PRL, NSG mice were inoculated subcutaneously with 5 × 106 Raji or K562 cell lines after the site was shaved and sterilized using an isopropyl wipe, for 9 d before ACT. For heterogenous expression of HER2, 5 × 105 MDA-MB-468 cells with a ratio of 1:3 HER2− to HER2+ were inoculated for 44 d before ACT. Engineered primary human T cells were injected via tail vein in 200 μl sterile saline. For B16 tumour models, 5 × 105 B16F10 melanoma cells were inoculated in the flanks of C57BL/6 mice. Mice were sublethally lymphodepleted by total body irradiation (100 cGy min−1 for 3 min and 51 s) 8 d after tumour-cell inoculation. Engineered Pmel-1 T cells (6 × 106 cells) were administered by intravenous injection at day 9. Mice were intraperitoneally dosed with 2 × 105 units recombinant human IL-2 (Peprotech 200-02) twice daily at least 10 h apart for a total of six doses. All mice received pegylated AuNRs intravenously via tail vein ~24 h before adoptive transfer of human T cells. At 25 h after ACT, photothermal heat treatments were administered and monitored as described above.
Software and statistical analysis.
All results are presented as mean, and error bars depict s.e.m. Statistical analysis was performed using GraphPad Prism 8.0.2 statistical software. Flow cytometry data were collected with BD FACSDIVA v8 (BD Biosciences) and analysed using FlowJo X (FlowJo). In vitro luminescent data were collected with Gen5 2.07 (Biotek). In vivo luminescence data were collected and analysed with Living Image 4.4.5 (PerkinElmer). Thermal imaging data were acquired and analysed using Research IR Max (FLIR). Figures were designed in Adobe Illustrator.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended data Fig. 1 |. Engineered Pmel-1 T cells enhance adoptive cell therapy in a high tumour burden setting.
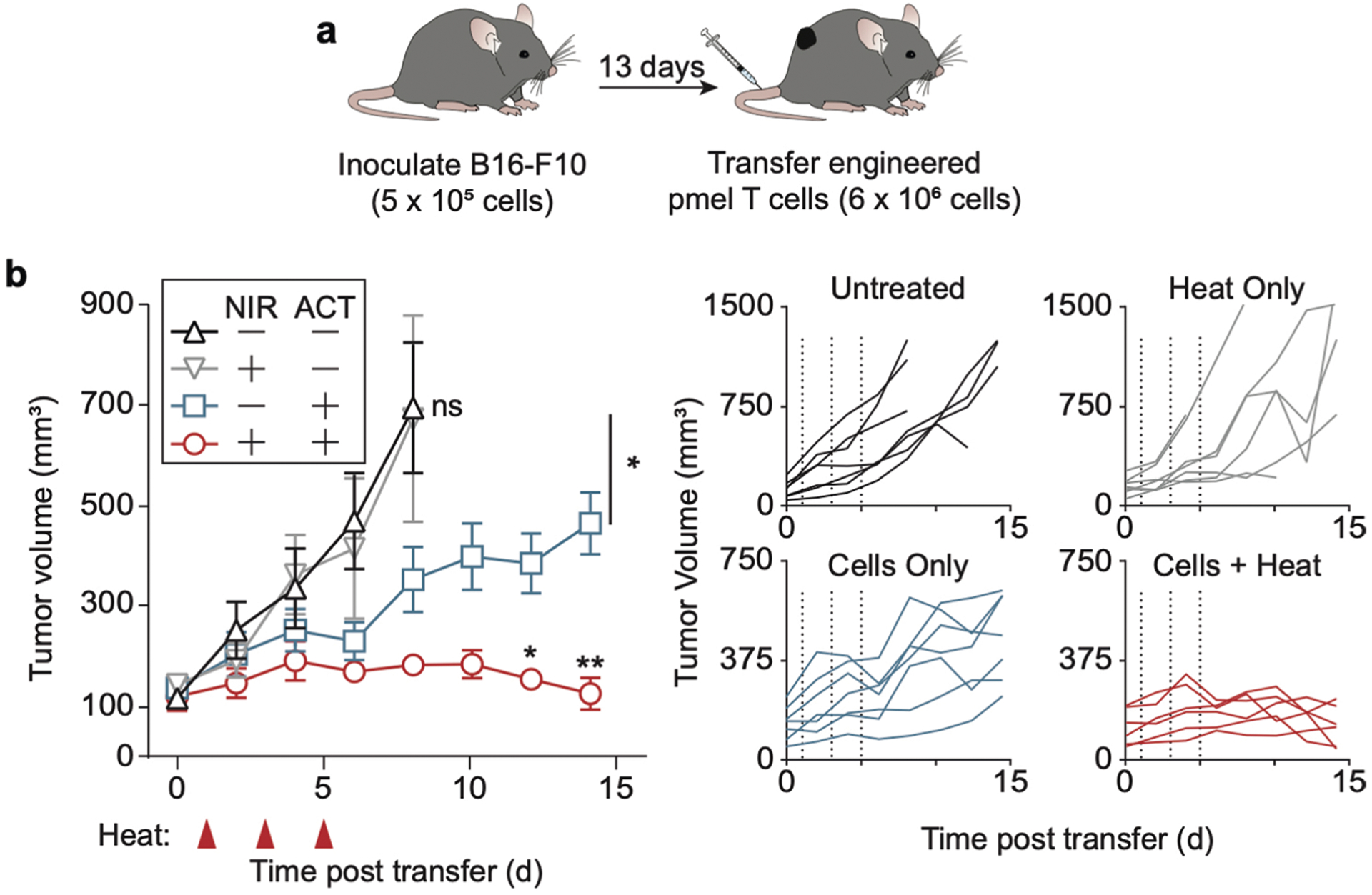
a, Schematic representation of large tumour B16-F10 bearing C57BL/6 J mice upon systemic T cell transfer. b, Tumour-growth curves following inoculation of B16F10 following transfer of engineered murine T cells on day 0 and heat treatments on days 1, 3, and 5 (*P = 0.0489 between untreated and cohorts which received cells only on day 8. *P = 0.0295 between cohorts receiving cells and heat versus cells only on day 12, **P = 0.0043 between cohorts receiving cells and heat versus cells only on day 14, two-way ANOVA and Tukey post-test and correction, mean ± SEM is depicted, n = 6–7 biologically independent mice).
Supplementary Material
Acknowledgements
We thank J. M. Brockman for their helpful insights during planning and experiments; K. Roy (Georgia Institute of Technology) for the constitutive αCD19 CAR (US9499629B2), wild-type K562s and Raji cells; and Y. Chen for the CD19+ K562. This work was funded by the NIH Director’s New Innovator Award (DP2HD091793), the National Centre for Advancing Translational Sciences (UL1TR000454) and the Shurl and Kay Curci Foundation. I.C.M. was supported by the Georgia Tech TI:GER programme. L.G. was supported by the Alfred P. Sloan Foundation, the National Institutes of Health GT BioMAT Training Grant under Award No. 5T32EB006343 and the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1451512. G.A.K. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542174). S.J.P. was supported by a STOP Cancer Foundation/Disrupt Seed and the Borstein Family Foundation for this work. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
I.C.M., L.G., A.Z. and G.A.K. are listed as inventors on a patent application pertaining to the results of this paper (US20200299686A1 and application no. 63/214,761). G.A.K. is co-founder and consultant at Glympse Bio and consults for Satellite Bio. This study could affect his personal financial status. S.J.P. is a scientific advisor to and receives royalties from Mustang Bio and Imugene Ltd. The terms of this arrangement have been reviewed and approved by Georgia Tech in accordance with its conflict-of-interest policies.
Extended data is available for this paper at https://doi.org/10.1038/s41551-021-00781-2.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-021-00781-2.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The data generated and analysed during the study are available from the corresponding author on reasonable request. Source data are provided with this paper.
References
- 1.Lim WA & June CH The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weber EW, Maus MV & Mackall CL The emerging landscape of immune cell therapies. Cell 181, 46–62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Priceman SJ, Forman SJ & Brown CE Smart CARs engineered for cancer immunotherapy. Curr. Opin. Oncol 27, 466–474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.John LB et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res 19, 5636–5646 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Klebanoff CA et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc. Natl Acad. Sci. USA 101, 1969–1974 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.John LB, Kershaw MH & Darcy PK Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2, e26286 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slaney CY, Wang P, Darcy PK & Kershaw MH CARs versus BiTEs: a comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. 8, 924–934 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Weber JS, Kahler KC & Hauschild A Management of immune-related adverse events and kinetics of response with ipilimumab. J. Clin. Oncol 30, 2691–2697 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Waldmann TA et al. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood 117, 4787–4795 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conlon KC et al. Redistribution, hyperproliferation, and activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol 33, 74–82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baumeister SH, Freeman GJ, Dranoff G & Sharpe AH Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol 34, 539–573 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Smith TT et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Invest 127, 2176–2191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephan SB et al. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol 33, 97–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stephan MT, Moon JJ, Um SH, Bershteyn A & Irvine DJ Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med 16, 1035–1041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang L et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol 36, 707–716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pegram HJ et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119, 4133–4141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rafiq S et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances antitumor efficacy in vivo. Nat. Biotechnol 36, 847–856 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi BD et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol 37, 1049–1058 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Kosti P et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumours. Cell Rep. Med 2, 100227 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao Q et al. Engineering T cells with hypoxia-inducible chimeric antigen receptor (HiCAR) for selective tumor killing. Biomark. Res 8, 56 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res 21, 2278–2288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmermann K et al. Design and characterization of an ‘all-in-one’ lentiviral vector system combining constitutive anti-GD2 CAR expression and inducible cytokines. Cancers 12, 375 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunert A et al. Intratumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 7, e1378842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roybal KT et al. Precision tumour recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kloss CC, Condomines M, Cartellieri M, Bachmann M & Sadelain M Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol 31, 71–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srivastava S et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 35, 489–503.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho JH et al. Engineering advanced logic and distributed computing in human CAR immune cells. Nat. Commun 12, 792 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Driel WJ et al. Hyperthermic intraperitoneal chemotherapy in ovarian cancer. N. Engl. J. Med 378, 230–240 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Chu KF & Dupuy DE Thermal ablation of tumours: biological mechanisms and advances in therapy. Nat. Rev. Cancer 14, 199–208 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Mitchell D et al. A heterogeneous tissue model for treatment planning for magnetic resonance-guided laser interstitial thermal therapy. Int. J. Hyperthermia 34, 943–952 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Lubner MG, Brace CL, Hinshaw JL & Lee FT Jr Microwave tumor ablation: mechanism of action, clinical results, and devices. J. Vasc. Interv. Radiol 21, S192–S203 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amin J, Ananthan J & Voellmy R Key features of heat shock regulatory elements. Mol. Cell. Biol 8, 3761–3769 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakurai H & Enoki Y Novel aspects of heat shock factors: DNA recognition, chromatin modulation and gene expression. FEBS J. 277, 4140–4149 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Jaeger AM, Makley LN, Gestwicki JE & Thiele DJ Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J. Biol. Chem 289, 30459–30469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitlock NA, Agarwal N, Ma JX & Crosson CE Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest. Ophthalmol. Vis. Sci 46, 1092–1098 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Wu BJ, Kingston RE & Morimoto RI Human HSP70 promoter contains at least two distinct regulatory domains. Proc. Natl Acad. Sci. USA 83, 629–633 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalmar B & Greensmith L Induction of heat shock proteins for protection against oxidative stress. Adv. Drug Deliv. Rev 61, 310–318 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Vilaboa NE et al. cAMP increasing agents prevent the stimulation of heat-shock protein 70 (HSP70) gene expression by cadmium chloride in human myeloid cell lines. J. Cell Sci 108, 2877–2883 (1995). [DOI] [PubMed] [Google Scholar]
- 39.Xu Q, Schett G, Li C, Hu Y & Wick G Mechanical stress-induced heat shock protein 70 expression in vascular smooth muscle cells is regulated by Rac and Ras small G proteins but not mitogen-activated protein kinases. Circ. Res 86, 1122–1128 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Kadonaga JT Perspectives on the RNA polymerase II core promoter. Wiley Interdiscip. Rev. Dev. Biol 1, 40–51 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flanagan SW, Ryan AJ, Gisolfi CV & Moseley PL Tissue-specific HSP70 response in animals undergoing heat stress. Am. J. Physiol 268, R28–R32 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Miller IC, Castro MG, Maenza J, Weis JP & Kwong GA Remote control of mammalian cells with heat-triggered gene switches and photothermal pulse trains. ACS Synth. Biol 7, 1167–1173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ede C, Chen X, Lin MY & Chen YY Quantitative analyses of core promoters enable precise engineering of regulated gene expression in mammalian cells. ACS Synth. Biol 5, 395–404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen J et al. Transplantation of prokaryotic two-component signaling pathways into mammalian cells. Proc. Natl Acad. Sci. USA 111, 15705–15710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klaassen CD, Liu J & Diwan BA Metallothionein protection of cadmium toxicity. Toxicol. Appl. Pharm 238, 215–220 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Safran M et al. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc. Natl Acad. Sci. USA 103, 105–110 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lokmic Z, Musyoka J, Hewitson TD & Darby IA Hypoxia and hypoxia signalling in tissue repair and fibrosis. Int. Rev. Cell. Mol. Biol 296, 139–185 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Daugaard M, Rohde M & Jaattela M The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett. 581, 3702–3710 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Yamaguchi M, Ito A, Ono A, Kawabe Y & Kamihira M Heat-inducible gene expression system by applying alternating magnetic field to magnetic nanoparticles. ACS Synth. Biol 3, 273–279 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Yin PT et al. Stem cell-based gene therapy activated using magnetic hyperthermia to enhance the treatment of cancer. Biomaterials 81, 46–57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakatsuji H et al. Surface chemistry for cytosolic gene delivery and photothermal transgene expression by gold nanorods. Sci. Rep 7, 4694 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gamboa L et al. Heat-triggered remote control of CRISPR-dCas9 for tunable transcriptional modulation. ACS Chem. Biol 15, 533–542 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muñoz-Sánchez J & Chánez-Cárdenas ME The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol 39, 556–570 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Fotakis G, Cemeli E, Anderson D & Timbrell JA Cadmium chloride-induced DNA and lysosomal damage in a hepatoma cell line. Toxicol. In Vitro 19, 481–489 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Phuagkhaopong S et al. Cadmium-induced IL-6 and IL-8 expression and release from astrocytes are mediated by MAPK and NF-κB pathways. Neurotoxicology 60, 82–91 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Rani A, Kumar A, Lal A & Pant M Cellular mechanisms of cadmium-induced toxicity: a review. Int. J. Environ 24, 378–399 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Mitchell RJ & Gu MB Construction and characterization of novel dual stress-responsive bacterial biosensors. Biosens. Bioelectron 19, 977–985 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Elias D et al. Optimization of hyperthermic intraperitoneal chemotherapy with oxaliplatin plus irinotecan at 43 degrees C after compete cytoreductive surgery: mortality and morbidity in 106 consecutive patients. Ann. Surg. Oncol 14, 1818–1824 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Yang XJ et al. Cytoreductive surgery and hyperthermic intraperitoneal chemotherapy improves survival of patients with peritoneal carcinomatosis from gastric cancer: final results of a phase III randomized clinical trial. Ann. Surg. Oncol 18, 1575–1581 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nikfarjam M, Muralidharan V & Christophi C Mechanisms of focal heat destruction of liver tumors. J. Surg. Res 127, 208–223 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Hellevik T & Martinez-Zubiaurre I Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front. Oncol 4, 1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jain PK, Lee KS, El-Sayed IH & El-Sayed MA Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: applications in biological imaging and biomedicine. J. Phys. Chem. B 110, 7238–7248 (2006). [DOI] [PubMed] [Google Scholar]
- 63.von Maltzahn G et al. Nanoparticles that communicate in vivo to amplify tumour targeting. Nat. Mater 10, 545–552 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.von Maltzahn G et al. Computationally guided photothermal tumor therapy using long-circulating gold nanorod antennas. Cancer Res. 69, 3892–3900 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mortier E et al. Soluble interleukin-15 receptor α (IL-15R α)-sushi as a selective and potent agonist of IL-15 action through IL-15R β/γ. Hyperagonist IL-15·IL-15Rα fusion proteins. J. Biol. Chem 281, 1612–1619 (2006). [DOI] [PubMed] [Google Scholar]
- 66.Robinson TO & Schluns KS The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol. Lett 190, 159–168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rhode PR et al. Comparison of the superagonist complex, ALT-803, to IL-15 as cancer immunotherapeutics in animal models. Cancer Immunol. Res 4, 49–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tomala J, Chmelova H, Mrkvan T, Rihova B & Kovar M In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J. Immunol 183, 4904–4912 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Watanabe K, Kuramitsu S, Posey AD & June CH Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front. Immunol 9, 2486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parihar R et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol. Res 7, 363–375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steinbacher J et al. An Fc-optimized NKG2D-immunoglobulin G fusion protein for induction of natural killer cell reactivity against leukemia. Int. J. Cancer 136, 1073–1084 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Xia Y et al. Treatment with a fusion protein of the extracellular domains of NKG2D to IL-15 retards colon cancer growth in mice. J. Immunother 37, 257–266 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Godbersen C et al. NKG2D ligand-targeted bispecific T-cell engagers lead to robust antitumor activity against diverse human tumors. Mol. Cancer Ther 16, 1335–1346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Evans SS, Repasky EA & Fisher DT Fever and the thermal regulation of immunity: the immune system feels the heat. Nat. Rev. Immunol 15, 335–349 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giavridis T et al. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med 24, 731–738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Norelli M et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med 24, 739–748 (2018). [DOI] [PubMed] [Google Scholar]
- 77.Gamboa L, Zamat AH & Kwong GA Synthetic immunity by remote control. Theranostics 10, 3652–3667 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Henderson TA & Morries LD Near-infrared photonic energy penetration: can infrared phototherapy effectively reach the human brain? Neuropsychiatr. Dis. Treat 11, 2191–2208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He L et al. Near-infrared photoactivatable control of Ca2+ signaling and optogenetic immunomodulation. Elife 4, e10024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pan Y et al. Mechanogenetics for the remote and noninvasive control of cancer immunotherapy. Proc. Natl Acad. Sci. USA 115, 992–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abedi MH, Lee J, Piraner DI & Shapiro MG Thermal control of engineered T-cells. ACS Synth. Biol 9, 1941–1950 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Piraner DI, Abedi MH, Moser BA, Lee-Gosselin A & Shapiro MG Tunable thermal bioswitches for in vivo control of microbial therapeutics. Nat. Chem. Biol 13, 75–80 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Kerkar SP et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 70, 6725 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giordano-Attianese G et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat. Biotechnol 38, 426–432 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Weber EW et al. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 3, 711–717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.June CH Remote controlled CARs: towards a safer therapy for leukemia. Cancer Immunol. Res 4, 643 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Jethwa PR, Barrese JC, Gowda A, Shetty A & Danish SF Magnetic resonance thermometry-guided laser-induced thermal therapy for intracranial neoplasms: initial experience. Neurosurgery 71, 133–145 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Rahmathulla G et al. MRI-guided laser interstitial thermal therapy in neuro-oncology: a review of its current clinical applications. Oncology 87, 67–82 (2014). [DOI] [PubMed] [Google Scholar]
- 89.Shah AH et al. The role of laser interstitial thermal therapy in surgical neuro-oncology: series of 100 consecutive patients. Neurosurgery 87, 266–275 (2020). [DOI] [PubMed] [Google Scholar]
- 90.Bevilacqua A, Fiorenza MT & Mangia F A developmentally regulated GAGA box-binding factor and Sp1 are required for transcription of the hsp70.1 gene at the onset of mouse zygotic genome activation. Development 127, 1541–1551 (2000). [DOI] [PubMed] [Google Scholar]
- 91.Ramirez VP, Stamatis M, Shmukler A & Aneskievich BJ Basal and stress-inducible expression of HSPA6 in human keratinocytes is regulated by negative and positive promoter regions. Cell Stress Chaperones 20, 95–107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gaestel M, Gotthardt R & Muller T Structure and organisation of a murine gene encoding small heat shock-protein Hsp25. Gene 128, 279–283 (1993). [DOI] [PubMed] [Google Scholar]
- 93.Priceman SJ et al. Regional delivery of chimeric antigen receptor–engineered T cells effectively targets HER2+ breast cancer metastasis to the brain. Clin. Cancer Res 24, 95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hwang LN, Yu Z, Palmer DC & Restifo NP The in vivo expansion rate of properly stimulated transferred CD8+ T cells exceeds that of an aggressively growing mouse tumor. Cancer Res. 66, 1132–1138 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. The data generated and analysed during the study are available from the corresponding author on reasonable request. Source data are provided with this paper.